Introduction
Preeclampsia (PE), a specific complication of
pregnancy characterized by hypertension and proteinuria (1), is a threat to maternal and pediatric
health and affects 5-7% of all pregnant women worldwide every year
(2-4). In severe cases, PE can lead to
multiorgan failure and even mortality (5,6).
Low-dose aspirin treatment is reported to decrease the prevalence
of hypertension in pregnancy (7-9).
Moreover, trophoblast complement inhibitory therapy with heparin
reduced the incidence of primary outcomes (such as severe
preeclampsia, newborn weight ≤5th percentile or major abruptio
placentae) in women with adverse pregnancy outcomes in antecedent
pregnancy (10). As PE is
life-threatening and lacks effective treatment, the development of
specific therapies targeting the pathogenesis of PE is urgently
required.
Although the precise mechanisms underlying the
occurrence of PE remain unknown, the leading hypotheses suggest the
involvement of inadequate uterine trophoblast invasion and poor
remodeling of placental spiral arteries (11-13). Importantly, the widespread
endothelial dysfunction caused by impaired remodeling of spiral
arteries accelerates the onset of symptomatic PE via the release of
anti-angiogenic factors, such as soluble fms-like tyrosine kinase 1
(sFLT-1) and soluble endoglin (sENG) (14-16). Metformin is an oral hypoglycaemic
agent that is widely accepted for the treatment of gestational
diabetes. Recently, Brownfoot et al (17) revealed that metformin decreased
sFLT-1 and sENG secretion, as well as and improved endothelial
dysfunction in patients with PE. Hence, a medication that
alleviates endothelial dysfunction may demonstrate promise for the
prevention and treatment of PE.
Numerous peptides have been reported to be
biologically active and have been researched for therapeutic
applications in various diseases, including cancer, hypertension
and diabetes (18-21). Apelin has been observed to
function in regulating fluid balance, local regulation of blood
vessels and cardiac contractility. For instance, Ho et al
(22) revealed that, in rodents,
apelin prevented the development of systemic hypertension and
preserved the cellular architecture of the kidney that is often
impaired in PE. Currently, increasing numbers of peptides have been
approved for clinical research, such as liraglutide and
abaloparatide (23,24), but specific peptides available for
PE treatment are limited. Our previous study systemically evaluated
the peptide profile in the serum from patients with PE (25). However, additional efforts are
required to examine the peptides with unknown functions.
Based on previous findings, the present study aimed
to investigated the biological functions of a novel peptide derived
from the complement C4 A chain (PDCC4), which has a high stability
and a low molecular weight (25).
Subsequently, the potential mechanism underlying the protective
effects provided by PDCC4 on endothelial dysfunction were
examined.
Materials and methods
Peptide synthesis and administration
The amino acid sequence of PDCC4 is
NGFKSHALQLNNRQIR. PDCC4 and N-terminal fluorescein
isothiocyanate-labelled PDCC4 (FITC-PDCC4) were chemically
synthesized with >95% purity by Shanghai Science Peptide
Biological Technology Co., Ltd. The peptide was dissolved in
sterile water to a concentration of 10 mM for storage and diluted
to the indicated concentration before use.
Cell culture and treatment
Human umbilical vein endothelial cells (HUVECs) were
purchased from ScienCell Research Laboratories, Inc. HUVECs were
cultured using DMEM (ScienCell Research Laboratories, Inc.)
supplemented with 10% FBS (ScienCell Research Laboratories, Inc.)
in a 37°C humidified incubator with 5% CO2. Cells were
randomly divided into four groups for treatment: Control group,
PDCC4 group, TNF-α group and TNF-α + PDCC4 group. The TNF-α and
TNF-α + PDCC4 groups were administered 20 ng/ml TNF-α (23-25), while the control group received an
equal volume of sterile water. A total of 30 min before TNF-α
treatment, the PDCC4 and TNF-α + PDCC4 groups were administered 50
µM PDCC4, while the TNF-α group received an equal volume of
sterile water.
HUVECs were administrated with 50 µM
FITC-PDCC4 and incubated for 1 h at 37°C in the dark, then imaged
with a laser confocal fluorescence microscopy (Zeiss AG;
magnification, ×400) to observe the cell penetrating capacity of
PDCC4. To investigate the function of PDCC4, various concentrations
(10, 20, 50 and 100 µM) of PDCC4 were administrated to
HUVECs and then cells were cultured for 30 min at 37°C.
Subsequently, HUVECs were incubated with 20 ng/ml TNF-α
(Sigma-Aldrich; Merck KGaA) for 24 h at 37°C to induce endothelial
dysfunction, as previously described (26-28). Additionally, to investigate the
effect of inhibiting PI3K on function of PDCC4, HUVECs were
preconditioned with 2 µM LY294002 (Sigma-Aldrich; Merck
KGaA) for 1 h at 37°C before administration of PDCC4.
5-Ethynyl-2′-deoxyuridine (EdU)
assay
HUVECs were seeded in 24-well plates at a
concentration of 5,000 cells/well, and were then incubated for 12 h
at 37°C. After cells attached, HUVECs were treated as
aforementioned for 24 h. Then, the proliferation of the HUVECs was
measured with an EdU assay kit (Beyotime Institute of
Biotechnology) as previously described (29). EdU was added to the culture medium
at a concentration of 10 µM. After incubation at 37°C for 2
h, the cells were fixed with 4% paraformaldehyde for 30 min at room
temperature. Cells were washed twice with PBS and treated with 0.3%
Triton X-100 for 10 min at room temperature. Finally, Alexa Fluor
488 azide and DAPI were used successively to stain the cells for 30
and 10 min in the dark at room temperature. Images were acquired
using a fluorescence microscope (magnification, ×200) (30,31).
Wound healing assays
HUVECs were seeded in 6-well plates at a
concentration of 5×105 cells per well and cultured until
they reached confluency. After the cells were treated as
aforementioned, a scratch was made down the center of the layers of
cells in each well using a sterile 1,000 µl micropipette
tip. Cells were then washed with PBS twice and cultured in medium
without FBS. Images of three random visual fields of each group
were captured using a bright-field microscope (magnification, ×40)
at 0 and 24 h after scratching. The width of the healed wound was
quantified using ImageJ software (v1.52a; National Institutes of
Health), and the migratory rate was calculated (32,33).
Tube formation assay
A total of 100 µl liquefied Matrigel (Becton,
Dickinson and Company) was added to the 24-well plate. The plate
was incubated at 37°C until the Matrigel solidified. Subsequently,
200 µl HUVECs (1×105 cells/ml) that were treated
as aforementioned were seeded in a 24-well plate. The preliminary
experiment indicated that untreated HUVECs completely formed
tubular structures on the Matrigel after 6-8 h of incubation (data
not shown). Thus, the tube formation of the HUVECs was observed
using a bright-field microscope (magnification, ×200) at 6-8 h, as
also previously described (34-36). The branch number and length of the
tubes were quantified using ImageJ software (v1.52a; National
Institutes of Health).
Mitochondrial membrane potential
assay
HUVECs were seeded in 6-well plates at a
concentration of 5×105 cells per well for treatment as
aforementioned. Then, a
5,5′,6,6′,tetra-chloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine
iodide (JC-1) staining kit (Beyotime Institute of Biotechnology)
was used according to the manufacturer's instructions to detect the
mitochondrial membrane potential of the HUVECs (37,38). Briefly, the culture medium in each
well was replaced with JC-1 staining medium (5 µg/ml). After
a 20 min incubation in the dark at 37°C, HUVECs were washed twice
with PBS and a fluorescence microscope (magnification, ×400) was
used to capture images of the mitochondria.
Cell Counting Kit-8 (CCK-8) assay
A CCK-8 assay kit (cat. no. CK04-13; Dojindo
Molecular Technologies, Inc.) was applied to evaluate the effect of
different concentrations of PDCC4 on the proliferation of HUVECs
according to the manufacturer's instructions. The cells were seeded
in 96-well plates at a concentration of 3,000 cells/well. After the
cells were attached, they were pretreated with PDCC4 (10, 20, 50
and 100 µM) for 30 min at 37°C and then cotreated with 20
ng/ml TNF-α in a 37°C humidified incubator with 5% CO2.
At 0, 24, 48 and 72 h, 10 µl CCK-8 solution was added to the
medium at 37°C (39,40). The absorbance at 450 nm was
measured using a multifunctional microplate reader (Hybrid
Technology; BioTek Instruments, Inc.).
Animal protocol
All animal studies were approved by the Animal Care
and Ethical Committee of Nanjing Medical University. Animal
experiments were performed according to the Guide for the Care and
Use of Laboratory Animals (41).
A total of 72 Sprague-Dawley (SD) rats (48 female, 24 male; age, 8
weeks; weight, 280-320 g) were purchased from the Animal Center of
Nanjing Medical University. The rats were housed with 60% humidity
and a 12-h light/dark cycle at 26°C and free to standard rat chow
and water. Female rats were mated with males after 7 days of
acclimatization. Gestational day (GD) 0 was defined via a vaginal
smear analysis of sperm. On GD14, the female SD rats were randomly
divided into four groups (n=12 rats in each): Saline group, Saline
+ PDCC4 group, LPS group and LPS + PDCC4 group. On GD14, the rats
in the LPS group and LPS + PDCC4 group were intravenously injected
with LPS (Sigma-Aldrich; Merck KGaA; 1 µg/kg dissolved in 2
ml sterile saline) via an infusion pump (infusion rate, 2 ml/h) as
previously described (42), while
the rats in the Saline group and Saline + PDCC4 group were injected
with an equal volume of saline via an infusion pump (infusion rate,
2 ml/h). Rats in the Saline + PDCC4 group and LPS + PDCC4 group
were intravenously injected with PDCC4 (10 mg/kg) once per day on
GD16, 17 and 18, while the rats in the Saline group and LPS group
were injected with an equal volume of saline.
