Introduction
Paraquat (PQ) is a highly effective and inexpensive
contact heterocyclic herbicide and one of the most widely used
pesticides in agriculture globally (1). PQ poisoning has a high mortality
rate, with no specific antidote or effective treatment option
(2). PQ can cause damage to the
lungs, liver, kidney, heart and other organs, with lung damage
being the most prominent. Due to the strong dopamine absorption
system in the lungs, PQ, which has a chemical structure similar to
polyamines, mainly accumulates in the lungs (3). Consequently, PQ poisoning leads to
acute lung injury (ALI) or even acute respiratory distress syndrome
(ARDS), which is responsible for early death (4). The mechanisms through which PQ
causes ALI are complex and unclear; however, the majority of
studies have reported that the biological processes of redox
response and inflammatory cascade reaction play important roles in
PQ poisoning (5-8).
Lipoxins (LXs) are a type of endogenous lipid media
produced by arachidonic acid (AA) during sequential catalysis by
different lipoxygenases. They can be divided into 4 types according
to their molecular conformation (9), among which lipoxin A4 (LXA4) is
generated via continuous oxidation catalysis. As a negative
regulator of inflammation, LXA4 functions as an anti-inflammatory
agent, promoting the resolution of inflammation, regulating immune
function, and stimulating tissue and cell damage repair (10,11). In various inflammation-related
diseases, LXA4 inhibits the release of pro-inflammatory factors and
the infiltration of inflammatory cells, and promotes the chemotaxis
and recruitment of macrophages, thus enhancing their
non-inflammatory phagocytotic function (12,13). Furthermore, LXA4 inhibits ROS
generation, preventing tissues or macrophages from oxidative
stress-mediated damage (14-16). LXA4 has also been found to
increase the activity of antioxidant enzymes in various organs, and
to play a protective role in restoring the oxidant/antioxidant
balance (17-19).
However, the biological effects of LXA4 on
PQ-induced ALI have yet to be confirmed, at least to the best of
our knowledge. Therefore, the aim of the present study was to
investigate the role of LXA4 in PQ-induced ALI and to elucidate the
possible underlying mechanisms.
Materials and methods
Animals
Specific pathogen-free male SD rats (aged 6-8 weeks;
body weight, 250-300 g; n=40), purchased from Beijing HFK
Bioscience Co. Ltd, were allowed to acclimatize in the laboratory
at the Experimental Animal Center of Shengjing Hospital of China
Medical University for 1 week with free access to food and drink.
The animals were maintained under a controlled temperature
(20-25°C) and constant humidity (40-70%), and were subjected to a
natural photoperiod (12/12 h light/dark cycle). The experiments
were approved by the Ethics Committee of Shengjing Hospital of
China Medical University (approval no. 2019PS656K). All procedures
involving animals were performed in accordance with the ARRIVE
guidelines and the National Institutes of Health Guidelines for the
Care and Use of Laboratory Animals (NIH Publications no. 8023,
revised in 1978).
Establishment of animal models
The rats were randomly divided into 4 groups as
follows: The control (CON; n=10), PQ poisoning (PQ; n=10), PQ
poisoning + lipoxin A4 (PQ + LXA4; n=10) and LXA4 (n=10) groups.
The rats in the PQ and PQ + LXA4 groups were intraperitoneally
injected with PQ (Sigma-Aldrich; Merck KGaA) at a dose of 20 mg/kg
(6,20), while an equivalent volume of
saline was injected into the rats in the CON and LXA4 groups. At 1
h after the PQ or saline injection, the rats in the PQ + LXA4 and
LXA4 groups were injected daily with LXA4 diluted to 0.1 mg/ml in
saline (Cayman Chemical Co.) at a dose of 2 µg/kg through
the tail vein (21,22), while the PQ and CON groups were
treated with an injection of equal volume of normal saline. The
rats were sacrificed by an overdose of sodium pentobarbital
(>150 mg/kg) injected intraperitoneally following exposure to PQ
for 48 h. When the cessation of the heartbeat and breathing of the
rats was confirmed, and there were no reflexes, the death of the
experimental animals was confirmed painlessly. Subsequently,
bronchoalveolar lavage fluid (BALF) and lung specimens were
collected.
Analysis of lung wet/dry weight
ratio
The upper lobe of the right lung was collected
following exposure to PQ and the wet weight was obtained. The lung
tissues were then placed in an oven at 80°C for 48 h and weighed
again to obtain the dry weight. Finally, the lung wet/dry (W/D)
weight ratio was assessed to assess the severity of pulmonary
edema.
H&E staining and scoring of lung
injuries
The middle lobe of the right lung was collected,
placed in 4% paraformaldehyde, and fixed at 4°C for at least 48 h.
Lung tissues were further subjected to concentration gradient
dehydration and wax dipping; the tissues were then embedded in
paraffin, cut into 3.5-µm-thick slices. The sections were
deparaffinized with xylene, hydrated, then incubated with
hematoxylin for 3 min, washed and stained with eosin for 1 min at
room temperature. Subsequently, the sections were dehydrated with
gradient ethanol for 20 min, immersed in xylene to be transparent
for 2 min, washed and finally sealed with a neutral gel. After
sealing the slides with neutral gum, pathological changes were
observed using a microscope (Nikon Corporation) and recorded using
NIS-Element software (version 4.6). Pulmonary injury was then
scored according to previously described methods (23). Briefly, the following 5-level
scale was applied to independently score the pathological changes
(alveolar congestion, hemorrhage, and wall thickening, coupled with
neutrophil infiltration or aggregation in the alveolar space or
blood vessel wall and transparent membrane formation) as follows:
0, 1, 2, 3 and 4 points represented no or minor, mild, moderate,
severe and extremely severe lesions, respectively. Finally, the
severity of pulmonary injury was evaluated by the sum of the scores
of each pathological feature.
Measurement of superoxide dismutase (SOD)
content in lung tissues
The lung tissues were weighed accurately; the
homogenates [10% (w/v)] were resuspended in normal saline and
centrifuged at 2,500-3,000 × g at 4°C for 10 min. Following
instructions provided with the SOD kit (Nanjing Jiancheng
Bioengineering Institute), the supernatants were added to a 96-well
plate, mixed, and incubated at 37°C for 20 min. The absorbance of
the sample was measured at 450 nm using a microplate reader
(Elx800, BioTek Instruments, Inc.).
Detection of malondialdehyde (MDA)
content in lung tissues
The thiobarbituric acid (TBA) method was used to
determine the MDA levels in lung tissue by measuring the absorbance
value at 532 nm using a microplate reader (Elx800, BioTek
Instruments, Inc.), according to the instructions provided with the
MDA kit (Nanjing Jiancheng Bioengineering Institute).
Cells and cell culture
RAW264.7 cells were obtained from the Cell Bank Type
Culture Collection of the Chinese Academy of Sciences. RAW264.7
mouse macrophages were cultured in high-glucose Dulbecco's modified
Eagle's medium (DMEM), containing 10% fetal bovine serum (FBS) and
incubated at 37°C with 5% CO2 in a humidified
atmosphere. The culture medium was changed daily.
Cell viability assay
Various concentrations of PQ (10, 50, 100, 250, 500
and 1,000 µm) or LXA4 (0, 1, 10, 25, 50, 100, 500 and 1,000
nm) were applied to determine the appropriate PQ and LXA4
experimental concentrations. The effects of PQ or LXA4 on cell
viability were assessed using a Cell Counting kit-8 (CCK-8) kit
(Dojindo Laboratories, Inc.).
Cell transfection
The GV417 vector-Toll-like receptor 4 (TLR4) plasmid
(Shanghai GeneChem Co., Ltd.) was transfected into the cells to
achieve the overexpression of TLR4. Specifically, when cells grew
to a density of 70-90%, Lipofectamine 3000 reagent (Thermo Fisher
Scientific, Inc.) and the TLR4 overexpression plasmid at the
concentration of 10 µg/ml were applied for transfection
according to the manufacturer's instructions. Following 24 h of
transfection, the expression level of TLR4 was evaluated by western
bot analysis and reverse transcription-quantitative polymerase
chain reaction (RT-qPCR).
Establishment of cell models
After selecting the optimal concentration of PQ or
LXA4, the RAW264.7 mouse macrophages were divided into 6 groups as
follows: i) The control (CON) group, cultured in normal medium; ii)
PQ poisoning (PQ) group, exposed to medium containing PQ (100
µm) for 24 h; iii) PQ poisoning + LXA4 (PQ + LXA4) group, at
the point of 1-h exposure to PQ (100 µm), the cells were
treated with 10 µl LXA4 (100 nm) for a further 24 h; iv) PQ
poisoning + LXA4 + GV417-TLR4 plasmid (PQ + LXA4 + TLR4) group,
after 24 h of TLR4-plasmid transfection, the cells were treated as
described above for the PQ + LXA4 group; v) PQ poisoning + LXA4 +
empty plasmid (PQ + LXA4 + mock) group, after 24 h of empty
vector-plasmid transfection, the cells were then treated as
described above for the PQ + LXA4 group; and vi) The LXA4 group,
cells were only treated with 10 µl LXA4 (100 nm) for 24 h.
