The Golgi apparatus is a processing and sorting hub
in the transport and targeting of soluble cargo proteins and lipids
to different destinations in the cell (1). Considering its central role in the
secretory pathway, alterations in the structure and function of the
Golgi apparatus are expected to affect the homeostasis of cellular
proteins and lipids. Increasing evidence suggests that structural
changes and functional disorder of the Golgi apparatus are involved
in many human diseases such as neurodegenerative diseases (2-4),
ischemic stroke (5,6), cardiovascular diseases (7,8),
pulmonary arterial hypertension (9,10), infectious diseases (11-13), and cancer (14). However, much work is still needed
to elucidate how the Golgi apparatus affects the progression of
these diseases.
In this review, we describe the central roles of the
Golgi apparatus in cells, and discuss diseases associated with
structural changes and functional disorder of the Golgi apparatus.
We highlight some of the studies that explore links between
mutation in genes encoding Golgi resident proteins and human
diseases. By analyzing their pathophysiology, we found that the
majority of genes leading to human diseases are involved in
membrane trafficking. Considering the mechanistic links between
Golgi resident proteins, membrane trafficking, and the development
of genetic diseases, we suggest a term for these disorders based on
their similar pathophysiology: Golgi apparatus membrane trafficking
disorders.
In 1898, the Italian anatomist Camillio Golgi
initially described the cell organelle that bears his name, the
Golgi apparatus (15). The Golgi
apparatus is characterized by a series of flattened, cisternal
membrane structures forming the so-called Golgi stack, which is
surrounded by vesicles. Based on the distribution of resident
proteins, the Golgi stack can be divided into three regions: The
cis-, medial-, and trans-Golgi cisternae (16). The Golgi stacks in vertebrate
cells are laterally interconnected by tubular membranes and exhibit
a twisted ribbon-like network known as the Golgi ribbon (17). The structure of the Golgi ribbon
is supported by the Golgi matrix (18). The Golgi matrix is believed to
comprise highly dynamic structural proteins, which is important for
structural integrity and vesicular trafficking.
The Golgi apparatus has two main functions. The
first is the post-translational protein modification. Similar to
glycosylation, it is a common post-translational modification
occurring in the endoplasmic reticulum (ER) and Golgi and the
glycan processing occurs throughout the Golgi stacks. The second is
the sorting, packing, routing and recycling of these modified
cargos to the appropriate cellular destinations (1). The main secretory pathway can be
divided into the following steps (19): First, newly synthesized proteins
or lipids enter the exit sites of the ER and are sorted into
budding vesicles that are dependent on the COPII. Second, vesicles
move to the ER-Golgi intermediate compartment (ERGIC) and forward
to the cis-Golgi networks (CGN). Third, proteins or lipids enter
cis-Golgi cisternae and move towards the trans-Golgi cisternae.
Vesicular transport and cisternal maturation are the two classical
models of intra-Golgi transport (20). The vesicular transport model
proposes that Golgi cisternae are static, and the cargos are
transported through them by COPI vesicles. The cisternal maturation
model suggests that cisternae are dynamic structures, while Golgi
enzymes are recycled via retrograde transport of COPI vesicles.
Fourth, vesicles reach the trans-Golgi networks (TGN), which are
involved in the sorting of products to their final destinations
such as lysosomes, endosomes, or the plasma membrane.
The structural integrity of the Golgi apparatus is
vital for its normal function, and Golgi fragmentation could result
in a wide range of diseases and disorders. Functional changes of
the Golgi Apparatus include perturbations in Golgi pH, aberrant
Golgi glycosylation, and membrane trafficking. Golgi fragmentation
has been found to often be an early causative event in the process
of cell apoptosis (21,22). With pharmacological or oxidative
stress, a series of changes occur in the Golgi apparatus, such as
cargo overloading, ionic imbalance, and abnormal luminal acidity.
These changes can lead to defects in membrane trafficking. We
previously presented 'Golgi stress' as a new concept to explain the
Golgi-specific stress response (23). The Golgi stress response
constitutes autoregulation to repair the Golgi apparatus and may
initiate signaling pathways to alleviate stress. The nucleus
signaling pathways of the Golgi stress response was identified in a
previous study: The procaspase-2/golgin-160, TFE3, HSP47, and the
CREB3-ARF4 pathways (24). If
these pathways fail to repair overstimulation, the Golgi is
completely disassembled, inducing cell apoptosis.
Apoptosis triggered by structural changes and
functional disorder of the Golgi contributes to the pathogenesis of
many diseases, such as neurodegenerative diseases (25), ischemic stroke (5,6),
cardiovascular diseases (26),
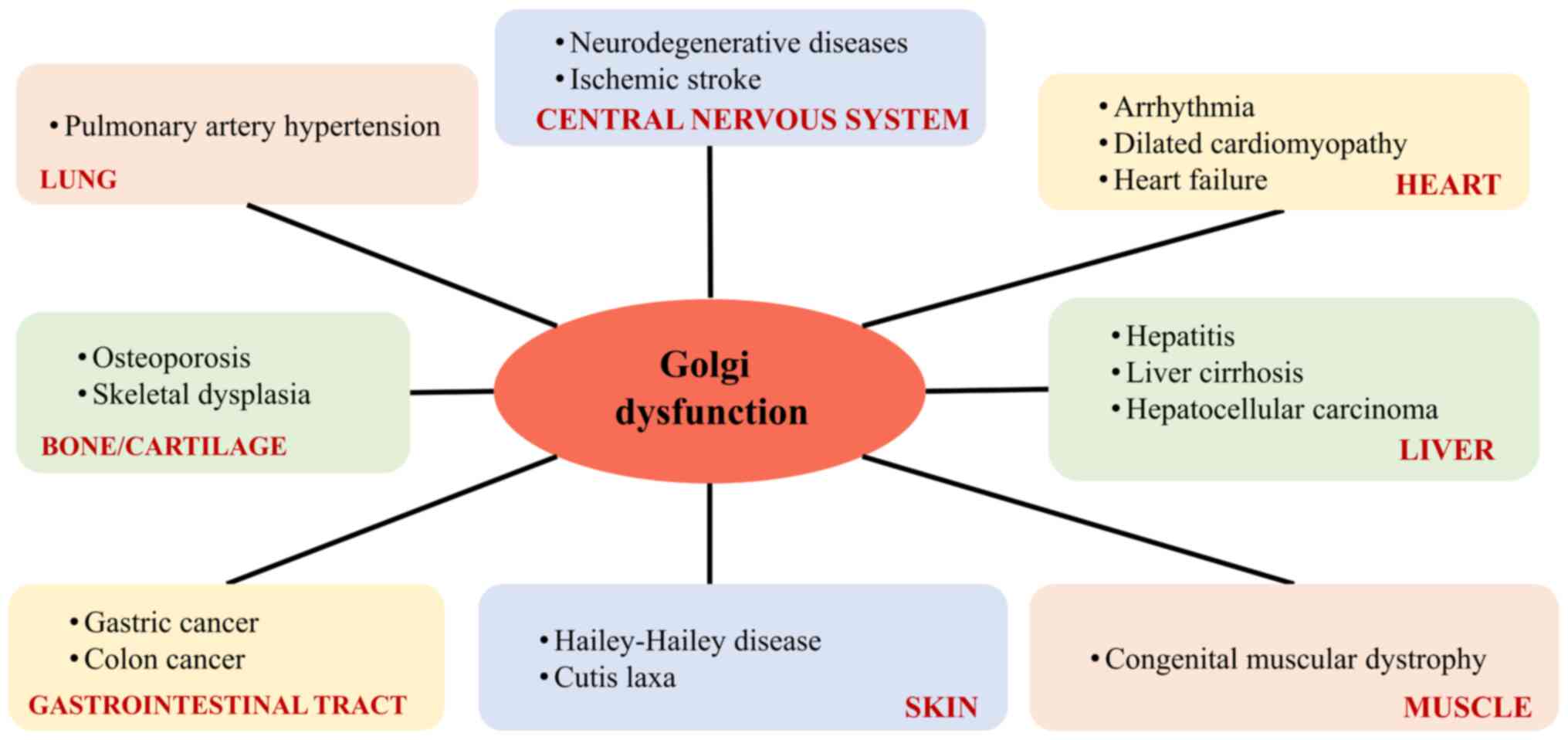
pulmonary arterial hypertension (9,10), infectious diseases (12,13), and cancer (27). A summary of diseases relating to
the Golgi apparatus, classified on the basis of the main organ
affected is shown in Fig. 1.
Structural and functional changes of the Golgi
apparatus are associated with several neurodegenerative diseases,
such as Amyotrophic lateral sclerosis (28), Alzheimer's disease (29), Parkinson's disease (3), Huntington's disease (30), Creutzfeldt-Jacob disease
(31) and multiple system
atrophy (32). Golgi
fragmentation is not a consequence of apoptosis, but a very early
event in the pathological cascade in neurodegenerative disorders
and precedes other pathological changes in the neuron (33). Golgi fragmentation may alter
neuronal physiology, and induce failures in transport to axons,
dendrites, and synapses (34).
Finally, Golgi alteration may trigger a stress response and, as
consequence, result in neuronal death. Furthermore, Golgi
fragmentation in neurodegenerative disease alters protein
trafficking and production, such as amyloid precursor protein in
Alzheimer's disease (35), and
sodium-dependent vitamin C transporter 2 in Huntington's disease
(36). The causes of Golgi
fragmentation in neurodegenerative diseases may be diverse. First,
alteration of the microtubule and microfilament stabilization may
also be the cause (37). In
Alzheimer's disease and other tauopathies, tau-induced
microtubule-bundling may result in Golgi fragmentation (38). Furthermore, perturbations in
Golgi pH are also responsible for Golgi fragmentation. The Purkinje
cells from the Golgi pH regulator conditional knockout mice
exhibited Golgi fragmentation, followed by axonal degeneration and
neuronal loss (39).
Golgi fragmentation has been identified in diseases
such as infection by Orf virus (12), Chlamydia trachomatis
(40,41), Hepatitis C virus (HCV) (42), Human Rhinovirus (HRV) (13), and Rickettsia rickettsii
(43). Golgi fragmentation in
these infectious diseases is mainly reflected in two aspects: i)
Escaping from the immune response. In infected cells, Golgi
fragmentation reduces MHC class I complex surface expression by
defective membrane trafficking (43,44), which may aid in escaping host
cellular immune recognition (12); ii) Enhancing viral replication.
In human rhinovirus-1A infection, the Golgi in host cells is
fragmented and rearranged into vesicles that appear to be used as
the membrane source for the assembly of viruses (45). Similarly, in Oropouche virus
replication, proteins in the endosomal sorting complex required for
transport in the host cell are hijacked in Golgi cisternae to
mediate remodeling of Golgi membranes, resulting in enlargement of
the Golgi stacks, where the endosomal sorting complex required for
transport participates in the assembly of viral factories (46). Thus, structural changes in the
Golgi apparatus may enhance viral replication in infectious
diseases by providing membranes.