Measurement of blood pressure, urinary
protein and creatinine
On GD7, 11, 14, 16 and 18, a non-invasive
volume-pressure recording blood pressure monitoring system
(Visitech BP2000; VisiTech International Ltd.) was employed to
measure the systolic blood pressure (SBP) of the rats.
Beginning on GD19, 5 ml of 24-h urine was collected
from the rats using individual metabolic cages. Then, the
concentration of urinary protein was determined using the
pyrogallol red method as previously described (43), and the concentration of creatinine
was assayed using a commercial kit (cat. no. C011-2; Nanjing
Jiancheng Bioengineering Institute), according to the
manufacturer's instructions.
Pathological examination
On GD20, the rats were anaesthetized and sacrificed.
Then, the fetuses, placentas and kidneys were isolated. Living and
dead fetuses were counted, and the live fetuses were weighed. The
placentas and kidneys were fixed with 4% paraformaldehyde overnight
at room temperature, embedded in paraffin and sliced into
3-µm thick sections. Then, tissue sections were subjected to
hematoxylin and eosin (H&E) staining, which was performed as
previously described (44,45),
and sections were imaged using light microscopy (magnification,
×100).
Immunohistochemistry
The protein expression levels of proinflammatory and
anti-inflammatory factors in rat placentas were estimated via
immunohistochemistry analysis. Paraffin-embedded sections of
placentas were washed with xylene and ascending alcohol, for
air-drying and hydration, and were then incubated in Tris EDTA
buffer for 15 min at 95°C for antigen retrieval. The paraffin
sections were incubated at room temperature in 3% hydrogen peroxide
solution for 20 min to eliminate endogenous peroxidase activity.
The sections were incubated in PBS containing 3% bovine serum
albumin (Sigma-Aldrich; Merck KGaA) for 15 min at room temperature
to block non-specific binding. The sections were incubated
overnight in a 4°C humidified incubator with primary antibodies
against IL-4 (1:200; cat. no. bs-20686R; BIOSS) and IL-6 (1:200;
cat. no. bs-0782R; BIOSS). Then, tissue sections were washed three
times with PBS, and incubated with a secondary antibody (1:100;
cat. no. bs-0295G-Bio; BIOSS) and a tertiary antibody (1:500; cat.
no. bs-0437P-HRP; BIOSS) in a 37°C incubator for 30 min. Finally,
the labelled antibody was visualized with a 3,3′-diaminobenzidine
kit (46). The stained sections
were imaged using light microscopy (magnification, ×100), and
images were loaded to ImageJ software (v1.52a; National Institutes
of Health) to quantify the expression of proteins.
ELISA
A total of 500 µl blood was drawn via the
tail vein before sacrifice. The supernatant serum was collected
after the blood samples centrifuged at 1,500 × g for 15 min at 4°C.
The sFlt-1 and sEng levels were determined according to the
manufacture's instruction of the ELISA kits (cat. nos. CSB-E07350r
and CSB-EL007667RA; Cusabio Technology LLC). The concentrations
were measured according to the absorbance of the samples and
standards at the 450 nm wavelength (47).
RNA sequencing (seq)
HUVECs were lysed using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) and total RNA was
extracted using RNAprep pure Cell/Bacteria kit (cat. no. DP430;
Tiangen Biotech Co., Ltd.), and then the integrity of total RNA was
inspected via a Bioanalyzer 2100 (Agilent Technologies, Inc.).
Qualified total RNA was further purified using RNA Clean XP kit
(Beckman Coulter, Inc.) and RNase-Free DNase Set (Qiagen AB)
according to the manu-facturer's instructions. Ribosomal RNA (rRNA)
was removed from total RNA using the Ribo-Zero™ Gold kit
(Epicentre; Illumina, Inc.), and fragmented rRNA-depleted RNA was
converted to double-stranded cDNAs using ScriptSeq™ v2 RNA-Seq
Library Preparation kit (Epicentre; Illumina, Inc.) according to
the manufacturer's instructions. cDNAs were used for end repair, 3′
adenylation, adapter ligation and cDNA enrichment using a VAHTS
Stranded mRNA-seq Library Prep kit (Illumina, Inc.). The quality of
libraries and quantification were inspected using Bioanalyzer 2100
and qPCR with a Library Quantification kit (KAPA Biosystems; Roche
Diagnostics, Inc.). Then, 4 nM cDNA libraries were run on HiSeq.
2500 platform (Illumina, Inc.) in rapid run mode for 2X 100 bp
paired-end reads using a TruSeq PE Cluster kit and a TruSeq SBS kit
(cat. nos. PE-401-3001 and FC-401-3001; Illumina, Inc.) and data
were summarized, normalized and quality controlled using HiSeq
Control software (v 2.2.68; Illumina, Inc.).
To analyze the differential expression of genes, the
raw reads were normalized to Fragments per Kilobase of exon per
Million mapped reads. The t-test results and P-values were adjusted
for the False Discovery Rate (FDR) using the Benjamini-Hochberg
procedure, and a FDR-adjusted P-value<0.05 was considered the
threshold for statistical significance. Differentially expressed
genes were uploaded to Database for Annotation, Visualization and
Integrated Discovery (v6.8; https://david.ncifcrf.gov/tools.jsp) for Gene Ontology
(GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway
enrichment analysis.
Reverse transcription-quantitative PCR
(RT-qPCR)
Cells and placenta tissues of rats were lysed using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). Total RNA was extracted with a Total RNA Isolation kit
(Tiangen Biotech Co., Ltd.) and then subjected to reverse
transcription using a SuperScript IV kit (Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. cDNA was
synthesized at 50°C for 10 min and 80°C for 10 min. RT-qPCR was
performed using PowerUp™ SYBR™ Green Master mix (Thermo Fisher
Scientific, Inc.) and an ABI ViiA 7 System (Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. The
following thermocycling conditions were used: Initial denaturation
at 95°C for 120 sec, denaturing at 95°C for 15 sec and extension at
60°C for 60 sec for 40 cycles. The primer sequences are listed in
Table SI. Expression values of
targeted genes were normalized to the values of GAPDH using the
2−ΔΔCq method (48).
Western blotting
HUVECs were lysed with RIPA buffer (Beyotime
Institute of Biotechnology), and total protein was extracted. The
protein concentration was measured using the BCA method (Beyotime
Institute of Biotechnology). After processing with loading buffer,
proteins (30 µg) were subjected to electrophoresis with a
10% polyacrylamide gel. Subsequently, the proteins were transferred
to PVDF (EMD Millipore) membranes. After blocking with 5% fat-free
milk for 1 h at room temperature, the membranes were incubated with
primary antibodies at 4°C overnight. The primary antibodies used in
this study were phosphorylated (p)-PI3K (1:2,000; cat. no.
bs-3332R; BIOSS), PI3K (1:2,000; cat. no. 4249; Cell Signaling
Technology, Inc.), p-mTOR (1:2,000; Ser2481; cat. no. ab137133;
Abcam), mTOR (1:2,000; cat. no. 2983; Cell Signaling Technology,
Inc.), HIF1α (1:2,000; cat. no. 36169; Cell Signaling Technology,
Inc.) and β-actin (1:5,000; cat. no. 20536-1-AP; ProteinTech Group,
Inc.) (49). After washing three
times with TBST containing 0.1% Tween-20 (10 min every time) at
room temperature, the membranes were incubated with goat
anti-rabbit IgG horseradish peroxidase (1:2,000; cat. no. ab205718;
Abcam) for 1 h at room temperature. After washing again with TBST,
the immunoblots were visualized using chemiluminescence reagents
(EMD Millipore) and grey value of immunoblots were semi-quantified
using ImageJ software (v1.52a; National Institutes of Health).
Statistical analysis
GraphPad Prism 7 software (GraphPad Software, Inc.)
was used for statistical analysis and data plot-ting. Quantitative
data are presented as the mean ± SD. Data with two groups were
compared using the two-tailed Student's t-test, and multiple group
comparisons were determined via one-way ANOVA and Bonferroni's
multiple comparison test. χ2 tests were used to compare
survival rate of fetal rats. P<0.05 was considered to indicate a
statistically significant difference. All experiments were
performed for three independent times with six duplications each
time.