Following 24 h of treatment, the cells and culture supernatants
were collected for use in subsequent experiments.
Reactive oxygen species (ROS) assays
After removing the cell culture solution, DCFH-DA
was added, which was diluted in serum-free medium to each plate
well, followed by incubation in an incubator at 37°C for 30 min.
The cells were then washed 3 times with serum-free medium. Images
were obtained under a fluorescence microscope (Nikon
Corporation).
Determination of pro-inflammatory
mediators
Commercially available ELISA kits (EK0526, EK0527,
EK0390 and EK0394, Wuhan Boster Biological Technology, Ltd.) were
used to measure the tumor necrosis factor (TNF)-α and interleukin
(IL)-1β levels in BALF and cell supernatants. The TNF-α and IL-1β
levels were calculated according to the standard curve established
by the concentration range.
RT-qPCR
Total RNA was extracted from the lung tissue and
RAW264.7 macrophages using RNAiso Plus reagent (9108, Takara Bio,
Inc.). Complementary DNA (cDNA) was synthesized using the
PrimeScript RT (with gDNA eraser) kit (RR047A, Takara Bio, Inc.).
Subsequently, qPCR was performed using the TB Green Premix Ex Taq™
II (Tli RNaseH Plus) kit (RR820A, Takara Bio, Inc.) on the Roche
Light Cycler 480 system. We carry out the thermocycling according
to the following procedure: Initial denaturation at 95°C for 30
sec, followed by 40 cycles of 5 sec denaturation at 95°C, annealing
and extension at 60°C for 20 sec. β-actin was used as an internal
control, and the 2−ΔΔCq formula was used to calculate
relative gene expression. The sequences of the primers used in the
present study are listed in Table
I.
 | Table IPrimer sequences of the genes
detected by RT-qPCR. |
Table I
Primer sequences of the genes
detected by RT-qPCR.
| Gene | Primer sequences
(5′-3′) |
|---|
| Rat TLR4 | F:
ACTTTATCCAGAGCCGTTGGTGTATC |
| R:
TCAAGGACAATGAAGATGATGCCAGAG |
| Rat MyD88 | F:
CGGCAACTAGAACAGACAGACTATCG |
| R:
TCGTCAGAAACAACCACCACCATG |
| Rat TNF-α | F:
CCACGCTCTTCTGTCTACTGAACTTC |
| R:
TGGGCTACGGGCTTGTCACTC |
| Rat IL-1β | F:
AATCTCACAGCAGCATCTCGACAAG |
| R:
TCCACGGGCAAGACATAGGTAGC |
| Rat TRAM | F:
TCGCAAGAAGAGCAACAAGAACCC |
| R:
AGAGCATCGCCAGACAGGAGAC |
| Rat TRIF | F:
GTCGCGGAAGTGTGACTAGAAGAG |
| R:
TGCAGCTACCAGAAACCCTCCTC |
| Rat β-actin | F:
TGTCACCAACTGGGACGATA |
| R:
GGGGTGTTGAAGGTCTCAAA |
| Mouse TLR4 | F:
GCCATCATTATGAGTGCCAATT |
| R:
AGGGATAAGAACGCTGAGAATT |
| Mouse | F:
CGGAACTTTTCGATGCCTTTAT |
| MyD88 | R:
CACACACAACTTAAGCCGATAG |
| Mouse | F:
ATGTCTCAGCCTCTTCTCATTC |
| TNF-α | R:
GCTTGTCACTCGAATTTTGAGA |
| Mouse IL-1β | F:
TCGCAGCAGCACATCAACAAGAG |
| R:
AGGTCCACGGGAAAGACACAGG |
| Mouse | F:
GACAGTGTGGATGCCGATCAAGAC |
| TRAM | R:
TGGTCCTGCTCTCCTGTTGGTG |
| Mouse TRIF | F:
ACGATCCTGCTCCTGACTGCTAG |
| R:
TCCTGCCTCCCAGACTGTGTAAG |
| Mouse Actb | F:
GTGCTATGTTGCTCTAGACTTCG |
| R:
ATGCCACAGGATTCCATACC |
Western blot analysis
RIPA lysis buffer mixed with protease inhibitor
(PMSF) and phosphatase inhibitor (PI) was prepared to extract total
protein from the lung tissues or RAW264.7 following exposure to PQ.
Nuclear and cytoplasmic protein extraction kits (P0028, Beyotime
Institute of Biotechnology) were used to isolate nuclear and
cytoplasmic proteins. The concentration of the extracted protein
was measured using a BCA assay kit (P0010, Beyotime Institute of
Biotechnology), and a total of 50 µg protein sample was
loaded onto each well of a 10% sodium dodecyl sulfate
polyacrylamide gel for electrophoresis. Following electrophoresis,
the proteins were transferred onto a polyvinylidene fluoride (PVDF)
membrane followed by incubation in 5% skim milk or 5% BSA blocking
solution on a rotary shaker at room temperature for 2 h.
Subsequently, the membranes with proteins were incubated on a
shaker at 4°C overnight with the following primary antibodies: TLR4
(1:1,000, #AF7017), myeloid differentiation primary response 88
(MyD88; 1:1,000, #AF5195), phosphoinositide 3-kinase (PI3K;
1:1,000, #AF6241), phosphorylated (p-)PI3K (1:1,000, #AF3242) (all
from Affinity Biosciences); AKT (1:1,000, #4691), p-AKT (1:2,000,
#4060), IκBα (1:1,000, #4814) and p-IκBα (1:1,000, #2859) (all from
Cell Signaling Technology, Inc.); nuclear factor (NF)-κB p65
(1:500, ab16502, Abcam); TATA box binding protein (TBP; 1:2,000,
#22006-1-AP) and β-actin (1:2000, #20536-1-AP) (both from
ProteinTech Group, Inc.). The following day, the membranes were
washed using Tris-buffered saline with Tween-20 (TBST;
Sigma-Aldrich; Merck KGaA) 3 times and incubated with the
HRP-conjugated secondary antibody (#ZB-5301, ZSJQB Co., Ltd.) for 1
h at room temperature. After washing a further 3 times, enhanced
chemiluminescence (ECL) (KF003-100, Affinity Biosciences) solution
was applied to visualize the immunoreactive bands, and ImageJ
(version 1.8.0) software was used for quantitative image
analysis.
Statistical analysis
The data are expressed as the means ± standard
deviation (SD). One-way analysis of variance (ANOVA) and Tukey's
test was performed to analyze the statistical differences between
each group. A P-value <0.05 was considered to indicate a
statistically significant difference. All statistical analyses were
performed using Graphpad Prism 7.0 software.
Results
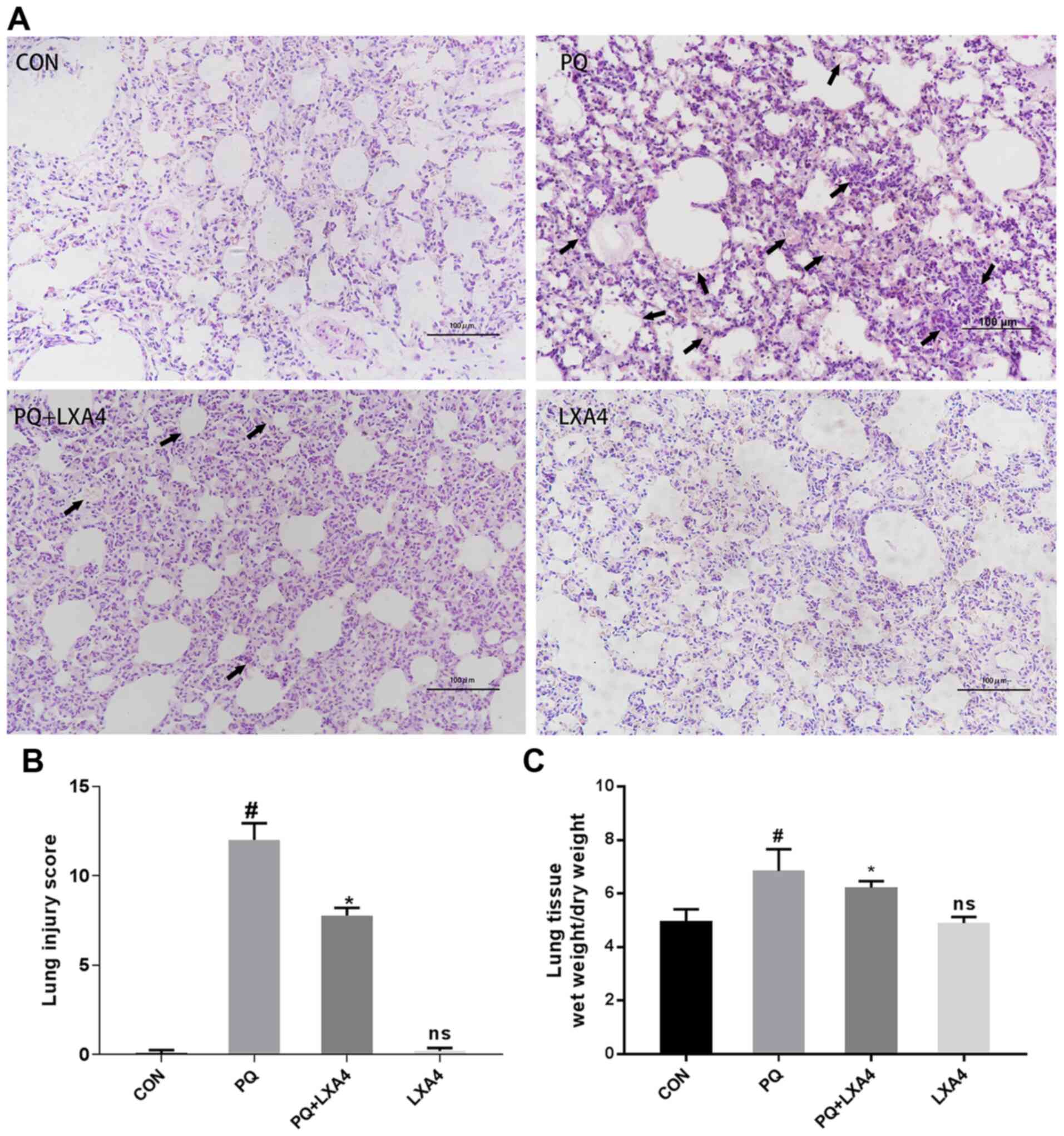
LXA4 alleviates lung histopathological
damage induced by PQ
As shown in Fig.
