Introduction
Bronchopulmonary dysplasia (BPD) is one of the most
common diseases in preterm infants with a gestational age of <30
weeks or a birth weight of <1,200 g, especially in those
requiring oxygen inhalation or mechanical ventilation during
treatment (1). The major
pathological characteristics of BPD are obstruction of
alveolarization and simplification of pulmonary vascular
development (2,3). Since its initial description in
1967, advances in the comprehensive management technology for BPD
in preterm new-borns have increased the survival rate substantially
(4,5); however, BPD still affects ~10,000
neonates each year in the United States (6). Thus, further improvements in
developmental outcomes and morbidity reduction of BPD are urgently
needed (7). The pathogenesis of
BPD is extremely complex and remains unclear (8-10),
emphasising the need for an improved understanding of alveolar
development under both normal and pathological conditions.
Mitochondrial autophagy (mitophagy) is an important
process in mitochondrial quality control (11), involving ubiquitin- and
receptor-dependent pathways (12). cells regulate biosynthetic
functions by selectively identifying and eliminating damaged or
excess mitochondria (11).
Abnormal mitophagy is closely associated with lung diseases, such
as COPD and pulmonary fibrosis (13); however, the role of mitophagy in
BPD is not well-established.
The classic process of mitophagy is primarily
mediated by the phosphatase and tensin homolog-induced putative
kinase 1 (PINK1)-Parkin pathway. PINK1 is a serine/threo-nine
protein kinase localised on the outer mitochondrial membrane
(14). When the mitochondria are
healthy in cells, PINK1 is degraded by proteasomes and maintained
at a low level (15). However,
upon a decrease in the mitochondrial membrane potential (MMP),
PINK1 is not transported into the mitochondria but rather
aggregates on the outer membrane and recruits Parkin, an E3
ubiquitin ligase (14).
Subsequent binding to p62 and microtubule-associated protein-1
light chain-3B (Lc3B) leads to the engulfment and degradation of
mitochondria by autophagosomes (16). Several previously identified
mitochondrial receptors, such as Nip3-like protein X (NIX),
BcL2/adenovirus E1B 19 kDa protein-interacting protein 3 and FUN14
domain-containing protein 1 (17), are located on the outer
mitochondrial membrane and directly recruit LC3B for mitochondrial
degradation (18). Mitophagy
contributes to stem cell differentiation, aging and other
pathological processes, such as neurodegenerative diseases, cancer
and metabolic diseases (19);
however, little is known about its roles in lung development and
BPD. Therefore, the present study aimed to assess the effects of
PINK1-Parkin and NIX on rat alveolar type II (AT-II) cells, which
play a crucial role in the pathogenesis of BPD (20).
Changes in metabolic activity are closely linked to
cell fate decisions and development (21). Mitochondria are involved in
various biological activities, such as energy synthesis,
reduction-oxidation reactions, calcium signalling, reactive oxygen
species generation and macromolecule synthesis (22). Therefore, it was hypothesised that
mitochondrial injuries induced by hyperoxia impair normal energy
synthesis and to contribute to BPD.
To test this hypothesis, the expression of
PINK1-Parkin and NIX was evaluated in an experimental rat model and
its association with mitochondrial damage and metabolic changes.
The present results may help to clarify the role of mitophagy in
lung epithelial cells and demonstrate the critical role of
mitophagy in lung developmental disorders.
Materials and methods
Model establishment and animal
treatment
In total, eight pregnant Sprague-dawley rats
(300-350 g) were purchased from the department of Animals,
Experimental centre, Shengjing Hospital of china Medical
University. Each pregnant rat was fed independently at 20-26°C, 12
h light/dark cycle and free to access to food and water. Eighty
offspring were born on days 21-23 of pregnancy and were randomly
divided into a model group (n=40) and control group (n=40) after
birth. Following our previously described methods (23), new-born rats in the model group
were exposed to a high oxygen environment (FiO2=0.85)
within 12 h after birth, and the control group was exposed to
normoxic air (FiO2=0.21). The CO2
concentration was controlled at <0.5% using soda lime, and
silica gel was used to remove water vapor from the oxygen tank. The
maternal rats were used to feed the new-born rats and were
exchanged among cages every 24 h to minimise the effect of
differences in nursing ability. The cage was opened for 30 min
every day and clean drinking water and food were provided. On
postnatal day (P)1, 3, 7 and 14 after modelling, 10 pup rats in
each group were anaesthetised using pentobarbital sodium
(intraperitoneal injection at the dose of 50 mg/kg) and euthanized
by cervical dislocation. Then the chest cavity was opened quickly
to dissect the lung tissue. All experimental procedures were
reviewed and approved by The Ethics Committee of China Medical
University (Shenyang, China).
Histopathology
After being resected, the left lung was cut to 0.5
cm thickness sections, fixed with 4% paraformaldehyde at room
temperature for 48 h and embedded in paraffin, and the right lung
was stored at -80°C for subsequent experiments. The sections were
dehydrated using graded ethyl alcohol solutions (75% ethanol
overnight, 95% ethanol for 4 h and 100% ethanol for 2 h), and
embedded with paraffin after treatment with xylene. The tissue
sections were stained with haematoxylin and eosin (haematoxylin for
5 min, rinsed with PBS for 30 min and eosin for 5 min) at room
temperature using an automatic dyeing machine (Leica Autostainer
XL). Histopathological changes were observed using a light
microscope (Eclipse Ci; Nikon Corporation) at x200 magnification.
Each slice was analysed in six random regions. The radial alveolar
count (RAC) and alveolar wall thickness were determined to evaluate
alveolar development using ImageJ software version 1.80 (National
Institutes of Health). These assessments were carried out
independently by two pathologists who were blinded to the
grouping.
Transmission electron microscopy
After treatment, 1 mm3 of fresh lung
tissue was fixed in 4% paraformaldehyde and 2.5% glutaraldehyde in
0.1 M phosphate buffer overnight at 4°C and dehydrated in graded
ethyl alcohol solutions (50-90%) at 4°C, before embedding in
acetone for 4 h at room temperature. Ultrathin sections (50-60 nm
thickness) were obtained and double-stained with uranyl acetate and
lead citrate for 30 min at room temperature. Finally, a
transmission electron microscope (JEM-1200EX; Hitachi
High-Technologies Corporation) was used to observe lung tissue
samples from the model group at P1, 3, 7 and 14 at 100 kV. Under a
magnification of x10,000 (for six fields of view at each time
point), AT-II cells with peculiar lamellar bodies were identified,
and the number and morphological changes of mitochondria were
recorded. According to previously described methods (24), mitochondrial size were measured.
Mitochondrial fragments that were <1 μm3 and
not divided (usually round) were identified and the average
percentage of mitochondrial fragments in the field of view was
counted using mitochondrial fragmentation index (MFI). A random
double-blind analysis of the mitochondrial structure in AT-II cells
was performed. ImageJ software version 1.80 (National Institutes of
Health) was used to analyse images obtained using electron
microscopy at different time points.
Western blotting
The lung tissue was harvested and homogenised in
RIPA lysis buffer with a protease inhibitor (P1045; Beyotime
Institute of Biotechnology). The protein concentration was measured
using the bicinchoninic acid method and samples were boiled with a
loading buffer for protein extraction. In total, 15 μg/lane
samples were loaded onto a 4-20% gel, resolved using SdS-PAGE and
subsequently transferred to polyvinylidene fluoride (PVDF)
membranes. The membranes were then sealed using 5% skimmed milk for
2 h to block non-specific binding at room temperature. The
following primary antibodies were used: PINK1 (1:500; cat. no.
Bc100494; Novus Biologicals), Parkin (1:100; cat. no. sc-32282;
Santa Cruz Biotechnology, Inc.), NIX (1:1,000; cat. no. ab8399,
Abcam), Cytochrome C oxidase subunit IV isoform I (COX4) (1:500;
cat. no. ab14744; Abcam), LC3B (1:1,000; cat. no. 2775; Cell
Signalling Technology, Inc.) and β-actin (1:1,000; cat. no.
60008-1-Ig, ProteinTech Group, Inc.). The PVDF membranes were mixed
with primary antibodies, diluted in 10% skimmed milk in
Tris-buffered saline and Tween-20 (TBST), and incubated overnight
at 4°C. The next day, after washing with TBST for three times, the
PVDF membranes were incubated with a horseradish
peroxidase-conjugated secondary antibody (1:10,000; cat. no.
