1. Introduction
Atherosclerosis (AS) is a chronic disease with
complex etiology, which involves early local injury of the arterial
intima, followed by lipid deposition, proliferation of the intimal
fibrous tissue, local thickening of the intima and, ultimately,
plaque formation (1). Vascular
plaque-induced stenosis by AS may lead to insufficient arterial
blood supply and cardiovascular diseases (2). The most serious complications caused
by plaque rupture are myocardial infarction, cerebral ischemia, and
ischemia of the surrounding tissue (3). The pathogenesis of AS has not been
fully elucidated to date. Previous studies have demonstrated that
AS is associated with lipid metabolism disorders, endothelial cell
damage, inflammation and immune dysfunction, involving macrophages,
endothelial cells, vascular smooth muscle cells and platelets
(4,5). In recent years, AS animal models
mainly include mice, rabbits, miniature pigs, non-human primates
and transgenic animals (6). AS is
a vicious circle combining multiple factors and long-term effects.
So, elucidating the underlying mechanism is crucial for the
treatment and prevention of the disease.
Despite not having been fully elucidated, it is
believed that lipid metabolism disorders, endothelial cell injury,
inflammation and immune dysfunction are the most important factors
implicated in the pathogenesis of AS (5,7).
Functional damage of endothelial cells is the initiating step in
the early stage of AS (8).
Endothelial cell damage in AS plaques leads to further plaque
instability, rupture (9) and
secondary thrombosis, thus accelerating disease progression and
affirming the important role of endothelial cell integrity
(10). Glycolysis, the most
important energy source for endothelial cells, is used to quickly
produce energy, enabling cells to respond to environmental changes
(11). Intermediate metabolic
products produced during glycolysis affect cell survival (12); therefore, glycolytic rates in
endothelial cells play a key role in maintaining their homeostasis
and reducing the risk for AS.
The aim of the present study was to review the
pathogenesis of AS, the role of endothelial cell damage and
glycolysis, and the role of associated target genes and the
involved signalling pathways, in order to indicate new approaches
to the research on AS pathogenesis and intervention methods, and
aid in the development of novel treatments for AS.
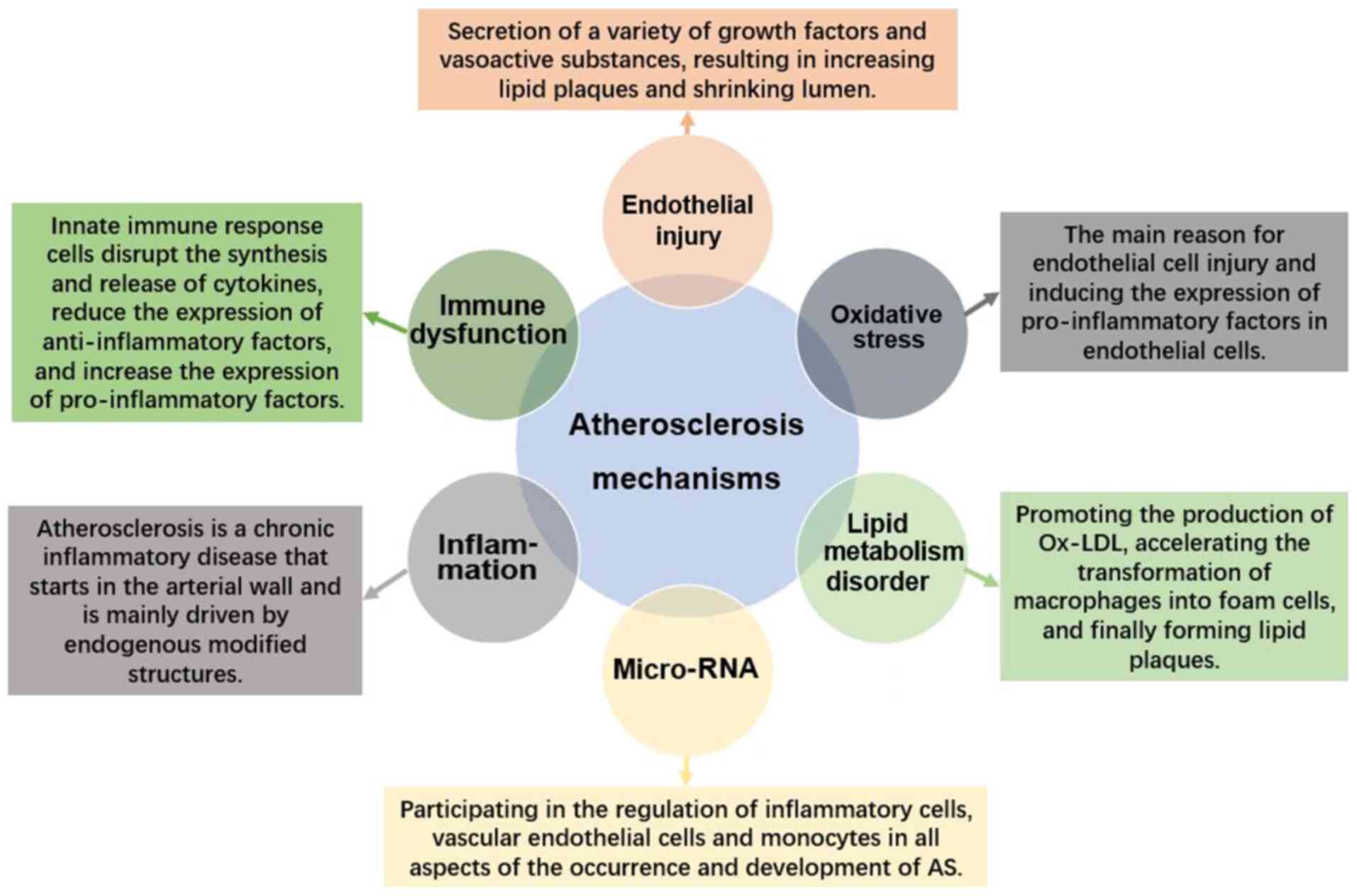
2. Factors implicated in AS
Pathogenesis
The pathogenesis of AS is extremely complicated
(Fig. 1). AS is currently
considered to be the result of the interaction among various
mechanisms, including lipid metabolism disorder, inflammatory cell
infiltration, oxidative stress, immune dysfunction and vascular
endothelial cell damage, the latter of which ultimately leads to
plaque rupture and thrombosis, leading to serious cardiovascular
and cerebrovascular diseases (1,4,13).
Role of oxidative stress in AS
Oxidative stress is the initiating factor of AS
inflammatory response, with reactive oxygen species (ROS) and
oxidized low-density lipoprotein (Ox-LDL; formed by oxidative
modification of LDL) being the main factors responsible for
endothelial cell damage and for inducing the expression of
pro-inflammatory factors in endothelial cells (14,15). When endothelial cells are
continuously exposed to external as well as endogenous oxidants,
oxidative stress is likely to induce production of various
biologically active substances, which may cause endothelial cell
functional damage and apoptosis (16,17). This process leads to the synthesis
and release of inflammatory factors, further aggravating vascular
inflammation (18). In addition,
oxidative stress regulates the expression of vascular wall genes by
acting on transcription factors of the vascular wall cells. For
example, intracellular ROS directly oxidatively modify the
transcription factor itself, thereby participating in the
occurrence and development of AS (14).
Role of lipid metabolism disorder in
AS
Excessive blood lipid levels are the main cause of
AS. In the hyperlipidemic state, the elevated plasma LDL
cholesterol (LDL-C) is deposited on the vascular intima and enters
macrophages via membrane receptors (19). In addition, LDL also undergoes
oxidative modification to form Ox-LDL, leading to changes in
endothelial cell function, and causing increased permeability and
lipid deposition in the inner membrane (20). Ox-LDL exhibits strong affinity for
scavenger receptors found on mononuclear macrophages, leading to
its quick internalisation (21).
However, Ox-LDL is toxic for macrophages, causing them to become
activated, rapidly proliferate, aggregate and degenerate (22). Finally, the macrophages undergo
apoptosis and become foam cells, which then aggregate to form AS
lipid plaques. Moreover, Ox-LDL binds to vascular endothelial cells
through lectin-like oxidized LDL receptor-1 to disrupt
intra-cellular signaling and cause endothelial cell dysfunction
(23). Ox-LDL can also promote
the continuous proliferation of vascular smooth muscle cells and
their outward migration to form plaques on the inner wall of blood
vessels.
Role of endothelial cell injury in
AS
Disruption of endothelial cell morphology and
function leads to vascular barrier function impairment, as well as
to changes in the intimal integrity and permeability (24). The apoptosis and shedding of
endothelial cells promote the adhesion and aggregation of platelets
from the blood (25).
Dysfunctional endothelial cells, macrophages and platelets secrete
a variety of growth factors and vasoactive substances, stimulating
the continuous proliferation of smooth muscle cells in the media,
and enter the intima, while also causing contractions of the
vascular wall (26). As a result,
the fatty plaques increase in size while the lumen becomes
progressively narrowed, promoting the formation of AS lesions
(8).
Role of inflammation in AS
AS involves not only lipid deposition in blood
vessel walls, but also chronic inflammation (27). Oxidative stress persists
throughout AS. AS has been proven to be a chronic inflammatory
disease initiated in the arterial wall, mainly driven by
modification of endogenous structures and dysfunction of the
vascular endothelium (28).