1A, histopathological injuries were observed in the PQ group,
reflected by alveolar structure destruction, alveolar congestion or
bleeding, alveolar cavity collapse, inflammatory cell infiltration
and the formation of transparent film in part of the alveolar
cavity. In the poisoned group treated with LXA4, the
histopathological injuries were all attenuated, together with
decreased pulmonary hemorrhage and edema. Significant
histopathological differences were not observed between the CON and
LXA4 groups. The histological scores of the PQ group were
significantly higher than those of the CON group, while the use of
LXA4 substantially attenuated the lung injury scores (Fig. 1B).
LXA4 reduces the lung W/D weight ratio in
rats exposed to PQ
The lung W/D weight ratio was measured in each group
of rats to confirm the protective effects of LXA4 on pulmonary
edema induced by PQ. As shown in Fig. 1C, the W/D ratio of the PQ-treated
rat lungs was higher compared to that of the CON group, but was
markedly reduced by LXA4.
LXA4 attenuates the inhibitory effects on
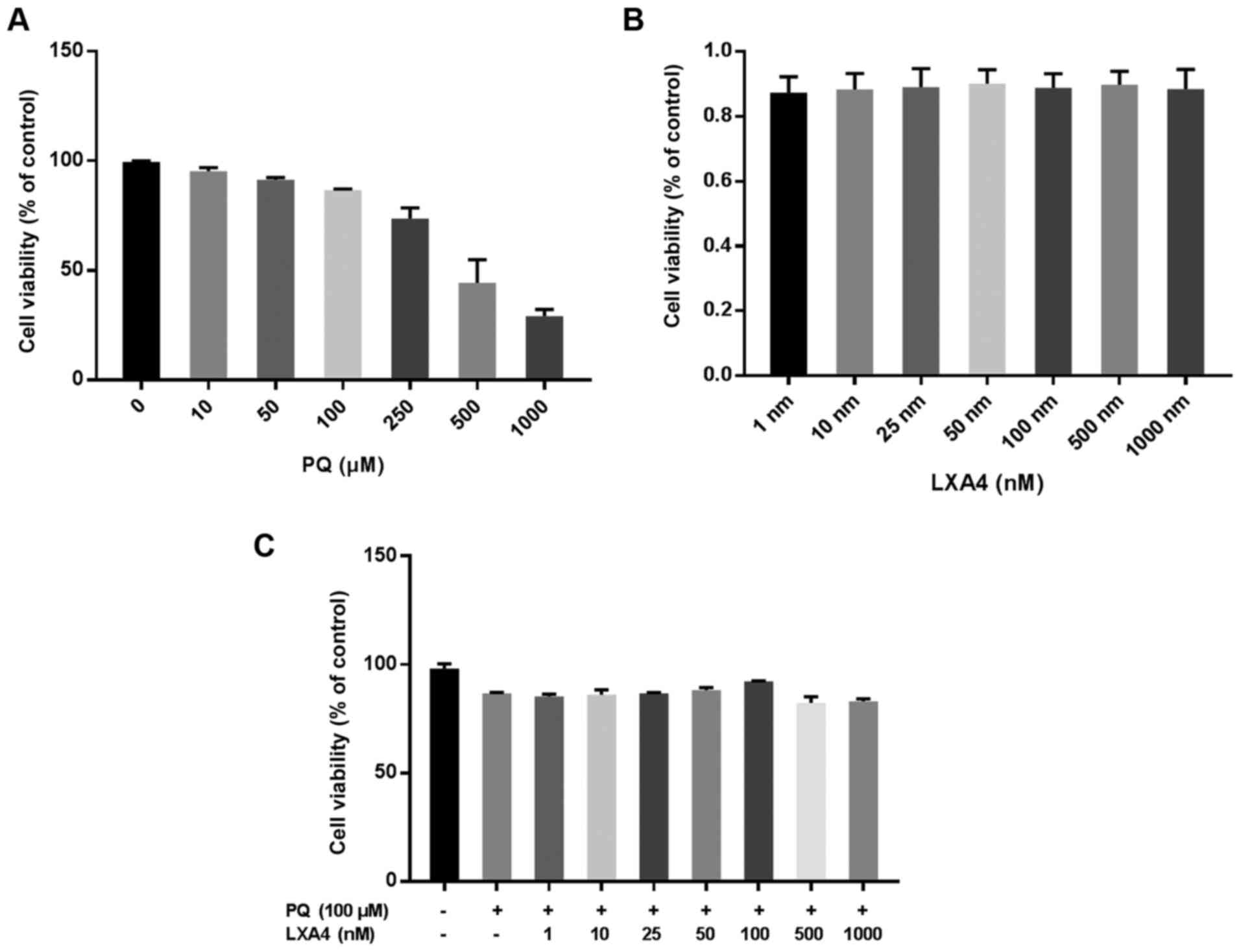
the viability of RAW264.7 cells induced by PQ
CCK-8 assay was used to evaluate the optimal
concentration of PQ for use in subsequent experiments. The results
suggested that the inhibitory effect of PQ on the viability of
RAW264.7 cells occurred in a concentration-dependent manner. When
the PQ concentration was ≥100 µM, RAW264.7 cell viability
was markedly inhibited (Fig.
2A). Therefore, PQ at 100 µM was used as the optimum
dose in subsequent in vitro experiments. The same method was
used to determine the possible toxic effects of LXA4 on RAW264.7
macrophages and the optimal concentration of LXA4 that could reduce
PQ cytotoxicity. Notably, in the present study, it was found that
treatment with LXA4 alone had no toxic effect on the RAW264.7 cells
(Fig. 2B), and when the LXA4
concentration was 100 nM, its ability to reduce PQ cytotoxicity was
optimal (Fig. 2C). Thus, the
concentration of 100 nM LXA4 was selected as the optimum dose for
use in subsequent studies.