Aberrant Golgi glycosylation is reported to regulate
invasion of cancer cells, such as in prostate (47), breast (48), and gastric cancer (49). Golgi glycosylation is involved in
basic molecular and cellular biology processes occurring in cancer,
such as cell signaling transduction and communication, cancer cell
dissociation and invasion, cell-matrix adhesion, cancer
angiogenesis, immune regulation and metastasis (50). Similar to epithelial cadherin, a
transmembrane glycoprotein, is involved in epithelial cell-cell
adhesion in tumors (51). The
Golgi glycosylation of N-linked glycans on epithelial cadherin can
affect the epithelial-mesenchymal transition, which is related to
the formation of metastatic lesions (49). This process is suggested to help
cancer cells leave their original position during wound healing and
other normal physiological processes, which is an essential
mechanism for metastasis and diffusion of cancer cells (52,53). The GOLPH3 complex is an important
molecular component in the process of Golgi-driven tumor
progression. The role of the GOLPH3 complex in cancer includes: i)
Regulating Golgi glycosylation, which is important in driving the
cancer phenotype (54); ii)
promoting the cellular DNA damage response that enhances cellular
survival under DNA damage (55);
iii) interacting with components of the retromer complex that
enhances growth-factor-induced mTOR signaling (56); and iv) regulating cell migration
by promoting reorientation of the Golgi apparatus towards the
leading edge (57). In addition
to GOLPH3, the Golgi protein GM130 is important in Golgi
glycosylation and protein membrane trafficking in cancer cells.
Downregulation of GM130 induces autophagy, inhibits glycosylation,
decreases angiogenesis, and suppresses tumorigenesis (58). In general, aberrant Golgi
glycosylation causes carcinogenesis, but may also be a consequence
of cancer progression.
Golgi dysfunction was also observed in pulmonary
arterial hypertension, and cardiovascular diseases. In an in
vivo model of pulmonary arterial hypertension, Golgi
dysfunction and intracellular trafficking with trapping of diverse
vesicle tethers, giantin, p115, and soluble
N-ethylmaleimide-sensitive factor attachment protein receptors
(SNAREs) were observed in the Golgi membranes of enlarged pulmonary
arterial endothelial cells and smooth muscle cells (9,10,59). Golgi-mediated membrane
trafficking dysfunctions play important roles in the pathogenesis
of pulmonary arterial hypertension (60).
Structural changes and functional disorder of the
Golgi apparatus have been identified in many cardiovascular
diseases, such as heart failure, dilated cardiomyopathy,
arrhythmia, and chronic arial fibrillation (61-64). A previous review clarified the
relationship between the Golgi apparatus and various cardiovascular
diseases (26). For example, in
dilated cardiomyopathy patients, morphological changes in Golgi
vesicle are consistent with the secretion of natriuretic peptide as
the rate of protein secretion affects the morphology and size of
Golgi vesicles (7). In addition,
the Golgi vesicle area is inversely proportional to the left
ventricular end-diastolic diameter and the end-systolic diameter,
and is proportional to the left ventricular ejection fraction
(65).
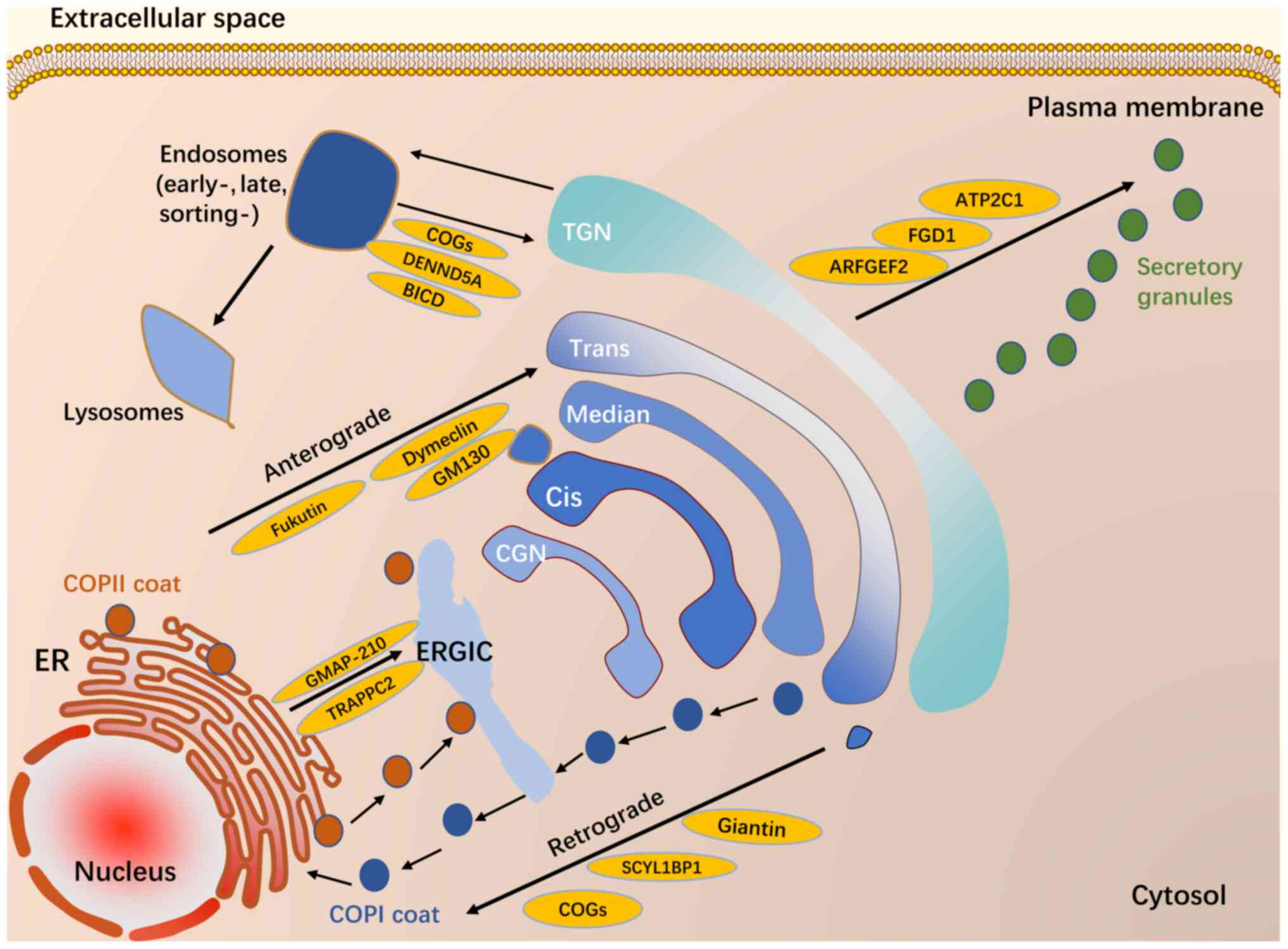
In addition to being an intermediate site in
pathogenic cascades in diseases, the Golgi apparatus can be the
primary target for diseases caused by genetic mutations in Golgi
resident proteins. Mutations in proteins localized to the Golgi
apparatus can be deleterious for the structure and function of this
organelle, impeding membrane trafficking pathways through it
(Fig. 2) and resulting in
disease. We highlight some of the studies that explore links
between Golgi resident proteins and disease.
Adjacent Golgi stacks are linked by tubules forming
a membrane network termed the Golgi ribbon (66). This structure is a highly ordered
and continuous structure that is adjacent to the nucleus. The Golgi
ribbon comprises proteins that mediate cisternal stacking and the
material supporting the Golgi ribbon is the Golgi matrix (67). The concept of the Golgi matrix
was introduced by Slusarewicz and colleagues, who isolated a
detergent-insoluble, salt-resistant Golgi fraction in 1994
(18). The main function of the
Golgi matrix is maintaining normal structure and mediating protein
trafficking through the Golgi cisternae. During cisternal
progression, the Golgi matrix must be dynamic to adapt to Golgi
structural changes.
Golgi matrix proteins include golgins and Golgi
reassembly stacking proteins (GRASPs) (67), both of which are important for
maintaining Golgi structure and regulating protein and lipid
trafficking through the stacks. Golgins are a family of conserved
coiled-coil proteins that were originally identified as a group of
Golgi-localized antigens (68,69). The golgins not only capture
incoming vesicles, but also clearly distinguish vesicles from
different origins (70). GRASPs
include GRASP65 (71) and
GRASP55 (72). The former
localizes to the cis-Golgi cisternae while the latter localizes to
the medial/trans-Golgi cisternae. The functions of GRASPs include
Golgi structure formation, specific cargo transport, apoptosis, and
cell migration (73).
Given the important multiple functions of Golgi
matrix proteins, mutation of Golgi matrix proteins has serious
consequences on health. Increasing studies support that the
mutation of Golgi matrix proteins including GM130, Bicaudal-D
(BICD), GMAP-210, giantin (74),
and SCYL1BP1 (also known as GORAB) (75), leads to diseases. The present
review included some proteins as examples to elaborate on the
pathogenic mechanism of Golgi matrix proteins.
The first example is GM130 (also known as GOLGA2),
the first identified Golgi matrix protein (76). GM130 is a peripheral membrane
protein attached to the Golgi membrane that is important in
maintaining the adaxial Golgi reticular structure (77). In neurodegenerative diseases,
GM130 knockout in hippocampal neurons is reported to cause
damage to dendritic structures (78). In mouse neuron experiments,
specific knockout of GM130 resulted in disruption of the
Golgi architecture and positioning in cerebellar Purkinje cells and
to deficient secretory cargo trafficking. As a consequence,
progressive cerebellar atrophy of Purkinje cells resulted in
delayed movement and ataxia in mice (79). This animal experimental study
indicates that GM130 mutations are causative in
neurodegenerative disease.
The fourth example is GORAB (also known as
SCYL1BP1). GORAB, localized to the trans-side of the Golgi, is a
member of the golgin family and interacts with Rab6. Mutation in
GORAB results in gerodermia osteodysplastica (GO)
characterized by wrinkly skin and osteoporosis (75). GORAB functions in COPI
trafficking, and acts as a scaffolding factor for COPI assembly at
the TGN by interacting with Scyl1. GORAB mutations perturb
COPI assembly at the TGN, and result in reduced recycling of
COPI-mediated retrieval of trans-Golgi enzymes and improper
glycosylation (90).