SA00001-1 and SA00001-2, ProteinTech Group, Inc.) at 20°C for 2 h
and washed in TBST. Finally, band density was determined using
image capture densitometry (GE Amersham Imager 600; Cyvita) using
an Enhanced chemiluminescence Substrate ECL kit (Santa Cruz
Biotechnology, Inc.).
Mitochondrial separation
Mitochondrial separation was performed using a
tissue mitochondrial isolation kit according to the manufacturer's
instructions (cat. no. C3606; Beyotime Institute of Biotechnology).
The mitochondrial lysis buffer consisted of 0.25 M sucrose, 10 mM
Tris-hydrochloric acid, 3 mM MgCl2, 0.1 mM EDTA and a
mixture of phosphatase and protease inhibitors. After homogenising
the fresh lung tissue with mitochondrial lysis buffer on ice, the
homogenate was centrifuged at 4°C and 600 x g for 5 min. The
supernatant was recovered and centrifuged at 11,000 x g at 4°C for
10 min. The precipitate was washed in the lysis buffer and
centrifuged again at 12,000 x g at 4°C for 15 min. Finally, the
mitochondrial precipitate was recovered and suspended in stock
solution for the MMP assay.
Primary AT-II cell isolation
AT-II cells of new-born rats were isolated on
postnatal days 1, 3, 7 and 14. Briefly, lung tissues of new-born
rats were perfused with normal saline, cut using scissors and
digested with 0.25% trypsin-EDTA (cat. no. 25200056; Gibco; Thermo
Fisher Scientific, Inc.) at 37°C. After 30 min, digestion was
terminated and samples were filtered through 60 and 200-μm
filters. The cell pellet obtained after centrifugation at 200 x g
at 20°C for 5 min and was resuspended in 0.1% collagenase I at 37°C
for digestion (cat. no. 17100017; Gibco; Thermo Fisher Scientific,
Inc.) The cells were harvested by centrifugation at 200 x g at 20°C
for 5 min, resuspended in Dulbecco's modified Eagle's medium (DMEM)
(Corning, Inc.) containing 1% penicillin streptomycin double
antibody and 10% fetal bovine serum (10099141; Gibco; Thermo Fisher
Scientific, Inc.) and incubated. The culture dish was replaced
every 30 min for a total of six times to remove fibroblasts and
unattached cells. Finally, an IgG-coated dish was used for
differential attachment to remove foreign cells, such as
fibroblasts and macrophages. After 12 h, the cells were collected
for subsequent experiments.
MMP assay
The
5,5',6,6'-tetrachloro-1,1',3,3'-tetraethyl-benz-imidazole carbon
iodide (JC-1) fluorescent probe (cat. no. C2006; Beyotime Institute
of Biotechnology) was used to detect MMP (mtΔΨ). In brief, the
medium in the six-well plate was discarded and 0.5 mM Jc-1 staining
working solution was added to primary AT-II cells for 15 min,
followed by incubation in the dark environment at 37°C for 20 min.
After washing three times with dMEM (10-013-CV, Corning, Inc.), the
mtΔΨ value for each sample was determined as the ratio of the red
fluorescence intensity to the green fluorescence intensity. In
normal cells, mitochondria have a higher membrane potential, which
can cause JC-1 to form orange-red fluorescent J-aggregates outside
the mitochondrial membrane (25).
In mitochondrial-damaged cells, sensor dyes appear as green
fluorescent monomers. Live cells were observed using 80x
fluorescent objective. The objective was connected to the Photon
Technologies dual Emission system (LSM880; Zeiss AG) and the
excitation wavelength was set to 490 and 525 nm. The fluorescence
was emitted and collected separately using Zen Imaging Software3.0.
Data are shown as ratios (F590/F530). A decrease in the red
(F590)/green (F530) fluorescence intensity ratio indicated a loss
of MMP.
Immunofluorescence double staining
The extracted primary AT-II cells were plated at
200,000 cells/ml for subsequent detection. The plated cells were
washed with PBS and fixed with 4% paraformaldehyde at 4°C for 30
min. Primary antibodies were added, including antibodies against
LC3B (1:200; cat. no. 2775; anti-rabbit, Cell Signalling
Technology, Inc.) and COX4 (1:100; cat. no. ab14744; anti-mouse,
Abcam), and incubated overnight at 4°C. After washing with PBS,
donkey anti-rabbit IgG H&L (Alexa Fluor® 594; cat.
no. ab150076, Abcam) and donkey anti-mouse IgG H&L (Alexa
Fluor® 488; cat. no. ab150105; Abcam) were added,
incubated for 2 h at room temperature, and counterstained with DAPI
for 5 min at room temperature. Similarly, tissue paraffin sections
were dehydrated using graded ethanol (75% overnight, 95% for 4 h
and 100% for 2 h) and subjected to fluorescent double staining.
Primary antibodies against PINK1 (1:100; cat. no. BC100494;
anti-rabbit; Novus Biologicals) and Parkin (1:100; cat. no.
sc-32282; anti-mouse; Santa Cruz Biotechnology, Inc.) were applied
at 4°C overnight. After washing with PBS, a secondary antibody
labelled with fluorescein isothiocyanate (Alexa Fluor®
594; cat. no. ab150076, and Alexa Fluor® 488; cat. no.
ab150105; both Abcam) was added and incubated for 2 h and
counterstained with DAPI for 5 min, both at room temperature. After
observation at x400 magnification using a two-photon confocal
microscope (LSM880; Zeiss AG), 3-dimensional reconstruction was
performed using ImageJ software1.80, which was also used to analyse
the changes in fluorescence intensity changes.
Seahorse XF energy analysis
The oxygen consumption rate (OCR) and extracellular
acidification rate (ECAR) were measured using the Seahorse XFe96
Flux Analyzer (Agilent Technologies, Inc.). According to methods
suggested by the manufacturer, rat primary AT-II cells were
extracted on P3, 7 and 14. cells were seeded on XF96 cell plates
(Agilent Technologies, Inc.) at a density of 20,000 cells/well.
On-board testing was performed 12 h after seeding. The original
medium in the cell plate was discarded and replaced with serum
-free bicarbonate-containing analysis medium (10 mM glucose, 2 mM
glutamine and 1 mM ammonium pyruvate). For the mitochondrial stress
test, based on pilot experiments with the oxidative phosphorylation
uncoupling agent Trifluoromethoxy carbonylcyanide phenylhydrazone
(FCCP) at four concentrations of 0.5, 1, 1.5 and 2 mM, the optimal
concentration was identified as 2 mM for the subsequent detection
in AT-II cells. Oligomycin, Rotentone and Antimycin A were used at
the concentrations specified in the manufacturer's instructions. No
glucose was added in the first stage to detect basal respiration.
In the second stage, oligomycin was added to detect the ATP-related
metabolic capacity of mitochondria. In the third stage, the
uncoupling agent FCCP was added to detect the maximum mitochondrial
pressure. Finally, rotenone and antimycin A were added to detect
the non-mitochondrial oxygen consumption. Similarly, metabolic
regulators, such as glucose, oligomycin and 2-deoxyglucose (2-DG),
were used for glycolytic stress according to the method described
by the manufacturer. No glucose was added and extracellular
acidification was tested in the first stage. In the second stage,
glycolysis was detected after the addition of glucose. In the third
stage, oligomycin was added to detect the maximum storage capacity
of glycolysis. In the fourth stage, 2-DG was added to detect the
residual capacity of glycolysis. Ten biological replicates were
established for each group.
Statistical analysis
Data analysis was performed using GraphPad Prism
version 8.0 (GraphPad Software). Parameters were compared between
groups using the unpaired Student's t-tests at each time point and
no multiple comparisons were made across timepoints or within a
timepoint. correlation analysis was performed using Pearson's
tests. All data are expressed as means ± standard error of the
mean. n represents the number of animals in each group or the
number of independent experiments. P<0.05 was considered to
indicate a statistically significant difference.
Results
Hyperoxia exposure leads to delayed
alveolarization
New-born lung tissues rats were collected at P1, 3,
7 and 14 after exposure to normoxic or hyperoxic conditions and
were evaluated with respect to alveolar development. In the two
groups, the number of alveoli and capillaries increased gradually.