Certain lipids act as signaling molecules, and bind to cell
receptors to activate the expression of specific genes and produce
a number of pro-inflammatory cytokines (19). This leads to an increase in the
numbers of inflammatory cells, including macrophages, increased
phagocytosis of Ox-LDL and production of pro-inflammatory factors,
such as tumor necrosis factor (TNF)-α, interleukin (IL)-1 and IL-8
(29). These factors further
aggravate the pathology of AS.
Role of immune dysfunction in AS
AS is also an autoimmune disease, stimulated by
accumulated lipoproteins, as well as specific T lymphocytes and
their antibodies, in the blood vessel wall (30). It was previously demonstrated that
T lymphocytes infiltrate the aorta, where they accumulate and
express restricted T-cell receptors, thereby promoting AS through
immune regulation (13). In
addition, the cells of innate immune response, such as monocytes
and neutrophils, play an important role in the occurrence and
development of AS (31). Upon
immune function impairment, these cells disrupt cytokine production
(27). Thus, the expression of
anti-inflammatory factors decreases and the expression of
pro-inflammatory factors increases, ultimately promoting AS
development (29).
Role of microRNAs (miRNA) of the
circulatory system in AS
miRNAs are found in non-coding regions of the genome
and play important roles in gene transcription,
post-transcriptional processing, cell proliferation, cell
differentiation, cell apoptosis, ontogeny, heredity and epigenetics
(21,23). It has been recently discovered
that miRNAs are involved in inflammatory cell regulation and, thus,
all aspects of AS pathology, including vascular endothelial cell
and monocyte development, differentiation and function (32). Studies have shown that miR-143 can
affect the formation of AS plaques by inhibiting endothelial cell
glycolysis, while miR-33 is closely associated with macrophage
metabolism (33,34).
Drug intervention for AS
The aim of currently available anti-AS treatments,
which mainly include statins, antithrombotic drugs and surgical
intervention, is to reduce serum LDL levels (35). Statins are methylglutaryl-CoA
reductase inhibitors that have been found to be effective at
halting disease progression and reducing the incidence of
cardiovascular and cerebrovascular complications. By inhibiting
methylglutaryl-CoA reductase, statins lower total cholesterol and
LDL-C, increase HDL-C, activate nitric oxide (NO) synthase,
increase endothelial NO levels, and prevent NO decrease caused by
Ox-LDL (36). Probucol, a strong
synthetic antioxidant, is a symmetrical di-tert-butylphenol
structure that is easily oxidized, thus reducing free oxygen
radicals and diminishing their oxidizing capacity (37).
The irreversible antioxidant effect of probucol is
attributed to its strong affinity for LDL-C, thus inhibiting LDL-C
and reducing the formation of Ox-LDL. Anti-platelet aggregation
drugs may also exert an anti-inflammatory effect on the injured
vascular intima (38).
The combination of statins and antioxidants may
prevent thrombosis and AS plaque formation (39). In addition, niacin, a
broad-spectrum lipid-modulating drug, acts by inhibiting the
expression of serum adhesion molecules and inflammatory cytokines
(40). The anti-inflammatory
effect of niacin is achieved by downregulating the NF-κB signaling
pathway.
In addition to Western medicine, Traditional Chinese
Medicine (TCM) has also achieved marked benefits in the treatment
of AS. In TCM, AS is commonly categorised as 'blood stasis' and
'phlegm turbidity' (41). Modern
TCM theories mostly attribute the pathogenesis of AS to phlegm,
toxins and stasis. TCM arrests AS progression by activating blood
and dredging collaterals, inhibiting inflammation and plaque
formation, and stabilizing plaques (42,43). Commonly used TCM agents include
Chaihu Shugan San, Yiqi Yangyin Recipe, Danhuang Tongmai Capsule
and Simiao Yongan Decoction (44-46). From a macro perspective, TCM is
guided by Chinese medicine principles, and the treatment is
performed holistically. In recent years, TCM has been found to add
unique advantages to the treatment of complex diseases, such as
AS.
3. Vascular endothelial cells
Physiological functions of vascular
endothelial cells
The inner vascular endothelium in the heart, blood
and lymphatic vessels is composed of squamous epithelial cells, and
is important for maintaining the complete structure and function of
the blood vessel wall (47).
Endothelial cells serve not onlyas a mechanical barrier, but also
as receptors and endocrine organs, as they are capable of
synthesizing and releasing a variety of endothelial-derived
vasoactive factors (9). When
damaged, vascular endothelial cells secrete a variety of molecules
to promote leukocyte adhesion. They also secrete
endothelium-dependent factors, such as endothelin, angiotensin, NO
and prostacyclin, regulating vasoconstriction and vasodilation
(48). Endothelium-dependent
dilation factors may also inhibit platelet activation and
aggregation. Plasma plasminogen activator synthesized by vascular
endothelial cells has high affinity for fibrin and can activate
plasminogen to dissolve thrombi (24). Under normal conditions, these
cells reduce inflammation by regulating the release of various
cytokines and inflammatory factors, such as intercellular adhesion
molecule-1, vascular cell adhesion molecule-1 and monocyte
chemokines (49). However, the
injured endothelium promotes a series of inflammatory reactions,
causing endothelial cell dysfunction and favouring AS
development.
Mechanism underlying the development of
AS caused by vascular endothelial cell injury
Vascular endothelial cell function is closely
associated with AS, which is a chronic inflammatory response of the
arterial wall to endothelial cell injury (8). Vascular endothelial cell injury and
functional damage serve as the initiating factor. The dysfunction
and morphological damage of endothelial cells may manifest as a
variety of endothelial abnormalities, causing leukocyte adhesion,
vasoconstriction, platelet activation, oxidative stress and
inflammation, followed by thrombus formation which, in turn,
promotes the formation of AS plaques (24,25,50,51). Endothelial progenitor cells
participate in reendothelialization and angiogenesis, and may play
a pivotal role in the occur-rence and development of AS (52).
Increased intimal permeability
Following endothelial cell injury, lipids accumulate
in the blood vessels, resulting in increased permeability of the
vascular intima. This causes monocytes and phagocytes to be
released into the bloodstream, increasing the adhesion to
endothelial cells (50). LDL is
then oxidized to the cytotoxic Ox-LDL, which is damaging to
endothelial and smooth muscle cells. Macrophage Ox-LDL uptake
occurs through scavenger receptors, accelerating their
transformation into foam cells that eventually form the AS plaques
(47).
Increased leukocyte adhesion
Leukocytes, particularly monocytes, adhere to the
endothelium through intercellular adhesion molecules, leukocyte
adhesion molecules, TNF-α and vascular cell adhesion molecules.
Subsequently, guided by chemokines, they migrate to the intima and
differentiate into macrophages (53). Macrophages secrete a variety of
inflammatory factors, such as TNF-α, IL-1 and IL-6, to promote the
formation of AS plaques. In addition, damage to endothelial cells
leads to increased expression of adhesion molecules, while also
increasing the susceptibility of endothelial cells to inflammatory
stimuli. This again increases the expression of adhesion molecules,
promoting the adhesion of monocytes to endothelial cells and
triggering AS development (34).
Promoting thrombosis
Stimulation or damage of endothelial cells causes a
decrease in their antithrombotic properties, which promotes
thrombus formation (25). Among
other factors, increased secretion of the von Willebrand factor and
thromboxane mediate platelet aggregation and adhesion to the
endothelium to promote thrombosis, while plasminogen activator
inhibitors inhibit thrombolysis (5). Thus, the micro-thrombi formed on the
endothelial inner membrane are difficult to dissolve, contributing
to the formation of plaques (8).
Regulation of endothelial progenitor
cells
Endothelial progenitor cells, a type of precursor
cell with a high proliferation potential, are a heterogeneous cell
population with multiple origins and different phenotypes, capable
of producing endothelial cells (54). Endothelial progenitor cells can
selectively be recruited to damaged or ischemic areas by stress and
inflammatory stimuli to form new blood vessels through
differentiation and proliferation, without relying on the original
vasculature (55). Inflammation
is the pathophysiological basis of various cardiovascular diseases.
Pro-inflammatory cytokines stimulate the expression of adhesion
molecules on the surface of endothelial cells and promote the onset
of AS (56). Inflammation can
stimulate the release of endothelial progenitor cells from the bone
marrow into peripheral blood, thus promoting tissue repair
(57). Therefore, the occurrence
of AS is associated with endothelial progenitor cells, which are
involved in repair and intimal hyperplasia following endothelial
cell injury, which also promote the formation and stability of AS
plaques (52).
4. Glycolysis
It was previously demonstrated that 85% of ATP in
endothelial cells is produced by glycolysis, of which ~60% is used
for homeostatic maintenance and 40% for proliferation (11). The high glycolytic properties of
endothelial cells result from their low content of mitochondria,
which comprise only 5% of the cell. Although endothelial cells
reside in a high-oxygen environment, they consume little oxygen and
can, therefore, deliver oxygen to nearby tissues (58). Endothelial cells use glycolysis to
quickly produce energy, which helps them adapt to changes in their
environment. More metabolic intermediate products are produced
through glycolysis, affecting cell survival (59). Compared with other cells, resting
endothelial cells have a high-efficiency glycolysis rate. When
cells undergo migration or proliferation, their glycolysis rates
double (60). Glycolysis is
required to meet the energy demands of blood vessel sprouting,
since the apical cells at the front must continuously migrate
forward to form filamentous or laminar pseudopodia, while stem
cells at the back increase their proliferation rate and form a
lumen (61). Endothelial cell
dysfunction caused by Ox-LDL and other factors is the main cause of
AS. It was previously demonstrated that excessive activation of
glycolysis is a key factor leading to endothelial cell dysfunction
and proliferation (12).