A final example of the effects of loss of expression
of a Golgi matrix protein is GMAP-210 (also known as TRIP11). This
CGN golgin acts in asymmetric membrane tethering (91). In animal experiments, a nonsense
mutation in Trip11 led to a loss of GMAP-210, which led to
abnormal Golgi-mediated glycosylation and cellular transport of
proteins in chondrocytes and osteoblasts of mice (92). Similarly, GMAP-210
mutations were found in patients with human chondrodysplasia
achondrogenesis 1A (92), and
odontochondrodysplasia (93).
In addition to matrix proteins, several proteins
that localize to Golgi membranes are also important for normal
Golgi structure and function such as the tethering factors Rab
GTPases and SNAREs, which regulate the specific targeting and
fusion of transport carriers with Golgi membranes. The maintenance
of Golgi luminal ion concentrations depends on the secretory
pathway Ca2+/Mn2+ ATPases and vacuolar
H+ ATPase (V-ATPase). Therefore, the impaired
performance of mutated Golgi resident proteins creates serious and
highly diverse pathologies in the Golgi. Emerging studies on
patient genetics have identified mutations in Golgi resident
protein-coding genes that are related to diseases. We focus on some
of these proteins, and discuss the activities of mutated Golgi
resident proteins that result in disease.
Golgi acidity is an important role for maintaining
the morphological integrity of the Golgi and transporting various
kinds of cargo (99,100). Under normal conditions, the
Golgi cavity is weakly acidic and the pH of the Golgi reticular
structure decreases gradually from the CGN to the TGN (101). The Golgi luminal pH is
regulated by V-ATPase (102),
AE2a HCO3-/Cl- exchanger, and Golgi pH
regulator (103). Luminal pH is
closely tied to Golgi function. Partial V-ATPase dysfunction is
related to multiple disease states (104). ATP6V1E1, ATP6V1A,
and ATP6V0A2 encode different subunits of the V-ATPase pump.
A study showed that Golgi subunit-isoform of the V-ATPase
(ATP6V0A2) mutations lead to structural changes in the
extracellular matrix that is responsible for skin elasticity
(105). Clinically, the
dysfunction of the Golgi-localized V-ATPase caused by mutations in
the ATP6VOA2 gene is directly related to cutis laxa.
Mutations in ATP6V1E1 or ATP6V1A also cause
autosomal-recessive cutis laxa (106). Autosomal recessive cutis laxa
type II is a heterogeneous condition characterized by sagging,
inelastic, and wrinkled skin (107,108). The mechanism may involve
impaired intracellular acidification of the Golgi and damaged
retrograde trafficking from the Golgi to the ER (100,108).
The Golgi apparatus is an important organelle for
the post-translational modification of cargos. The
post-translational modification of secreted and membrane proteins
is mediated by the Golgi resident enzymes such as
glycosyltransferases, glycosidases, and kinases. Glycosylation is
an enzymatic reaction that chemically links monosaccharides or
polysaccharides (glycans) to other saccharides, proteins, or lipids
(111). Golgi glycosylation is
a modification by Golgi-resident glycosylation enzymes including
glycosidases and glycosyltransferases (112). The normal function of Golgi
glycosylation depends on the precise Golgi localization and normal
activities of Golgi resident enzymes. The proper localization of
Golgi resident enzymes is controlled by finely regulated vesicular
trafficking in the Golgi. If the balance between anterograde and
retrograde trafficking is defective, Golgi glycosylation is
affected, resulting in Golgi glycosylation abnormalities (113). Mutations in Golgi resident
putative glycosyltransferases are directly linked to human
congenital muscular dystrophies:
Like-acetylglucosaminyl-transferase (LARGE) in congenital muscular
dystrophy syndrome (114),
fukutin in Fukuyama-type congenital muscular dystrophy (115), and fukutin-related protein in
band muscular dystrophy syndrome (116). These mutations appear to affect
cell migration in the developing brain, resulting in combined
clinical manifestations in muscle and brain development. In an
animal model, mutations in Golgi resident glycosyltransferases are
also associated with the neurodegenerative disease, such as
ST3GAL5,β1,4-gala ctosyltransferase 4 (B4GalT4) (117), and glycosyltransferase 8 domain
containing 1 (GLT8D1). GLT8D1 is a glycosyltransferase enzyme
located in the Golgi apparatus. A recent study reported that
mutated GLT8D1 induces motor deficits in zebrafish embryos
consistent with amyotrophic lateral scle- rosis (118). However, another study suggested
that GLT8D1 is not likely the causative gene for ALS in mainland
China (119).
Rab proteins are members of the small Ras-like
GTPase family that regulate the four steps of membrane transport by
recruiting effector molecules. Golgi-associated Rab proteins
including Rab1, Rab2, Rab6, Rab18, Rab33B, and Rab43 have a central
role in Golgi organization and membrane trafficking (120). Rab33B is localized to
medial-Golgi cisternae and is important in Golgi-to-ER retrograde
trafficking. Rab39B, a neuronal-specific protein, is a novel Rab
GTPase that localizes to the Golgi and is related to synapse
formation. Mutations in the Rab33B coding gene cause
Smith-McCort dysplasia (121)
and mutations in the Rab39B gene cause X-linked mental
retardation (122).
SNAREs are proteins involved in docking and fusion
of transport to intermediate membranes. Golgi SNAP receptor complex
member 2 (GOSR2) is a member of the SNAREs family that localizes to
the CGN and is involved in ER-to-Golgi trafficking (123). Homozygous mutations in
GOSR2 lead to progressive myoclonus epilepsy (124). Clinical manifestations include
early ataxia, myoclonus, and convulsive seizures. A possible
mechanism involves GOSR2 mutations leading to GOSR2 protein
that cannot be localized to the CGN and blocks SNAREs complex
formation. SNAREs complex dysfunction could lead to the impaired
fusion of vesicles with cis-Golgi cisternae, hindering ER-to-Golgi
membrane trafficking. The perturbation of early ER-to-Golgi
transport may result in changes in the regulated release of
neurotransmitters and proper sorting of neurotransmitter receptors
at synapses in neurons, potentially leading to epilepsy (125,126).
In the above section, we introduced the
pathophysiology of some diseases related to Golgi resident
proteins. A summary of genetic diseases caused by mutations in
genes encoding Golgi resident proteins is presented in Table I. By analyzing the
pathophysiology of these diseases, we found that the majority of
genes leading to human diseases are involved in defects in membrane
trafficking (Fig. 2). For
example, TRAPPC2 mutation, involving the membrane
trafficking pathway between ER-to-Golgi in bone cells and
chondrocytes, results in X-linked spondyloepiphyseal dysplasia
tarda (127). The conserved
oligomeric Golgi (COG) complex is a conserved, hetero-octameric
protein complex localized in the Golgi cis/medial cisternae
(128). In addition to the COG3
subunit, mutations in seven other COG subunits result in human
congenital disorders of glycosylation (CD G) type II, which is
mainly marked by misregulation of protein glycosylation, and
defects in retrograde trafficking through the Golgi (129,130). The mutation in FGD1
resulting in Aarskog-Scott syndrome may lead to the obstruction of
post-Golgi trafficking, such as the Golgi-to-plasma membrane
trafficking pathway (131).
Mutation in TRIP11 mainly involves ER to ERGIC and
anterograde trafficking (132).
Therefore, membrane trafficking defects play a major role in the
pathogenic process of mutation in genes encoding Golgi resident
protein. Intracellular membrane trafficking is a fundamental
process responsible for compartmentalization of the biosynthesis
pathway and secretion cargos, including hormones, growth factors,
antibodies, matrix and serum proteins, digestive enzymes, and many
more. Defective membrane trafficking results in protein sorting
defects, undegraded proteins due to defective Golgi-to-lysosome
trafficking, downregulation of protein secretion, and
mislocalization of proteins.
Considering the mechanistic links between Golgi
resident proteins, membrane trafficking, and the development of
genetic diseases, we suggest a term for these disorders based on
their similar pathophysiology: Golgi apparatus membrane trafficking
disorders. It is a group of genetic diseases in which the mutation
of the gene encoding Golgi resident protein results in membrane
trafficking defects within the cells. Golgi apparatus membrane
trafficking defects typically result in the accumulation of
undegraded proteins, mislocalization of proteins, and impaired
glycosylation of proteins. However, the cascade events following
the Golgi apparatus and defective membrane trafficking, ultimately
leading to human diseases, remain to be clarified in further
research.
Although the Golgi apparatus-mediated membrane
trafficking pathway exists in all kinds of tissues and organs in
human, the trafficking defects on tissues is often selective. The
most sensitive to membrane trafficking defects is the nervous
system, skin, bone, cartilage, and skeletal muscle and the reasons
for mutations occurring in these genes mostly affecting these
tissues remain to be elucidated. Firstly, neurons are
extraordinarily polarized cells, the extension of dendrites and
axons requires a significant expansion of the cell surface area,
and new plasma membrane proteins must be delivered through the
membrane trafficking. For the nervous system, intracellular
trafficking functionally impacts neuronal development, homeostasis,
as well as neurodegeneration (133). Secondly, it is generally known
that skin, bone, cartilage, and skeletal muscle fiber comprise
large amounts of the extracellular matrix which define the
structure and physical properties. Almost all extracellular matrix
components are transported by intra- cellular trafficking systems.
Alterations in Golgi apparatus membrane trafficking can lead to
glycosylation abnormalities. The assembly and maintenance of the
extracellular matrix are susceptible to impairment of matrix
protein glycosylation. Thus, the skin, bone, cartilage, and
skeletal muscle are most sensitive to impaired glycosylation of
cargo proteins, and membrane trafficking defects. Therefore, the
loss of some Golgi resident proteins, such as ATP6V1A, ATP6V1E1
(106), ATP6VOA2 (108), TMEM165 (134), GOLGB1 (88), SCYL1BP1 (75), TRAPPC11 (135), TRAPPC2 (136), and TRIP11 (92), manifest primarily in these
matrix-rich tissues.
The Golgi apparatus participates in the occurrence
and development of disease and could be the key to finding new
targets for disease diagnosis and therapy.
Golgi glycoprotein 73 (GP73, also referred to as
GOLPH2), a resident Golgi membrane protein, is predominantly
expressed in biliary epithelial cells in the normal human liver
(137). GP73 expression is
upregulated in chronic Hepatitis B virus (HBV) infection (138), chronic HCV infection (139), non-alcoholic fatty liver
disease (140), and
hepatocellular carcinoma (HCC) (141,142). Serum GP73, a new marker for
HCC, is reported to appear earlier than serum α-fetoprotein. The
combined detection of serum α-fetoprotein and GP73 can improve
sensitivity and specificity for HCC diagnosis (143,144). However, several studies showed
GP73 levels were not higher in HCC patients than in patients with
other liver diseases such as cirrhosis (145,146). In addition to being a marker,
the expression of GP73 is critical for chemo- therapeutic
resistance in HCC cell lines (147).