With a decrease in the alveolar cavity diameter and an increase in
the alveolar ridge structure, the thickness of the alveolar septum
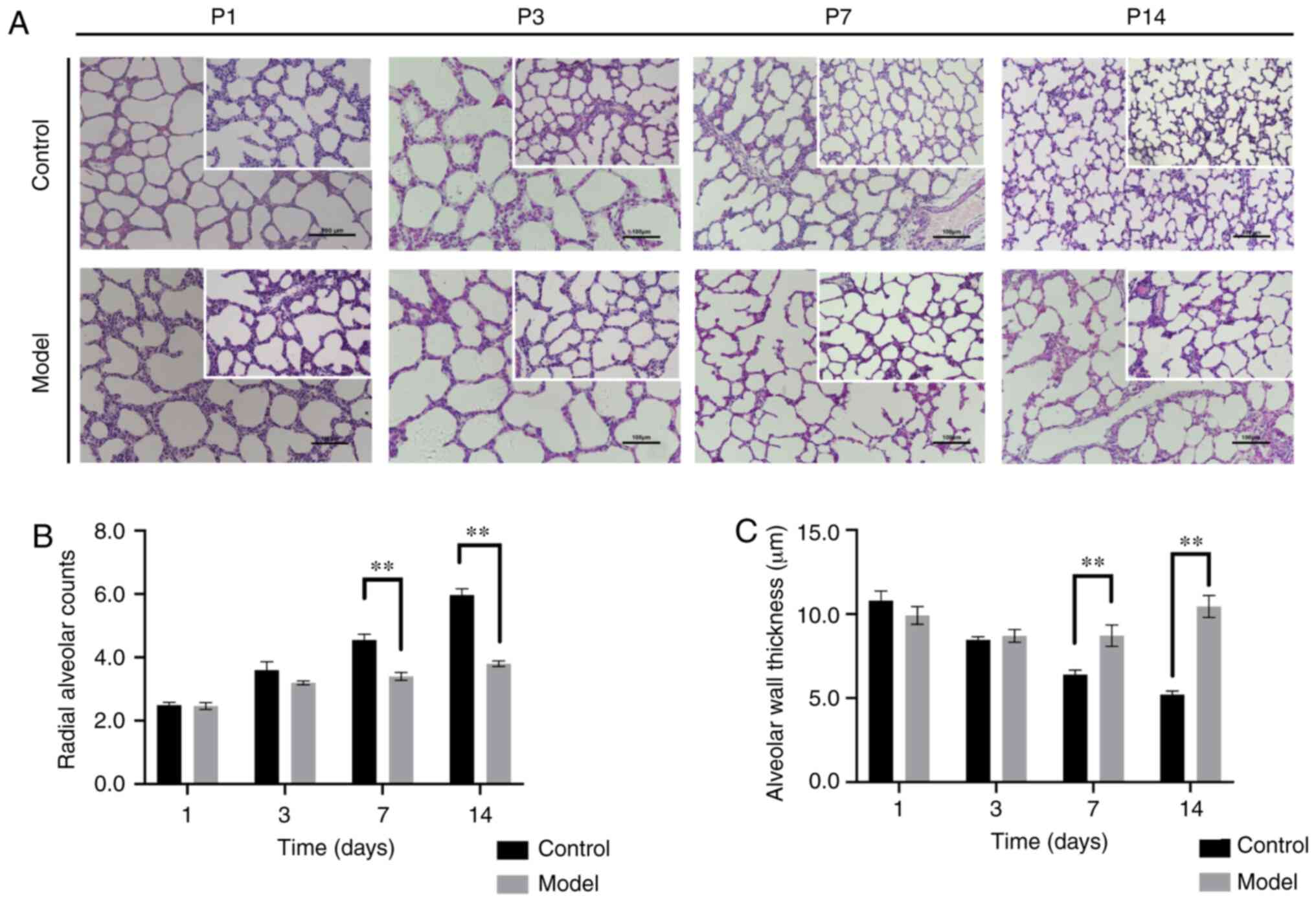
gradually decreased. compared with the control group (n=10), the
model group (n=10) showed a widened alveolar septum, oedema and
inflammatory cell infiltration in some fields at P3 (Fig. 1A). There was no statistically
significant difference in alveolar RAC values between the control
and model groups at P1 and P3 (both P>0.05). Similarly, there
was no significant difference in alveolar wall thickness between
the groups at P1 and P3 (both P>0.05). However, from P7, the
alveolar diameter in the model group was larger compared with that
in the control group, and the alveolar ridge structure was reduced
and blunt. The RAc was lower in the model group (P<0.01;
Fig. 1B), whereas the thickness
of the lung septum was higher (P<0.01; Fig. 1c) in the model group compared with
the control group. These results showed that the model group had
alveolar growth delay at P7 and P14.
Hyperoxia exposure leads to mitochondrial
morphological disorders in AT-II cells
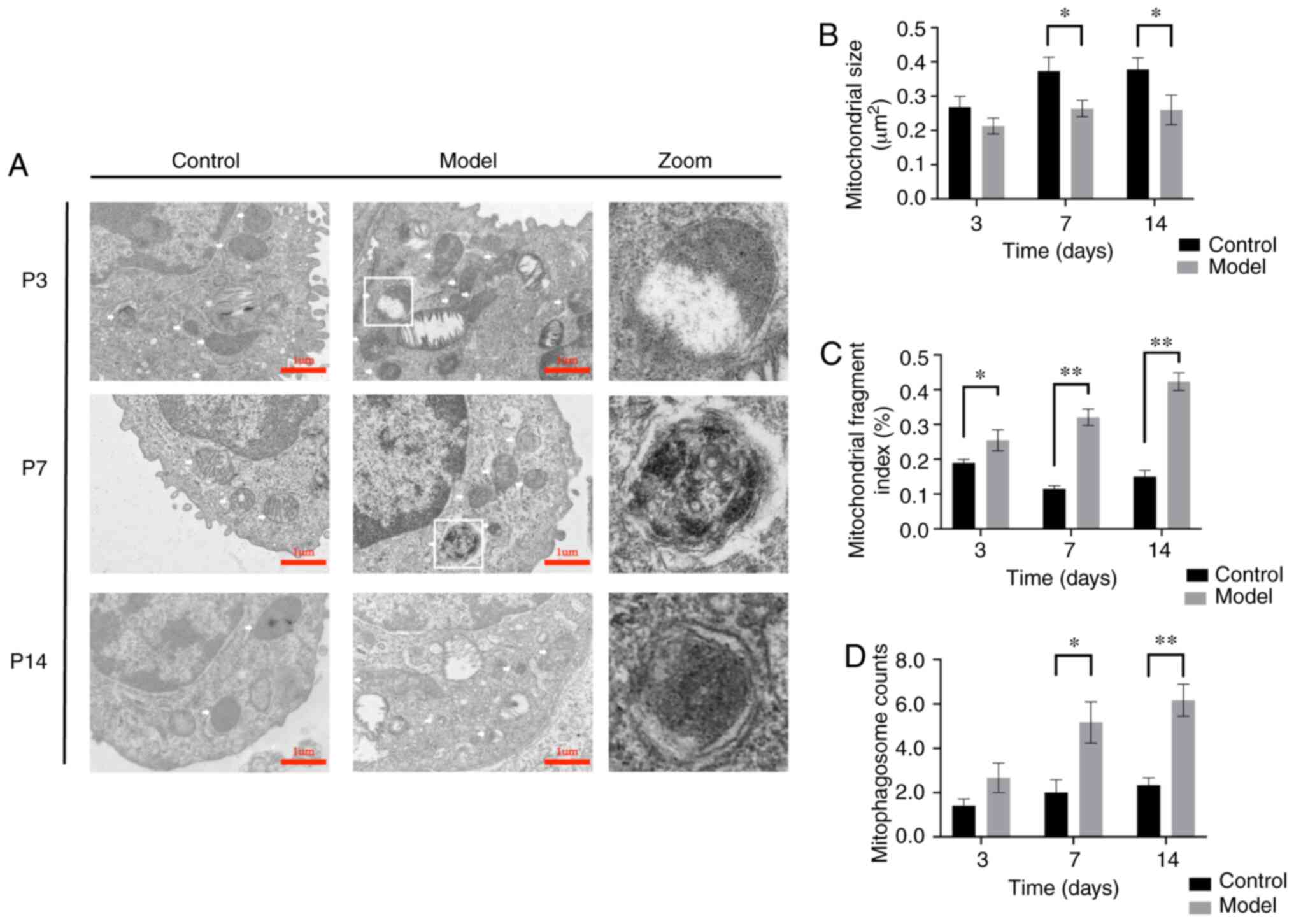
The mitochondria of AT-II cells in each group were
observed using transmission electron microscopy and the
ultrastructures were compared. In the control group, there were
compact mitochondria, abundant lipid lamellar bodies and a normal
basic cell substructure. The volume of the mitochondria gradually
increased and the crista gradually became denser in the control
group. The mitochondrial structure was disrupted in the model group
as of P3. This was manifested as mitochondrial swelling and
fracture, destruction of the mitochondrial double membrane
structure, mitochondrial cristae disappeared, and the mitochondria
became longer and increasingly divided. Peculiar lamellar corpuscle
structural damage. After oxygen exposure, mitochondria were
ruptured and mitophagosomes wrapped in autophagosome membranes were
observed in the zoomed-in images (Fig. 2A). At P3, the MFI was
significantly increased (P<0.05), but swelling also increased,
and the average mitochondrial area did not differ from that in the
control group (P>0.05). However, from P7, the MFI and
mitophagosomes in the model group were significantly increased
(P<0.01) and the average mitochondria area was significantly
lower compared with that in the control group (P<0.01) (Fig. 2A-d). In summary, mitochondrial
damage in AT-II cells of the model group increased gradually over
time, with increases in swelling and fragmentation becoming more
obvious over time.