Maintaining the metabolic balance of endothelial cells by
inhibiting glycolysis to reduce dysfunction and inflammation may
represent a novel treatment strategy for AS (11). The rate-limiting enzymes of
glycolysis may be a bridge to elucidating the association between
endothelial cell injury and AS.
Under physiological conditions, cells metabolize
glucose mainly through oxidative phosphorylation, while the
glycolytic pathway is activated only under hypoxic conditions
(62). The glycolytic pathway
comprises a series of enzymatic reactions in which glucose from
tissues is degraded (63).
Glucose is first transported into the cell via glucose transporters
(GLUTs), and is then phosphorylated by hexokinase (HK) into
glucose-6-phosphate, which cannot penetrate the cell membrane
(64). Glucose-6-phosphate is
then converted into pyruvate by hexose phosphate isomerase and
phosphofructokinase (PFK) (65).
The pyruvate produced through glycolysis may directly enter the
tricarboxylic acid cycle or be converted into lactic acid by
lactate dehydrogenase (LDHA) (66). The lactic acid is then transported
outside the cell via the monocarboxylic acid transporter ¼
(67). The ATP produced by this
process provides energy for the cell.
Regulation of HK in endothelial
glycolysis
HK is the first rate-limiting enzyme in the
glycolytic pathway. This enzyme catalyses the conversion of glucose
to glucose-6-phosphate, and produces ATP through oxidative
phosphorylation or glycolysis (68). It was previously demonstrated that
the AKT/mTOR signalling pathway is associated with HK production.
High HK expression in endothelial cells facilitates efficient
glycolysis and promotes rapid cell proliferation (69). In addition, HK can bind to
voltage-dependent anion channels in the outer mitochondrial
membrane to prevent binding of the pro-apoptotic protein Bax. This
prevents the release of cytochrome c from the mitochondria,
thereby exerting an anti-apoptotic effect (70). Therefore, HK not only promotes
cell proliferation, but also inhibits cell apoptosis.
Regulation of PFK in endothelial
glycolysis
PFK, the second glycolytic rate-limiting enzyme in
the glycolytic pathway, catalyses the conversion of fructose
6-phosphate to fructose 1,6-diphosphate. The expression of PFK is
regulated by a number of factors, such as c-Src activation and
hypoxia-inducible factor (HIF)-1α, promoting PFK2 expression
(71). TP53-induced glycolysis
and apoptosis regulator (TIGAR) may reduce PFK expression and
inhibit glycolysis. The kinase activity of PFKFB3, a subtype of
PFK2, in vascular endothelial cells, is affected by the RAS and
AMP-activated protein kinase (AMPK) signalling pathways (72). In addition, PFKFB3 can also
promote endothelial cell inflammation through TNF-α, thereby
promoting AS development (73).
Regulation of PK in endothelial
glycolysis
PK, the third rate-limiting enzyme in the glycolytic
pathway, specifically catalyses the conversion of
phosphoenolpyruvate to pyruvate. This irreversible reaction is a
crucial regulatory step in the glycolytic pathway. PKM2 is the only
PK subtype that can switch between high-activity (tetramer) and
low-activity (dimer) forms. Following phosphorylation, PKM2 is
converted to a dimer, promoting the upstream glycolytic products of
PK to enter the biosynthetic pathway (74). In addition, the expression of PKM2
is differentially regulated by the PI3K/AKT signal-ling pathway
(75). PKM2-regulated glycolysis
contributes to the proliferation and migration of vascular smooth
muscle cells, and is positively correlated with AS development
(12).
Regulation of LDHA in endothelial
glycolysis
LDHA, the fourth rate-limiting enzyme in the
glycolytic pathway, catalyses the production of lactic acid from
pyruvate. This enzyme enables recycling of pyruvate and reduced
nicotinamide purine dinucleotide, and plays a key role in promoting
efficient cell glycolysis (76).
SRC can also phosphorylate LDHA and promote conversion enzyme
activity. HIF-1α is the upstream regulator of LDHA, and both HIF-1α
and LDHA stimulate the inflammatory response (77).
5. Prediction and analysis of targets
associated with endothelial cell glycolysis in AS
Target prediction
Although endothelial cell glycolysis plays an
important role in AS, related research is scarce. Therefore, in the
present study, the term 'NCBI-gene' was used to search for genes
associated with endothelial cell glycolysis in AS, and 76 genes
were identified (Table I). The
Comparative Toxicogenomics Database (CTD) is a database used to
describe the association between chemical substances, genes and
human diseases. CTD was used to conduct Kyoto Encyclopedia of Genes
and Genomes pathway enrichment analysis (Table II); the pathways included in the
analysis were the immune system pathway, the signal transduction
pathway, pathways in cancer, the PI3K/AKT, IL-4, IL-13, HIF-1, NGF
and FoxO signalling pathways, the platelet activation, signalling
and aggregation pathway, as well as the fluid shear stress and AS
pathways.
 | Table IGene information on atherosclerosis
and endothelial cell glycolysis. |
Table I
Gene information on atherosclerosis
and endothelial cell glycolysis.
| No. | Gene name | Full name | Chromosome | Location | ID |
|---|
| 1 | VEGFA | Vascular
endothelial growth factor A | 6 | 6p21.1 | 7422 |
| 2 | CXCL8 | C-X-C motif
chemokine ligand 8 | 4 | 4q13.3 | 3576 |
| 3 | PPARG | Peroxisome
proliferator activated receptor gamma | 3 | 3p25.2 | 5468 |
| 4 | IL33 | Interleukin 33 | 9 | 9p24.1 | 90865 |
| 5 | IL6 | Interleukin 6 | 7 | 7p15.3 | 3569 |
| 6 | HIF1A | Hypoxia-inducible
factor 1 subunit alpha | 14 | 14q23.2 | 3091 |
| 7 | TP53 | Tumor protein
p53 | 17 | 17p13.1 | 7157 |
| 8 | KDR | Kinase insert
domain receptor | 4 | 4q12 | 3791 |
| 9 | TGFB1 | Transforming growth
factor beta 1 | 19 | 19q13.2 | 7040 |
| 10 | miR21 | MicroRNA 21 | 17 | 17q23.1 | 406991 |
| 11 | CDKN2A | Cyclin-dependent
kinase inhibitor 2A | 9 | 9p21.3 | 1029 |
| 12 | IL1B | Interleukin 1
beta | 2 | 2q14.1 | 3553 |
| 13 | AKT1 | AKT
serine/threonine kinase 1 | 14 | 14q32.33 | 207 |
| 14 | BCL2 | BCL2 apoptosis
regulator | 18 | 18q21.33 | 596 |
| 15 | SIRT1 | Sirtuin 1 | 10 | 10q21.3 | 23411 |
| 16 | STAT3 | Signal transducer
and activator of transcription 3 | 17 | 17q21.2 | 6774 |
| 17 | TLR4 | Toll-like receptor
4 | 9 | 9q33.1 | 7099 |
| 18 | PTGS2 |
Prostaglandin-endoperoxide synthase 2 | 1 | 1q31.1 | 5743 |