Transmembrane protein 165 (TMEM165) functions in ion
homeostasis, membrane trafficking, and glycosylation in the Golgi
apparatus (148). Findings of a
study showed that mutations in TMEM165 cause CDG type II in humans
(134). Other research has
found that expression of TMEM165 mRNA and protein is apparently
increased in HCC patient tissues and contributes to the invasive
activity of cancer cells (149). This result indicates that
TMEM165 is a possible biomarker for HCC. GS28 is a member of the
SNAREs protein family. GS28 protein immunoreactivity was observed
in both nuclear and cytoplasmic compartments of cancer cells. High
nuclear expression of GS28 is associated with poor prognosis for
colorectal (150) and cervical
cancer patients (151).
Anti-Golgi antibodies (AGAs) were first found in
1982 in the serum of patients with Sjogren's syndrome complicated
with lymphoma (152). AGAs have
also been found in other immunological diseases (153-155). Currently, at least 20 Golgi
autoantigens are known, including golgin-97, golgin-67, golgin-245,
golgin-95, golgin-160, and giantin. AGA positivity is commonly
found in connective tissue diseases such as Sjogren's syndrome,
rheumatoid arthritis, and systemic lupus erythematosus (154,156); cerebellar malignant disease
such as idiopathic late-onset cerebellar ataxia (157); infectious diseases such as
HBV/HCV infection, Epstein-Barr virus infection and HIV infection
(155,158,159); and tumors, such as HCC and lung
cancer (160). Although AGAs
are not specific to any disease, their clinical detection may be
helpful for classifying and following the progress of some
connective tissue diseases. For example, compared to
anti-BICD2-negative patients, single specificity anti-BICD2
patients may be more associated with inflammatory myopathy and
interstitial lung disease (161).
Biomarkers are crucial for early diagnosis,
assessing response to treatment, and classifying diseases into
subtypes. Biomarker discovery involves many critical steps such as
clinical study design, sample collection, data integration, and
protein/peptide identification and preservation. These steps should
be carefully controlled before confirmation and verification.
Therefore, in clinical applications, these biomarkers are potential
diagnostic markers. Large-scale investigations are needed and more
sensitive and specific detection methods need to be researched.
In addition to biomarker discovery, the functions
of the Golgi apparatus and its associated molecules in maintaining
cell structural integrity and its central role in membrane
trafficking pathways provide possible targets for disease therapy.
These targets may be direct, due to genetic disease (Table I), or indirect, as in cancer.
Compared to non-transformed and normal cells, cancer cells have
morphological and functional changes in the Golgi apparatus that
drive invasion and migration in a unique microenvironment. These
changes provide therapeutic targets for interventions. A research
team developed a bovine serum albumin pH-responsive photothermal
ablation agent that preferentially accumulates in the Golgi of
cancer cells compared to normal cells due to morphological changes
in the Golgi apparatus (162).
The agent is activated by the weakly acidic microenvironment of the
Golgi in cancer cells for photothermal therapy. In this method, a
photothermal ablation agent converts light energy into heat and
kills cancer cells with high specificity and minimal invasiveness
by hyper-pyrexia (162).
Another research team developed a prodrug nanoparticle system,
which appeared to target the Golgi apparatus and realized retinoic
acid release under an acidic environment. The retinoic
acid-conjugated chondroitin sulfate could reduce the expression of
metastasis-associated proteins by inducing Golgi fragmentation
(163). Those findings suggest
that the Golgi apparatus is a promising target for the development
of novel drugs. A review summarized small molecules as drugs
targeting the Golgi apparatus for the treatment of diseases
(164), such as LTX-401,
inhibitors of Golgi-associated lipid transfer proteins,
glucosylceramide synthase inhibitors, O-glycosylation inhibitors,
PI4KIIIb inhibitors and inhibitors of ARF activation. Whether these
drugs that target the Golgi apparatus can be applied in clinical
practice needs to be determined.
The central role of the Golgi apparatus in critical
cell processes such as the transport, processing, and sorting of
proteins and lipids has placed it at the forefront of cell science.
Several previous studies have suggested that the Golgi apparatus
plays a critical role in diseases, particularly in
neurodegenerative diseases. However, few studies focus on human
diseases caused by mutations in genes encoding Golgi resident
proteins and summarize the common features of these genetic
diseases. In the present review, we summed up the genetic diseases
caused by mutations in genes encoding Golgi resident proteins. By
analyzing their pathophysiology, we identified that the majority of
genes are involved in membrane trafficking. The nervous system,
skin, bone, cartilage, and skeletal muscle are the most sensitive
tissues to defective membrane trafficking. It is reasonable to hope
that our basic knowledge of Golgi-mediated membrane trafficking
will continue to provide insights into the pathogenesis of genetic
diseases and that studies of these diseases will continue to
enhance our under- standing of the critical role of the Golgi
apparatus in diseases. In addition, the finding of Golgi-related
biomarker and Golgi-based therapeutics further emphasize the
importance of Golgi apparatus in human pathology. Taken together,
advances in Golgi apparatus biology provide opportunities to
translate discoveries into clinical medicine. Thus, we highlighted
the importance of underlying clinical insights and provided a new
direction for future research.
The present study was supported by grants from the
National Natural Science Foundation of China (grant no.
81974213).
Not applicable.
JL and YH were mainly responsible for collecting
relevant information and completing this review. ZJ, LZ and TL were
mainly responsible for consulting literature materials and revising
the manuscript. ZH was responsible for the conception of this
review and the assignment of tasks. There was no additional
assistance with manuscript preparation. All authors read and
approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
|
1
|
Rios RM and Bornens M: The Golgi apparatus
at the cell centre. Curr Opin Cell Biol. 15:60–66. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gautam M, Jara JH, Sekerkova G, Yasvoina
MV, Martina M and Özdinler PH: Absence of alsin function leads to
corticospinal motor neuron vulnerability via novel disease
mechanisms. Hum Mol Genet. 25:1074–1087. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rendón WO, Martínez-Alonso E, Tomás M,
Martínez-Martínez N and Martínez-Menárguez JA: Golgi fragmentation
is Rab and SNARE dependent in cellular models of Parkinson's
disease. Histochem Cell Biol. 139:671–684. 2013. View Article : Google Scholar
|
|
4
|
Brandstaetter H, Kruppa AJ and Buss F:
Huntingtin is required for ER-to-Golgi transport and for secretory
vesicle fusion at the plasma membrane. Dis Model Mech. 7:1335–1340.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yuan D, Liu C and Hu B: Dysfunction of
membrane trafficking leads to ischemia-reperfusion injury after
transient cerebral ischemia. Transl Stroke Res. 9:215–222. 2018.
View Article : Google Scholar :
|
|
6
|
Li T, You H, Mo X, He W, Tang X, Jiang Z,
Chen S, Chen Y, Zhang J and Hu Z: GOLPH3 mediated Golgi stress
response in modulating N2A cell death upon oxygen-glucose
deprivation and reoxygenation injury. Mol Neurobiol. 53:1377–1385.
2016. View Article : Google Scholar
|
|
7
|
Tarazón E, Roselló-Lletí E, Ortega A,
Gil-Cayuela C, González-Juanatey JR, Lago F, Martínez-Dolz L,
Portolés M and Rivera M: Changes in human Golgi apparatus reflect
new left ventricular dimensions and function in dilated
cardiomyopathy patients. Eur J Heart Fail. 19:280–282. 2017.
View Article : Google Scholar
|
|
8
|
Stancu CS, Toma L and Sima AV: Dual role
of lipoproteins in endothelial cell dysfunction in atherosclerosis.
Cell Tissue Res. 349:433–446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee J, Reich R, Xu F and Sehgal PB: Golgi,
trafficking, and mitosis dysfunctions in pulmonary arterial
endothelial cells exposed to monocrotaline pyrrole and NO
scavenging. Am J Physiol Lung Cell Mol Physiol. 297:L715–L728.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sehgal PB, Mukhopadhyay S, Xu F, Patel K
and Shah M: Dysfunction of Golgi tethers, SNAREs, and SNAPs in
monocrotaline-induced pulmonary hypertension. Am J Physiol Lung
Cell Mol Physiol. 292:L1526–L1542. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu H, Li H, Wang P, Chen M, Huang Z, Li
K, Li Y, He J, Han J and Zhang Q: Persistent and acute chlamydial
infections induce different structural changes in the Golgi
apparatus. Int J Med Microbiol. 304:577–585. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rohde J, Emschermann F, Knittler MR and
Rziha HJ: Orf virus interferes with MHC class I surface expression
by targeting vesicular transport and Golgi. BMC Vet Res. 8:1142012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mousnier A, Swieboda D, Pinto A, Guedán A,
Rogers AV, Walton R, Johnston SL and Solari R: Human rhinovirus 16
causes Golgi apparatus fragmentation without blocking protein
secretion. J Virol. 88:11671–11685. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tan X, Banerjee P, Guo HF, Ireland S,
Pankova D, Ahn YH, Nikolaidis IM, Liu X, Zhao Y, Xue Y, et al:
Epithelial-to- mesenchymal transition drives a pro-metastatic Golgi
compaction process through scaffolding protein PAQR11. J Clin
Invest. 127:117–131. 2017. View Article : Google Scholar
|
|
15
|
Golgi C: On the structure of nerve cells.
1898. J Microsc. 155:3–7. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mollenhauer HH and Morré DJ: Perspectives
on Golgi apparatus form and function. J Electron Microsc Tech.
17:2–14. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Storrie B, White J, Röttger S, Stelzer EH,
Suganuma T and Nilsson T: Recycling of golgi-resident
glycosyltransferases through the ER reveals a novel pathway and
provides an explanation for nocodazole-induced Golgi scattering. J
Cell Biol. 143:1505–1521. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Slusarewicz P, Nilsson T, Hui N, Watson R
and Warren G: Isolation of a matrix that binds medial Golgi
enzymes. J Cell Biol. 124:405–413. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Papanikou E and Glick BS: Golgi
compartmentation and identity. Curr Opin Cell Biol. 29:74–81. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Glick BS and Luini A: Models for Golgi
traffic: A critical assessment. Cold Spring Harb Perspect Biol.
3:a0052152011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sundaramoorthy V, Sultana JM and Atkin JD:
Golgi fragmentation in amyotrophic lateral sclerosis, an overview
of possible triggers and consequences. Front Neurosci. 9:4002015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gonatas NK, Stieber A and Gonatas JO:
Fragmentation of the Golgi apparatus in neurodegenerative diseases
and cell death. J Neurol Sci. 246:21–30. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jiang Z, Hu Z, Zeng L, Lu W, Zhang H, Li T
and Xiao H: The role of the Golgi apparatus in oxidative stress: Is
this organelle less significant than mitochondria? Free Radic Biol
Med. 50:907–917. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu JY, He JL, Huang Y, Xiao H, Jiang Z
and Hu ZP: The Golgi apparatus in neurorestoration. J
Neuroresstoratology. 7:116–128. 2019. View Article : Google Scholar
|
|
25
|
Fan J, Hu Z, Zeng L, Lu W, Tang X, Zhang J
and Li T: Golgi apparatus and neurodegenerative diseases. Int J Dev
Neurosci. 26:523–534. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lu L, Zhou Q, Chen Z and Chen L: The
significant role of the Golgi apparatus in cardiovascular diseases.