Hyperoxia exposure leads to decreased MMP
in AT-II cells
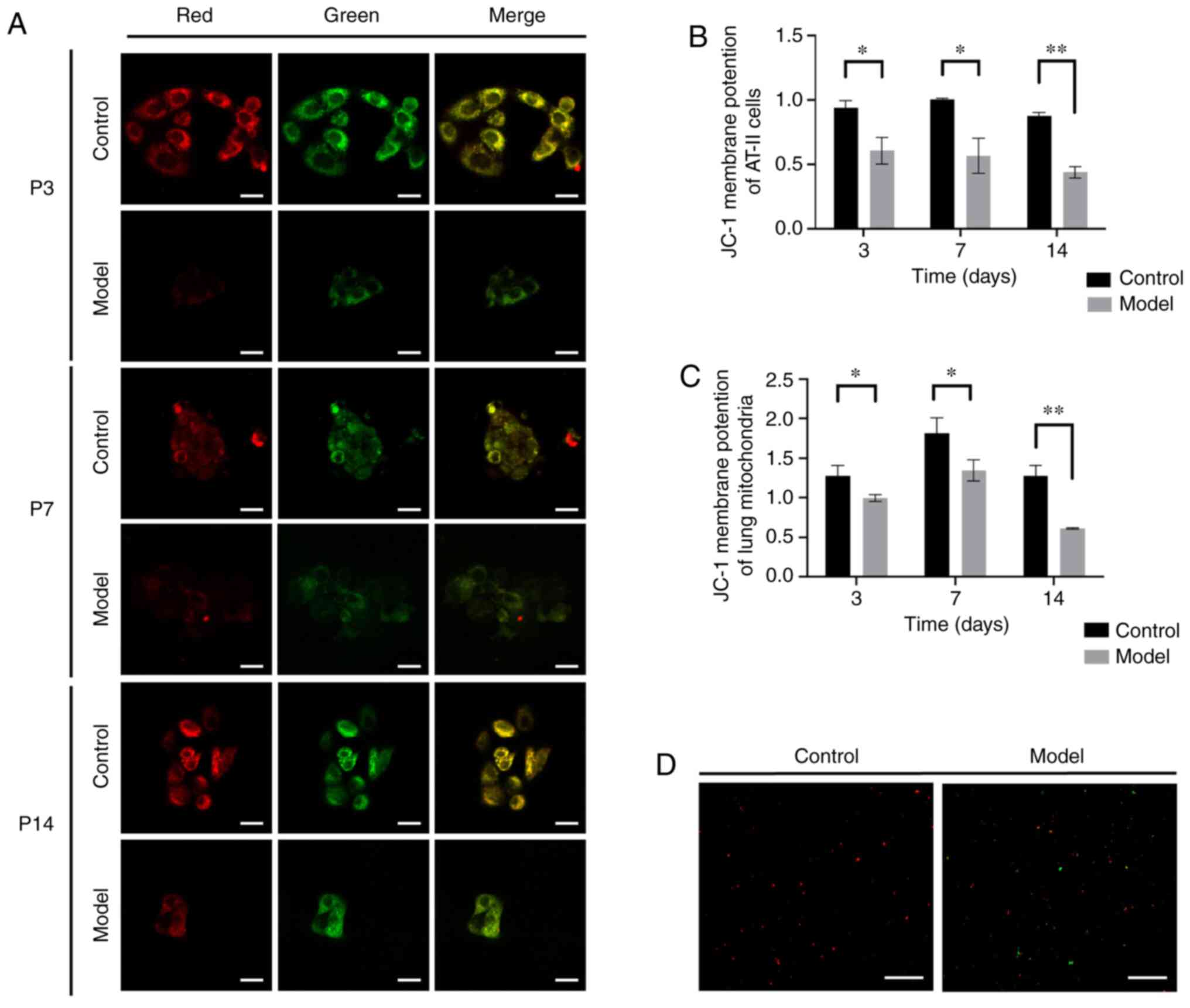
A decrease in MMP is a sensitive signal of
mitochondrial damage and autophagy activation (26). Accordingly, MMP was measured in
primary AT-II cells (n=6) using the fluorescence intensity
(Fig. 3A) and ratio of JC-1
polymer levels (red)/JC-1 monomer levels (green) to evaluate
mitochondrial function damage in the model group at each time
point. At P3, the mtΔΨ was 0.605 in the model group, while the mtΔΨ
ratio was 0.937 in the control (P<0.05). And the mtΔΨ ratio
decreased to 0.564 at P7 (P<0.05) and 0.437 at P14 (P<0.01),
while the mtΔΨ fluorescence intensity ratio was 1.004 in the
control group at P7 and 0.876 at P14 (Fig. 3B). The mtΔΨ ratio of the model
group was lower compared with that of the control group in AT-II
cells. The membrane potential of mitochondria extracted from the
lung tissues was also detected using JC-1 staining within 30 min
and measured fluorescence intensity using a microplate reader. The
mtΔΨ was 1.280 in the control and 0.994 in the model group at P3,
the difference from the control group was statistically significant
(P<0.05). The mtΔΨ in the model group was 0.742 times less
compared with the control group at P7 and 0.478 times less compared
with the control group at P14 (P<0.01; Fig. 3C and D). These results showed that
the MMP in the model group decreased under hyperoxia exposure.
Increase in mitophagy protein expression
in hyper-oxia-exposed lung tissues of rats with simple lung
structures
PINK1-Parkin double staining was evaluated in lung
tissue sections to assess the ability of PINK1 to recruit Parkin
(Fig. 4A). The two proteins were
distributed in the cytoplasm of lung cells. The co-localisation of
PINK1 and Parkin fluorescence was lower at P3 and higher at P7 and
P14 in the model group compared with the control group. Next, the
protein levels of PINK1 and Parkin in lung tissues were
investigated using western blotting (Fig. 4B-E). compared with that in the
control group, the expression of PINK1 in the model group was
significantly elevated at P7 and P14 (P<0.05 and P<0.01,
respectively; Fig. 4C).
consistently, the expression level of Parkin in the model group was
not different at P1 or P3 compared with that in the control group;
however, a significant difference was observed at P7 and P14
(P<0.01 and P<0.05, respectively; Fig. 4E). The transformation of LC3B from
LC3BI (the free form) to LC3BII (the conjugated form of
phosphatidyl ethanolamine) is an important process in the formation
of autophagosomes (27). LC3BII/I
expression levels in the model group were significantly elevated
compared with the control group at P1, 3, 4 and 7 (P<0.05 or
P<0.01; Fig. 4D). These
results suggested that mitophagy occurred more frequently in the
model group compared with the control group.