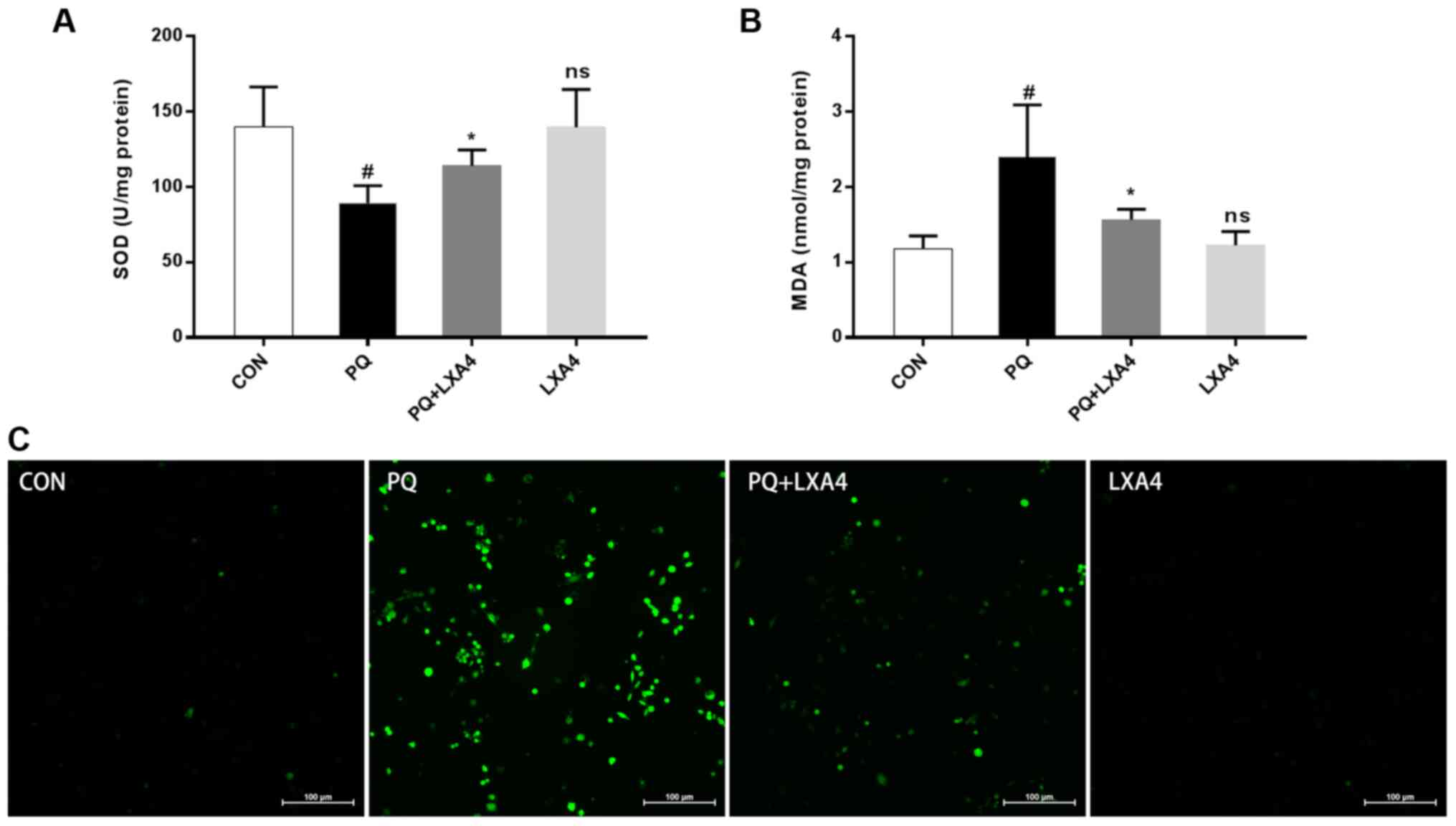
LXA4 reduces oxidative stress in lung
tissues and ROS generation in RAW264.7 macrophages
As shown in Fig. 3A
and B, lung SOD activity significantly decreased following
exposure to PQ, while the MDA content markedly increased; however,
LXA4 treatment significantly restored SOD activity and decreased
the MDA content. The ROS expression level in the RAW264.7
macrophages increased significantly following exposure to PQ,
whereas this decreased upon treatment with LXA4 (Fig. 3C). These results indicated that
LXA4 exerted beneficial effects on PQ-induced oxidative damage.
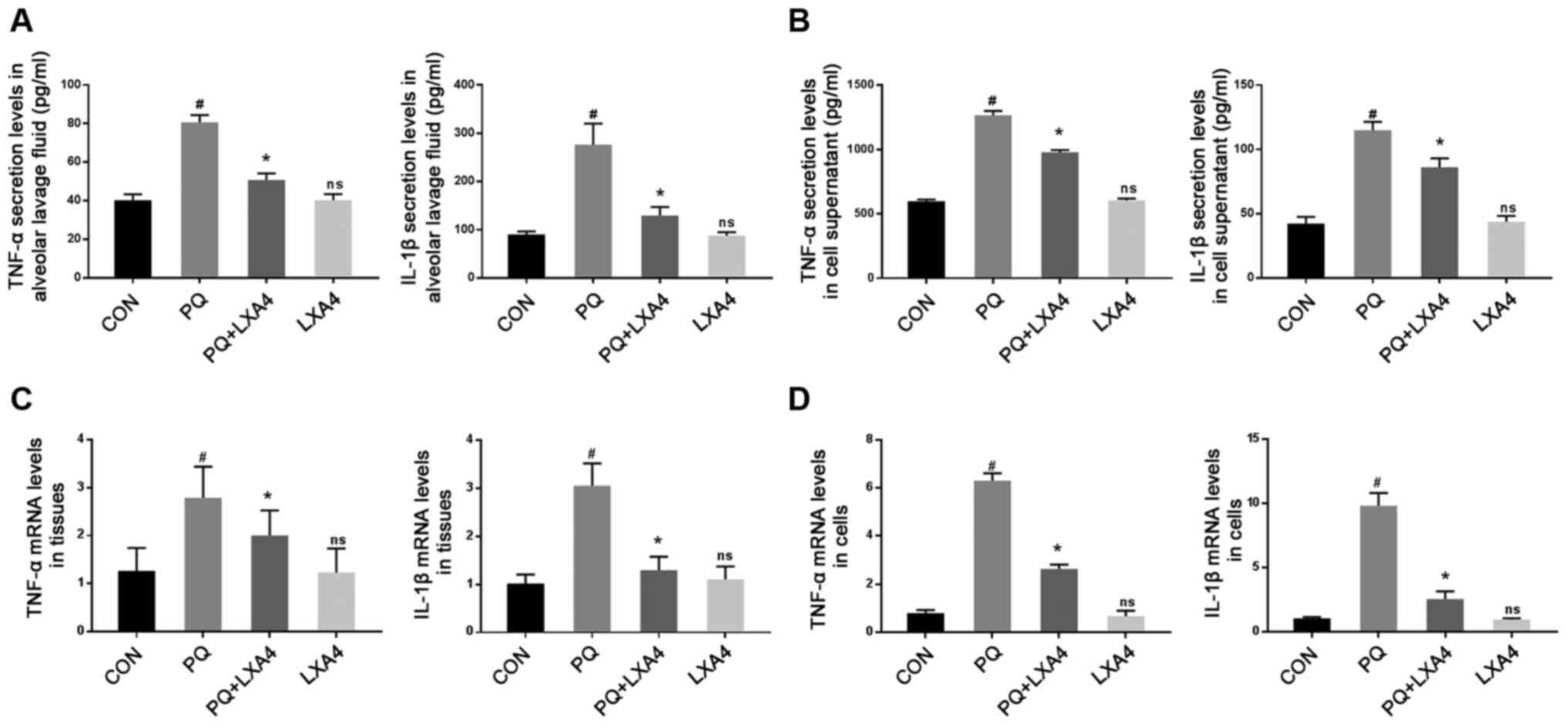
LXA4 inhibits the PQ-induced increase in
the levels of pro-inflammatory cytokines
ELISA was used to measure the levels of
pro-inflammatory factors in BALF and RAW264.7 cell supernatants.
The results revealed that PQ markedly increased TNF-α and IL-1β
secretion, while LXA4 treatment significantly reduced these levels
(Figs. 4A and B).
Simultaneously, the mRNA expression of inflammatory factors was
determined to further evaluate the effects of LXA4 on PQ. The
results revealed that the expression levels of TNF-α and IL-1β were
significantly upregulated in lung tissues and RAW264.7 cells
following PQ exposure, whereas LXA4 administration reversed these
effects (Fig. 4C and D).