Results
PDCC4 at a concentration of 50 µM has a
significant effect on the expression of biomarkers of endothelial
dysfunction
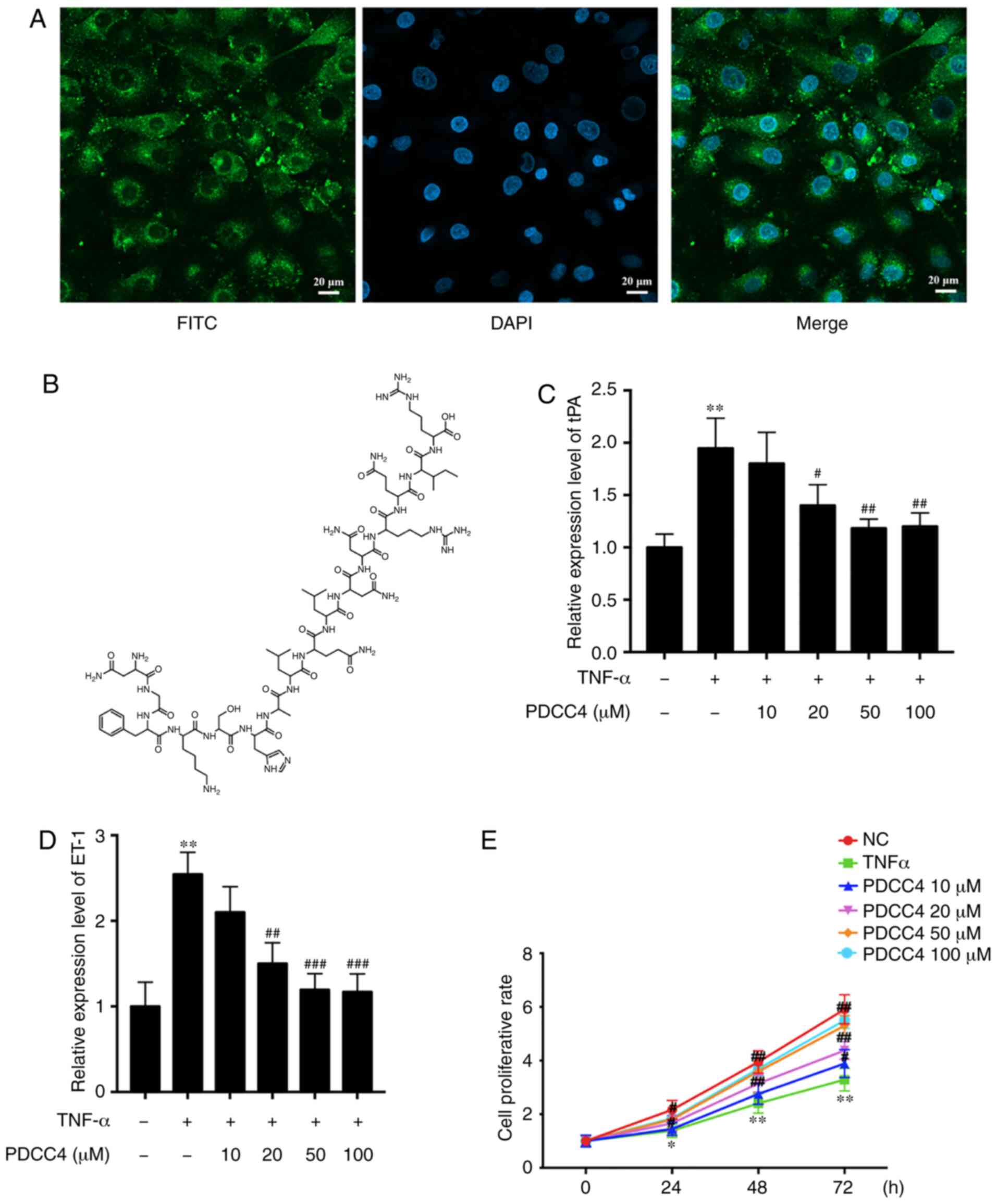
The ability of PDCC4 to penetrate cells was first
evaluated, before analysis of its function in HUVECs. To track the
delivery of the peptide, PDCC4 was conjugated to FITC. HUVECs were
treated with 50 µM FITC-labelled PDCC4 for 1 h. The
fluorescent images demonstrated that PDCC4 crossed the membrane,
and the majority was located in the cytoplasm, suggesting a
favorable cell-penetrating ability of PDCC4 (Fig. 1A). The chemical formula of PDCC4
is presented in Fig. 1B.
To investigate the biological function of PDCC4 in
HUVECs, biomarkers of endothelial dysfunction were detected via
RT-qPCR following treatment of cells with different concentrations
of PDCC4. A dose-dependent decline in the mRNA expression of
relevant markers, including tissue plasminogen activator (tPA) and
endothelin-1 (ET-1), was observed after PDCC4 treatment (Fig. 1C and D). Compared with the TNF-α
treatment alone group, the group treated with 50 µM PDCC4
had the most significant effect on the mRNA expression levels of
tPA and ET-1.
PDCC4 moderates the suppressed
proliferation of TNF-α-induced HUVECs
A CCK-8 assay was conducted to determine whether
PDCC4 (10, 20, 50 and 100 µM) reversed the decreased
proliferation capacity of HUVECs. The results demonstrated that
PDCC4 moderated proliferation inhibition induced by TNF-α in a
dose-dependent manner (Fig. 1E).
PDCC4 had the most significant effect on the proliferation capacity
of HUVECs at 50 µM. Therefore, a concentration of 50
µM was selected as the appropriate concentration for further
study. An EdU assay was performed to measure the effect of PDCC4 on
HUVEC proliferation. EdU was used to label newly synthesized DNA
with green fluorescence. The total DNA was determined via staining
with DAPI (blue fluorescence). Proliferation is indicated as the
green/blue ratio. It was found that the proliferation of HUVECs was
significantly inhibited in the TNF-α group, but that this
inhibition was reversed in the TNF-α + PDCC4 group (Fig. 2A).
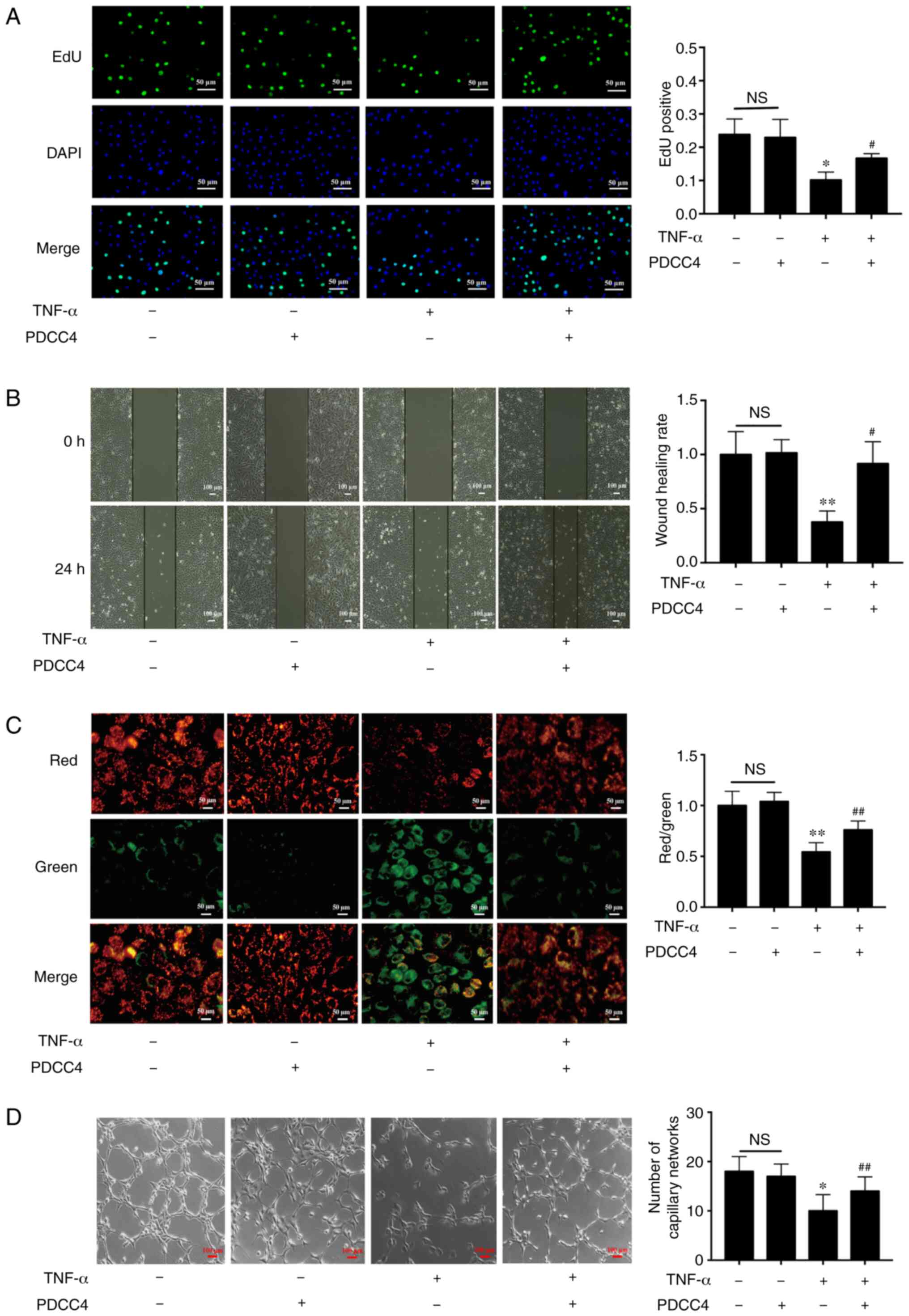 | Figure 2PDCC4 rescues the impaired
proliferation, migration, mitochondrial function and tube formation
of HUVECs treated with TNF-α. (A) Proliferative HUVECs were treated
with EdU, representative fluorescent images were captured and the
rate of EdU labelling was calculated. The proliferation of HUVECs
was inhibited by TNF-α, and this change was rescued by PDCC4 (n=3
per group). Scale bar, 50 µm. (B) Representative images of
wound healing were captured at 0 and 24 h after scratching the
cells, and the wound healing rate was calculated. PDCC4 prevented
the migration of HUVECs from inhibition by TNF-α (n=3 per group).