J Cell Physiol. 233:2911–2919. 2018. View Article : Google Scholar
|
|
27
|
Millarte V and Farhan H: The Golgi in cell
migration: Regulation by signal transduction and its implications
for cancer cell metastasis. ScientificWorldJournal.
2012:4982782012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mourelatos Z, Gonatas NK, Stieber A,
Gurney ME and Dal Canto MC: The Golgi apparatus of spinal cord
motor neurons in transgenic mice expressing mutant Cu, Zn
superoxide dismutase becomes fragmented in early, preclinical
stages of the disease. Proc Natl Acad Sci USA. 93:5472–5477. 1996.
View Article : Google Scholar
|
|
29
|
Joshi G, Bekier ME II and Wang Y: Golgi
fragmentation in Alzheimer's disease. Front Neurosci. 9:3402015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Strehlow AN, Li JZ and Myers RM: Wild-type
huntingtin participates in protein trafficking between the Golgi
and the extracellular space. Hum Mol Genet. 16:391–409. 2007.
View Article : Google Scholar
|
|
31
|
Sakurai A, Okamoto K, Fujita Y, Nakazato
Y, Wakabayashi K, Takahashi H and Gonatas NK: Fragmentation of the
Golgi apparatus of the ballooned neurons in patients with
corticobasal degeneration and Creutzfeldt-Jakob disease. Acta
Neuropathol. 100:270–274. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sakurai A, Okamoto K, Yaguchi M, Fujita Y,
Mizuno Y, Nakazato Y and Gonatas NK: Pathology of the inferior
olivary nucleus in patients with multiple system atrophy. Acta
Neuropathol. 103:550–554. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
van Dis V, Kuijpers M, Haasdijk ED,
Teuling E, Oakes SA, Hoogenraad CC and Jaarsma D: Golgi
fragmentation precedes neuromuscular denervation and is associated
with endosome abnormalities in SOD1-ALS mouse motor neurons. Acta
Neuropathol Commun. 2:382014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pottorf T, Mann A, Fross S, Mansel C and
Vohra BPS: Nicotinamide mononucleotide adenylyltransferase 2
maintains neuronal structural integrity through the maintenance of
golgi structure. Neurochem Int. 121:86–97. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Joshi G and Wang Y: Golgi defects enhance
APP amyloidogenic processing in Alzheimer's disease. Bioessays.
37:240–247. 2015. View Article : Google Scholar
|
|
36
|
Covarrubias-Pinto A, Parra AV,
Mayorga-Weber G, Papic E, Vicencio I, Ehrenfeld P, Rivera FJ and
Castro MA: Impaired intracellular trafficking of sodium-dependent
vitamin C transporter 2 contributes to the redox imbalance in
Huntington's disease. J Neurosci Res. 99:223–235. 2021. View Article : Google Scholar
|
|
37
|
Mani M, Thao DT, Kim BC, Lee UH, Kim DJ,
Jang SH, Back SH, Lee BJ, Cho WJ, Han IS and Park JW: DRG2 knock-
down induces Golgi fragmentation via GSK3β phosphorylation and
microtubule stabilization. Biochim Biophys Acta Mol Cell Res.
1866:1463–1474. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rodríguez-Cruz F, Torres-Cruz FM,
Monroy-Ramírez HC, Escobar-Herrera J, Basurto-Islas G, Avila J and
García-Sierra F: Fragmentation of the Golgi apparatus in
neuroblastoma cells is associated with tau-induced ring-shaped
microtubule bundles. J Alzheimers Dis. 65:1185–1207. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sou YS, Kakuta S, Kamikubo Y, Niisato K,
Sakurai T, Parajuli LK, Tanida I, Saito H, Suzuki N, Sakimura K, et
al: Cerebellar neurodegeneration and neuronal circuit remodeling in
Golgi pH regulator-deficient mice. eNeuro. 6:ENEURO.0427-18.2019.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Heuer D, Rejman Lipinski A, Machuy N,
Karlas A, Wehrens A, Siedler F, Brinkmann V and Meyer TF: Chlamydia
causes fragmentation of the Golgi compartment to ensure
reproduction. Nature. 457:731–735. 2009. View Article : Google Scholar
|
|
41
|
Pruneda JN, Bastidas RJ, Bertsoulaki E,
Swatek KN, Santhanam B, Clague MJ, Valdivia RH, Urbé S and Komander
D: A chlamydia effector combining deubiquitination and acetylation
activities induces Golgi fragmentation. Nat Microbiol. 3:1377–1384.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hansen MD, Johnsen IB, Stiberg KA,
Sherstova T, Wakita T, Richard GM, Kandasamy RK, Meurs EF and
Anthonsen MW: Hepatitis C virus triggers Golgi fragmentation and
autophagy through the immunity-related GTPase M. Proc Natl Acad Sci
USA. 114:E3462–E3471. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Aistleitner K, Clark T, Dooley C and
Hackstadt T: Selective frag- mentation of the trans-Golgi apparatus
by Rickettsia rickettsii. PLoS Pathog. 16:e10085822020. View Article : Google Scholar
|
|
44
|
Ganesan M, Mathews S, Makarov E, Petrosyan
A, Kharbanda KK, Kidambi S, Poluektova LY, Casey CA and Osna NA:
Acetaldehyde suppresses HBV-MHC class I complex presentation on
hepatocytes via induction of ER stress and Golgi fragmentation. Am
J Physiol Gastrointest Liver Physiol. 319:G432–G442. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Quiner CA and Jackson WT: Fragmentation of
the Golgi apparatus provides replication membranes for human
rhinovirus 1A. Virology. 407:185–195. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Barbosa NS, Mendonça LR, Dias MVS,
Pontelli MC, da Silva EZM, Criado MF, da Silva-Januário ME,
Schindler M, Jamur MC, Oliver C, et al: ESCRT machinery components
are required for orthobunyavirus particle production in Golgi
compartments. PLoS Pathog. 14:e10070472018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Petrosyan A, Holzapfel MS, Muirhead DE and
Cheng PW: Restoration of compact Golgi morphology in advanced
prostate cancer enhances susceptibility to galectin-1-induced
apoptosis by modifying mucin O-glycan synthesis. Mol Cancer Res.
12:1704–1716. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tokuda E, Itoh T, Hasegawa J, Ijuin T,
Takeuchi Y, Irino Y, Fukumoto M and Takenawa T:
Phosphatidylinositol 4-phosphate in the Golgi apparatus regulates
cell-cell adhesion and invasive cell migration in human breast
cancer. Cancer Res. 74:3054–3066. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhao J, Yang C, Guo S and Wu Y: GM130
regulates epithelial-to-mesenchymal transition and invasion of
gastric cancer cells via snail. Int J Clin Exp Pathol.
8:10784–10791. 2015.PubMed/NCBI
|
|
50
|
Pinho SS and Reis CA: Glycosylation in
cancer: Mechanisms and clinical implications. Nat Rev Cancer.
15:540–555. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pinho SS, Seruca R, Gärtner F, Yamaguchi
Y, Gu J, Taniguchi N and Reis CA: Modulation of E-cadherin function
and dysfunction by N-glycosylation. Cell Mol Life Sci.
68:1011–1020. 2011. View Article : Google Scholar
|
|
52
|
Baschieri F, Confalonieri S, Bertalot G,
Di Fiore PP, Dietmaier W, Leist M, Crespo P, Macara IG and Farhan
H: Spatial control of Cdc42 signalling by a GM130-RasGRF complex
regulates polarity and tumorigenesis. Nat Commun. 5:48392014.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Taniguchi N and Kizuka Y: Glycans and
cancer: Role of N-glycans in cancer biomarker, progression and
metastasis, and therapeutics. Adv Cancer Res. 126:11–51. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rizzo R, Parashuraman S, D'Angelo G and
Luini A: GOLPH3 and oncogenesis: What is the molecular link? Tissue
Cell. 49:170–174. 2017. View Article : Google Scholar
|
|
55
|
Farber-Katz SE, Dippold HC, Buschman MD,
Peterman MC, Xing M, Noakes CJ, Tat J, Ng MM, Rahajeng J, Cowan DM,
et al: DNA damage triggers Golgi dispersal via DNA-PK and GOLPH3.
Cell. 156:413–427. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Scott KL, Kabbarah O, Liang MC, Ivanova E,
Anagnostou V, Wu J, Dhakal S, Wu M, Chen S, Feinberg T, et al:
GOLPH3 modulates mTOR signalling and rapamycin sensitivity in
cancer. Nature. 459:1085–1090. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xing M, Peterman MC, Davis RL, Oegema K,
Shiau AK and Field SJ: GOLPH3 drives cell migration by promoting
Golgi reorientation and directional trafficking to the leading
edge. Mol Biol Cell. 27:3828–3840. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chang SH, Hong SH, Jiang HL, Minai-Tehrani
A, Yu KN, Lee JH, Kim JE, Shin JY, Kang B, Park S, et al:
GOLGA2/GM130, cis-Golgi matrix protein, is a novel target of
anticancer gene therapy. Mol Ther. 20:2052–2063. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sehgal PB, Mukhopadhyay S, Patel K, Xu F,
Almodóvar S, Tuder RM and Flores SC: Golgi dysfunction is a common
feature in idiopathic human pulmonary hypertension and vascular
lesions in SHIV-nef-infected macaques. Am J Physiol Lung Cell Mol
Physiol. 297:L729–L737. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sehgal PB and Lee JE: Protein trafficking
dysfunctions: Role in the pathogenesis of pulmonary arterial
hypertension. Pulm Circ. 1:17–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Muhammad E, Levitas A, Singh SR, Braiman
A, Ofir R, Etzion S, Sheffield VC, Etzion Y, Carrier L and Parvari
R: PLEKHM2 mutation leads to abnormal localization of lysosomes,
impaired autophagy flux and associates with recessive dilated
cardiomyopathy and left ventricular noncompaction. Hum Mol Genet.
24:7227–7240. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hatt PY: Cellular changes and damage in
mechanically over-loaded hearts. Recent Adv Stud Cardiac Struct
Metab. 6:325–333. 1975.
|
|
63
|
Satoh H: Sino-atrial nodal cells of
mammalian hearts: Ionic currents and gene expression of pacemaker
ionic channels. J Smooth Muscle Res. 39:175–193. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Jungk L, Franke H, Salameh A and Dhein S:
Golgi fragmentation in human patients with chronic atrial
fibrillation: A new aspect of remodeling. Thorac Cardiovasc Surg.