| 19 | ADIPOQ | Adiponectin C1Q and
collagen domain containing | 3 | 3q27.3 | 9370 |
| 20 | NFE2L2 | Nuclear factor
erythroid 2 like 2 | 2 | 2q31.2 | 4780 |
| 21 | NOTCH1 | Notch receptor
1 | 9 | 9q34.3 | 4851 |
| 22 | MTOR | Mechanistic target
of rapamycin kinase | 1 | 1p36.22 | 2475 |
| 23 | CTNNB1 | Catenin beta 1 | 3 | 3p22.1 | 1499 |
| 24 | PTEN | Phosphatase and
tensin homolog | 10 | 10q23.31 | 5728 |
| 25 | LEP | Leptin | 7 | 7q32.1 | 3952 |
| 26 | ESR1 | Estrogen receptor
1 | 6 | 6q25.1-q25.2 | 2099 |
| 27 | SOD1 | Superoxide
dismutase 1 | 21 | 21q22.11 | 6647 |
| 28 | MAPK1 | Mitogen-activated
protein kinase 1 | 22 | 22q11.22 | 5594 |
| 29 | RELA | RELA proto-oncogene
NF-kB subunit | 11 | 11q13.1 | 5970 |
| 30 | miR34A | MicroRNA 34a | 1 | 1p36.22 | 407040 |
| 31 | IFNG | Interferon
gamma | 12 | 12q15 | 3458 |
| 32 | AGTR1 | Angiotensin II
receptor type 1 | 3 | 3q24 | 185 |
| 33 | ABCB1 | ATP-binding
cassette subfamily B member 1 | 7 | 7q21.12 | 5243 |
| 34 | CD44 | CD44 molecule
(Indian blood group) | 11 | 11p13 | 960 |
| 35 | STAT1 | Signal transducer
and activator of transcription 1 | 2 | 2q32.2 | 6772 |
| 36 | HGF | Hepatocyte growth
factor | 7 | 7q21.11 | 3082 |
| 37 | NAMPT | Nicotinamide
phosphoribosyl transferase | 7 | 7q22.3 | 10135 |
| 38 | CYBB | Cytochrome b-245
beta chain | X | Xp21.1-p11.4 | 1536 |
| 39 | KL | Klotho | 13 | 13q13.1 | 9365 |
| 40 | SRC | SRC proto-oncogene
non-receptor tyrosine kinase | 20 | 20q11.23 | 6714 |
| 41 | PPARA | Peroxisome
proliferator activated receptor alpha | 22 | 22q13.31 | 5465 |
| 42 | BSG | Basigin (Ok blood
group) | 19 | 19p13.3 | 682 |
| 43 | TXN | Thioredoxin | 9 | 9q31.3 | 7295 |
| 44 | NOS2 | Nitric oxide
synthase 2 | 17 | 17q11.2 | 4843 |
| 45 | FOXO3 | Forkhead box
O3 | 6 | 6q21 | 2309 |
| 46 | PRKCE | Protein kinase C
epsilon | 2 | 2p21 | 5581 |
| 47 | NR4A1 | Nuclear receptor
subfamily 4 group A member 1 | 12 | 12q13.13 | 3164 |
| 48 | NOX4 | NADPH oxidase
4 | 11 | 11q14.3 | 50507 |
| 49 | PRKAA1 | Protein kinase
AMP-activated catalytic subunit alpha 1 | 5 | 5p13.1 | 5562 |
| 50 | miR210 | MicroRNA 210 | 11 | 11p15.5 | 406992 |
| 51 | MALAT1 |
Metastasis-associated lung adenocarcinoma
transcript 1 | 11 | 11q13.1 | 378938 |
| 52 | IGF1R | Insulin-like growth
factor 1 receptor | 15 | 15q26.3 | 3480 |
| 53 | AHR | Aryl hydrocarbon
receptor | 7 | 7p21.1 | 196 |
| 54 | CD36 | CD36 molecule | 7 | 7q21.11 | 948 |
| 55 | EZH2 | Enhancer of zeste 2
polycomb repressive complex 2 subunit | 7 | 7q36.1 | 2146 |
| 56 | SIRT6 | Sirtuin 6 | 19 | 19p13.3 | 51548 |
| 57 | IKBKB | Inhibitor of
nuclear factor kappa B kinase subunit beta | 8 | 8p11.21 | 3551 |
| 58 | miR122 | MicroRNA 122 | 18 | 18q21.31 | 406906 |
| 59 | UCP2 | Uncoupling protein
2 | 11 | 11q13.4 | 7351 |
| 60 | ENO1 | Enolase 1 | 1 | 1p36.23 | 2023 |
| 61 | PPARGC1A | PPARG coactivator 1
alpha | 4 | 4p15.2 | 10891 |
| 62 | SNAI1 | Snail family
transcriptional repressor 1 | 20 | 20q13.13 | 6615 |
| 63 | PPBP | Pro-platelet basic
protein | 4 | 4q13.3 | 5473 |
| 64 | DICER1 | Dicer 1
ribonuclease III | 14 | 14q32.13 | 23405 |
| 65 | TXNIP |
Thioredoxin-interacting protein | 1 | 1q21.1 | 10628 |
| 66 | IL22 | Interleukin 22 | 12 | 12q15 | 50616 |
| 67 | TNFSF13B | TNF superfamily
member 13b | 13 | 13q33.3 | 10673 |
| 68 | SREBF2 | Sterol regulatory
element-binding transcription factor 2 | 22 | 22q13.2 | 6721 |
| 69 | miR497 | MicroRNA 497 | 17 | 17p13.1 | 574456 |
| 70 | miR206 | MicroRNA 206 | 6 | 6p12.2 | 406989 |
| 71 | TUG1 | Taurine upregulated
1 | 22 | 22q12.2 | 55000 |
| 72 | miR135B | MicroRNA 135b | 1 | 1q32.1 | 442891 |
| 73 | miR142 | MicroRNA 142 | 17 | 17q22 | 406934 |
| 74 | miR135A1 | MicroRNA
135a-1 | 3 | 3p21.2 | 406925 |
| 75 | miR148B | MicroRNA 148b | 12 | 12q13.13 | 442892 |
| 76 | miR30C1 | MicroRNA 30c-1 | 1 | 1p34.2 | 407031 |
| Table IIPotential pathways of atherosclerosis
and endothelial cell glycolysis. |
Table II
Potential pathways of atherosclerosis
and endothelial cell glycolysis.
| Pathway | No. | Genes |
|---|
| Immune system | 36 | AKT1, BCL2, CD36,
CD44, CTNNB1, CXCL8, CYBB, FOXO3, HGF, HIF1A, IFNG, IKBKB, IL1B,
IL22, IL33, IL6, KL, MAPK1, MTOR, NOS2, NR4A1, PPBP, PRKCE, PTEN,
PTGS2, RELA, SRC, STAT1, STAT3, TGFB1, TLR4, TNFSF13B, TP53, TXN,
TXNIP, VEGFA |
| Signal
transduction | 30 | AGTR1, AKT1,
CTNNB1, CXCL8, CYBB, ESR1, FOXO3, HGF, HIF1A, IGF1R, IKBKB, IL6,
KDR, KL, LEP, MAPK1, MTOR, NOTCH1, NR4A1, PPBP, PRKAA1, PRKCE,
PTEN, RELA, SRC, STAT1, STAT3, TGFB1, TP53, VEGFA |
| Pathways in
cancer | 23 | AGTR1, AKT1, BCL2,
CDKN2A, CTNNB1, CXCL8, HGF, HIF1A, IGF1R, IKBKB, IL6, MAPK1, MTOR,
NOS2, PPARG, PTEN, PTGS2, RELA, STAT1, STAT3, TGFB1, TP53,
VEGFA |
| PI3K-AKT signalling
pathway | 17 | AKT1, BCL2, FOXO3,
HGF, IGF1R, IKBKB, IL6, KDR, MAPK1, MTOR, NR4A1, PRKAA1, PTEN,
RELA, TLR4, TP53, VEGFA |
| Cellular responses
to stress | 17 | CDKN2A, CXCL8,
CYBB, EZH2, HIF1A, IL6, MAPK1, MTOR, NOX4, PRKAA1, RELA, SIRT1,
SOD1, STAT3, TP53, TXN, VEGFA |
| Interleukin-4 and
13 signalling | 16 | AKT1, BCL2, CD36,
CXCL8, FOXO3, HGF, HIF1A, IL1B, IL6, NOS2, PTGS2, STAT1, STAT3,
TGFB1, TP53, VEGFA |
| HIF-1 signalling
pathway | 15 | AKT1, BCL2, CYBB,
ENO1, HIF1A, IFNG, IGF1R, IL6, MAPK1, MTOR, NOS2, RELA, STAT3,
TLR4, VEGFA |
| Platelet
activation, signaling and aggregation | 10 | AKT1, CD36, HGF,
MAPK1, PPBP, PRKCE, SOD1, SRC, TGFB1, VEGFA |
| NGF signalling via
TRKA from the plasma membrane | 12 | AKT1, FOXO3, HGF,
KL, MAPK1, MTOR, NR4A1, PRKCE, PTEN, SRC, STAT3, TP53 |
| Fluid shear stress
and atherosclerosis | 15 | AKT1, BCL2, CTNNB1,
CYBB, IFNG, IKBKB, IL1B, KDR, NFE2L2, PRKAA1, RELA, SRC, TP53, TXN,
VEGFA |
| FoxO signalling
pathway | 11 | AKT1, FOXO3, IGF1R,
IKBKB, IL6, MAPK1, PRKAA1, PTEN, SIRT1, STAT3, TGFB1 |
| PI3K/AKT
activation | 10 | AKT1, FOXO3, HGF,
KL, MAPK1, MTOR, NR4A1, PTEN, SRC, TP53 |
| LAT2/NTAL/LAB on
calcium mobilization | 10 | AKT1, FOXO3, HGF,
KL, MAPK1, MTOR, NR4A1, PTEN, SRC, TP53 |
The screened genes play important regulatory roles
in multiple signalling pathways, indicating that such interactions
should also be present among their corresponding proteins. The
STRING database, used to study the protein interaction networks,
helps to mine core regulatory genes. Based on that, a
protein-protein interaction network was built and Cytoscape 3.2.1
software (https://cytoscape.org/download_old_versions.html)
was used to visualize it (Fig.