Scale bar, 100 µm. (C) Representative fluorescent images of
HUVECs after staining with JC-1; the mitochondrial membrane
potential is presented as the ratio of red to green fluorescent
density. PDCC4 increased the mitochondrial membrane potential of
HUVECs exposed to TNF-α (n=3 per group). Scale bar, 50 µm.
(D) Representative images of the tube formation assays indicated
that PDCC4 preserved the HUVEC tube formation capacity, which was
impaired by TNF-α (n=3 per group). Scale bar, 100 µm.
*P<0.05 and **P<0.01 vs. negative
control group; #P<0.05 and ##P<0.01 vs.
TNF-α (+) group. HUVEC, human umbilical vein endothelial cells;
PDCC4, peptide derived from complement C4 A chain; EdU,
5-ethynyl-2′-deoxyuridine; NS, not significant. |
PDCC4 alleviates the damage to HUVEC
migration and tube formation
The results suggested that the migratory capacity of
TNF-α-induced HUVECs was suppressed compared with that of the
control group. After treatment with PDCC4, the impaired migratory
function was significantly reversed (Fig. 2B). Representative images indicated
that the number of branch points of the tube-like structures of the
TNF-α + PDCC4 group was greater compared with that of the TNF-α
group, suggesting that tube formation was enhanced by PDCC4
treatment (Fig. 2D).
PDCC4 relieves the mitochondrial
depolarization induced by TNF-α
By examining mitochondrial depolarization, the
protective effect of PDCC4 on TNF-α-induced cell death in HUVECs
were further determined (Fig.
2C). Control HUVECs stained with JC-1 emitted mitochondrial
orange-red fluorescence and exhibited little green fluorescence.
The fluorescence images in the TNF-α group demonstrate obvious
green fluorescence and little orange-red fluorescence. Compared
with the TNF-α group, the TNF-α + PDCC4 group had decreased
mitochondrial depolarization and notable orange-red
fluorescence.
PI3K/mTOR/HIF1α pathway may be involved
in the PDCC4-mediated rescue of endothelial dysfunction
To determine the underlying mechanism of PDCC4
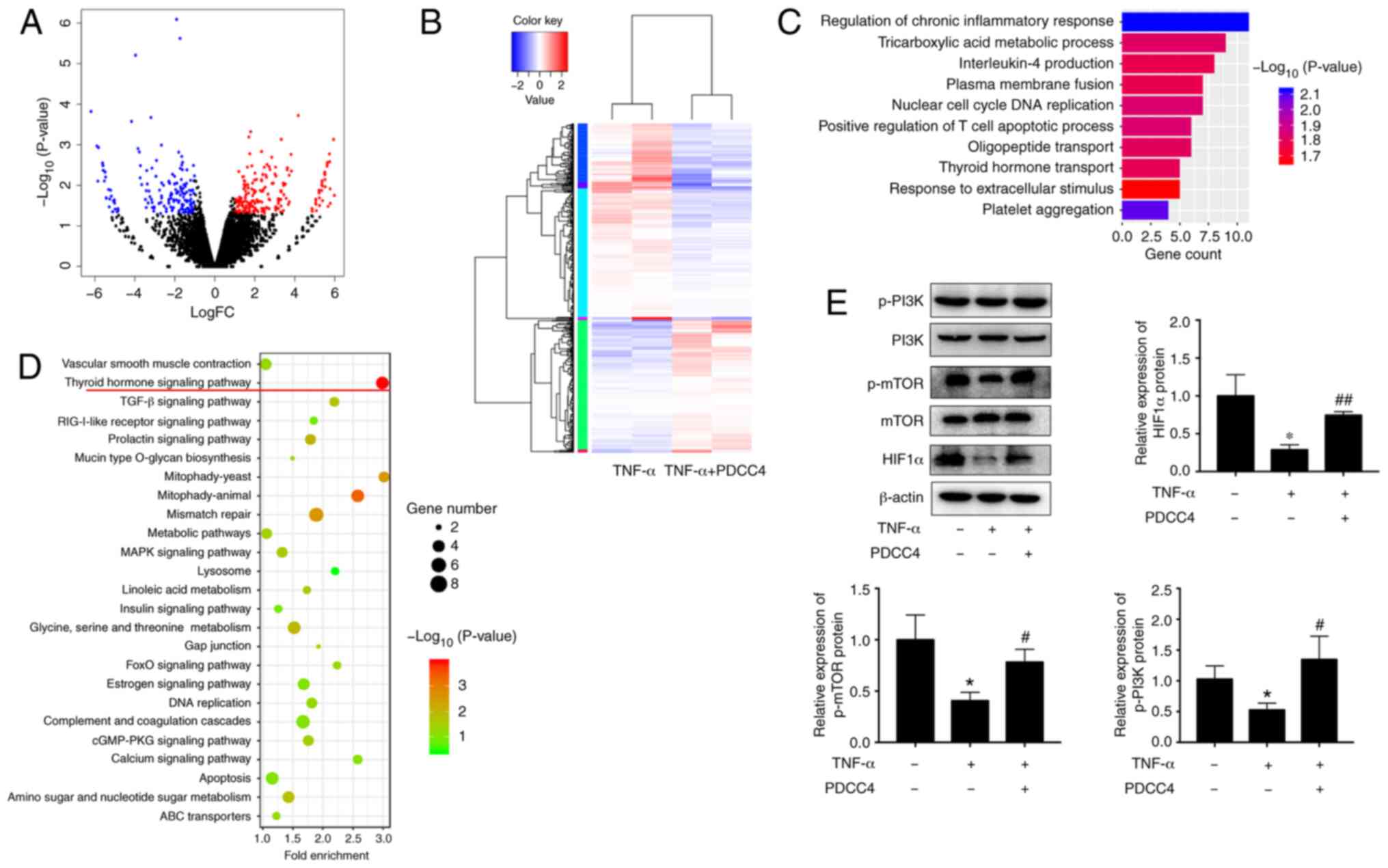
function, the transcriptome after PDCC4 treatment was examine via
RNA-seq. A total of 442 genes, including 178 downregulated genes
and 264 upregulated genes, had significant differences when
comparing the group treated with PDCC4 and the control group
(Fig. 3A and B). GO analysis
identified enrichment in important biological processes, which
included the 'regulation of chronic inflammatory response' and
'thyroid hormone transport', indicating that the relevant genes may
be involved in these processes (Fig.
3C). KEGG pathway analysis was also applied to analyze the
dysregulated genes involved in various signaling pathways, and it
was observed that the 'thyroid hormone signaling pathway' had the
highest enrichment (Fig. 3D).
Previous studies have revealed that key proteins of
the thyroid hormone signaling pathway, including PI3K, mTOR and
HIF1α, can be involved in the pathological process of PE
development (50,51). Therefore, it was hypothesized that
PDCC4 may function via the PI3K/mTOR/HIF1α signaling pathway.
Western blot analyses were conducted to assess the key proteins
involved in these pathways. After treatment with PDCC4, increase in
protein expression levels were observed for p-PI3K, mTOR and total
HIF1α (Fig. 3E). These results
suggested that the activation of the PI3K/mTOR/HIF1α signaling
pathway may be associated with the functions of PDCC4.
Relationship between the PI3K/mTOR/HIF1α
pathway and the role of PDCC4 in relieving endothelial
dysfunction
To further determine the relationship between the
PI3K/mTOR/HIF1α pathway and PDCC4 functions, the highly selective
PI3K inhibitor LY294002 was used to treat HUVECs. The results
demonstrated that administration of 2 µM LY294002 inhibited
the proliferation and migration promoted by PDCC4 (Fig. 4A and B). Moreover, LY294002
dampened the reversal effect of PDCC4 on impaired mitochondrial
membrane potential induced by TNF-α (Fig. 4C). However, treatment of LY294002
alone had no effect on mitochondrial membrane potential of HUVECs
(data not shown). The number of branch points of tube-like
structures was also diminished following treatment with the
inhibitor LY294002 in comparison with the TNF-α + PDCC4 group
(Fig. 4D).