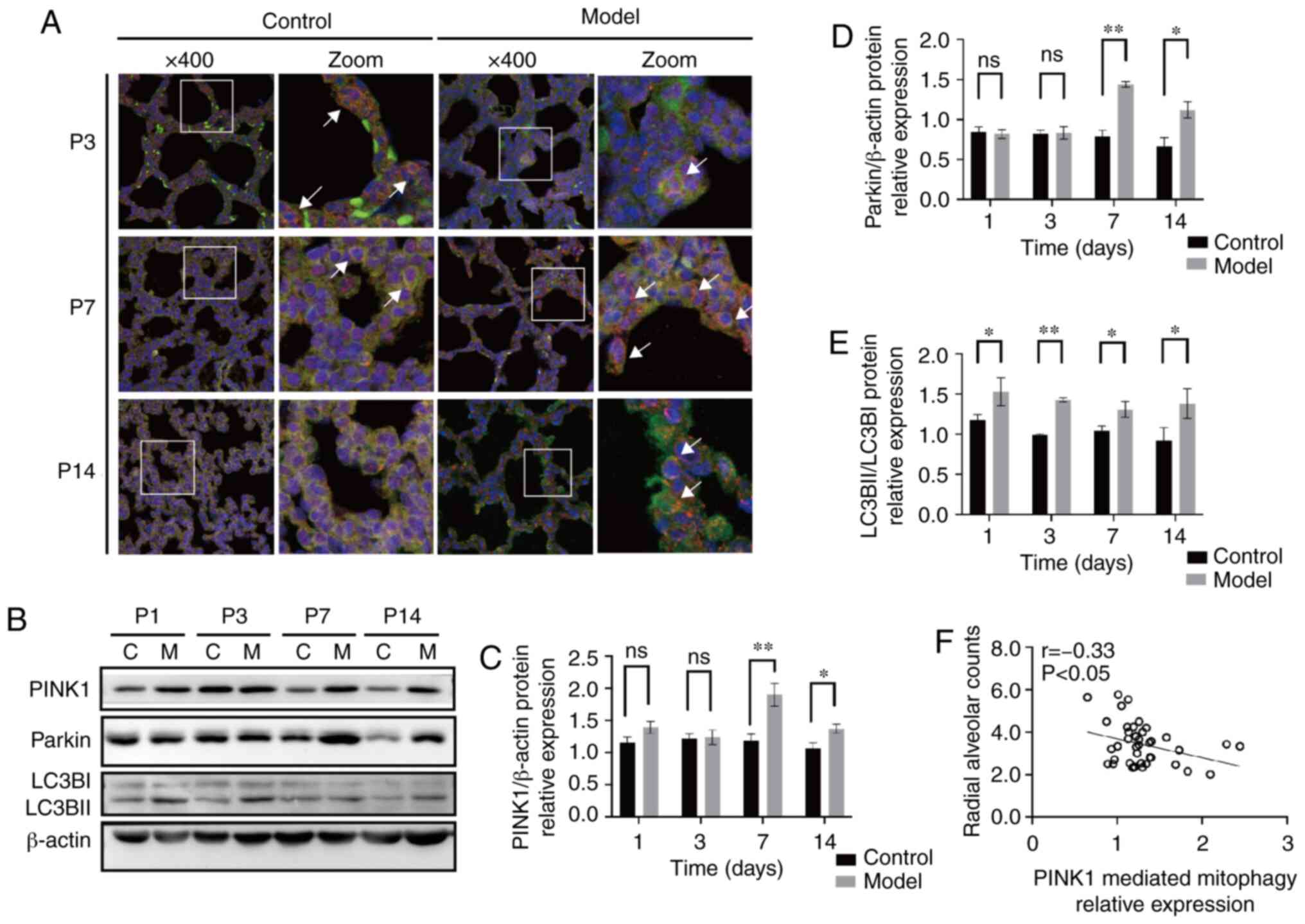 | Figure 4Expression changes of PINK1 and
Parkin in the lung tissues of rats with bronchopulmonary dysplasia.
(A) Representative immunostaining of lung sections from the two
groups using anti-PINK1 (red) and anti-Parkin (green) antibodies.
Yellow puncta denote co-localisation. Magnification, x400. (B)
Western blot analyses of PINK1, Parkin and LC3BI/LC3BII in the
lungs in the C group and M group at P1, 3, 7 and 14. (C) density
analyses of PINK1. (D) Density analyses of Parkin. (E) Density
analyses of LC3BII/LC3BI. Blots were stripped and reblotted using
an anti-β-actin antibody as a loading control. (F) Correlation
analyses between radial alveolar counts and PINK1 mediated
mitophagy relative expression in rat lungs. PINK1 mediated
mitophagy was negatively correlated with the alveolar development
index (r=-0.33; P<0.05). The data are expressed as mean ± SEM
from at least six different experiments. *P<0. 05,
**P<0. 01 vs. control. P, postnatal day; PINK1,
putative kinase 1; C, control; M, model; ns, not significant; LC3B,
microtubule-associated protein-1 light chain-3B. |
Mitophagy in AT-II cells increases in
hyperoxia-exposed lung tissues of rats with simple lung
structures
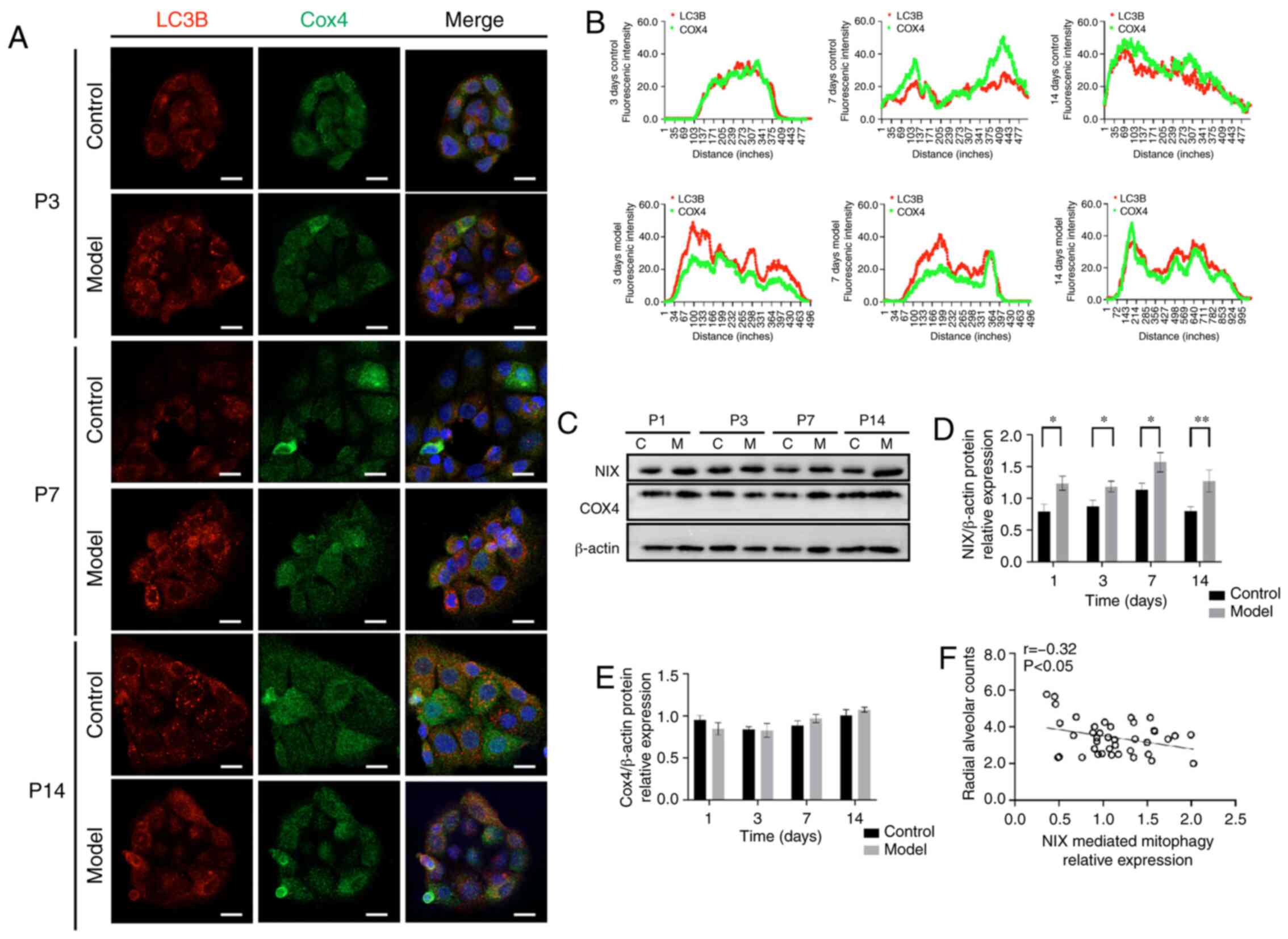
After clarifying the overall protein expression
levels of PINK1 and Parkin during lung development, the
co-localisation between the mitochondrial marker protein COX4 and
autophagy marker protein LC3B was evaluated in primary AT-II cells
in the postnatal period. A hallmark of the mitophagy process is the
co-localisation of mitochondria with autophagosomes for elimination
(28). Immunofluorescence
revealed higher LC3B protein levels in the model group compared
with in the control group, reaching a peak at P7 (Fig. 5A and B). In addition, the COX4
expression level was no significantly different between the two
groups (P>0.05) at each time point (Fig. 5C and E), but the NIX expression
level was higher in the model group compared with in the control
group (P<0.05) from P1 (Fig. 5C
and D) and it was negatively correlated with radial alveolar
counts (r=-0.32, P<0.05) (Fig.