67:98–106. 2019. View Article : Google Scholar
|
|
65
|
Prasad K and Singal PK: Ultrastructure of
failing myocardium due to induced chronic mitral insufficiency in
dogs. Br J Exp Pathol. 58:289–300. 1977.PubMed/NCBI
|
|
66
|
Rambourg A, Clermont Y and Hermo L:
Three-dimensional architecture of the golgi apparatus in sertoli
cells of the rat. Am J Anat. 154:455–476. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Xiang Y and Wang Y: New components of the
Golgi matrix. Cell Tissue Res. 344:365–379. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kooy J, Toh BH, Pettitt JM, Erlich R and
Gleeson PA: Human autoantibodies as reagents to conserved Golgi
components. Characterization of a peripheral, 230-kDa
compartment-specific Golgi protein. J Biol Chem. 267:20255–20263.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Fritzler MJ, Hamel JC, Ochs RL and Chan
EK: Molecular characterization of two human autoantigens: Unique
cDNAs encoding 95- and 160-kD proteins of a putative family in the
Golgi complex. J Exp Med. 178:49–62. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wong M and Munro S: Membrane trafficking.
The specificity of vesicle traffic to the Golgi is encoded in the
golgin coiled-coil proteins. Science. 346:12568982014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Barr FA, Puype M, Vandekerckhove J and
Warren G: GRASP65, a protein involved in the stacking of Golgi
cisternae. Cell. 91:253–262. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Shorter J and Warren G: A role for the
vesicle tethering protein, p115, in the post-mitotic stacking of
reassembling Golgi cisternae in a cell-free system. J Cell Biol.
146:57–70. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Vinke FP, Grieve AG and Rabouille C: The
multiple facets of the Golgi reassembly stacking proteins. Biochem
J. 433:423–433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Bergen DJM, Stevenson NL, Skinner REH,
Stephens DJ and Hammond CL: The Golgi matrix protein giantin is
required for normal cilia function in zebrafish. Biol Open.
6:1180–1189. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Hennies HC, Kornak U, Zhang H, Egerer J,
Zhang X, Seifert W, Kühnisch J, Budde B, Nätebus M, Brancati F, et
al: Gerodermia osteodysplastica is caused by mutations in SCYL1BP1,
a Rab-6 interacting golgin. Nat Genet. 40:1410–1412. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Nakamura N, Rabouille C, Watson R, Nilsson
T, Hui N, Slusarewicz P, Kreis TE and Warren G: Characterization of
a cis-Golgi matrix protein, GM130. J Cell Biol. 131:1715–1726.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Alvarez C, Garcia-Mata R, Hauri HP and
Sztul E: The p115-inter- active proteins GM130 and giantin
participate in endoplasmic reticulum-Golgi traffic. J Biol Chem.
276:2693–2700. 2001. View Article : Google Scholar
|
|
78
|
Huang W, She L, Chang XY, Yang RR, Wang L,
Ji HB, Jiao JW and Poo MM: Protein kinase LKB1 regulates polarized
dendrite formation of adult hippocampal newborn neurons. Proc Natl
Acad Sci USA. 111:469–474. 2014. View Article : Google Scholar
|
|
79
|
Liu C, Mei M, Li Q, Roboti P, Pang Q, Ying
Z, Gao F, Lowe M and Bao S: Loss of the golgin GM130 causes Golgi
disruption, Purkinje neuron loss, and ataxia in mice. Proc Natl
Acad Sci USA. 114:346–351. 2017. View Article : Google Scholar
|
|
80
|
Matanis T, Akhmanova A, Wulf P, Del Nery
E, Weide T, Stepanova T, Galjart N, Grosveld F, Goud B, De Zeeuw
CI, et al: Bicaudal-D regulates COPI-independent Golgi-ER transport
by recruiting the dynein-dynactin motor complex. Nat Cell Biol.
4:986–992. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
81
|
Hoogenraad CC, Akhmanova A, Howell SA,
Dortland BR, De Zeeuw CI, Willemsen R, Visser P, Grosveld F and
Galjart N: Mammalian Golgi-associated Bicaudal-D2 functions in the
dynein-dynactin pathway by interacting with these complexes. EMBO
J. 20:4041–4054. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Jaarsma D, van den Berg R, Wulf PS, van
Erp S, Keijzer N, Schlager MA, de Graaff E, De Zeeuw CI, Pasterkamp
RJ, Akhmanova A and Hoogenraad CC: A role for bicaudal-D2 in radial
cerebellar granule cell migration. Nat Commun. 5:34112014.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Will L, Portegies S, van Schelt J, van
Luyk M, Jaarsma D and Hoogenraad CC: Dynein activating adaptor
BICD2 controls radial migration of upper-layer cortical neurons in
vivo. Acta Neuropathol Commun. 7:1622019. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Storbeck M, Horsberg Eriksen B, Unger A,
Hölker I, Aukrust I, Martínez-Carrera LA, Linke WA, Ferbert A,
Heller R, Vorgerd M, et al: Phenotypic extremes of BICD2-opathies:
From lethal, congenital muscular atrophy with arthrogryposis to
asymptomatic with subclinical features. Eur J Hum Genet.
25:1040–1048. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Neveling K, Martinez-Carrera LA, Hölker I,
Heister A, Verrips A, Hosseini-Barkooie SM, Gilissen C, Vermeer S,
Pennings M, Meijer R, et al: Mutations in BICD2, which encodes a
golgin and important motor adaptor, cause congenital
autosomal-dominant spinal muscular atrophy. Am J Hum Genet.
92:946–954. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Oates EC, Rossor AM, Hafezparast M,
Gonzalez M, Speziani F, MacArthur DG, Lek M, Cottenie E, Scoto M,
Foley AR, et al: Mutations in BICD2 cause dominant congenital
spinal muscular atrophy and hereditary spastic paraplegia. Am J Hum
Genet. 92:965–973. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Sonnichsen B, Lowe M, Levine T, Jämsä E,
Dirac-Svejstrup B and Warren G: A role for giantin in docking COPI
vesicles to Golgi membranes. J Cell Biol. 140:1013–1021. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Katayama K, Kuriki M, Kamiya T, Tochigi Y
and Suzuki H: Giantin is required for coordinated production of
aggrecan, link protein and type XI collagen during chondrogenesis.
Biochem Biophys Res Commun. 499:459–465. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Stevenson NL, Bergen DJM, Skinner REH,
Kague E, Martin-Silverstone E, Robson Brown KA, Hammond CL and
Stephens DJ: Giantin-knockout models reveal a feedback loop between
Golgi function and glycosyltransferase expression. J Cell Sci.
130:4132–4143. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Witkos TM, Chan WL, Joensuu M, Rhiel M,
Pallister E, Thomas-Oates J, Mould AP, Mironov AA, Biot C,
Guerardel Y, et al: GORAB scaffolds COPI at the trans-Golgi for
efficient enzyme recycling and correct protein glycosylation. Nat
Commun. 10:1272019. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Sato K, Roboti P, Mironov AA and Lowe M:
Coupling of vesicle tethering and Rab binding is required for in
vivo functionality of the golgin GMAP-210. Mol Biol Cell.
26:537–553. 2015. View Article : Google Scholar :
|
|
92
|
Smits P, Bolton AD, Funari V, Hong M,
Boyden ED, Lu L, Manning DK, Dwyer ND, Moran JL, Prysak M, et al:
Lethal skeletal dysplasia in mice and humans lacking the golgin
GMAP-210. N Engl J Med. 362:206–216. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wehrle A, Witkos TM, Unger S, Schneider J,
Follit JA, Hermann J, Welting T, Fano V, Hietala M, Vatanavicharn
N, et al: Hypomorphic mutations of TRIP11 cause
odontochondrodysplasia. JCI Insight. 4:e1247012019. View Article : Google Scholar :
|
|
94
|
West DW: Energy-dependent calcium
sequestration activity in a Golgi apparatus fraction derived from
lactating rat mammary glands. Biochim Biophys Acta. 673:374–386.
1981. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Shull GE, Miller ML and Prasad V:
Secretory pathway stress responses as possible mechanisms of
disease involving Golgi Ca2+ pump dysfunction.
Biofactors. 37:150–158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Sudbrak R, Brown J, Dobson-Stone C, Carter
S, Ramser J, White J, Healy E, Dissanayake M, Larrègue M, Perrussel
M, et al: Hailey-Hailey disease is caused by mutations in ATP2C1
encoding a novel Ca(2+) pump. Hum Mol Genet. 9:1131–1140. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Hu Z, Bonifas JM, Beech J, Bench G,
Shigihara T, Ogawa H, Ikeda S, Mauro T and Epstein EH Jr: Mutations
in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease.
Nat Genet. 24:61–65. 2000. View
Article : Google Scholar
|
|
98
|
Okunade GW, Miller ML, Azhar M, Andringa
A, Sanford LP, Doetschman T, Prasad V and Shull GE: Loss of the
Atp2c1 secretory pathway Ca(2+)-ATPase (SPCA1) in mice causes Golgi
stress, apoptosis, and midgestational death in homozygous embryos
and squamous cell tumors in adult heterozygotes. J Biol Chem.
282:26517–26527. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Lázaro-Diéguez F, Jiménez N, Barth H,
Koster AJ, Renau-Piqueras J, Llopis JL, Burger KN and Egea G: Actin
filaments are involved in the maintenance of Golgi cisternae
morphology and intra-Golgi pH. Cell Motil Cytoskeleton. 63:778–791.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Huang C and Chang A: pH-dependent cargo
sorting from the Golgi. J Biol Chem. 286:10058–10065. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Rivinoja A, Pujol FM, Hassinen A and
Kellokumpu S: Golgi pH, its regulation and roles in human disease.
Ann Med. 44:542–554. 2012. View Article : Google Scholar
|
|
102
|
Drory O and Nelson N: The emerging
structure of vacuolar ATPases. Physiology (Bethesda). 21. pp.
317–325. 2006
|
|
103
|
Maeda Y, Ide T, Koike M, Uchiyama Y and
Kinoshita T: GPHR is a novel anion channel critical for
acidification and functions of the Golgi apparatus. Nat Cell Biol.
10:1135–1145. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Banerjee S and Kane PM: Regulation of
V-ATPase activity and organelle pH by phosphatidylinositol
phosphate lipids. Front Cell Dev Biol. 8:5102020. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Morava E, Guillard M, Lefeber DJ and
Wevers RA: Autosomal recessive cutis laxa syndrome revisited. Eur J
Hum Genet. 17:1099–1110. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Van Damme T, Gardeitchik T, Mohamed M,
Guerrero-Castillo S, Freisinger P, Guillemyn B, Kariminejad A,
Dalloyaux D, van Kraaij S, Lefeber DJ, et al: Mutations in ATP6V1E1
or ATP6V1A cause autosomal-recessive cutis laxa. Am J Hum Genet.