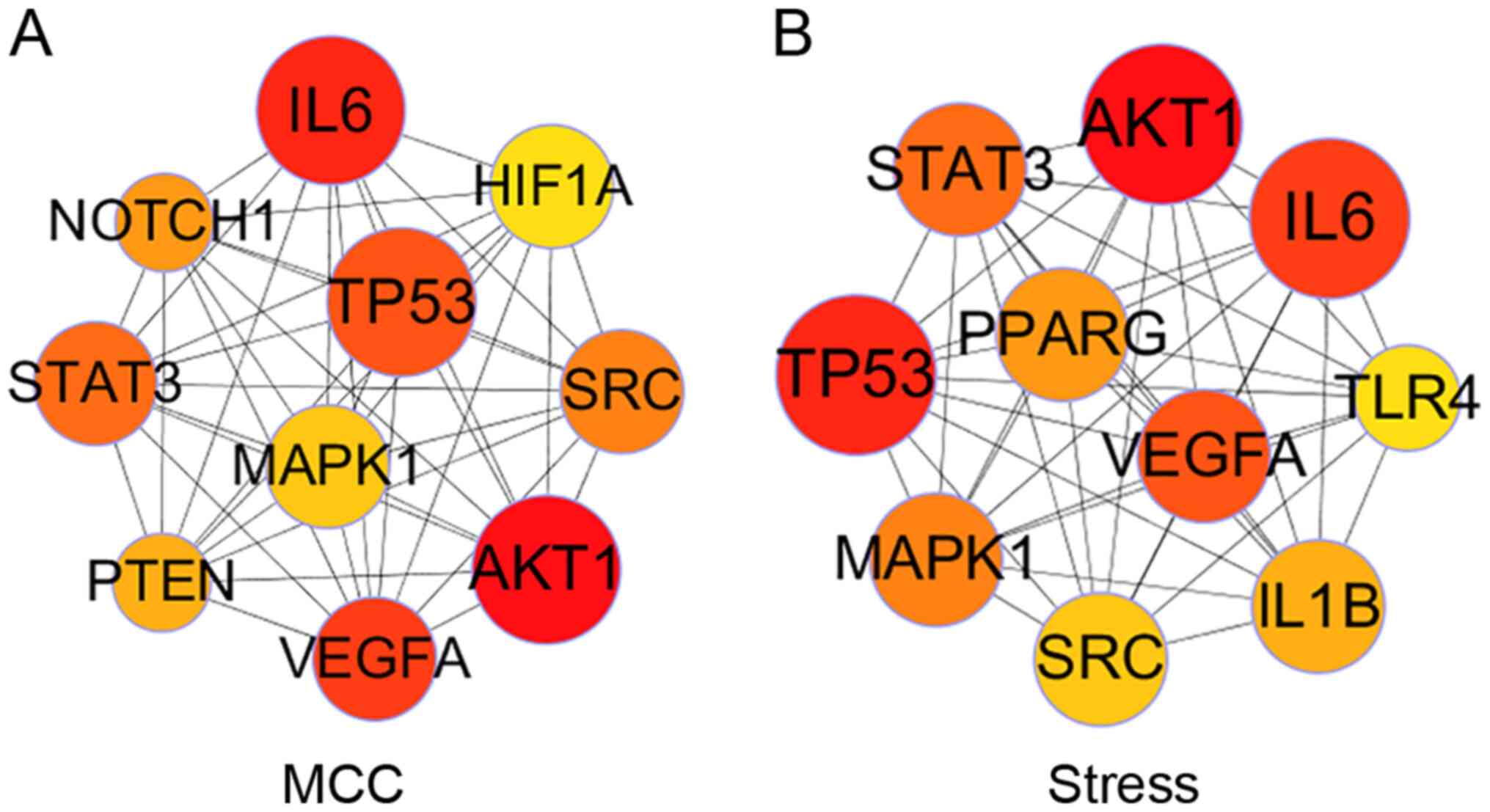
2). The node size in Fig. 1
reflects its importance in the network. CytoHubba is a network
analysis plug-in specifically used to identify key nodes in the
Cytoscape software, providing a variety of key node identification
algorithms, which focus on different network topology
characteristics. To make the identification of key targets as
accurate as possible, the top 10 key targets were identified based
on two algorithms, namely Matthews correlation coefficient (MCC)
and Stress (Fig. 3). Next, the
intersection of the two results was considered as the key targets
for AS endothelial glycolysis, including AKT1, IL-6, vascular
endothelial growth factor (VEGF)A, TP53, signal transducer and
activator of transcription 3 (STAT3), SRC and mitogen-activated
protein kinase (MAPK)1.
Target analysis
All 7 predicted targets are factors proven to
promote or inhibit the process of AS. AKT1 and TP53 can directly
regulate glycolysis, thereby affecting AS. In addition to the
regulation of glycolysis, IL-6 can also regulate AS through
inflammation; VEGFA, STAT3 and SRC are also closely associated with
immune system dysfunction. The regulatory effect of MAPK1 on AS is
mainly mediated through immune response, but the association
between glycolysis and MAPK1 has not been explored. The available
research on these 7 targets is currently not sufficient, and
further experiments are required to verify the results. The purpose
of the present review was also to provide a direction for future AS
studies.
Evaluation of AKT1 involvement in
endothelial cell glycolysis in AS
AKT, including AKT1, AKT2 and AKT3, play an
important role in cell proliferation, survival, metabolism and
migration (78). AKT can activate
mTOR, another major regulatory gene of cell metabolism, thereby
promoting the expression of HIF-1α. HIF-1α then regulates the
expression of glycolysis-related proteins, such as GLUTs, PK, HK2
and LDHA (79). These pathways
promote glucose uptake and lactic acid production. In addition,
endothelial glycolysis can be regulated by the FKBP51/AKT1 and
AKT1/GSK/3β signalling pathways (80,81). Therefore, as regards energy
metabolism, AKT1 may be a key target in AS.
Evaluation of IL-6 involvement in
endothelial cell glycolysis in AS
IL-6 is an important pro-inflammatory cytokine
(82). Chronic inflammation is
also considered as part of AS pathology, suggesting a possible role
for IL-6 (83). It has been shown
that IL-6 affects AS development through the acute phase response,
and exerts an effect on insulin resistance (84), while it also promotes glycolysis
through PFKFB3 (85). The
IL-6/STAT3 pathway is also a pathway involved in glycolysis
regulation (86).
Evaluation of VEGFA involvement in
endothelial cell glycolysis in AS
VEGFA promotes angiogenesis and regulates
endothelial cell proliferation, macrophage infiltration and foam
cell formation through signal transduction (87); therefore, it plays a key
regulatory role in the formation and stabilization of AS plaques
(88). In addition, it was
previously demonstrated that endothelial glycolysis may be promoted
by enhancing VEGFA expression, making it an important contributor
to AS development and endothelial glycolysis (89). Therefore, VEGFA is an important
target in AS and endothelial glycolysis.
Evaluation of TP53 involvement in
endothelial cell glycolysis in AS
TP53, also referred to as 'the guardian of the
genome', maintains gene stability. As it is mutated in >50% of
tumour cells, TP53 is used for cancer prediction and risk
assessment (90). In addition,
TP53 regulates energy metabolism through the AKT/mTOR and NF-κB
signalling pathways, and it inhibits glycolysis by downregulating
the expression of rate-limiting enzymes and activating TP53-induced
glycolysis and TIGAR (91,92).
It was previously demonstrated that endothelial autophagy is
inhibited in advanced AS by regulating mTOR and TIGAR (93). However, there is insufficient
research on AS endothelial glycolysis, and the TP53 regulatory
effects in AS require further investigation.
Evaluation of STAT3 involvement in
endothelial cell glycolysis in AS
STAT3, a transcription factor, regulates cell
growth, apoptosis and inflammation, and plays an important role in
cancer, AS, as well as cardiovascular and cerebrovas-cular diseases
(94). STAT3 is also strongly
associated with endothelial cell dysfunction, macrophage
polarization and inflammatory responses, thereby promoting AS
development (95). In addition,
it can enhance cell metabolism through HK2 upregulation. Previous
studies have demonstrated that the IL-6/STAT3 and JAK2/STAT3
pathways can promote glycolysis, confirming the crucial role of
STAT3 in AS and glycolysis (96,97). Therefore, STAT3 was proven to be
an important factor in AS and glycolysis.
Evaluation of SRC involvement in
endothelial cell glycolysis in AS
SRC is an oncogene that regulates intracellular
signal transduction, cell proliferation and cell migration
(98). SRC phosphorylation and
activation also participates in intracellular metabolic processes
(99). It was previously
demonstrated that the SRC/AKT/LKB1/AMPKα signalling pathway can
shift the intracellular metabolic pathway from glycolysis to
aerobic metabolism (100). In
addition, SRC-related pathways in endothelial cells promote AS by
affecting leukocyte adhesion and monocyte transport (101). Thus, SRC is another important
target in AS and endothelial cell glycolysis.
Evaluation of MAPK1 involvement in
endothelial cell glycolysis in AS
MAPK is an important intracellular enzyme and the
upstream signalling transduction molecule of several pathways
(102). It was previously
demonstrated that the P38/MAPK signalling pathway can aggravate AS,
while MAPK1 affects cell adhesion and the immune response to
promote AS development (103).
However, further research is required to confirm the regulatory
role of MAPK1 in AS and endothelial cell glycolysis.
6. Discussion
Although some progress has been made in the
treatment of AS and its complications, with a consequent
improvement in patient survival (1), long-term illness puts pressure on
the heart and is associated with a major socioeconomic burden
(3). AS is currently considered
as a chronic inflammatory disease, and the underlying mechanisms
mainly involve oxidative stress, lipid metabolism disorders,
endothelial cell damage, inflammation and immune dysfunction
(5,27). Eventually, plaque rupture and
thrombosis causes acute life-threatening cardiovascular and
cerebrovascular diseases (1).
Vascular endothelium inflammation and cell dysfunction are
considered as the initiating and central events in AS (29). Since glycolysis is the main
metabolic pathway in endothelial cells, an in-depth study of the
association between endothelial cell glycolysis and AS may further
elucidate AS pathophysiology and provide clues for its prevention
(11).
Through mining and analysis of endothelial cell
glycolysis and AS-related genes, 8 key targets were identified,
namely AKT1, IL-6, VEGFA, TP53, STAT3, SRC and MAPK1. These targets
were found to directly or indirectly affect the expression of key
rate-limiting enzymes in endothelial cell glycolysis. In addition,
the pathways obtained by the enrichment of these genes included the
immune system pathway, the signal transduction pathway, pathways in
cancer, the PI3K/AKT, IL-4, IL-13, HIF-1, NGF and FoxO signalling
pathways, the platelet activation, signalling and aggregation
pathway, as well as the fluid shear stress and AS pathways. The
aforementioned results are based on existing research and
correlation prediction (104).