5F). These results indicated that NIX mediated mitophagy also
occurred more frequently in the model group compared with in the
control group and participated in the pathological process of
BPD.
Hyperoxia leads to metabolic changes in
primary AT-II cells of model rats with simple lung structures
Real-time analyses of ECAR and OCR were performed
with primary AT-II cells using the Seahorse XF96 metabolic
extracellular flux analyser. Anaerobic metabolism levels were
evaluated. At P7, non-glycolytic acidification, glycolytic capacity
and the glycolytic reserve were significantly lower in the model
group compared with in the control group (P<0.05 or P<0.01)
(Fig. 6A and B). There were no
significant differences in the cellular glycolysis levels between
groups under basic conditions. However, as for the mitochondrial
pressure and aerobic metabolism levels, the basal respiration
capacity, ATP generation capacity (both P<0.05) and remaining
respiration capacity of the model group (P<0.01) were higher at
P3 in the model group compared with in the control group. At P7,
the maximal respiration and spare respiratory capacity were
significantly lower in the model group compared with in the control
group (P<0.01) and the results showed no difference at P14
(Fig. 6c and d). These results
suggested that there was a change in the metabolic capacity after
hyperoxia exposure at P3 and P7.
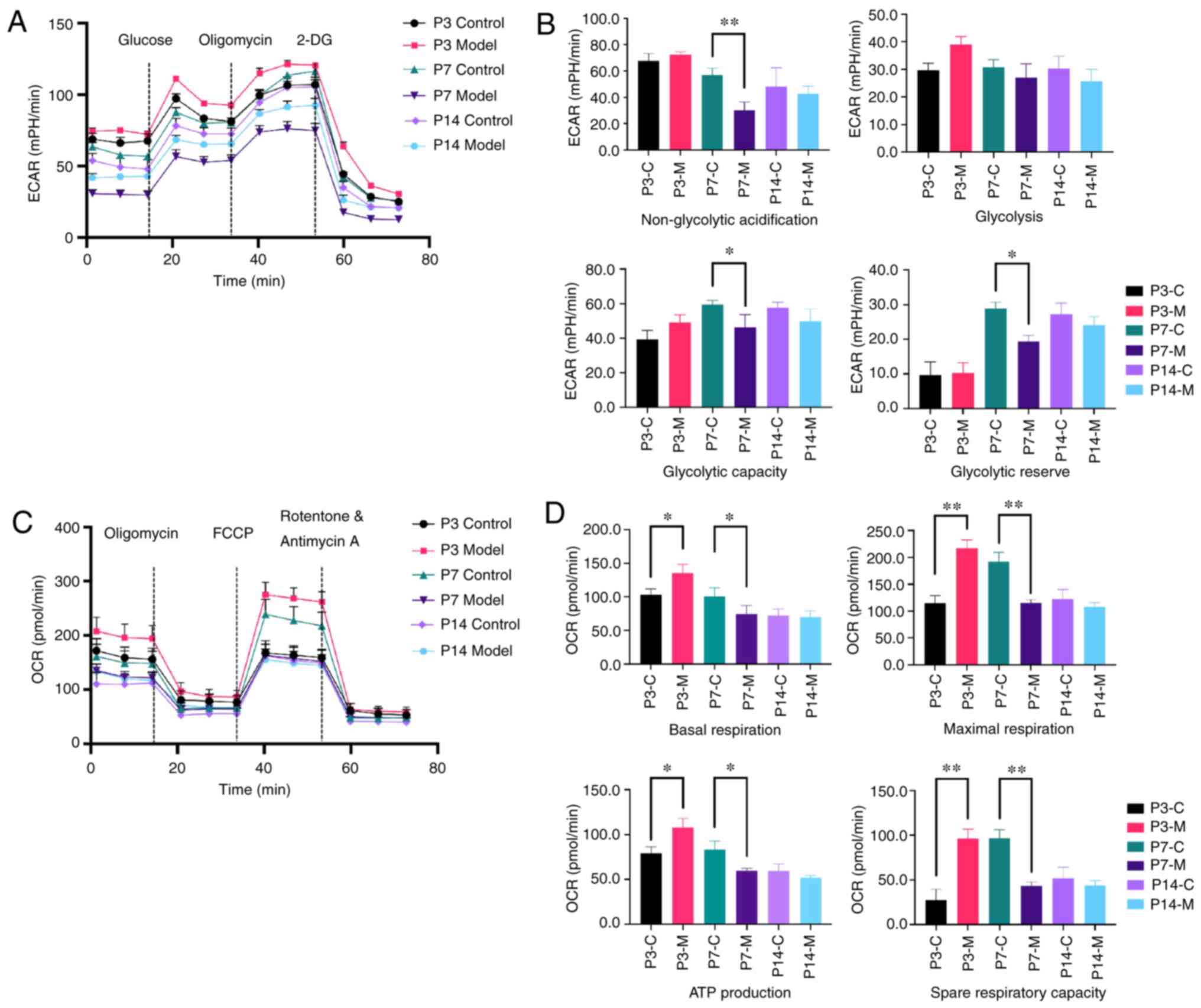 | Figure 6Changes in metabolic activity in
primitive AT-II cells. Seahorse XF96 was used to test the ECAR and
OCR in primary AT-II cells using real-time analysis. (A)
Representative graph of the ECAR output of AT-II cells and
responses to glucose, oligomycin and 2-DG. (B) Anaerobic metabolism
levels were evaluated. (C) Representative graph of the OCR output
of AT-II cells and responses to oligomycin, FCCP and
rotenone/antimycin A. (D) Mitochondrial pressure and aerobic
metabolism levels were tested. The assay was run in one plate with
10 replicates and data are presented as mean ± SEM.
*P<0.05, **P<0.01 vs. control. ECAR,
extracellular acidification rate; OCR, oxygen consumption rate;
AT-II, alveolar type II cells; FCCP, trifluoromethoxy
carbonylcyanide phenylhydrazone; P, postnatal day. C, control; M,
model; 2-DG, 2-deoxyglucose. |
Discussion
Supplemental oxygen is the most common therapeutic
agent used in neonatal treatment worldwide; however, prolonged
exposure to high oxygen alters the development of lung tissues and
vascular beds, resulting in BPD in preterm infants (29-31). The overall incidence of BPD in
infants born at <28 weeks of gestational age is estimated to be
48-68% according to a study performed at the National Institute of
child Health and Human development Neonatal Research Network
(3). BPD is a multifactorial
disease; however, one of its major risk factors is premature
exposure of the lungs to a hyperoxic environment that leads to the
production of superfluous reactive oxygen species (32). Mitochondria are not only the main
source of reactive oxygen species but are also critical for the
maintenance of homeostasis and a wide range of biological
activities, such as producing energy, buffering cytosolic calcium
and regulating signal transduction (33). However, the role of mitophagy in
BPD is not well-established. Therefore, it was hypothesised that
mitochondrial injuries induced by hyperoxia impair normal
biological mitophagy functions, thereby leading to the onset of
BPD.
To adapt to extrauterine life after birth and for
postnatal energy production during lung development, various lung
cell types undergo mitochondrial morphological changes, including
changes to volume density, size, number and distribution (34). In the present study, oxygen
exposure in new-born rats resulted in a relatively large alveolar
cavity, irregularly spaced lungs and a decreased radial alveolar
count. In addition to alveolar morphological changes, the MMP in
the model group decreased at the early stage of hyperoxia exposure.
The volume of the mitochondria gradually increased and the crista
gradually became denser in the control group, whereas in the model
group, the level of mitochondrial disruption and fragments
increased over time. This suggests that hyperoxia exposure leads to
delayed alveolarization and mitochondrial morphological disorders
in AT-II cells.
As a major mechanism of mitochondria quality
control, mitophagy is responsible for the degradation and recycling
of damaged mitochondria (35).
PINK1 can recruit Parkin and the two proteins act synergistically
to mark damaged mitochondria for clearance by the activation of
downstream mitophagy (36).