100:216–227. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Kariminejad A, Afroozan F, Bozorgmehr B,
Ghanadan A, Akbaroghli S, Khorram Khorshid HR, Mojahedi F, Setoodeh
A, Loh A, Tan YX, et al: Discriminative features in three autosomal
recessive cutis laxa syndromes: Cutis laxa IIA, cutis laxa IIB, and
geroderma osteoplastica. Int J Mol Sci. 18:6352017. View Article : Google Scholar :
|
|
108
|
Kornak U, Reynders E, Dimopoulou A, van
Reeuwijk J, Fischer B, Rajab A, Budde B, Nürnberg P, Foulquier F;
ARCL Debré-type Study Group; et al: Impaired glycosylation and
cutis laxa caused by mutations in the vesicular H+-ATPase subunit
ATP6V0A2. Nat Genet. 40:32–34. 2008. View Article : Google Scholar
|
|
109
|
Tümer Z: An overview and update of ATP7A
mutations leading to Menkes disease and occipital horn syndrome.
Hum Mutat. 34:417–429. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Huster D, Hoppert M, Lutsenko S, Zinke J,
Lehmann C, Mössner J, Berr F and Caca K: Defective cellular
localization of mutant ATP7B in Wilson's disease patients and
hepatoma cell lines. Gastroenterology. 124:335–345. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Guan JL, Machamer CE and Rose JK:
Glycosylation allows cell-surface transport of an anchored
secretory protein. Cell. 42:489–496. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Roth J: Protein N-glycosylation along the
secretory pathway: Relationship to organelle topography and
function, protein quality control, and cell interactions. Chem Rev.
102:285–303. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Rosnoblet C, Peanne R, Legrand D and
Foulquier F: Glycosylation disorders of membrane trafficking.
Glycoconj J. 30:23–31. 2013. View Article : Google Scholar
|
|
114
|
Grewal PK, McLaughlan JM, Moore CJ,
Browning CA and Hewitt JE: Characterization of the LARGE family of
putative glycosyltransferases associated with dystroglycanopathies.
Glycobiology. 15:912–923. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Kobayashi K, Nakahori Y, Miyake M,
Matsumura K, Kondo-Iida E, Nomura Y, Segawa M, Yoshioka M, Saito K,
Osawa M, et al: An ancient retrotransposal insertion causes
Fukuyama-type congenital muscular dystrophy. Nature. 394:388–392.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Brockington M, Blake DJ, Prandini P, Brown
SC, Torelli S, Benson MA, Ponting CP, Estournet B, Romero NB,
Mercuri E, et al: Mutations in the fukutin-related protein gene
(FKRP) cause a form of congenital muscular dystrophy with secondary
laminin alpha2 deficiency and abnormal glycosylation of
alpha-dystroglycan. Am J Hum Genet. 69:1198–1209. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Desplats PA, Denny CA, Kass KE, Gilmartin
T, Head SR, Sutcliffe JG, Seyfried TN and Thomas EA: Glycolipid and
ganglioside metabolism imbalances in Huntington's disease.
Neurobiol Dis. 27:265–277. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Cooper-Knock J, Moll T, Ramesh T, Castelli
L, Beer A, Robins H, Fox I, Niedermoser I, Van Damme P, Moisse M,
et al: Mutations in the glycosyltransferase domain of GLT8D1 are
associated with familial amyotrophic lateral sclerosis. Cell Rep.
26:2298–2306.e5. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Li W, Liu Z, Sun W, Yuan Y, Hu Y, Ni J,
Jiao B, Fang L, Li J, Shen L, et al: Mutation analysis of GLT8D1
and ARPP21 genes in amyotrophic lateral sclerosis patients from
mainland China. Neurobiol Aging. 85:156.e1–156.e4. 2020. View Article : Google Scholar
|
|
120
|
Liu S and Storrie B: Are Rab proteins the
link between Golgi organization and membrane trafficking? Cell Mol
Life Sci. 69:4093–4106. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Salian S, Cho TJ, Phadke SR, Gowrishankar
K, Bhavani GS, Shukla A, Jagadeesh S, Kim OH, Nishimura G and
Girisha KM: Additional three patients with Smith-McCort dysplasia
due to novel RAB33B mutations. Am J Med Genet A. 173:588–595. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Giannandrea M, Bianchi V, Mignogna ML,
Sirri A, Carrabino S, D'Elia E, Vecellio M, Russo S, Cogliati F,
Larizza L, et al: Mutations in the small GTPase gene RAB39B are
responsible for X-linked mental retardation associated with autism,
epilepsy, and macrocephaly. Am J Hum Genet. 86:185–195. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Bock JB, Matern HT, Peden AA and Scheller
RH: A genomic perspective on membrane compartment organization.
Nature. 409:839–841. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Corbett MA, Schwake M, Bahlo M, Dibbens
LM, Lin M, Gandolfo LC, Vears DF, O'Sullivan JD, Robertson T, Bayly
MA, et al: A mutation in the Golgi Qb-SNARE gene GOSR2 causes
progressive myoclonus epilepsy with early ataxia. Am J Hum Genet.
88:657–663. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Lowe SL, Peter F, Subramaniam VN, Wong SH
and Hong W: A SNARE involved in protein transport through the Golgi
apparatus. Nature. 389:881–884. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
126
|
Malsam J and Söllner TH: Organization of
SNAREs within the Golgi stack. Cold Spring Harb Perspect Biol.
3:a0052492011. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Gedeon AK, Colley A, Jamieson R, Thompson
EM, Rogers J, Sillence D, Tiller GE, Mulley JC and Gécz J:
Identification of the gene (SEDL) causing X-linked
spondyloepiphyseal dysplasia tarda. Nat Genet. 22:400–404. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Willett R, Ungar D and Lupashin V: The
Golgi puppet master: COG complex at center stage of membrane
trafficking interactions. Histochem Cell Biol. 140:271–283. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Climer LK, Dobretsov M and Lupashin V:
Defects in the COG complex and COG-related trafficking regulators
affect neuronal Golgi function. Front Neurosci. 9:4052015.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Miller VJ and Ungar D: Re'COG'nition at
the Golgi. Traffic. 13:891–897. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Egorov MV, Capestrano M, Vorontsova OA, Di
Pentima A, Egorova AV, Mariggiò S, Ayala MI, Tetè S, Gorski JL,
Luini A, et al: Faciogenital dysplasia protein (FGD1) regulates
export of cargo proteins from the golgi complex via Cdc42
activation. Mol Biol Cell. 20:2413–2427. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Roboti P, Sato K and Lowe M: The golgin
GMAP-210 is required for efficient membrane trafficking in the
early secretory pathway. J Cell Sci. 128:1595–1606. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Zhang H, Winckler B and Cai Q:
Introduction to the special issue on membrane trafficking in
neurons. Dev Neurobiol. 78:167–169. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Rosnoblet C, Legrand D, Demaegd D,
Hacine-Gherbi H, de Bettignies G, Bammens R, Borrego C, Duvet S,
Morsomme P, Matthijs G and Foulquier F: Impact of disease-causing
mutations on TMEM165 subcellular localization, a recently
identified protein involved in CDG-II. Hum Mol Genet. 22:2914–2928.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Larson AA, Baker PR II, Milev MP, Press
CA, Sokol RJ, Cox MO, Lekostaj JK, Stence AA, Bossler AD, Mueller
JM, et al: TRAPPC11 and GOSR2 mutations associate with
hypoglycosylation of α-dystroglycan and muscular dystrophy. Skelet
Muscle. 8:172018. View Article : Google Scholar
|
|
136
|
Davis EE, Savage JH, Willer JR, Jiang YH,
Angrist M, Androutsopoulos A and Katsanis N: Whole exome sequencing
and functional studies identify an intronic mutation in TRAPPC2
that causes SEDT. Clin Genet. 85:359–364. 2014. View Article : Google Scholar
|
|
137
|
Riener MO: Diagnosis of tumours of the
liver and the biliary tract: New tissue and serum markers.
Pathologe. 32(Suppl 2): S304–S309. 2011.In German. View Article : Google Scholar
|
|
138
|
Xu Z, Liu L, Pan X, Wei K, Wei M, Liu L,
Yang H and Liu Q: Serum Golgi protein 73 (GP73) is a diagnostic and
prognostic marker of chronic HBV liver disease. Medicine
(Baltimore). 94:e6592015. View Article : Google Scholar
|
|
139
|
Liu Y, Zou Z, Zhu B, Hu Z and Zeng P:
CXCL10 decreases GP73 expression in hepatoma cells at the early
stage of hepatitis C virus (HCV) infection. Int J Mol Sci.
14:24230–24241. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Zheng KI, Liu WY, Pan XY, Ma HL, Zhu PW,
Wu XX, Targher G, Byrne C, Wang XD, Chen YP, et al: Combined and
sequential non-invasive approach to diagnosing non-alcoholic
steatohepatitis in patients with non-alcoholic fatty liver disease
and persistently normal alanine aminotransferase levels. BMJ Open
Diabetes Res Care. 8:e0011742020. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Hou SC, Xiao MB, Ni RZ, Ni WK, Jiang F, Li
XY, Lu CH and Chen BY: Serum GP73 is complementary to AFP and
GGT-II for the diagnosis of hepatocellular carcinoma. Oncol Lett.
6:1152–1158. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Zhao J, Guo LY, Yang JM and Jia JW:
Sublingual vein parameters, AFP, AFP-L3, and GP73 in patients with
hepatocellular carcinoma. Genet Mol Res. 14:7062–7067. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Liu X, Wan X, Li Z, Lin C, Zhan Y and Lu
X: Golgi protein 73(GP73), a useful serum marker in liver diseases.
Clin Chem Lab Med. 49:1311–1316. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Morota K, Nakagawa M, Sekiya R, Hemken PM,
Sokoll LJ, Elliott D, Chan DW and Dowell BL: A comparative
evaluation of Golgi protein-73, fucosylated hemopexin,
α-fetoprotein, and PIVKA-II in the serum of patients with chronic
hepatitis, cirrhosis, and hepatocellular carcinoma. Clin Chem Lab
Med. 49:711–718. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Gu Y, Chen W, Zhao Y, Chen L and Peng T:
Quantitative analysis of elevated serum Golgi protein-73 expression
in patients with liver diseases. Ann Clin Biochem. 46:38–43. 2009.
View Article : Google Scholar
|
|
146
|
Tian L, Wang Y, Xu D, Gui J, Jia X, Tong
H, Wen X, Dong Z and Tian Y: Serological AFP/Golgi protein 73 could
be a new diagnostic parameter of hepatic diseases. Int J Cancer.