At present, research on AS mainly uses genetically modified animal
models, such as ApoE−/− and db/db mice, and the findings
cannot yet be translated into clinical application. Therefore,
these targets must be further verified before they reach the
clinical stage. The aim of the present review was to contribute to
the study of AS mechanisms and provide novel suggestions, since
future AS therapies should be explored from a new perspective.
Advanced science and technology, including high-content technology,
single cell transcriptomics, lipometa-bolic technology and
epigenetic modification technology, will hopefully accelerate the
search for novel AS treatments.
Funding
This review was supported by the National Natural
Science Foundation of China (grant no. 81973514), the Central
Public-Interest Scientific Institution Basal Research Fund (grant
no. 2018PT35030) and the Drug Innovation Major Project (grant no.
2018ZX09711001-009).
Availability of data and materials
Not applicable.
Authors' contributions
GS and XS contributed to the conception and design
of the study. RW performed the literature search and wrote the
manuscript. MW prepared the figures. JY revised the manuscript. All
the authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Kobiyama K and Ley K: Atherosclerosis.
Circ Res. 123:1118–1120. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Spinelli FR, Barone F, Cacciapaglia F,
Pecani A and Piga M: Atherosclerosis and autoimmunity. Mediators
Inflamm. 2018:67304212018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu H and Daugherty A: Atherosclerosis.
Arterioscler Thromb Vasc Biol. 35:485–491. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Libby P, Buring JE, Badimon L, Hansson GK,
Deanfield J, Bittencourt MS, Tokgözoğlu L and Lewis EF:
Atherosclerosis. Nat Rev Dis Primers. 5:562019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bories GFP and Leitinger N: Macrophage
metabolism in athero-sclerosis. FEBS Lett. 591:3042–3060. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao Y, Qu H, Wang Y, Xiao W, Zhang Y and
Shi D: Small rodent models of atherosclerosis. Biomed Pharmacother.
129:1104262020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goonewardena SN, Prevette LE and Desai AA:
Metabolomics and atherosclerosis. Curr Atheroscler Rep. 12:267–272.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sitia S, Tomasoni L, Atzeni F, Ambrosio G,
Cordiano C, Catapano A, Tramontana S, Perticone F, Naccarato P,
Camici P, et al: From endothelial dysfunction to atherosclerosis.
Autoimmun Rev. 9:830–834. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen PY, Schwartz MA and Simons M:
Endothelial-to-mesenchymal transition, vascular inflammation, and
atherosclerosis. Front Cardiovasc Med. 7:532020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gao F, Chen J and Zhu H: A potential
strategy for treating athero-sclerosis: Improving endothelial
function via AMP-activated protein kinase. Sci China Life Sci.
61:1024–1029. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pircher A, Treps L, Bodrug N and Carmeliet
P: Endothelial cell metabolism: A novel player in atherosclerosis?
Basic principles and therapeutic opportunities. Atherosclerosis.
253:247–257. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao X, Tan F, Cao X, Cao Z, Li B, Shen Z
and Tian Y: PKM2-dependent glycolysis promotes the proliferation
and migration of vascular smooth muscle cells during
atherosclerosis. Acta Biochim Biophys Sin (Shanghai). 52:9–17.
2020. View Article : Google Scholar
|
|
13
|
Ilhan F and Kalkanli ST: Atherosclerosis
and the role of immune cells. World J Clin Cases. 3:345–352. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiao W, Jia Z, Zhang Q, Wei C, Wang H and
Wu Y: Inflammation and oxidative stress, rather than hypoxia, are
predominant factors promoting angiogenesis in the initial phases of
atherosclerosis. Mol Med Rep. 12:3315–3322. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dinh QN, Chrissobolis S, Diep H, Chan CT,
Ferens D, Drummond GR and Sobey CG: Advanced atherosclerosis is
associated with inflammation, vascular dysfunction and oxida-tive
stress, but not hypertension. Pharmacol Res. 116:70–76. 2017.
View Article : Google Scholar
|
|
16
|
Yang K, Zhang H, Luo Y, Zhang J, Wang M,
Liao P, Cao L, Guo P, Sun G and Sun X: Gypenoside XVII prevents
atherosclerosis by attenuating endothelial apoptosis and oxidative
stress: Insight into the ERα-Mediated PI3K/Akt Pathway. Int J Mol
Sci. 18:772017. View Article : Google Scholar
|
|
17
|
Feletou M, Cohen RA, Vanhoutte PM and
Verbeuren TJ: TP receptors and oxidative stress hand in hand from
endothelial dysfunction to atherosclerosis. Adv Pharmacol.
60:85–106. 2010.PubMed/NCBI
|
|
18
|
Armstrong AW, Voyles SV, Armstrong EJ,
Fuller EN and Rutledge JC: Angiogenesis and oxidative stress:
Common mechanisms linking psoriasis with atherosclerosis. J
Dermatol Sci. 63:1–9. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gupta M, Blumenthal C, Chatterjee S,
Bandyopadhyay D, Jain V, Lavie CJ, Virani SS, Ray KK, Aronow WS and
Ghosh RK: Novel emerging therapies in atherosclerosis targeting
lipid metabolism. Expert Opin Investig Drugs. 29:611–622. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee J, Jung S, Kim N, Shin MJ, Ryu DH and
Hwang GS: Myocardial metabolic alterations in mice with
diet-induced atherosclerosis: Linking sulfur amino acid and lipid
metabolism. Sci Rep. 7:135972017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Novák J, Olejníčková V, Tkáčová N and
Santulli G: Mechanistic role of MicroRNAs in coupling lipid
metabolism and atherosclerosis. Adv Exp Med Biol. 887:79–100. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shao D, Lian Z, Di Y, Zhang L, Rajoka MSR,
Zhang Y, Kong J, Jiang C and Shi J: Dietary compounds have
potential in controlling atherosclerosis by modulating macrophage
cholesterol metabolism and inflammation via miRNA. NPJ Sci Food.
2:132018. View Article : Google Scholar
|
|
23
|
Giral H, Kratzer A and Landmesser U:
MicroRNAs in lipid metabolism and atherosclerosis. Best Pract Res
Clin Endocrinol Metab. 30:665–676. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gimbrone MA Jr and García-Cardeña G:
Endothelial cell dysfunction and the pathobiology of
atherosclerosis. Circ Res. 118:620–636. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mudau M, Genis A, Lochner A and Strijdom
H: Endothelial dysfunction: The early predictor of atherosclerosis.
Cardiovasc J Afr. 23:222–231. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brix B, Mesters JR, Pellerin L and Johren
O: Endothelial cell-derived nitric oxide enhances aerobic
glycolysis in astrocytes via HIF-1α-mediated target gene
activation. J Neurosci. 32:9727–9735. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Legein B, Temmerman L, Biessen EA and
Lutgens E: Inflammation and immune system interactions in
atherosclerosis. Cell Mol Life Sci. 70:3847–3869. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Libby P and Hansson GK: Taming immune and
inflammatory responses to treat atherosclerosis. J Am Coll Cardiol.
71:173–176. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wolf D and Ley K: Immunity and
Inflammation in atherosclerosis. Circ Res. 124:315–327. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hansson GK and Hermansson A: The immune
system in athero-sclerosis. Nat Immunol. 12:204–212. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Iwata H and Nagai R: Novel immune signals
and atherosclerosis. Curr Atheroscler Rep. 14:484–490. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kuosmanen SM, Kansanen E, Kaikkonen MU,
Sihvola V, Pulkkinen K, Jyrkkänen HK, Tuoresmäki P, Hartikainen J,
Hippeläinen M, Kokki H, et al: NRF2 regulates endothelial
glycolysis and proliferation with miR-93 and mediates the effects
of oxidized phospholipids on endothelial activation. Nucleic Acids
Res. 46:1124–1138. 2018. View Article : Google Scholar :
|
|
33
|
Ouimet M, Ediriweera HN, Gundra UM, Sheedy
FJ, Ramkhelawon B, Hutchison SB, Rinehold K, van Solingen C,
Fullerton MD, Cecchini K, et al: MicroRNA-33-dependent regulation
of macrophage metabolism directs immune cell polarization in
atherosclerosis. J Clin Invest. 125:4334–4348. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kumar S, Kim CW, Simmons RD and Jo H: Role
of flow-sensitive microRNAs in endothelial dysfunction and
atherosclerosis: Mechanosensitive athero-miRs. Arterioscler Thromb
Vasc Biol. 34:2206–2216. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Barton M: Mechanisms and therapy of
atherosclerosis and its clinical complications. Curr Opin
Pharmacol. 13:149–153. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gonzalez L and Trigatti BL: Macrophage
apoptosis and necrotic core development in atherosclerosis: A
rapidly advancing field with clinical relevance to imaging and
therapy. Can J Cardiol. 33:303–312. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen C, Wang Y, Cao Y, Wang Q, Anwaier G,
Zhang Q and Qi R: Mechanisms underlying the inhibitory effects of
probucol on elastase-induced abdominal aortic aneurysm in mice. Br
J Pharmacol. 177:204–216. 2020. View Article : Google Scholar
|
|
38
|
Guo X, Wang L, Xia X, Wang P and Li X:
Effects of atorvastatin and/or probucol on recovery of
atherosclerosis in high-fat-diet-fed apolipoprotein E-deficient
mice. Biomed Pharmacother. 109:1445–1453. 2019. View Article : Google Scholar
|
|
39
|
Profumo E, Buttari B, D'Arcangelo D,
Tinaburri L, Dettori MA, Fabbri D, Delogu G and Riganò R: The
nutraceutical dehydroz-ingerone and its dimer counteract
inflammation- and oxidative stress-induced dysfunction of in vitro
cultured human endothelial cells: A novel perspective for the
prevention and therapy of atherosclerosis. Oxid Med Cell Longev.