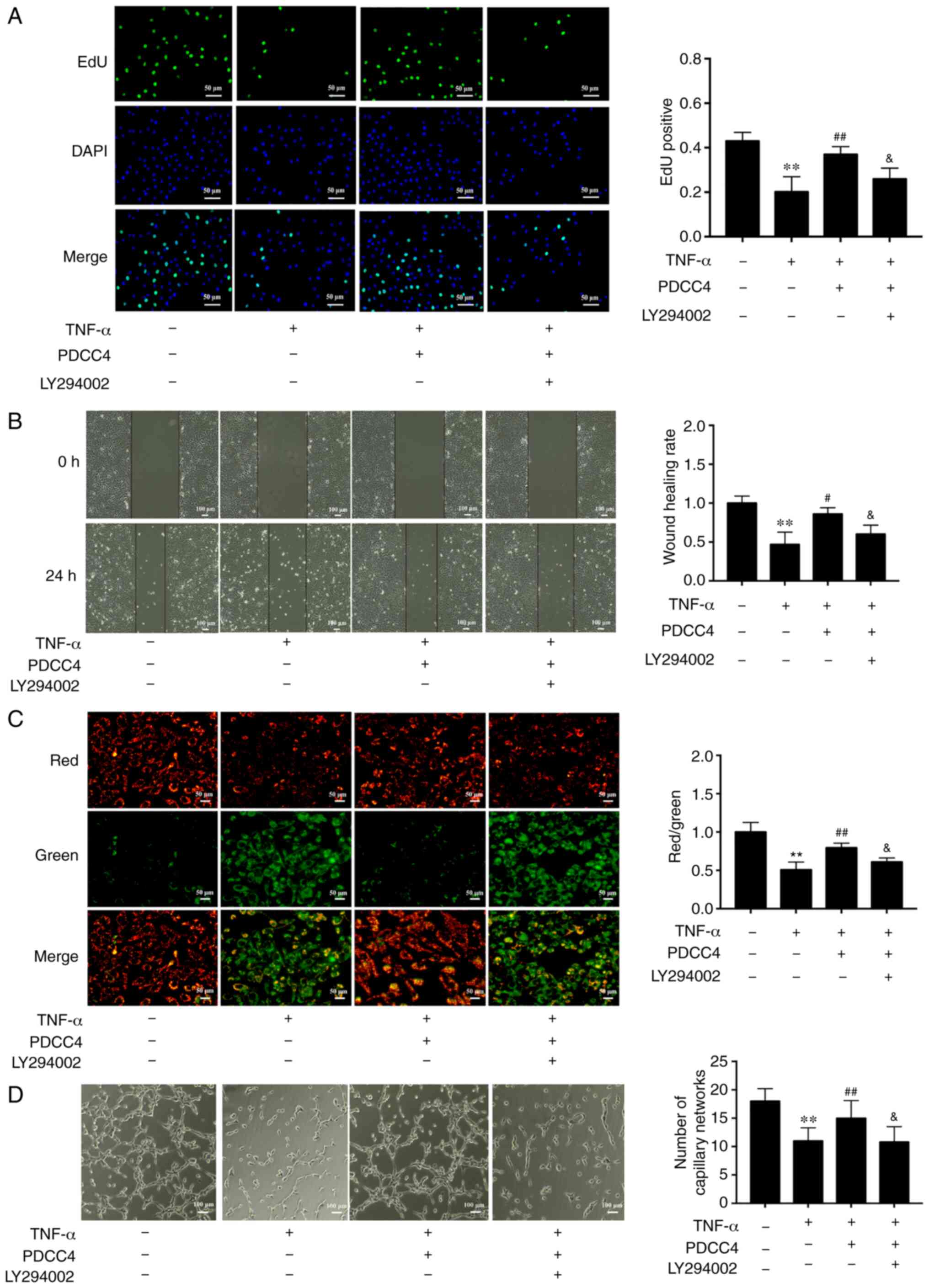 | Figure 4PDCC4 attenuates the endothelial
dysfunction induced by TNF-α via the PI3K/mTOR/HIF1α pathway. (A)
Proliferative HUVECs were treated with EdU, representative
fluorescent images were captured, and the rate of EdU labelling
calculated. PDCC4 attenuated the antiproliferative effect of TNF-α,
which was inhibited by LY294002 (n=3 per group). Scale bar, 50
µm. (B) Representative images of wound healing were captured
at 0 and 24 h after scratching, and the wound healing rate was
calculated. PDCC4 attenuated the antimigration effect of TNF-α,
which was inhibited by LY294002 (n=3 per group). Scale bar, 100
µm. (C) Representative fluorescent images of HUVECs after
staining with JC-1 are illustrated, and the mitochondrial membrane
potential is presented as the ratio of red to green fluorescent
density. LY294002 inhibited PDCC4′s ability to increase the
mitochondrial membrane potential of HUVECs exposed to TNF-α (n=3
per group). Scale bar, 50 µm. (D) Representative images of
the tube formation assay indicated that PDCC4 prevented HUVEC tube
formation from being impaired by TNF-α, which was inhibited by
LY294002 (n=3 per group). Scale bar, 100 µm.
**P<0.01 vs. negative control group;
#P<0.05 and ##P<0.01 vs. TNF-α (+)
group; &P<0.05 vs. TNF-α (+) + PDCC4 group.
HUVEC, human umbilical vein endothelial cells; PDCC4, peptide
derived from complement C4 A chain; EdU,
5-ethynyl-2′-deoxyuridine. |
PDCC4 moderates the pathological process
of LPS-induced PE in a rat model
To evaluate the effects of PDCC4 on LPS-stimulated
rat, a three treatment protocol (5, 10 and 20 mg/kg) was
established in the preliminary experiment. It was found that 5
mg/kg had no effect on SBP and the urinary protein of rat, while 10
and 20 mg/kg significantly attenuated LPS induced hypertension and
proteinuria. However, no obvious difference between the effect of
10 and 20 mg/kg was observed (data not shown). Thus, 10 mg/kg PDCC4
was used in the following experiments. There were significant
differences in the SBP values among the four groups on GD16 after
treatment with LPS. The high SBP values in the LPS group persisted
after treatment with only LPS during pregnancy. Treatment with 10
mg/kg PDCC4 significantly alleviated the SBP values compared with
the LPS group (Fig. 5A). The
ratio of urinary protein and creatinine in the LPS group was
increased, while PDCC4 treatment decreased the urinary protein
level (Fig. 5B). There were more
live fetuses in the LPS + PDCC4 group compared with in the LPS
group (Fig. 5C). In addition, the
LPS + PDCC4 group had an increase in the birth weight of survivors
compared with the LPS group (Fig.
5D). The numbers of live fetuses and resorptions were
significantly different among the groups (Fig. 5E). Stillbirth was induced by LPS,
resulting in an increased fetal mortality rate, and the LPS + PDCC4
group had a decreased rate of stillbirth (Fig. 5E).
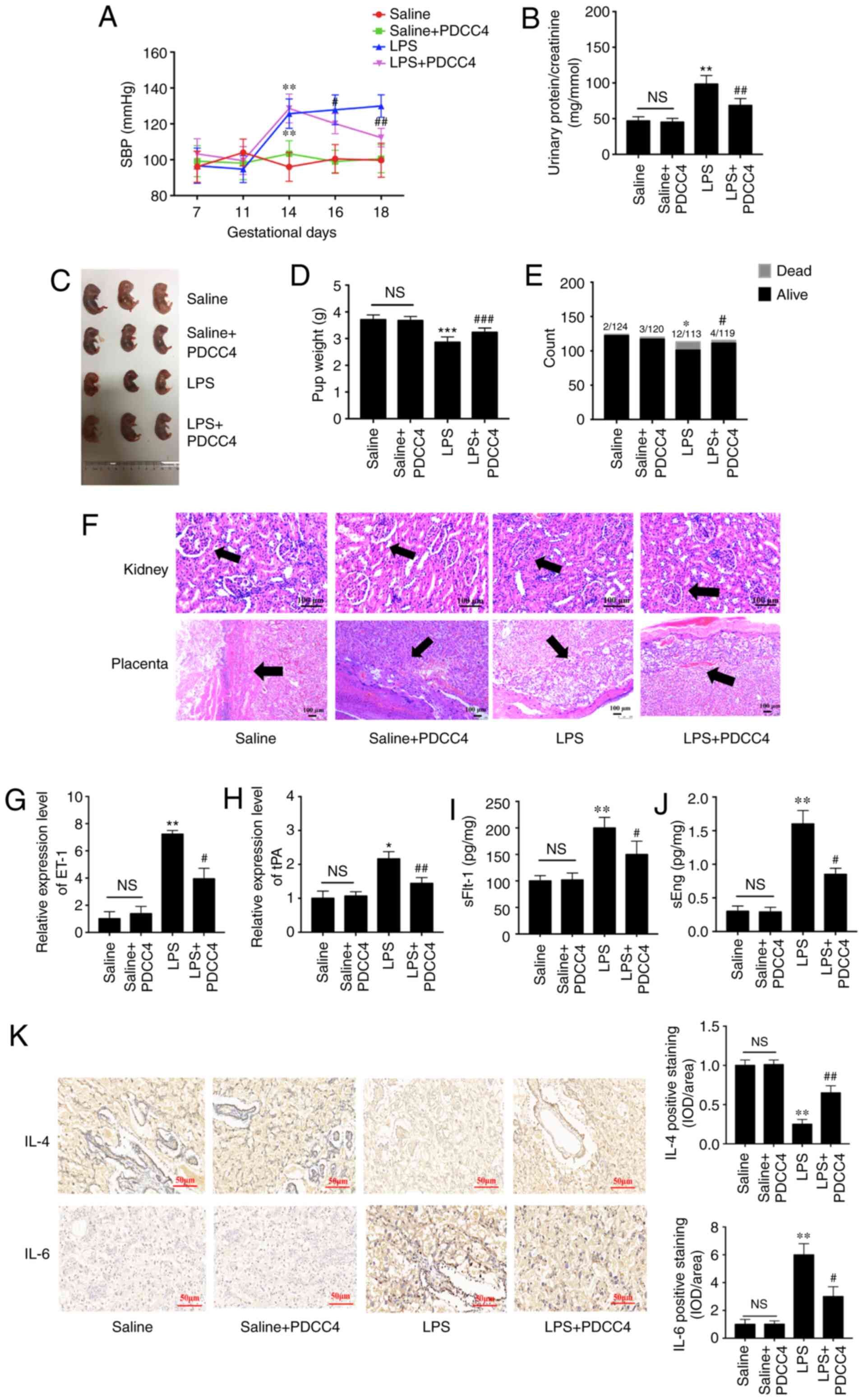 | Figure 5PDCC4 relieves the pathological
manifestations of preeclampsia in a rat model. (A) SBP of the four
groups was evaluated on GD7, 11, 14, 16 and 18. The SBP of the rats
increased significantly after treatment with LPS, and this change
was rescued by PDCC4 (n=18 per group). (B) Ratio of urinary protein
to creatinine in the four groups was analyzed (n=12 per group). (C)
Representative pictures of fetal appearance were captured for the
four groups (n=12 per group). (D) Fetal weight and the (E) numbers
of live and dead fetuses in each group were determined (n=12 per
group). (F) Pathological alterations of the kidneys and placentas
were observed after hematoxylin and eosin staining. Black arrows
indicated glomerulus in kidney (upper) and basal zone in placenta
(lower). The PDCC4-treated group exhibited mild inflammation
compared with the LPS-treated group in a microscopic analysis of
morphology. Scale bar, 100 µm. PDCC4 decreased the
expression levels of markers of endothelial dysfunction, (G) ET-1
and (H) tPA, in the rat placentas (n=12 per group). Increasing
serum levels of (I) sFlt-1 and (J) sEng in each group were declined
by PDCC4 (n=12 per group). (K) Immunohistochemical staining
demonstrated that PDCC4 decreased proinflammatory cytokines (IL-6)
and increased anti-inflammatory cytokines (IL-4) in PE rats (n=12
per group). Scale bar, 50 µm. *P<0.05,
**P<0.01 and ***P<0.001 vs. negative
control group; #P<0.05, ##P<0.01 and
###P<0.001 vs. PE group. SBP, systolic blood
pressure; GD, gestational day; PDCC4, peptide derived from
complement C4 A chain; tPA, tissue plasminogen activator; ET-1,
endothelin; sFlt-1, soluble fms-like tyrosine kinase 1; sEng,
soluble endoglin; NS, not significant; LPS, lipopolysaccharide; OD,
optical density. |
Changes in kidney structure and function in the
pregnant rats injected with LPS exhibited an increased glomerular
area, capillary structure disorder and abnormal protein production.