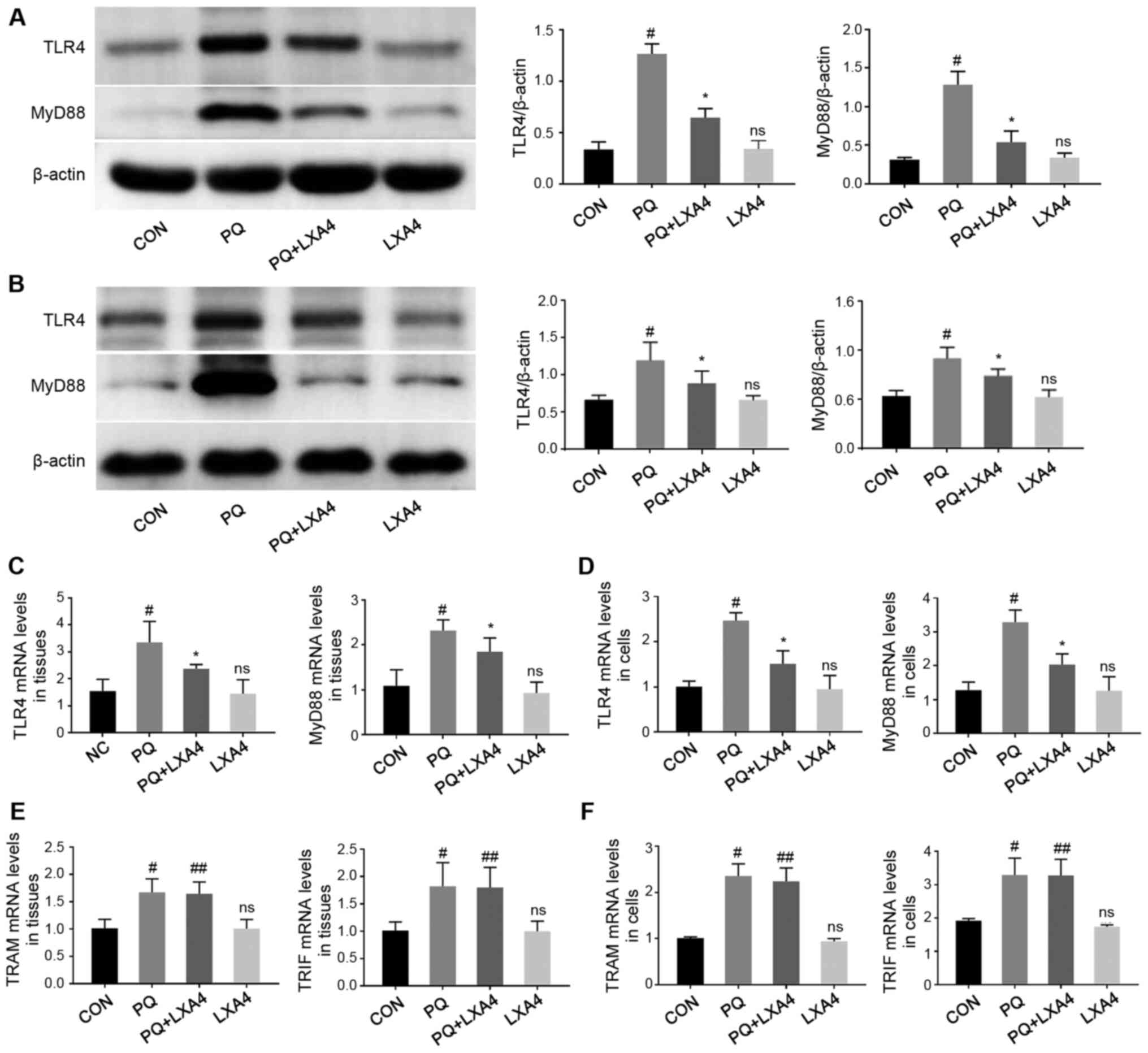
LXA4 inhibits PQ-induced TLR4 and MyD88
overexpression
In order to explore the possible immune mechanisms
responsible for the protective effects of LXA4 against PQ-induced
ALI, RT-qPCR and western blot analysis were performed to detect the
mRNA and protein levels of key molecules participating in the TLR4
signaling pathway in rat lung tissues and RAW264.7 cells. As shown
in Fig. 5, LXA4 administration
significantly reduced the elevated levels of TLR4 and MyD88 induced
by PQ. However, as for the other pathway mediated by TLR4, also
known as the Myd88-independent pathway, there was no significant
difference in the mRNA expression levels of TRAM and TRIF between
the PQ and PQ + LXA4 group. These results indicated that PQ
poisoning activated the TLR4 signaling pathway and that the
protective effects of LXA4 were mediated by the MyD88-dependent
pathway of TLR4.
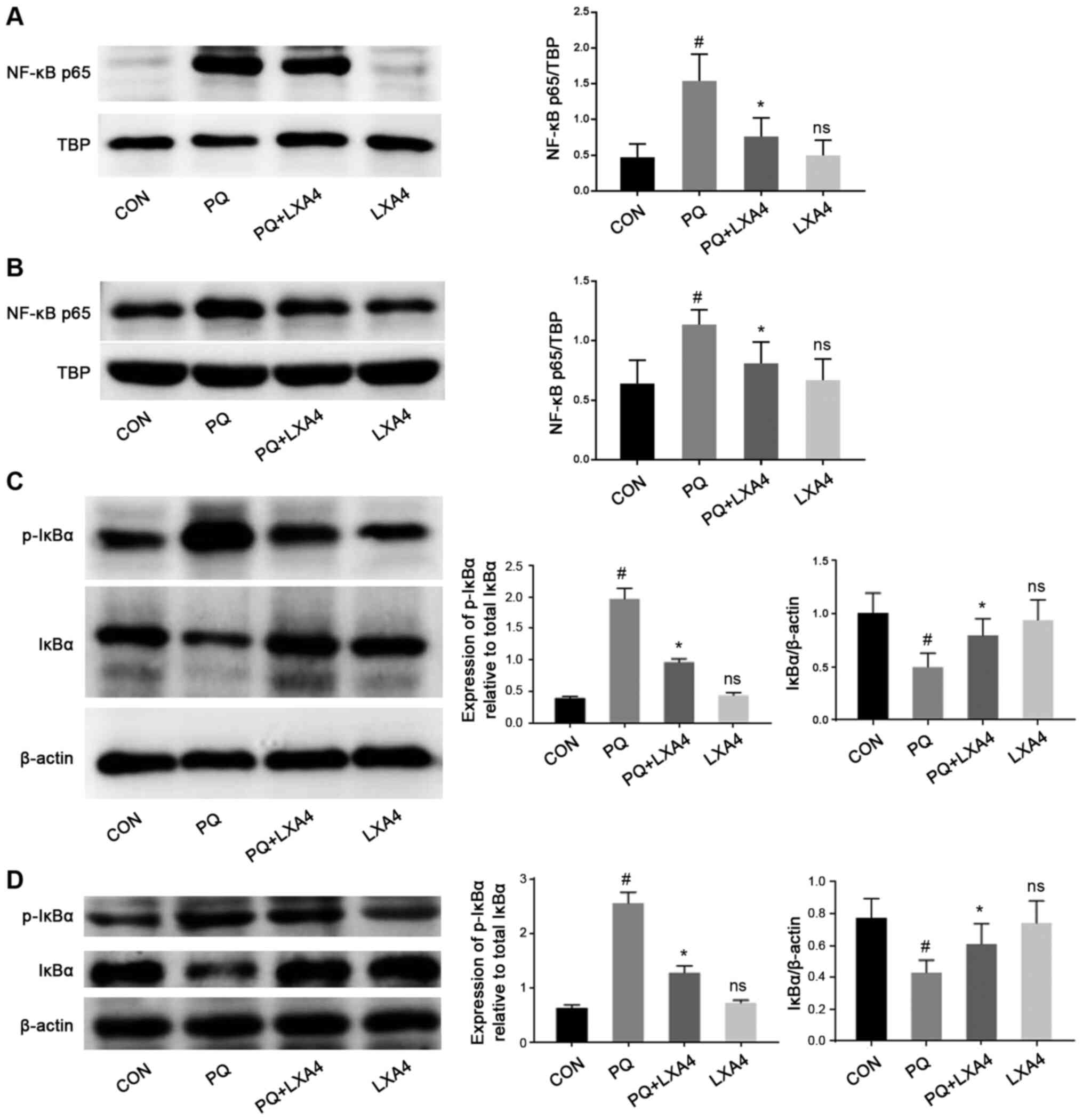
LXA4 suppresses the activation of the
NF-κB pathway
The activation of TLR4/MyD88 stimulates IκBα
phosphorylation and the nuclear transfer of NF-κB p65, thereby
activating the release of pro-inflammatory cytokines (24,25). Therefore, the present study
examined the effects of PQ and LXA4 on the NF-κB signaling pathway
to elucidate the mechanisms through which LXA4 inhibits PQ-induced
inflammation. The results from the in vitro and in
vivo experiments indicated that PQ significantly promoted the
process of IκBα degradation to p-IκBα, leading to an increase in
the transfer of NF-κB p65 to the nucleus. However, in the PQ + LXA4
group, the PQ-induced overactivation of the NF-κB signaling pathway
was markedly attenuated (Fig.
6), indicating that inflammatory responses could be inhibited
by LXA4 via the suppression of the NF-κB pathway.