2016:12464852016. View Article : Google Scholar
|
|
40
|
Jiang Y, Jin M, Chen J, Yan J, Liu P, Yao
M, Cai W and Pi R: Discovery of a novel niacin-lipoic acid dimer
N2L attenuating atherosclerosis and dyslipidemia with non-flushing
effects. Eur J Pharmacol. 868:1728712020. View Article : Google Scholar
|
|
41
|
Ren Y, Qiao W, Fu D, Han Z, Liu W, Ye W
and Liu Z: Traditional Chinese medicine protects against cytokine
production as the potential immunosuppressive agents in
atherosclerosis. J Immunol Res. 2017:74243072017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li TT, Wang ZB, Li Y, Cao F, Yang BY and
Kuang HX: The mechanisms of traditional Chinese medicine underlying
the prevention and treatment of atherosclerosis. Chin J Nat Med.
17:401–412. 2019.PubMed/NCBI
|
|
43
|
Tian F, Gu L, Si A, Yao Q, Zhang X, Zhao J
and Hu D: Metabolomic study on the faecal extracts of
atherosclerosis mice and its application in a traditional Chinese
Medicine. J Chromatogr B Analyt Technol Biomed Life Sci.
1007:140–148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fan Q, Liu Y, Rao J, Zhang Z, Xiao W, Zhu
T, Chai X, Ye K, Ning N, Yin Z, et al: Anti-atherosclerosis effect
of angong niuhuang pill via regulating Th17/Treg immune balance and
inhibiting chronic inflammatory on ApoE−/− mice model of
early and mid-term atherosclerosis. Front Pharmacol. 10:15842020.
View Article : Google Scholar
|
|
45
|
Zhu ZB, Song K, Huang WJ, Li H, Yang H,
Bai YQ, Guo KT, Yang RB, Lou WJ, Xia CH, et al: Si-Miao-Yong-An
(SMYA) decoction may protect the renal function through regulating
the autophagy-mediated degradation of ubiquitinated protein in an
atherosclerosis model. Front Pharmacol. 11:8372020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li L, Yu AL, Wang ZL, Chen K, Zheng W,
Zhou JJ, Xie Q, Yan HB, Ren P and Huang X: Chaihu-Shugan-San and
absorbed meranzin hydrate induce anti-atherosclerosis and
behavioral improvements in high-fat diet ApoE−/− mice
via anti-inflammatory and BDNF-TrkB pathway. Biomed Pharmacother.
115:1088932019. View Article : Google Scholar
|
|
47
|
Haskard DO, Boyle JJ, Evans PC, Mason JC
and Randi AM: Cytoprotective signaling and gene expression in
endothelial cells and macrophages-lessons for atherosclerosis.
Microcirculation. 20:203–216. 2013. View Article : Google Scholar
|
|
48
|
Dong Y, Fernandes C, Liu Y, Wu Y, Wu H,
Brophy ML, Deng L, Song K, Wen A, Wong S, et al: Role of
endoplasmic reticulum stress signalling in diabetic endothelial
dysfunction and athero-sclerosis. Diab Vasc Dis Res. 14:14–23.
2017. View Article : Google Scholar
|
|
49
|
Chrysohoou C, Kollia N and Tousoulis D:
The link between depression and atherosclerosis through the
pathways of inflammation and endothelium dysfunction. Maturitas.
109:1–5. 2018. View Article : Google Scholar
|
|
50
|
Jensen HA and Mehta JL: Endothelial cell
dysfunction as a novel therapeutic target in atherosclerosis.
Expert Rev Cardiovasc Ther. 14:1021–1033. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Theodorou K and Boon RA: Endothelial cell
metabolism in atherosclerosis. Front Cell Dev Biol. 6:822018.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Verma I, Syngle A, Krishan P and Garg N:
Endothelial progenitor cells as a marker of endothelial dysfunction
and atherosclerosis in Ankylosing Spondylitis: A cross-sectional
study. Int J Angiol. 26:36–42. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lee DY and Chiu JJ: Atherosclerosis and
flow: Roles of epigenetic modulation in vascular endothelium. J
Biomed Sci. 26:562019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Georgescu A, Alexandru N, Andrei E, Dragan
E, Cochior D and Dias S: Effects of transplanted circulating
endothelial progenitor cells and platelet microparticles in
atherosclerosis development. Biol Cell. 108:219–243. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xiang W, Hu ZL, He XJ and Dang XQ:
Intravenous transfusion of endothelial progenitor cells that
overexpress vitamin D receptor inhibits atherosclerosis in
apoE-deficient mice. Biomed Pharmacother. 84:1233–1242. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kong M, Zhao Y, Chen A and Lin A: The
importance of physiologic ischemia training in preventing the
development of atherosclerosis: The role of endothelial progenitor
cells in athero-sclerotic rabbits. Coron Artery Dis. 30:377–383.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Pákozdi A, Besenyei T, Paragh G, Koch AE
and Szekanecz Z: Endothelial progenitor cells in
arthritis-associated vasculogenesis and atherosclerosis. Joint Bone
Spine. 76:581–583. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hu X, Cai X, Ma R, Fu W, Zhang C and Du X:
Iron-load exacerbates the severity of atherosclerosis via inducing
inflammation and enhancing the glycolysis in macrophages. J Cell
Physiol. 234:18792–18800. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Flynn MC, Kraakman MJ, Tikellis C, Lee
MKS, Hanssen NMJ, Kammoun HL, Pickering RJ, Dragoljevic D,
Al-Sharea A, Barrett TJ, et al: Transient intermittent
hyperglycemia accelerates atherosclerosis by promoting
myelopoiesis. Circ Res. 127:877–892. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Matsuura Y, Yamashita A, Zhao Y, Iwakiri
T, Yamasaki K, Sugita C, Koshimoto C, Kitamura K, Kawai K, Tamaki
N, et al: Altered glucose metabolism and hypoxic response in
alloxan-induced diabetic atherosclerosis in rabbits. PLoS One.
12:e01759762017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Vojinovic D, van der Lee SJ, van Duijn CM,
Vernooij MW, Kavousi M, Amin N, Demirkan A, Ikram MA, van der Lugt
A and Bos D: Metabolic profiling of intra- and extracranial carotid
artery atherosclerosis. Atherosclerosis. 272:60–65. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Akins NS, Nielson TC and Le HV: Inhibition
of glycolysis and glutaminolysis: An emerging drug discovery
approach to combat cancer. Curr Top Med Chem. 18:494–504. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Deng F, Zhou R, Lin C, Yang S, Wang H, Li
W, Zheng K, Lin W, Li X, Yao X, et al: Tumor-secreted dickkopf2
accelerates aerobic glycolysis and promotes angiogenesis in
colorectal cancer. Theranostics. 9:1001–1014. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Godfrey R and Quinlivan R: Skeletal muscle
disorders of glycogenolysis and glycolysis. Nat Rev Neurol.
12:393–402. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Schoors S, De Bock K, Cantelmo AR,
Georgiadou M, Ghesquière B, Cauwenberghs S, Kuchnio A, Wong BW,
Quaegebeur A, Goveia J, et al: Partial and transient reduction of
glycolysis by PFKFB3 blockade reduces pathological angiogenesis.
Cell Metab. 19:37–48. 2014. View Article : Google Scholar
|
|
66
|
Fernie AR, Carrari F and Sweetlove LJ:
Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial
electron transport. Curr Opin Plant Biol. 7:254–261. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tang BL: Glucose, glycolysis, and
neurodegenerative diseases. J Cell Physiol. 235:7653–7662. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Paik JY, Lee KH, Ko BH, Choe YS, Choi Y
and Kim BT: Nitric oxide stimulates 18F-FDG uptake in human
endothelial cells through increased hexokinase activity and GLUT1
expression. J Nucl Med. 46:365–370. 2005.PubMed/NCBI
|
|
69
|
Kim JW, Gao P, Liu YC, Semenza GL and Dang
CV: Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively
induce vascular endothelial growth factor and metabolic switches
hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol.
27:7381–7393. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Song J, Li Y, Song J, Hou F, Liu B and Li
A: Mangiferin protects mitochondrial function by preserving
mitochondrial hexokinase-II in vessel endothelial cells. Biochim
Biophys Acta Mol Basis Dis. 1863:1829–1839. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Wang Y, Han X, Fu M, Wang J, Song Y, Liu
Y, Zhang J, Zhou J and Ge J: Qiliqiangxin attenuates
hypoxia-induced injury in primary rat cardiac microvascular
endothelial cells via promoting HIF-1α-dependent glycolysis. J Cell
Mol Med. 22:2791–2803. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wik JA, Lundbäck P, la Cour Poulsen L,
Haraldsen G, Skålhegg BS and Hol J: 3PO inhibits inflammatory NFκB
and stress-activated kinase signaling in primary human endothelial
cells independently of its target PFKFB3. PLoS One.
15:e02293952020. View Article : Google Scholar
|
|
73
|
Zhang R, Li R, Liu Y, Li L and Tang Y: The
glycolytic enzyme PFKFB3 controls TNF-α-induced endothelial
proinflammatory responses. Inflammation. 42:146–155. 2019.
View Article : Google Scholar
|
|
74
|
Lu S, Deng J, Liu H, Liu B, Yang J, Miao
Y, Li J, Wang N, Jiang C, Xu Q, et al: PKM2-dependent metabolic
reprogramming in CD4+ T cells is crucial for
hyperhomocysteinemia-accelerated atherosclerosis. J Mol Med (Berl).