After treatment with PDCC4, the renal histology of pregnant rats
was significantly relieved compared with that of the LPS group
(Fig. 5F). For instance, the
placentas of the LPS-treated rats mostly displayed severe
inflammation, whereas those of the LPS + PDCC4 group generally
exhibited less severe inflammation (Fig. 5F).
The expression levels of markers of endothelial
dysfunction, including tPA and ET-1, in the placentas were
detected. The results demonstrated that LPS increased the
expression levels of these marker, while PDCC4 decreased the mRNA
expression levels (Fig. 5G and
H). ELISA was conducted to investigate the effect of different
treatments on circulating angiogenic factors, such as sFlt-1 and
sEng. The LPS group had a significant increase in the sFlt-1 and
sEng levels compared with the Saline group, and this increase was
attenuated by PDCC4 (Fig. 5I and
J). The expression of the proinflammatory cytokine IL-6 in the
placentas was increased in the LPS-treated group compared with the
Saline group. By constant, this expression level was significantly
decreased by PDCC4 (Fig. 5K). The
anti-inflammatory cytokine IL-4 was significantly downregulated in
the LPS group but upregulated in the LPS + PDCC4 group (Fig. 5K).
Discussion
PE is unique to human pregnancy, and the most
effective treatment for this complication is delivery (52). It is widely accepted that
disturbances in placental blood vessel function and in placental
development contribute to the pathophysiology of PE (53-55). However, the etiology of PE is yet
to be fully elucidated. In the present study, PDCC4 was identified
as a novel peptide that moderated endothelial dysfunction by
regulating the PI3K/mTOR/HIF1α pathway. The present findings
indicated that PDCC4 was a promising candidate for PE
treatment.
Various peptides have been researched for potential
applications in the diagnosis and treatment of PE. For example,
Szabo et al (56) reported
that increased B-type natriuretic peptide (BNP) levels serve as
biomarkers for severe PE. Additionally, BNP levels in patients with
early-onset PE are significantly higher compared with those in
patients late-onset PE (57).
VG1177, a competitive antagonist peptide for MHC class II invariant
chain peptide, could also both prevent and treat PE-like features
in mice (58).
It has been reported that immune system activation
serves a major role in the pathophysiology of PE, characterized by
imbalanced proinflammatory and anti-inflammatory cytokines
(49). In patients with PE,
circulating proinflammatory cytokines, such as TNF-α, are increased
in maternal and cord blood, resulting in endothelial dysfunction
via diverse mechanisms, such as oxidative stress (59). Other proinflammatory cytokines,
such as IL-6, IL-8 and monocyte chemotactic protein 1, are secreted
by endothelial cells in response to TNF-α (60). Additionally, there is an increase
in the expression levels of tPA and ET-1, two biomarkers of
endothelial dysfunction (61,62). In the present study, treatment
with PDCC4 resulted in a decline in the expression of IL-6 in
placental tissue. The peptide PDCC4 also attenuated endothelial
dysfunction, as demonstrated by a decrease in the expression levels
of tPA and ET-1 both in vitro and in vivo. Several
clinical studies have revealed that serum and placental
anti-inflammatory cytokines, IL-4 and IL-10, are decreased in
patients with PE. Consistently, the present results indicated that
the expression of IL-4 in the placentas of PE-like rats was
decreased, but it was upregulated in the PDCC4-treated group.
Therefore, PDCC4 may participate in the regulation of immune
responses and moderate endothelial dysfunction.
PE is a complicated pathological process involving
various molecules and signaling pathways. For instance, PI3Ks
modulate cell metabolism, proliferation and survival by
phosphorylating the 3-OH group of inositol membrane lipids to
activate or deactivate intracellular proteins (63,64). mTOR, one of the conserved PI3Ks,
integrates various biochemical and growth factor signals, such as
glucose, insulin, amino acids and ATP (65). Previous studies have revealed a
decline in the phosphorylation of PI3K and mTOR in response to
oxidative stress in PE (47,50,66). Consistently, the present findings
indicated that the expression levels of p-PI3K and p-mTOR were
downregulated in response to endothelial dysfunction induced by
TNF-α, which was attenuated by PDCC4. The expression of HIF1α is
shown to be upregulated in response to activation of the PI3K/mTOR
signaling pathway, which contributes to neovascularization in
benign and malignant diseases (51,67,68). In the present study, the
expression of HIF1α was downregulated in response to TNF-α
treatment and was partially rescued by PDCC4 in vivo or
in vitro, which suggested that neovascularization promotes
PDCC4. Additionally, inhibition of PI3K decreased the
proliferation, migration and tube formation of HUVECs. It was also
identified that the mitochondrial potential of HUVECs increased
after PI3K inhibition. Therefore, the current results suggested
that PDCC4 relieved endothelial dysfunction by influencing the
PI3K/mTOR/HIF1α signaling pathway.
To investigate the anti-PE effect of PDCC4 in
vivo, SD rats were injected with LPS on GD14 to establish a
model of PE. To the best of our knowledge, PDCC4 is a novel peptide
discovered for the first time by our previous study, and its
pharmacokinetic parameters mostly remain unknown. PDCC4 treatment
decreased SBP and urinary protein levels in rats with PE, which was
similar to the major clinical characteristics observed in patients
with PE (4). As later clinical
manifestations include poor placental vessel remodeling and
placental ischemia, there is an increased prevalence of fetal
growth restriction and low birth weight (69). In the present study, the low fetal
weight and fetal length in rats with PE were attenuated by PDCC4
treatment. Aberrant immune responses during pregnancy are
considered a major pathogenic characteristic of PE (70). In the current study, not only were
the relative expression levels of inflammatory cytokines and
chemokines elevated, as described previously (71), but the placenta and kidney tissues
from the rats exhibited inflammatory cell infiltration and tissue
necrosis, which were relieved by PDCC4 treatment.
In the current research, the anti-PE effect of PDCC4
was verified both in vitro and in vivo, and an
underlying mechanism was proposed. However, due to the
unavailability of liquid chromatography and mass spectrum
equipment, it was not possible to detect the exact amounts of PDCC4
in rats. Moreover, short duration between first PDCC4 intervention
and SBP detection may partially interfere the evaluation of the
effects of PDCC on LPS induced hypertension. In a future
investigation, the effects of PDCC on SBP will be investigated for
a longer duration. Additionally, the direct target of PDCC4 remains
unknown and requires further research.
In conclusion, the present study provides insight
into a newly identified peptide that moderates vascular endothelial
cell injury by activating the PI3K/mTOR/HIF1α signaling pathway,
which is promising for PE treatment.
Supplementary Data
Funding
This study was supported by grants from the National
Natural Science Foundation of China (grant nos. 81771604, 81571444
and 81971393), the Nanjing Science and Technology Development
Project (grant no. 201715052) and The Project of Invigorating
Health Care through Science, Technology and Education-Jiangsu
Provincial Medical Youth Talent (grant no. QNRC2016112).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LX, RJ and HD designed the present study, which was
performed by LX, KX, LW and XY. LX, KX, LW and XY made substantial
contributions to acquisition and analysis of data. WL and CL also
made contributions to interpretation of data. LX wrote the initial
draft of the manuscript. KX revised it critically for important
intellectual content. RJ and HD gave final approval of the version
to be published. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal studies were approved by the Animal Care
and Ethical Committee of Nanjing Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Mol BWJ, Roberts CT, Thangaratinam S,
Magee LA, de Groot CJM and Hofmeyr GJ: Preeclampsia. Lancet.