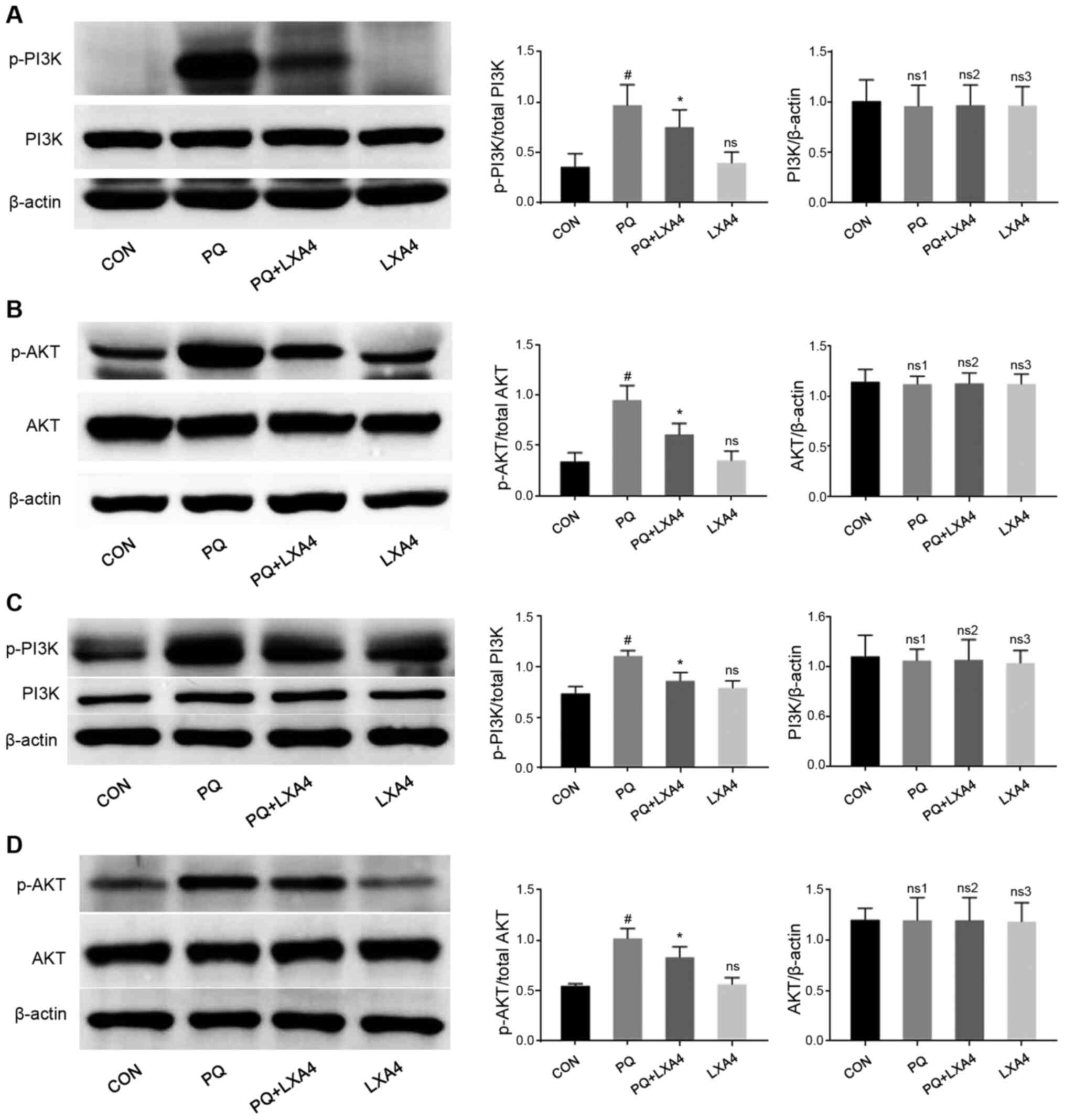
LXA4 suppresses the activation of the
PI3K/AKT pathway
Previous studies (26-28) have demonstrated that activated
TLR4/MyD88 can also activate the PI3K/AKT signaling pathway and
induce inflammatory responses. Therefore, the present study
investigated whether the PI3K/AKT signaling pathway was also
involved in the anti-inflammatory effects of LXA4 to further
clarify the cellular mechanisms responsible for the protective
effects of LXA4 on PQ-induced ALI. As shown in Fig. 7, PQ stimulation significantly
upregulated the phosphorylation of PI3K and AKT, both of which were
suppressed by LXA4 intervention. These results indicated that the
activation fo the PI3K/AKT pathway induced by PQ was inhibited by
LXA4.
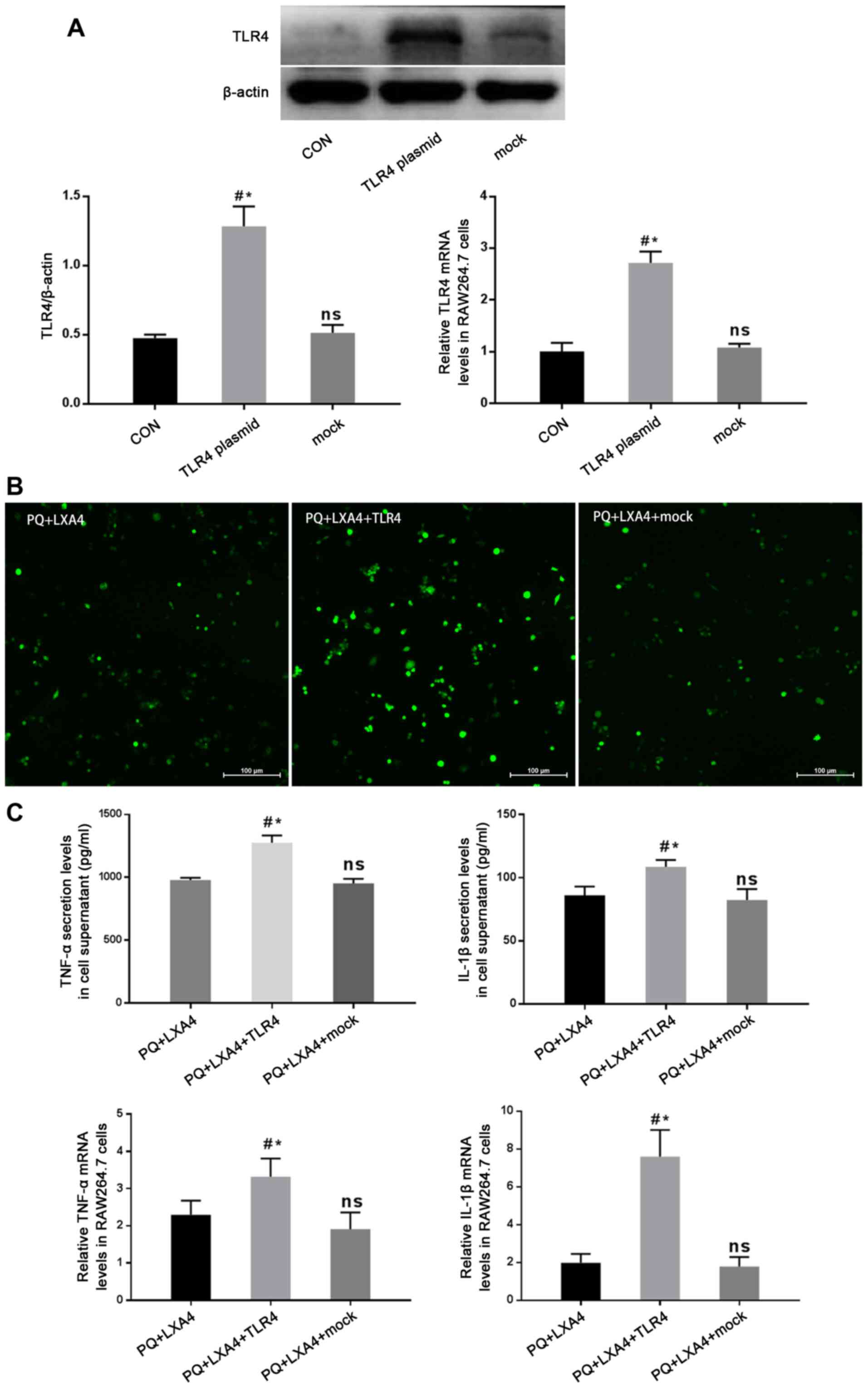
Mitigated protection of LXA4 following
the enforced overexpression of TLR4
In order to determine whether the protective effects
of LXA4 PQ poisoning are mediated by the suppression of TLR4, a
GV417-TLR4 plasmid was constructed, and rescue experiments were
conducted in vitro. As shown in Fig. 8A, the protein and mRNA expression
of TLR4 was significantly higher in the TLR4 overexpression group
than that in the CON and mock-transfection groups. The results also
revealed that the enforced overexpression of TLR4 reversed the
protective effects of LXA4 against PQ-induced ROS production and
pro-inflammatory cytokine secretion (Fig. 8B and C). In addition, the Myd88,
NF-κB and PI3K/AKT pathways inhibited by LXA4 were all re-activated
following transfection with TLR4 overexpression plasmid (Fig. 9). Therefore, these findings
verified that LXA4 exerted protective effects against PQ poisoning
through the suppression of TLR4 and its downstream MyD88-mediated
NF-κB/PI3K/AKT signaling pathway.