However, the involvement of mitophagy in chronic lung disease is
controversial. Disruptions in mitophagy caused by PINK1 deficiency
were reported to promote the occurrence of pulmonary fibrosis
(37). However, an increase in
PINK1 expression has also been observed and the disturbance of
mitophagy appears to be pathogenic in chronic obstructive pulmonary
disease (38,39). In the present study, the levels of
PINK1 and Parkin were not significantly higher on days P1 and P3,
but were higher on days P7 and P14, in the model group compared
with those in the control group. correlation analysis between RAC
and PINK1-Parkin mediated mitophagy revealed a negative
correlation. Fluorescence double staining indicated the
co-localisation of PINK1 and Parkin was also higher in the model
group compared with that in the control group, and the combination
was most significant at P7. Our previous study showed that the use
of rapamycin, a classic autophagy inducer, can improve alveolar
morphology development in a rat-based BPd model (40), as used in the present study.
Mitophagy is a type of macro-autophagy; however, traditional
autophagic inhibition does not prevent PINK1-Parkin-mediated
mitophagy and the inhibition of Parkin can affect mitochondrial
clearance (41), which suggests
that insufficient mitophagy mediated by PINK1-Parkin in the early
stage and the recruitment capacity does not continue to increase
linearly as the duration of hyperoxia exposure increases. It was
therefore hypothesised that PINK1-Parkin-mediated mitophagy
disorders may participate in the pathological process of BPD.
NIX is involved in the differentiation and
development of stem cells, such as the development of the nervous
system (18), retinal ganglion
cells (42) and macrophages
(42,43). Additionally, NIX may be a
substrate of PARK2 in the PINK1-Parkin pathway (44). In the present study, the NIX
expression level was higher in the model group compared with that
in the control group at all timepoints. Moreover, the negative
correlation between RAC and NIX-mediated mitophagy suggests that
NIX may also participate in the pathological process of BPD.
Mitochondrial and metabolic changes in cells and
their regulatory processes in BPD have been a focus of recent
research (21,45). Unlike other cells, AT-II cells
tend to use lactic acid as a substrate for mitochondrial
respiration (46). Accordingly,
premature oxygen exposure may change the glucose-oxygen metabolism
balance and affect the development of AT-II cells. The present
study the tested mitochondrial metabolic function of these cells to
confirm the increase of mitochondrial damage in hyperoxia-exposed
neonatal rats. After 3 days, the mitochondrial pressure and aerobic
function in the model group were higher and, after 7 days, the
mitochondrial pressure and glycolytic function were lower compared
with those in the control group. This indicated that the model
group exhibited metabolic changes. These findings demonstrated that
metabolic changes are only significant at the early phase before
BPd is established, suggesting that they may be a cause of
mitophagy disorders rather than a direct contributor to alveolar
morphological changes. Ratner et al (47) exposed neonatal rats to 75%
O2 for 72 h, followed by recovery in room air for 3
weeks or 2 weeks of hyperoxia exposure. The study showed that
mitochondrial OCR and ATP production decreased in the lung tissue,
suggesting that the inhibition of mitochondrial oxidative
phosphorylation (OXPHOS) is associated with bronchopulmonary
developmental arrest. das (48)
conducted metabolic tests in vitro in hyperoxia-induced
MLE-12 cells. das observed that the glycolytic reserve capacity and
glycolytic residual capacity were reduced and the OXPHOS ability
was impaired. Simon et al (49) demonstrated that glycolytic
capacity exhibits a compensatory increase in AT-II cells at 95%
O2; however, this was not accompanied by altered
activities of critical mitochondrial (cytochrome oxidase) or
glycolytic (pyruvate kinase and phosphofructokinase) enzymes. The
present results define the metabolic changes in the early stages of
BPD. It is worth noting that mitophagy is also closely related to
the metabolic transformation (50). McCoy et al (51) reported that the recruitment of
Parkin requires hexokinase activity and Liu et al reported
that Parkin can regulate pyruvate kinase M2 (52). The present study did not fully
explore the mechanism underlying the regulation of metabolic
targets, therefore this should be a focus of future research.
In conclusion, the present study demonstrated that
there was mitochondrial structural damage and decreased MMP, as
well as elevated levels of PINK1-Parkin and the mitophagy receptor
protein NIX, in rat AT-II cells under hyperoxia, and that the
mitochondrial damage gradually increased over time. corresponding
changes in mitochondrial metabolic capacity were also observed.
Thus, the accumulation of dysfunctional mitochondria and
abnormalities in mitophagy in the lung tissues may be key factors
in the pathogenesis of BPD and could serve as therapeutic
targets.
Acknowledgments
The authors would like to thank the technical
support by Professor Dongyan Liu from Department of Immunology,
Shengjing Hospital of China Medical University and the technical
support in im munofluorescence double staining provided by Mr.
Ziying Li, Miss Yue Zhan and Mr. Jianing Miao from the Department
of Research and Development Centre, Shengjing Hospital of China
Medical University.
Funding
This study was supported by grants from The National
Natural Science Foundation of China (grant no. 81571479) and The
345 Talent Project of the Shengjing Hospital (grant no. M0428).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XY and JF conceived and designed the study. XY, YS,
QC, XZ and ZL performed the experiments. XY, YS and XX analysed the
data and JF reviewed the data. XY wrote the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by The Ethics
Committee of China Medical University (Shenyang, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Abman SH, Bancalari E and Jobe A: The
evolution of bronchopulmonary dysplasia after 50 years. Am J Respir
Crit Care Med. 195:421–424. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Surate Solaligue DE, Rodriguez-Castillo
JA, Ahlbrecht K and Morty RE: Recent advances in our understanding
of the mechanisms of late lung development and bronchopulmonary
dysplasia. Am J Physiol Lung Cell Mol Physiol. 313:L1101–L1153.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stoll BJ, Hansen NI, Bell EF, Shankaran S,
Laptook AR, Walsh MC, Hale EC, Newman NS, Schibler K, Carlo WA, et
al: Neonatal outcomes of extremely preterm infants from the NICHD
neonatal research network. Pediatrics. 126:443–456. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
García-Muñoz Rodrigo F, Losada Martínez A,
Elorza Fernández MD, Moreno Hernando J, Figueras Aloy J and Vento
Torres M: The burden of respiratory disease in
very-low-birth-weight infants: Changes in perinatal care and
outcomes in a decade in Spain. Neonatology. 112:30–39. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dumpa V and Bhandari V: Surfactant,
steroids and non-invasive ventilation in the prevention of BPD.
Semin Perinatol. 42:444–452. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kalikkot Thekkeveedu R, Guaman MC and
Shivanna B: Bronchopulmonary dysplasia: A review of pathogenesis
and pathophysiology. Respir Med. 132:170–177. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jobe AH and Abman SH: Bronchopulmonary
dysplasia: A continuum of lung disease from the fetus to the adult.
Am J Respir Crit Care Med. 200:659–660. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen Y, Chang L, Li W, Rong Z, Liu W, Shan
R and Pan R: Thioredoxin protects fetal type II epithelial cells
from hyperoxia-induced injury. Pediatr Pulmonol. 45:1192–1200.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
McGrath-Morrow SA and Stahl J: Apoptosis
in neonatal murine lung exposed to hyperoxia. Am J Respir Cell Mol
Biol. 25:150–155. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
O'Reilly MA, Staversky RJ, Finkelstein JN
and Keng PC: Activation of the G2 cell cycle checkpoint enhances
survival of epithelial cells exposed to hyperoxia. Am J Physiol
Lung cell Mol Physiol. 284:L368–L375. 2003. View Article : Google Scholar
|
|
11
|
Bayne AN and Trempe JF: Mechanisms of
PINK1, ubiquitin and Parkin interactions in mitochondrial quality
control and beyond. Cell Mol Life Sci. 76:4589–4611. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Liu N and Lu B: Mechanisms and
roles of mitophagy in neurodegenerative diseases. cNS Neurosci
Ther. 25:859–875. 2019.PubMed/NCBI
|
|
13
|
Sureshbabu A and Bhandari V: Targeting
mitochondrial dysfunction in lung diseases: Emphasis on mitophagy.
Front Physiol. 4:3842013. View Article : Google Scholar
|
|
14
|
Meissner C, Lorenz H, Weihofen A, Selkoe
DJ and Lemberg MK: The mitochondrial intramembrane protease PARL
cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem.