129:1923–1931. 2011. View Article : Google Scholar
|
|
147
|
Ye JZ, Yan SM, Yuan CL, Wu HN, Zhang JY,
Liu ZH, Li YQ, Luo XL, Lin Y and Liang R: GP73 level determines
chemo-therapeutic resistance in human hepatocellular carcinoma
cells. J Cancer. 9:415–423. 2018. View Article : Google Scholar :
|
|
148
|
Lebredonchel E, Houdou M, Potelle S, de
Bettignies G, Schulz C, Krzewinski Recchi MA, Lupashin V, Legrand
D, Klein A and Foulquier F: Dissection of TMEM165 function in Golgi
glycosylation and its Mn2+ sensitivity. Biochimie.
165:123–130. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Lee JS, Kim MY, Park ER, Shen YN, Jeon JY,
Cho EH, Park SH, Han CJ, Choi DW, Jang JJ, et al: TMEM165, a Golgi
transmembrane protein, is a novel marker for hepatocellular
carcinoma and its depletion impairs invasion activity. Oncol Rep.
40:1297–1306. 2018.PubMed/NCBI
|
|
150
|
Lee SH, Yoo HJ, Rim DE, Cui Y, Lee A, Jung
ES, Oh ST, Kim JG, Kwon OJ, Kim SY and Jeong SW: Nuclear expression
of GS28 protein: A novel biomarker that predicts prognosis in
colorectal cancers. Int J Med Sci. 14:515–522. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Cho U, Kim HM, Park HS, Kwon OJ, Lee A and
Jeong SW: Nuclear expression of GS28 protein: A novel biomarker
that predicts worse prognosis in cervical cancers. PLoS One.
11:e01626232016. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Rodriguez JL, Gelpi C, Thomson TM, Real FJ
and Fernández J: Anti-golgi complex autoantibodies in a patient
with Sjögren syndrome and lymphoma. Clin Exp Immunol. 49:579–586.
1982.
|
|
153
|
Fritzler MJ, Etherington J, Sokoluk C,
Kinsella TD and Valencia DW: Antibodies from patients with
autoimmune disease react with a cytoplasmic antigen in the Golgi
apparatus. J Immunol. 132:2904–2908. 1984.PubMed/NCBI
|
|
154
|
Hong HS, Morshed SA, Tanaka S, Fujiwara T,
Ikehara Y and Nishioka M: Anti-Golgi antibody in rheumatoid
arthritis patients recognizes a novel antigen of 79 kDa (doublet)
by western blot. Scand J Immunol. 36:785–792. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Gentric A, Blaschek M, Julien C, Jouquan
J, Pennec Y, Berthelot JM, Mottier D, Casburn-Budd R and Youinou P:
Nonorgan-specific autoantibodies in individuals infected with type
1 human immunodeficiency virus. Clin Immunol Immunopathol.
59:487–494. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Griffith KJ, Chan EK, Lung CC, Hamel JC,
Guo X, Miyachi K and Fritzler MJ: Molecular cloning of a novel
97-kd Golgi complex autoantigen associated with Sjögren's syndrome.
Arthritis Rheum. 40:1693–1702. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Gaspar ML, Marcos MA, Gutierrez C, Martin
MJ, Bonifacino JS and Sandoval IV: Presence of an autoantibody
against a Golgi cisternal membrane protein in the serum and
cerebrospinal fluid from a patient with idiopathic late onset
cerebellar ataxia. J Neuroimmunol. 17:287–299. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Huidbüchel E, Blaschek M, Seigneurin JM,
Lamour A, Berthelot JM and Youinou P: Anti-organelle and
anti-cytoskeletal autoantibodies in the serum of Epstein-Barr
virus-infected patients. Ann Med Interne (Paris). 142:343–346.
1991.
|
|
159
|
Paraná R, Schinoni MI, de Freitas LA,
Codes L, Cruz M, Andrade Z and Trepo C: Anti-Golgi complex
antibodies during pegylated-interferon therapy for hepatitis C.
Liver Int. 26:1148–1154. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Mozo L, Simó A, Suarez A, Rodrigo L and
Gutiérrez C: Autoantibodies to Golgi proteins in hepatocellular
carcinoma: Case report and literature review. Eur J Gastroenterol
Hepatol. 14:771–774. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Fritzler MJ, Hudson M, Choi MY, Mahler M,
Wang M, Bentow C, Milo J and Baron M; Canadian Scleroderma Research
Group: Bicaudal D2 is a novel autoantibody target in systemic
sclerosis that shares a key epitope with CENP-A but has a distinct
clinical phenotype. Autoimmun Rev. 17:267–275. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Xue F, Wen Y, Wei P, Gao Y, Zhou Z, Xiao S
and Yi T: A smart drug: A pH-responsive photothermal ablation agent
for Golgi apparatus activated cancer therapy. Chem Commun (Camb).
53:6424–6427. 2017. View Article : Google Scholar
|
|
163
|
Li H, Zhang P, Luo J, Hu D, Huang Y, Zhang
ZR, Fu Y and Gong T: Chondroitin sulfate-linked prodrug
nanoparticles target the Golgi apparatus for cancer metastasis
treatment. ACS Nano. 13:9386–9396. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Zappa F, Failli M and De Matteis MA: The
Golgi complex in disease and therapy. Curr Opin Cell Biol.
50:102–116. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Borck G, Mollà-Herman A, Boddaert N,
Encha-Razavi F, Philippe A, Robel L, Desguerre I, Brunelle F,
Benmerah A, Munnich A and Colleaux L: Clinical, cellular, and
neuropathological consequences of AP1S2 mutations: Further
delineation of a recognizable X-linked mental retardation syndrome.
Hum Mutat. 29:966–974. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Ammann S, Schulz A, Krägeloh-Mann I,
Dieckmann NM, Niethammer K, Fuchs S, Eckl KM, Plank R, Werner R,
Altmüller J, et al: Mutations in AP3D1 associated with
immunodeficiency and seizures define a new type of Hermansky-Pudlak
syndrome. Blood. 127:997–1006. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Lu J, Tiao G, Folkerth R, Hecht J, Walsh C
and Sheen V: Overlapping expression of ARFGEF2 and Filamin A in the
neuroependymal lining of the lateral ventricles: Insights into the
cause of periventricular heterotopia. J Comp Neurol. 494:476–484.
2006. View Article : Google Scholar
|
|
168
|
Vogt G, El Choubassi N, Herczegfalvi Á,
Kölbel H, Lekaj A, Schara U, Holtgrewe M, Krause S, Horvath R,
Schuelke M, et al: Expanding the clinical and molecular spectrum of
ATP6V1A related metabolic cutis laxa. J Inherit Metab Dis. Dec
15–2020.Online ahead of print. PubMed/NCBI
|
|
169
|
Pflieger LT, Dansithong W, Paul S, Scoles
DR, Figueroa KP, Meera P, Otis TS, Facelli JC and Pulst SM: Gene
co-expression network analysis for identifying modules and
functionally enriched pathways in SCA2. Hum Mol Genet.
26:3069–3080. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Shestakova A, Zolov S and Lupashin V: COG
complex-mediated recycling of Golgi glycosyltransferases is
essential for normal protein glycosylation. Traffic. 7:191–204.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Jensson BO, Hansdottir S, Arnadottir GA,
Sulem G, Kristjansson RP, Oddsson A, Benonisdottir S, Jonsson H,
Helgason A, Saemundsdottir J, et al: COPA syndrome in an Icelandic
family caused by a recurrent missense mutation in COPA. BMC Med
Genet. 18:1292017. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Han C, Alkhater R, Froukh T, Minassian AG,
Galati M, Liu RH, Fotouhi M, Sommerfeld J, Alfrook AJ, Marshall C,
et al: Epileptic encephalopathy caused by mutations in the guanine
nucleotide exchange factor DENND5A. Am J Hum Genet. 99:1359–1367.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Dupuis N, Fafouri A, Bayot A, Kumar M,
Lecharpentier T, Ball G, Edwards D, Bernard V, Dournaud P, Drunat
S, et al: Dymeclin deficiency causes postnatal microcephaly,
hypomyelination and reticulum-to-Golgi trafficking defects in mice
and humans. Hum Mol Genet. 24:2771–2783. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Utine GE, Taşkıran EZ, Koşukcu C,
Karaosmanoğlu B, Güleray N, Doğan ÖA, Kiper PÖ, Boduroğlu K and
Alikaşifoğlu M: HERC1 mutations in idiopathic intellectual
disability. Eur J Med Genet. 60:279–283. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Xing G, Yao J, Wu B, Liu T, Wei Q, Liu C,
Lu Y, Chen Z, Zheng H, Yang X and Cao X: Identification of OSBPL2
as a novel candidate gene for progressive nonsyndromic hearing loss
by whole-exome sequencing. Genet Med. 17:210–218. 2015. View Article : Google Scholar
|
|
176
|
Dupuis N, Lebon S, Kumar M, Drunat S,
Graul-Neumann LM, Gressens P and El Ghouzzi V: A novel RAB33B
mutation in Smith-McCort dysplasia. Hum Mutat. 34:283–286. 2013.
View Article : Google Scholar
|
|
177
|
Kondo Y, Fu J, Wang H, Hoover C, McDaniel
JM, Steet R, Patra D, Song J, Pollard L, Cathey S, et al: Site-1
protease deficiency causes human skeletal dysplasia due to
defective inter-organelle protein trafficking. JCI Insight.
3:e1215962018. View Article : Google Scholar :
|
|
178
|
Carvalho DR, Speck-Martins CE, Brum JM,
Ferreira CR and Sobreira NLM: Spondyloepimetaphyseal dysplasia with
elevated plasma lysosomal enzymes caused by homozygous variant in
MBTPS1. Am J Med Genet A. 182:1796–1800. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Ng BG, Asteggiano CG, Kircher M,
Buckingham KJ, Raymond K, Nickerson DA, Shendure J, Bamshad MJ;
University of Washington Center for Mendelian Genomics; Ensslen M
and Freeze HH: Encephalopathy caused by novel mutations in the
CMP-sialic acid transporter, SLC35A1. Am J Med Genet A.
173:2906–2911. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Dörre K, Olczak M, Wada Y, Sosicka P,
Grüneberg M, Reunert J, Kurlemann G, Fiedler B, Biskup S, Hörtnagel
K, et al: A new case of UDP-galactose transporter deficiency
(SLC35A2-CDG): Molecular basis, clinical phenotype, and therapeutic
approach. J Inherit Metab Dis. 38:931–940. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Wei J, Zhang YY, Luo J, Wang JQ, Zhou YX,
Miao HH, Shi XJ, Qu YX, Xu J, Li BL and Song BL: The GARP complex
is involved in intracellular cholesterol transport via targeting
NPC2 to lysosomes. Cell Rep. 19:2823–2835. 2017. View Article : Google Scholar : PubMed/NCBI
|