387:999–1011. 2016. View Article : Google Scholar
|
|
2
|
Khan KS, Wojdyla D, Say L, Gulmezoglu AM
and Van Look PF: WHO analysis of causes of maternal death: A
systematic review. Lancet. 367:1066–1074. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Duley L: The global impact of preeclampsia
and eclampsia. Semin Perinatol. 33:130–137. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rana S, Lemoine E, Granger JP and
Karumanchi SA: Preeclampsia: Pathophysiology, challenges, and
perspectives. Circ Res. 124:1094–1112. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wirka RC and Quertermous T: Circulating
peptide prevents preeclampsia. Science. 357:643–644. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ghulmiyyah L and Sibai B: Maternal
mortality from preeclampsia/eclampsia. Semin Perinatol. 36:56–59.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rolnik DL, Wright D, Poon LC, O'Gorman N,
Syngelaki A, de Paco Matallana C, Akolekar R, Cicero S, Janga D,
Singh M, et al: Aspirin versus placebo in pregnancies at high risk
for preterm preeclampsia. N Engl J Med. 377:613–622. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
LeFevre ML; U.S. Preventive Services Task
Force: Low-dose aspirin use for the prevention of morbidity and
mortality from preeclampsia: U.S. Preventive Services Task Force
recommendation statement. Ann Intern Med. 161:819–826. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Duley L, Meher S and Abalos E: Management
of preeclampsia. BMJ. 332:463–468. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hossain N, Schatz F and Paidas MJ: Heparin
and maternal fetal interface: Why should it work to prevent
pregnancy complications? Thromb Res. 124:653–655. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lowe SA, Brown MA, Dekker GA, Gatt S,
McLintock CK, McMahon LP, Mangos G, Moore MP, Muller P, Paech M, et
al: Guidelines for the management of hypertensive disorders of
pregnancy 2008. Aust N Z J Obstet Gynaecol. 49:242–246. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pijnenborg R, Vercruysse L and Hanssens M:
The uterine spiral arteries in human pregnancy: Facts and
controversies. Placenta. 27:939–958. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jeyabalan A: Epidemiology of preeclampsia:
Impact of obesity. Nutr Rev. 71(Suppl 1): S18–S25. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Powe CE, Levine RJ and Karumanchi SA:
Preeclampsia, a disease of the maternal endothelium: The role of
antiangiogenic factors and implications for later cardiovascular
disease. Circulation. 123:2856–2869. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Young BC, Levine RJ and Karumanchi SA:
Pathogenesis of preeclampsia. Annu Rev Pathol. 5:173–192. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Roberts JM, Taylor RN, Musci TJ, Rodgers
GM, Hubel CA and McLaughlin MK: Preeclampsia: An endothelial cell
disorder. Am J Obstet Gynecol. 161:1200–1204. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brownfoot FC, Hastie R, Hannan NJ, Cannon
P, Tuohey L, Parry LJ, Senadheera S, Illanes SE, Kaitu'u-Lino TJ
and Tong S: Metformin as a prevention and treatment for
preeclampsia: Effects on soluble fms-like tyrosine kinase 1 and
soluble endoglin secretion and endothelial dysfunction. Am J Obstet
Gynecol. 214:356.e1–356.e15. 2016. View Article : Google Scholar
|
|
18
|
Papo N and Shai Y: Host defense peptides
as new weapons in cancer treatment. Cell Mol Life Sci. 62:784–790.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ellerby HM, Arap W, Ellerby LM, Kain R,
Andrusiak R, Rio GD, Krajewski S, Lombardo CR, Rao R, Ruoslahti E,
et al: Anti-cancer activity of targeted pro-apoptotic peptides. Nat
Med. 5:1032–1038. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim SJ, Xiao J, Wan J, Cohen P and Yen K:
Mitochondrially derived peptides as novel regulators of metabolism.
J Physiol. 595:6613–6621. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Petkov V, Mosgoeller W, Ziesche R, Raderer
M, Stiebellehner L, Vonbank K, Funk GC, Hamilton G, Novotny C,
Burian B and Block LH: Vasoactive intestinal peptide as a new drug
for treatment of primary pulmonary hypertension. J Clin Invest.
111:1339–1346. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ho L, van Dijk M, Chye STJ, Messerschmidt
DM, Chng SC, Ong S, Yi LK, Boussata S, Goh GH, Afink GB, et al:
ELABELA deficiency promotes preeclampsia and cardiovascular
malformations in mice. Science. 357:707–713. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jacobsen LV, Flint A, Olsen AK and
Ingwersen SH: Liraglutide in type 2 diabetes mellitus: Clinical
pharmacokinetics and pharmacodynamics. Clin Pharmacokinet.
55:657–672. 2016. View Article : Google Scholar :
|
|
24
|
Sleeman A and Clements JN: Abaloparatide:
A new pharmacological option for osteoporosis. Am J Health Syst
Pharm. 76:130–135. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dai X, Song X, Rui C, Meng L, Xue X, Ding
H, Shen R, Li J, Li J, Lu Y and Long W: Peptidome analysis of human
serum from normal and preeclamptic pregnancies. J Cell Biochem.
118:4341–4348. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen TG, Zhong ZY, Sun GF, Zhou YX and
Zhao Y: Effects of tumour necrosis factor-alpha on activity and
nitric oxide synthase of endothelial progenitor cells from
peripheral blood. Cell Prolif. 44:352–359. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gerhardt S, König V, Doll M,
Hailemariam-Jahn T, Hrgovic I, Zöller N, Kaufmann R, Kippenberger S
and Meissner M: Dimethylfumarate protects against TNF-α-induced
secretion of inflammatory cytokines in human endothelial cells. J
Inflamm (Lond). 12:492015. View Article : Google Scholar
|
|
28
|
Fratantonio D, Speciale A, Molonia MS,
Bashllari R, Palumbo M, Saija A, Cimino F, Monastra G and Virgili
F: Alpha-lipoic acid, but not di-hydrolipoic acid, activates Nrf2
response in primary human umbilical-vein endothelial cells and
protects against TNF-α induced endothelium dysfunction. Arch
Biochem Biophys. 655:18–25. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zeng RJ, Zheng CW, Gu JE, Zhang HX, Xie L,
Xu LY and Li EM: RAC1 inhibition reverses cisplatin resistance in
esophageal squamous cell carcinoma and induces downregulation of
glycolytic enzymes. Mol Oncol. 13:2010–2030. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cai S, Zhu Q, Guo C, Yuan R, Zhang X, Nie
Y, Chen L, Fang Y, Chen K, Zhang J, et al: MLL1 promotes myogenesis
by epigenetically regulating Myf5. Cell Prolif. 53:e127442020.
View Article : Google Scholar :
|
|
31
|
Sun Q, Li Q and Xie F: LncRNA-MALAT1
regulates proliferation and apoptosis of ovarian cancer cells by
targeting miR-503-5p. Onco Targets Ther. 12:6297–6307. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lv Y, Lu X, Li C, Fan Y, Ji X, Long W,
Meng L, Wu L, Wang L, Lv M and Ding H: miR-145-5p promotes
trophoblast cell growth and invasion by targeting FLT1. Life Sci.
239:1170082019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lip SV, Boekschoten MV, Hooiveld GJ, van
Pampus MG, Scherjon SA, Plösch T and Faas MM: Early-onset
preeclampsia, plasma microRNAs, and endothelial cell function. Am J
Obstet Gynecol. 222:497.e1–497.e12. 2020. View Article : Google Scholar
|
|
34
|
Liu L, Bi N, Wu L, Ding X, Men Y, Zhou W,
Li L, Zhang W, Shi S, Song Y and Wang L: MicroRNA-29c functions as
a tumor suppressor by targeting VEGFA in lung adenocarcinoma. Mol
Cancer. 16:502017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang C, Tao W, Wang Y, Bikow J, Lu B,
Keating A, Verma S, Parker TG, Han R and Wen XY: Rosuvastatin,
identified from a zebrafish chemical genetic screen for
antiangiogenic compounds, suppresses the growth of prostate cancer.
Eur Urol. 58:418–426. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen J, Zhang W, Xu Q, Zhang J, Chen W, Xu
Z, Li C, Wang Z, Zhang Y, Zhen Y, et al: Ang-(17) protects HUVECs
from high glucose-induced injury and inflammation via inhibition of
the JAK2/STAT3 pathway. Int J Mol Med. 41:2865–2878.
2018.PubMed/NCBI
|
|
37
|
Jin J, Qiu S, Wang P, Liang X, Huang F, Wu
H, Zhang B, Zhang W, Tian X, Xu R, et al: Cardamonin inhibits
breast cancer growth by repressing HIF-1α-dependent metabolic
reprogramming. J Exp Clin Cancer Res. 38:3772019. View Article : Google Scholar
|
|
38
|
Mi L, Zhang Y, Xu Y, Zheng X, Zhang X,
Wang Z, Xue M and Jin X: HMGB1/RAGE pro-inflammatory axis promotes
vascular endothelial cell apoptosis in limb ischemia/reperfusion
injury. Biomed Pharmacother. 116:1090052019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xiang Y, Yao X, Wang X, Zhao H, Zou H,
Wang L and Zhang QX: Houshiheisan promotes angiogenesis via
HIF-1α/VEGF and SDF-1/CXCR4 pathways: In vivo and in vitro. Biosci
Rep. 39:BSR201910062019. View Article : Google Scholar
|
|
40
|
Liu Y, Yang J, Bao J, Li X, Ye A, Zhang G
and Liu H: Activation of the cholinergic anti-inflammatory pathway
by nicotine ameliorates lipopolysaccharide-induced
preeclampsia-like symptoms in pregnant rats. Placenta. 49:23–32.