96:585–600. 2018. View Article : Google Scholar
|
|
75
|
Zhang X, Chen B, Wu J, Sha J, Yang B, Zhu
J, Sun J, Hartung J and Bao E: Aspirin enhances the protection of
Hsp90 from heat-stressed injury in cardiac microvascular
endothelial cells through PI3K-Akt and PKM2 pathways. Cells.
9:2432020. View Article : Google Scholar :
|
|
76
|
Serganova I, Cohen IJ, Vemuri K, Shindo M,
Maeda M, Mane M, Moroz E, Khanin R, Satagopan J, Koutcher JA and
Blasberg R: LDH-A regulates the tumor microenvironment via
HIF-signaling and modulates the immune response. PLoS One.
13:e02039652018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Chen SF, Pan MX, Tang JC, Cheng J, Zhao D,
Zhang Y, Liao HB, Liu R, Zhuang Y, Zhang ZF, et al: Arginine is
neuroprotective through suppressing HIF-1α/LDHA-mediated
inflammatory response after cerebral ischemia/reperfusion injury.
Mol Brain. 13:632020. View Article : Google Scholar
|
|
78
|
Fernández-Hernando C, József L, Jenkins
D, Di Lorenzo A and Sessa WC: Absence of Akt1 reduces vascular
smooth muscle cell migration and survival and induces features of
plaque vulnerability and cardiac dysfunction during
atherosclerosis. Arterioscler Thromb Vasc Biol. 29:2033–2040. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Pan C, Liu Q and Wu X:
HIF1α/miR-520a-3p/AKT1/mTOR feedback promotes the proliferation and
glycolysis of gastric cancer cells. Cancer Manag Res.
11:10145–10156. 2019. View Article : Google Scholar :
|
|
80
|
Zhong ZW, Zhou WC, Sun XF, Wu QC, Chen WK
and Miao CH: Dezocine regulates the malignant potential and aerobic
glycolysis of liver cancer targeting Akt1/GSK-3β pathway. Ann
Transl Med. 8:4802020. View Article : Google Scholar
|
|
81
|
Zhao X, Wu X, Wang H, Yu H and Wang J:
USP53 promotes apoptosis and inhibits glycolysis in lung
adenocarcinoma through FKBP51-AKT1 signaling. Mol Carcinog.
59:1000–1011. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Song L and Schindler C: IL-6 and the acute
phase response in murine atherosclerosis. Atherosclerosis.
177:43–51. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhang P, Chen X, Zhang Y, Su H, Zhang Y,
Zhou X, Sun M, Li L and Xu Z: Tet3 enhances IL-6 expression through
up-regulation of 5-hmC in IL-6 promoter in chronic hypoxia induced
athero-sclerosis in offspring rats. Life Sci. 232:1166012019.
View Article : Google Scholar
|
|
84
|
Bozic M, Alvarez A, de Pablo C,
Sanchez-Niño MD, Ortiz A, Dolcet X, Encinas M, Fernandez E and
Valdivielso JM: Impaired vitamin D signaling in endothelial cell
leads to an enhanced leukocyte-endothelium interplay: Implications
for atheroscle-rosis development. PLoS One. 10:e01368632015.
View Article : Google Scholar
|
|
85
|
Han J, Meng Q, Xi Q, Zhang Y, Zhuang Q,
Han Y, Jiang Y, Ding Q and Wu G: Interleukin-6 stimulates aerobic
glycolysis by regulating PFKFB3 at early stage of colorectal
cancer. Int J Oncol. 48:215–224. 2016. View Article : Google Scholar
|
|
86
|
Li H, Liang Q and Wang L: Icaritin
inhibits glioblastoma cell viability and glycolysis by blocking the
IL-6/Stat3 pathway. J Cell Biochem. Nov 2–2018.Epub ahead of print.
View Article : Google Scholar
|
|
87
|
Zhao N and Zhang J: Role of alternative
splicing of VEGF-A in the development of atherosclerosis. Aging
(Albany NY). 10:2695–2708. 2018. View Article : Google Scholar
|
|
88
|
Wang X, Hu Z, Wang Z, Cui Y and Cui X:
Angiopoietin-like protein 2 is an important facilitator of tumor
proliferation, metastasis, angiogenesis and glycolysis in
osteosarcoma. Am J Transl Res. 11:6341–6355. 2019.PubMed/NCBI
|
|
89
|
Peek CB, Levine DC, Cedernaes J, Taguchi
A, Kobayashi Y, Tsai SJ, Bonar NA, McNulty MR, Ramsey KM and Bass
J: Circadian clock interaction with HIF1α mediates oxygenic
metabolism and anaerobic glycolysis in skeletal muscle. Cell Metab.
25:86–92. 2017. View Article : Google Scholar
|
|
90
|
Zhao M, Fan J, Liu Y, Yu Y, Xu J, Wen Q,
Zhang J, Fu S, Wang B, Xiang L, et al: Oncogenic role of the
TP53-induced glycolysis and apoptosis regulator in nasopharyngeal
carcinoma through NF-κB pathway modulation. Int J Oncol.
48:756–764. 2016. View Article : Google Scholar
|
|
91
|
Ko YH, Domingo-Vidal M, Roche M, Lin Z,
Whitaker-Menezes D, Seifert E, Capparelli C, Tuluc M, Birbe RC,
Tassone P, et al: TP53-inducible glycolysis and apoptosis regulator
(TIGAR) metabolically reprograms carcinoma and stromal cells in
breast cancer. J Biol Chem. 291:26291–26303. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Xiong Y, Yepuri G, Forbiteh M, Yu Y,
Montani JP, Yang Z and Ming XF: ARG2 impairs endothelial autophagy
through regulation of MTOR and PRKAA/AMPK signaling in advanced
atherosclerosis. Autophagy. 10:2223–2238. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Li X, Wu L, Zopp M, Kopelov S and Du W:
p53-TP53-Induced glycolysis regulator mediated glycolytic
suppression attenuates DNA damage and genomic instability in
fanconi anemia hematopoietic stem cells. Stem Cells. 37:937–947.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Chen Q, Lv J, Yang W, Xu B, Wang Z, Yu Z,
Wu J, Yang Y and Han Y: Targeted inhibition of STAT3 as a potential
treatment strategy for atherosclerosis. Theranostics. 9:6424–6442.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Zhou X, Li D, Yan W and Li W: Pravastatin
prevents aortic atherosclerosis via modulation of signal
transduction and activation of transcription 3 (STAT3) to attenuate
interleukin-6 (IL-6) action in ApoE knockout mice. Int J Mol Sci.
9:2253–2264. 2008. View Article : Google Scholar
|
|
96
|
Li Y, Wang Y, Liu Z, Guo X, Miao Z and Ma
S: Atractylenolide I induces apoptosis and suppresses glycolysis by
blocking the JAK2/STAT3 signaling pathway in colorectal cancer
cells. Front Pharmacol. 11:2732020. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Zheng M, Cao MX, Yu XH, Li L, Wang K, Wang
SS, Wang HF, Tang YJ, Tang YL and Liang XH: STAT3 promotes invasion
and aerobic glycolysis of human oral squamous cell carcinoma via
inhibiting FoxO1. Front Oncol. 9:11752019. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Seki N, Hashimoto N, Taira M, Yagi S,
Yoshida Y, Ishikawa K, Suzuki Y, Sano H, Horiuchi S, Yoshida S, et
al: Regulation of Src homology 2-containing protein tyrosine
phosphatase by advanced glycation end products: The role on
atherosclerosis in diabetes. Metabolism. 56:1591–1598. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Park JS, Lee S, Jeong AL, Han S, Ka HI,
Lim JS, Lee MS, Yoon DY, Lee JH and Yang Y: Hypoxia-induced IL-32β
increases glycolysis in breast cancer cells. Cancer Lett.
356:800–808. 2015. View Article : Google Scholar
|
|
100
|
Nam K, Oh S and Shin I: Ablation of CD44
induces glycolysis-to-oxidative phosphorylation transition via
modulation of the c-Src-Akt-LKB1-AMPKα pathway. Biochem J.
473:3013–3030. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Byun S, Jung H, Chen J, Kim YC, Kim DH,
Kong B, Guo G, Kemper B and Kemper JK: Phosphorylation of hepatic
farnesoid X receptor by FGF19 signaling-activated Src maintains
cholesterol levels and protects from atherosclerosis. J Biol Chem.
294:8732–8744. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Lovren F, Pan Y, Shukla PC, Quan A, Teoh
H, Szmitko PE, Peterson MD, Gupta M, Al-Omran M and Verma S:
Visfatin activates eNOS via Akt and MAP kinases and improves
endothelial cell function and angiogenesis in vitro and in vivo:
Translational implications for atherosclerosis. Am J Physiol
Endocrinol Metab. 296:1440–1449. 2009. View Article : Google Scholar
|
|
103
|
Gupta A, Mohanty P and Bhatnagar S:
Integrative analysis of ocular complications in atherosclerosis
unveils pathway convergence and crosstalk. J Recept Signal
Transduct Res. 35:149–164. 2015. View Article : Google Scholar
|
|
104
|
Perrotta P, Emini Veseli B, Van der Veken
B, Roth L, Martinet W and De Meyer GRY: Pharmacological strategies
to inhibit intra-plaque angiogenesis in atherosclerosis. Vascul
Pharmacol. 112:72–78. 2019. View Article : Google Scholar
|