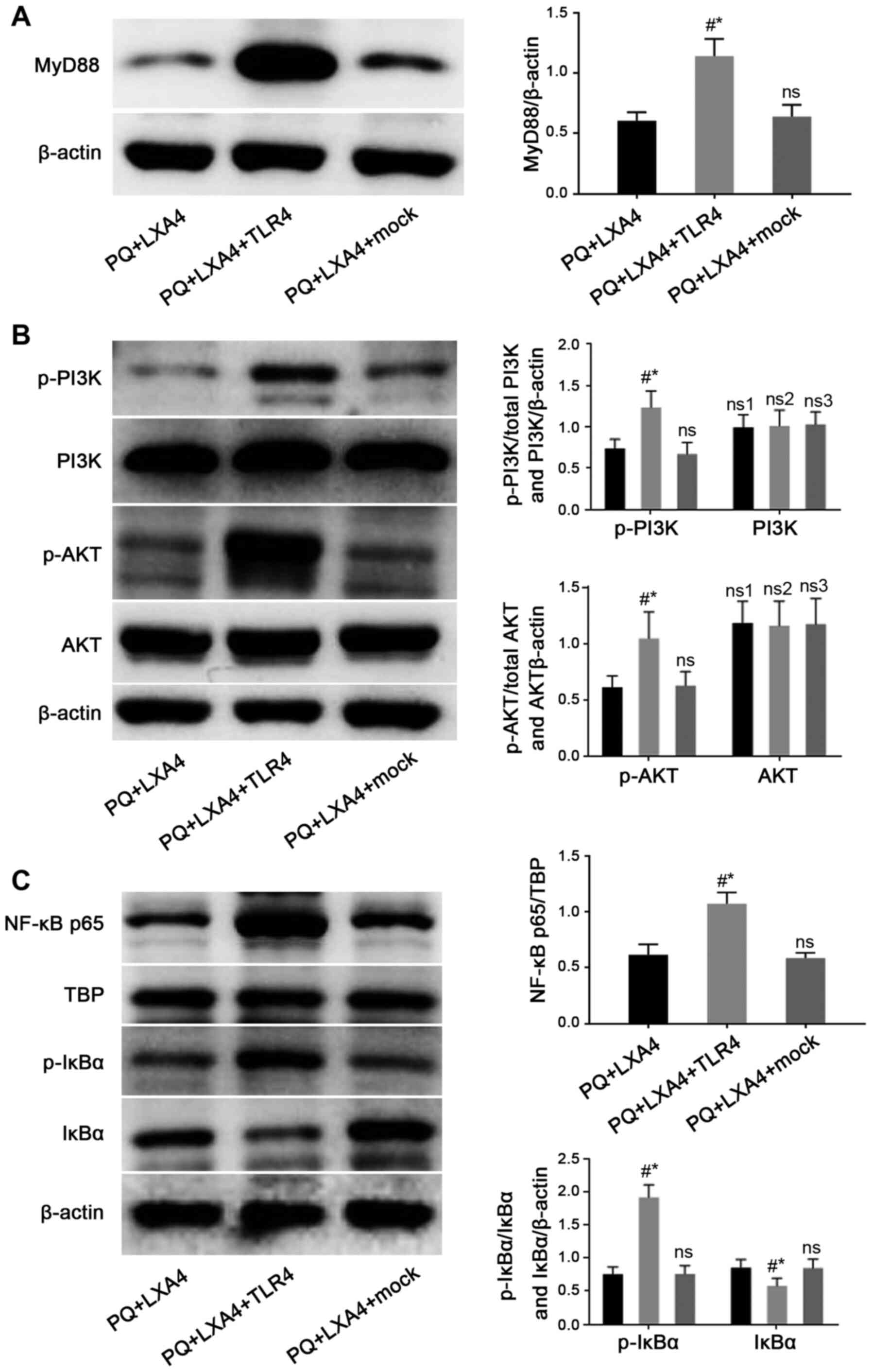 | Figure 9Mitigated protection of LXA4
following the enforced overexpression of TLR4. Detection of (A)
MyD88, and (B) total PI3K, p-PI3K, total AKT and p-AKT protein
expression, and (C) observation of NF-κB p65 nuclear protein, total
IκBα and p-IκBα protein levels in RAW264.7 cells by western blot
analysis. Data are presented as the means ± SD (n=6).
#P<0.05 vs. the PQ + LXA4 group.
*P<0.05 vs. the PQ + LXA4 + mock group. ns, not
significant; ns3, P>0.05 vs. the PQ + LXA4 group; ns1, P>0.05
vs. the PQ + LXA4 + TLR4 group; ns2, P>0.05 vs. the PQ + LXA4 +
mock group. LXA4, lipoxin A4; PQ, paraquat. |
Discussion
PQ is a highly toxic chemical herbicide and one of
the most common drugs that causes acute poisoning and severe damage
to multiple organs. Previous studies have proven that the lungs are
a specific target organ for pathological damages caused by PQ
(29). After PQ enters the body,
it rapidly becomes transported into the lung tissue through the
polyamine transportation system, resulting in ALI or even ARDS in
the early stage of poisoning. Currently, the mechanism of
PQ-induced ALI is mainly attributed to oxidative stress and
inflammatory response. In the present study, LXA4 was confirmed for
the first time to effectively attenuate lung damage caused by PQ,
and this protective effect was reflected by the inhibition of
pulmonary edema, inflammatory responses and oxidative stress.
Simultaneously, it was found that this beneficial effect of LXA4
was associated with the suppression of the TLR4/MyD88-mediated
NF-κB/PI3K/AKT signaling pathway.
LXA4 is an anti-inflammatory mediator produced by
the body itself during the late stage of acute inflammation.
Previous studies have confirmed its protective effects against
inflammation in some diseases, such as neuromyelitis optical
spectrum disorders (30),
obesity-induced adipose inflammation (31) and cerebrovascular endothelial
dysfunction (32). The results
from the present study suggested that PQ caused typical ALI and
induced severe inflammatory responses in lung tissues and RAW264.7
macrophages, mainly manifested by the destruction of alveolar
integrity, inflammatory cell infiltration, alveolar wall
thickening, pulmonary edema and upregulated levels of related
pro-inflammatory cytokines. Following LXA4 intervention, these
injuries and overactive inflammatory responses were alleviated.
SOD activity reflects the ability of the body to
scavenge oxygen-free radicals and is often used as a marker of the
antioxidant status of the body (33), while MDA levels reflect the
degree of oxidative stress damage to the body (34). PQ poisoning not only induces
excessive ROS production and impairs ROS clearance, leading to
oxidative stress reactions, but also interrupts the balance between
SOD and MDA, which is important for the antioxidant capacity of the
body. However, in the present study, following LXA4 treatment,
these conditions improved, as LXA4 restored the balance between SOD
and MDA, thus enhancing the ability of the body to resist oxidative
stress damage.
TLRs serve as significant pattern recognition
receptors in the host defense system and recognize
damage-associated molecular patterns (DAMPs) released by
non-infectious stimuli (35,36). Due to the ability to recognize
PQ-induced DAMP, TLR4 activates a downstream cascade of
inflammatory signals through the MyD88-dependent pathway (37-39). Consistent with the study by Shen
et al (24), the present
study found that TLR4 and MyD88 expression significantly increased
following exposure to PQ, and similar trends were observed in the
downstream molecules of TLR4 and MyD88 (such as NF-κB p65, TNF-α
and IL-1β). Owing to the beneficial effects of inhibiting the
inflammatory factors, TLR4 and MyD88, the present study
investigated whether the protective effects of LXA4 were closely
related to TLR4 and MyD88 suppression. The results of the present
study suggested that LXA4 inhibited the expression levels of TLR4,
MyD88, and their downstream inflammatory cytokines, whereas had no
effect on TRAM and TRIF, indicating that the anti-inflammatory
effects of LXA4 on PQ may be attributed to the suppression of the
TLR4 and MyD88-dependent signaling pathway.
The NF-κB transcription factor, a typical
pro-inflammatory signaling factor, has long been considered a
downstream transduction factor of TLR4/MyD88, and this feature is
based on its critical effect on promoting the release of
pro-inflammatory cytokines (40-42). Ideally, the RelA/p65 and p50
subunits of NF-κB constitute heterodimers, which bind to IκBα (the
NF-κB inhibitor), keeping NF-κB inactive in the cytoplasm.
Stimulated by external injuries, IκBα becomes phosphorylated and
degraded, thereby promoting NF-κB activation and translocation to
the nucleus, where activated NF-κB promotes the occurrence and
development of downstream inflammatory reactions (43-45). The results of the present study
demonstrated that LXA4 decreased the PQ-induced IκBα
phosphorylation and facilitated IκBα degradation, resulting in the
reduced nuclear transfer of NF-κB p65. Therefore, it was confirmed
that the beneficial effects of LXA4 on PQ poisoning were associated
with the inhibition of the NF-κB pathway.
The PI3K/AKT pathway, which is closely related to
TLR4/MyD88 (46) and reported as
a regulator of NF-κB, can enhance the transcriptional activity and
nuclear translocation of NF-κB, participate in multiple cell signal
activations and regulate the expression of several inflammatory
mediators (47,48). Similarly, the results of the
present study indicated that PQ stimulation increased the p-PI3K
and p-AKT expression levels, which were downregulated following
treatment with LXA4. Since the PI3K/AKT signaling pathway is
demonstrated as a key regulator of NF-κB activation by targeting
the transactivation domain of NF-κB p65 in an IKKβ-dependent manner
(49), nuclear NF-κB
downregulation may be partly due to the inhibitory effects of LXA4
on the PI3K/AKT pathway.
To further verify the effect of LXA4 on
TLR4/MyD88-mediated NF-κB/PI3K/AKT signaling pathway, the present
study constructed a GV417-TLR4 plasmid and explored the related
changes. The results suggested that the enforced overexpression of
TLR4 abrogated the protective effects of LXA4, with the emphasis on
re-elevated ROS production, re-enhanced pro-inflammatory cytokine
levels and re-activated the MyD88 and NF-κB/PI3K/AKT signaling
pathways.