2017. View Article : Google Scholar
|
|
41
|
National Research Council Committee for
the Update of the Guide for the Care and use of Laboratory Animals:
The National Academies Collection: Reports funded by National
Institutes of Health. Guide for the Care and Use of Laboratory
Animals. 8th edition. National Academies Press; Washington, DC:
2011
|
|
42
|
Shu W, Li H, Gong H, Zhang M, Niu X, Ma Y,
Zhang X, Cai W, Yang G, Wei M, et al: Evaluation of blood vessel
injury, oxidative stress and circulating inflammatory factors in an
L-NAME-induced preeclampsia-like rat model. Exp Ther Med.
16:585–594. 2018.PubMed/NCBI
|
|
43
|
Yalamati P, Bhongir AV, Karra M and Beedu
SR: comparative analysis of urinary total proteins by bicinchoninic
acid and pyrogallol red molybdate methods. J Clin Diagn Res.
9:Bc01–04. 2015.PubMed/NCBI
|
|
44
|
Lai WS and Ding YL: GNG7 silencing
promotes the proliferation and differentiation of placental
cytotrophoblasts in preeclampsia rats through activation of the
mTOR signaling pathway. Int J Mol Med. 43:1939–1950.
2019.PubMed/NCBI
|
|
45
|
Feldman AT and Wolfe D: Tissue processing
and hematoxylin and eosin staining. Methods Mol Biol. 1180:31–43.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Matsumoto K, Arao T, Tanaka K, Kaneda H,
Kudo K, Fujita Y, Tamura D, Aomatsu K, Tamura T, Yamada Y, et al:
mTOR signal and hypoxia-inducible factor-1 alpha regulate CD133
expression in cancer cells. Cancer Res. 69:7160–7164. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yuan Y, Shan N, Tan B, Deng Q, Liu Y, Wang
H, Luo X, He C, Luo X, Zhang H, et al: SRC-3 plays a critical role
in human umbilical vein endothelial cells by regulating the
PI3K/Akt/mTOR pathway in preeclampsia. Reprod Sci. 25:748–758.
2018. View Article : Google Scholar
|
|
48
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
49
|
Jonsson Y, Rubèr M, Matthiesen L, Berg G,
Nieminen K, Sharma S, Ernerudh J and Ekerfelt C: Cytokine mapping
of sera from women with preeclampsia and normal pregnancies. J
Reprod Immunol. 70:83–91. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen J, Yue C, Xu J, Zhan Y, Zhao H, Li Y
and Ye Y: Downregulation of receptor tyrosine kinase-like orphan
receptor 1 in preeclampsia placenta inhibits human trophoblast cell
proliferation, migration, and invasion by PI3K/AKT/mTOR pathway
accommodation. Placenta. 82:17–24. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Harati-Sadegh M, Kohan L, Teimoori B,
Mehrabani M and Salimi S: The association of the placental
Hypoxia-inducible factor1-α polymorphisms and HIF1-α mRNA
expression with preeclampsia. Placenta. 67:31–37. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Grotegut CA: Prevention of preeclampsia. J
Clin Invest. 126:4396–4398. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cohen JM, Kramer MS, Platt RW, Basso O,
Evans RW and Kahn SR: The association between maternal antioxidant
levels in midpregnancy and preeclampsia. Am J Obstet Gynecol. 213.
pp. 695.e1–613. 2015, View Article : Google Scholar
|
|
54
|
Bahado-Singh RO, Syngelaki A, Akolekar R,
Mandal R, Bjondahl TC, Han B, Dong E, Bauer S, Alpay-Savasan Z,
Graham S, et al: Validation of metabolomic models for prediction of
early-onset preeclampsia. Am J Obstet Gynecol. 213:530.e1–530.e10.
2015. View Article : Google Scholar
|
|
55
|
Ray JG, Booth GL, Alter DA and Vermeulen
MJ: Prognosis after maternal placental events and
revascularization: PAMPER study. Am J Obstet Gynecol.
214:106.e1–106.e14. 2016. View Article : Google Scholar
|
|
56
|
Szabo G, Molvarec A, Nagy B and Rigo J Jr:
Increased B-type natriuretic peptide levels in early-onset versus
late-onset preeclampsia. Clin Chem Lab Med. 52:281–288. 2014.
View Article : Google Scholar
|
|
57
|
Borges VTM, Zanati SG, Peraçoli MTS,
Poiati JR, Romão-Veiga M, Peraçoli JC and Thilaganathan B: Maternal
left ventricular hypertrophy and diastolic dysfunction and brain
natriuretic peptide concentration in early- and late-onset
pre-eclampsia. Ultrasound Obstet Gynecol. 51:519–523. 2018.
View Article : Google Scholar
|
|
58
|
Chatterjee P, Chiasson VL, Seerangan G, De
Guzman E, Milad M, Bounds KR, Gasheva O, Tobin RP, Hatahet M,
Kopriva S, et al: Depletion of MHC class II invariant chain peptide
or γ-δ T-cells ameliorates experimental preeclampsia. Clin Sci
(Lond). 131:2047–2058. 2017. View Article : Google Scholar
|
|
59
|
Kim J, Lee KS, Kim JH, Lee DK, Park M,
Choi S, Park W, Kim S, Choi YK, Hwang JY, et al: Aspirin prevents
TNF-α-induced endothelial cell dysfunction by regulating the
NF-κB-dependent miR-155/eNOS pathway: Role of a miR-155/eNOS axis
in preeclampsia. Free Radic Biol Med. 104:185–198. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Shaw J, Tang Z, Schneider H, Salje K,
Hansson SR and Guller S: Inflammatory processes are specifically
enhanced in endothelial cells by placental-derived TNF-α:
Implications in preeclampsia (PE). Placenta. 43:1–8. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Barone Gibbs B, Dobrosielski DA, Bonekamp
S, Stewart KJ and Clark JM: A randomized trial of exercise for
blood pressure reduction in type 2 diabetes: Effect on
flow-mediated dilation and circulating biomarkers of endothelial
function. Atherosclerosis. 224:446–453. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Onda K, Tong S, Nakahara A, Kondo M,
Monchusho H, Hirano T, Kaitu'u-Lino T, Beard S, Binder N, Tuohey L,
et al: Sofalcone upregulates the nuclear factor (erythroid-derived
2)-like 2/heme oxygenase-1 pathway, reduces soluble fms-like
tyrosine kinase-1, and quenches endothelial dysfunction: Potential
therapeutic for preeclampsia. Hypertension. 65:855–862. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Vanhaesebroeck B, Stephens L and Hawkins
P: PI3K signalling: The path to discovery and understanding. Nat
Rev Mol Cell Biol. 13:195–203. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Bartholomeusz C and Gonzalez-Angulo AM:
Targeting the PI3K signaling pathway in cancer therapy. Expert Opin
Ther Targets. 16:121–130. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kawahara T, Asthana S and Kneteman NM:
m-TOR inhibitors: What role in liver transplantation? J Hepatol.
55:1441–1451. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang X, Li Q, Jiang W, Xiong X, Li H,
Zhao J and Qi H: LAMA5 promotes human umbilical vein endothelial
cells migration, proliferation, and angiogenesis and is decreased
in preeclampsia. J Matern Fetal Neonatal Med. 33:1114–1124. 2020.
View Article : Google Scholar
|
|
67
|
Oladipupo S, Hu S, Kovalski J, Yao J,
Santeford A, Sohn RE, Shohet R, Maslov K, Wang LV and Arbeit JM:
VEGF is essential for hypoxia-inducible factor-mediated
neovascularization but dispensable for endothelial sprouting. Proc
Natl Acad Sci USA. 108:13264–13269. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Fukushima K, Murata M, Hachisuga M,
Tsukimori K, Seki H, Takeda S, Asanoma K and Wake N: Hypoxia
inducible factor 1 alpha regulates matrigel-induced endovascular
differentiation under normoxia in a human extravillous trophoblast
cell line. Placenta. 29:324–331. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kovo M, Schreiber L, Elyashiv O,
Ben-Haroush A, Abraham G and Bar J: Pregnancy outcome and placental
findings in pregnancies complicated by fetal growth restriction
with and without preeclampsia. Reprod Sci. 22:316–321. 2015.
View Article : Google Scholar
|
|
70
|
Cotechini T, Komisarenko M, Sperou A,
Macdonald-Goodfellow S, Adams MA and Graham CH: Inflammation in rat
pregnancy inhibits spiral artery remodeling leading to fetal growth
restriction and features of preeclampsia. J Exp Med. 211:165–179.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Szarka A, Rigó J Jr, Lázár L, Beko G and
Molvarec A: Circulating cytokines, chemokines and adhesion
molecules in normal pregnancy and preeclampsia determined by
multiplex suspension array. BMC immunol. 11:592010. View Article : Google Scholar : PubMed/NCBI
|