117:856–867. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nardin A, Schrepfer E and Ziviani E:
Counteracting PINK/Parkin deficiency in the activation of
mitophagy: A potential therapeutic intervention for Parkinson's
disease. Curr Neuropharmacol. 14:250–259. 2016. View Article : Google Scholar :
|
|
16
|
Liu H, Dai C, Fan Y, Guo B, Ren K, Sun T
and Wang W: From autophagy to mitophagy: The roles of P62 in
neurodegenerative diseases. J Bioenerg Biomembr. 49:413–422. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wei H, Liu L and Chen Q: Selective removal
of mitochondria via mitophagy: Distinct pathways for different
mitochondrial stresses. Biochim Biophys Acta. 1853:2784–2790. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Deczkowska A and Schwartz M: NIX-ing
mitochondria: From development to pathology. EMBO J. 36:1650–1652.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Um JH and Yun J: Emerging role of
mitophagy in human diseases and physiology. BMB Rep. 50:299–307.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yee M, Vitiello PF, Roper JM, Staversky
RJ, Wright TW, McGrath-Morrow SA, Maniscalco WM, Finkelstein JN and
O'Reilly MA: Type II epithelial cells are critical target for
hyperoxia-mediated impairment of postnatal lung development. Am J
Physiol Lung Cell Mol Physiol. 291:L1101–L1111. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cliff TS and Dalton S: Metabolic switching
and cell fate decisions: Implications for pluripotency,
reprogramming and development. Curr Opin Genet dev. 46:44–49. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Aravamudan B, Thompson MA, Pabelick CM and
Prakash YS: Mitochondria in lung diseases. Expert Rev Respir Med.
7:631–646. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hou A, Fu J, Yang H, Zhu Y, Pan Y, Xu S
and Xue X: Hyperoxia stimulates the transdifferentiation of type II
alveolar epithelial cells in newborn rats. Am J Physiol Lung Cell
Mol Physiol. 308:L861–L872. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hara H, Araya J, Ito S, Kobayashi K,
Takasaka N, Yoshii Y, Wakui H, Kojima J, Shimizu K, Numata T, et
al: Mitochondrial fragmentation in cigarette smoke-induced
bronchial epithelial cell senescence. Am J Physiol Lung Cell Mol
Physiol. 305:L737–L746. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Resseguie EA, Brookes PS and O'Reilly MA:
SMG-1 kinase attenuates mitochondrial ROS production but not cell
respiration deficits during hyperoxia. Exp Lung Res. 43:229–239.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Song SB, Jang SY, Kang HT, Wei B, Jeoun
UW, Yoon GS and Hwang ES: Modulation of mitochondrial membrane
potential and ROS generation by nicotinamide in a manner
independent of SIRT1 and mitophagy. Mol cells. 40:503–514.
2017.PubMed/NCBI
|
|
27
|
Peixoto P, Grandvallet C, Feugeas JP,
Guittaut M and Hervouet E: Epigenetic control of autophagy in
cancer cells: A key process for cancer-related phenotypes. Cells.
8:16562019. View Article : Google Scholar
|
|
28
|
Zachari M and Ktistakis NT: Mammalian
mitophagosome formation: A focus on the early signals and steps.
Front Cell Dev Biol. 8:1712020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Walsh BK, Brooks TM and Grenier BM: Oxygen
therapy in the neonatal care environment. Respir Care.
54:1193–1202. 2009.PubMed/NCBI
|
|
30
|
Jobe AH and Kallapur SG: Long term
consequences of oxygen therapy in the neonatal period. Semin Fetal
Neonatal Med. 15:230–235. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang J and Dong W: Oxidative stress and
bronchopulmonary dysplasia. Gene. 678:177–183. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bancalari E, Claure N and Sosenko IR:
Bronchopulmonary dysplasia: Changes in pathogenesis, epidemiology
and definition. Semin Neonatol. 8:63–71. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Scherz-Shouval R and Elazar Z: Regulation
of autophagy by ROS: Physiology and pathology. Trends Biochem Sci.
36:30–38. 2011. View Article : Google Scholar
|
|
34
|
El-Merhie N, Baumgart-Vogt E, Pilatz A,
Pfreimer S, Pfeiffer B, Pak O, Kosanovic D, Seimetz M, Schermuly
RT, Weissmann N and Karnati S: differential alterations of the
mitochondrial morphology and respiratory chain complexes during
postnatal development of the mouse lung. Oxid Med Cell Longev.
2017:91691462017. View Article : Google Scholar
|
|
35
|
Anzell AR, Maizy R, Przyklenk K and
Sanderson TH: Mitochondrial quality control and disease: Insights
into ischemia-reperfusion injury. Mol Neurobiol. 55:2547–2564.
2018. View Article : Google Scholar
|
|
36
|
Eiyama A and Okamoto K:
PINK1/Parkin-mediated mitophagy in mammalian cells. Curr Opin Cell
Biol. 33:95–101. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ahmad T, Sundar IK, Lerner CA, Gerloff J,
Tormos AM, Yao H and Rahman I: Impaired mitophagy leads to
cigarette smoke stress-induced cellular senescence: Implications
for chronic obstructive pulmonary disease. FASEB J. 29:2912–2929.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Patel AS, Song JW, Chu SG, Mizumura K,
Osorio JC, Shi Y, El-chemaly S, Lee CG, Rosas IO, Elias JA, et al:
Epithelial cell mitochondrial dysfunction and PINK1 are induced by
transforming growth factor-beta1 in pulmonary fibrosis. PLoS One.
10:e01212462015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Araya J, Tsubouchi K, Sato N, Ito S,
Minagawa S, Hara H, Hosaka Y, Ichikawa A, Saito N, Kadota T, et al:
PRKN-regulated mitophagy and cellular senescence during COPD
pathogenesis. Autophagy. 15:510–526. 2019. View Article : Google Scholar :
|
|
40
|
Zhang D, Wu L, Du Y, Zhu Y, Pan B, Xue X
and Fu J: Autophagy inducers restore impaired autophagy, reduce
apoptosis, and attenuate blunted alveolarization in
hyperoxia-exposed newborn rats. Pediatr Pulmonol. 53:1053–1066.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Durcan TM and Fon EA: The three 'P's of
mitophagy: PARKIN, PINK1, and post-translational modifications.
Genes dev. 29:989–999. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Esteban-Martinez L, Sierra-Filardi E,
McGreal RS, Salazar-Roa M, Mariño G, Seco E, Durand S, Enot D,
Graña O, Malumbres M, et al: Programmed mitophagy is essential for
the glycolytic switch during cell differentiation. EMBO J.
36:1688–1706. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Esteban-Martinez L and Boya P:
BNIP3L/NIX-dependent mitophagy regulates cell differentiation via
metabolic reprogramming. Autophagy. 14:915–917. 2018. View Article : Google Scholar :
|
|
44
|
Gao F, Chen D, Si J, Hu Q, Qin Z, Fang M
and Wang G: The mitochondrial protein BNIP3L is the substrate of
PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum Mol Genet.
24:2528–2538. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhao H, Dennery PA and Yao H: Metabolic
reprogramming in the pathogenesis of chronic lung diseases,
including BPD, COPD, and pulmonary fibrosis. Am J Physiol Lung Cell
Mol Physiol. 314:L544–L554. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lottes RG, Newton DA, Spyropoulos DD and
Baatz JE: Lactate as substrate for mitochondrial respiration in
alveolar epithelial type II cells. Am J Physiol Lung cell Mol
Physiol. 308:L953–L961. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ratner V, Starkov A, Matsiukevich D, Polin
RA and Ten VS: Mitochondrial dysfunction contributes to alveolar
developmental arrest in hyperoxia-exposed mice. Am J Respir Cell
Mol Biol. 40:511–518. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Das KC: Hyperoxia decreases glycolytic
capacity, glycolytic reserve and oxidative phosphorylation in
MLE-12 cells and inhibits complex I and II function, but not
complex IV in isolated mouse lung mitochondria. PLoS One.
8:e733582013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Simon LM, Raffin TA, Douglas WH, Theodore
J and Robin ED: Effects of high oxygen exposure on bioenergetics in
isolated type II pneumocytes. J Appl Physiol Respir Environ Exerc
Physiol. 47:98–103. 1979.PubMed/NCBI
|
|
50
|
Naik PP, Birbrair A and Bhutia SK:
Mitophagy-driven metabolic switch reprograms stem cell fate. cell
Mol Life Sci. 76:27–43. 2019. View Article : Google Scholar
|
|
51
|
McCoy MK, Kaganovich A, Rudenko IN, Ding J
and Cookson MR: Hexokinase activity is required for recruitment of
parkin to depolarized mitochondria. Hum Mol Genet. 23:145–156.
2014. View Article : Google Scholar
|
|
52
|
Liu K, Li F, Han H, Chen Y, Mao Z, Luo J,
Zhao Y, Zheng B, Gu W and Zhao W: Parkin regulates the activity of
pyruvate kinase M2. J Biol chem. 291:10307–10317. 2016. View Article : Google Scholar : PubMed/NCBI
|