In conclusion, the in vitro and in
vivo experiments of the present study demonstrated that LXA4
negatively regulated PQ-induced inflammation and oxidative stress
through the PI3K/AKT/NF-κB pathway mediated by TLR4 and MyD88,
possibly providing a novel therapeutic strategy against PQ-induced
toxicity. However, in the present study, it was not determined
whether other signaling pathways, such as FPR2, HMGB1, MAPK and
NLRP3, are also involved in the protective effects of LXA4 on
PQ-induced ALI. Therefore, further research is required on the
functions of LXA4.
Availability of data and materials
The analyzed datasets generated in the present study
are available from the corresponding author upon reasonable
request.
Authors' contributions
YL and MZ performed the majority of the entire
research, and together with the other authors, ensure the integrity
of the entire research. All authors (YL, NW, ZM, YW, YY, ZZ, YH and
MZ) participated in the study concept and the design and the
definition of knowledge content of this research. YL, NW and MZ
contributed to the experimental research, data collection and
statistical analysis. YL analyzed and interpreted the results and
drafted the manuscript. YL, ZM, YW, YY, ZZ and YH contributed to
the writing, review and revision of the manuscript. MZ contributed
to the preparation of the final version of manuscript. All authors
were responsible for the final content and read and approved the
final manuscript.
Ethics approval and consent to
participate
The experiments were approved by the Ethics
Committee of Shengjing Hospital of China Medical University
(approval no. 2019PS656K). All procedures involving animals were
performed in accordance with the ARRIVE guidelines and the National
Institutes of Health Guidelines for the Care and Use of Laboratory
Animals (NIH Publications no. 8023, revised in 1978).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Natural Science
Foundation of Liaoning Province, China (grant no. 201602879).
References
|
1
|
Baltazar T, Dinis-Oliveira RJ, Duarte JA,
de Lourdes Bastos M and Carvalho F: Paraquat research: Do recent
advances in limiting its toxicity make its use safer? Br J
Pharmacol. 168:44–45. 2013. View Article : Google Scholar :
|
|
2
|
Moon JM, Chun BJ and Cho YS: The
characteristics of emergency department presentations related to
acute herbicide or insecticide poisoning in South Korea between
2011-2014. J Toxicol Environ Health A. 79:466–476. 2016. View Article : Google Scholar
|
|
3
|
Sun B and Chen YG: Advances in the
mechanism of paraquat-induced pulmonary injury. Eur Rev Med
Pharmacol Sci. 20:1597–1602. 2016.PubMed/NCBI
|
|
4
|
Li G, Yuzhen L, Yi C, Xiaoxiang C, Wei Z,
Changqing Z and Shuang Y: DNaseI protects against paraquat-induced
acute lung injury and pulmonary fibrosis mediated by mitochondrial
DNA. Biomed Res Int. 2015:3869522015.PubMed/NCBI
|
|
5
|
Liu ZN, Zhao M, Zheng Q, Zhao HY, Hou WJ
and Bai SL: Inhibitory effects of rosiglitazone on paraquat-induced
acute lung injury in rats. Acta Pharmacol Sin. 34:1317–1324. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu Z, Sun M, Wang Y, Zhang L, Zhao H and
Zhao M: Silymarin attenuated paraquat-induced cytotoxicity in
macrophage by regulating Trx/TXNIP complex, inhibiting NLRP3
inflammasome activation and apoptosis. Toxicol In Vitro.
46:265–272. 2018. View Article : Google Scholar
|
|
7
|
Toygar M, Aydin I, Agilli M, Aydin FN,
Oztosun M, Gul H, Macit E, Karslioglu Y, Topal T, Uysal B and Honca
M: The relation between oxidative stress, inflammation, and
neopterin in the paraquat-induced lung toxicity. Hum Exp Toxicol.
34:198–1204. 2015. View Article : Google Scholar
|
|
8
|
Chen T, Wang R, Jiang W, Wang H, Xu A, Lu
G, Ren Y, Xu Y, Song Y, Yong S, et al: Protective effect of
astragaloside IV against paraquat-induced lung injury in mice by
suppressing rho signaling. Inflammation. 39:483–492. 2016.
View Article : Google Scholar
|
|
9
|
Levy BD and Serhan CN: Resolution of acute
inflammation in the lung. Annu Rev Physiol. 76:467–492. 2014.
View Article : Google Scholar
|
|
10
|
Shimizu S, Ogawa T, Seno S, Kouzaki H and
Shimizu T: Pro-Resolution mediator lipoxin A4 and its receptor in
upper airway inflammation. Ann Otol Rhinol Laryngol. 122:683–689.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Romano M, Cianci E, Simiele F and
Recchiuti A: Lipoxins and aspirin-triggered lipoxins in resolution
of inflammation. Eur J Pharmacol. 5:49–63. 2015. View Article : Google Scholar
|
|
12
|
Zhang L, Wu P, Jin Sw, Yuan P, Wan Jy,
Zhou Xy, Xiong W, Fang F and Ye Dy: Lipoxin A4 negatively regulates
lipopolysaccharide-induced differentiation of RAW264.7 murine
macrophages into dendritic-like cells. Chin Med J (Engl).
5:981–987. 2007. View Article : Google Scholar
|
|
13
|
Martini AC, Forner S, Bento AF and Rae GA:
Neuroprotective effects of lipoxin A4 in central nervous system
pathologies. Biomed Res Int. 2014:3162042014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu L, Li HH, Wu Q, Miao S, Liu ZJ, Wu P
and Ye DY: Lipoxin A4 activates Nrf2 pathway and ameliorates cell
damage in cultured cortical astrocytes exposed to oxygen-glucose
deprivation/reperfusion insults. J Mol Neurosci Aug. 56:848–857.
2015. View Article : Google Scholar
|
|
15
|
Zong L, Li J, Chen X, Chen K, Li W, Li X,
Zhang L, Duan W, Lei J, Xu Q, et al: Lipoxin A4 attenuates cell
invasion by inhibiting ROS/ERK/MMP pathway in pancreatic cancer.
Oxid Med Cell Longev. 2016:68157272016. View Article : Google Scholar
|
|
16
|
Zhou XY, Wu P, Zhang L, Xiong W, Li YS,
Feng YM and Ye DY: Effects of lipoxin A(4) on lipopolysaccharide
induced proliferation and reactive oxygen species production in
RAW264.7 macrophages through modulation of G-CSF secretion. Inflamm
Res. 56:324–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zong H, Li X, Lin H, Hou C and Ma F:
Lipoxin A4 pretreatment mitigates skeletal muscle
ischemia-reperfusion injury in rats. Am J Transl Res. 9:1139–1150.
2017.PubMed/NCBI
|
|
18
|
Zhao Q, Shao L, Hu X, Wu G, Du J, Xia J
and Qiu H: Lipoxin a4 preconditioning and postconditioning protect
myocardial ischemia/reperfusion injury in rats. Mediators Inflamm.
2013:2313512013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen XQ, Wu SH, Zhou Y and Tang YR:
Lipoxin A4-induced heme oxygenase-1 protects cardiomyocytes against
hypoxia/reoxygenation injury via p38 MAPK activation and Nrf2/ARE
complex. PLoS One. 24:e671202013. View Article : Google Scholar
|
|
20
|
Liu ZN, Zhao H, Liu W, Li T, Wang Y and
Zhao M: NLRP3 inflammasome activation is essential for
paraquat-induced acute lung injury. Inflammation. 38:433–444. 2015.
View Article : Google Scholar
|
|
21
|
Wang Q, Lian QQ, Li R, Ying BY, He Q, Chen
F, Zheng X, Yang Y, Wu DR, Zheng SX, et al: Lipoxin A(4) activates
alveolar epithelial sodium channel, na, K-ATPase, and increases
alveolar fluid clearance. Am J Resp Cell Mol Biol. 48:610–618.
2013. View Article : Google Scholar
|
|
22
|
Qi W, Li H, Cai XH, Gu JQ, Meng J, Xie HQ,
Zhang JL, Chen J, Jin XG, Tang Q, et al: Lipoxin A(4) activates
alveolar epithelial sodium channel gamma via the
microRNA-21/PTEN/AKT pathway in lipopolysaccharide-induced
inflammatory lung injury. Lab Invest. 95:1258–1268. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mikawa K, Nishina K, Takao Y and Obara H:
ONO-1714, a nitric oxide synthase inhibitor, attenuates
endotoxin-induced acute lung injury in rabbits. Anesth Analg.
97:1751–1755. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shen H, Wu N, Wang Y, Guo F, Chen L, Zhang
Z, Jia D and Zhao M: MyD88 gene knockout attenuates
paraquat-induced acute lung injury. Toxicol Lett. 5:41–46. 2017.
View Article : Google Scholar
|
|
25
|
Kayama H, Ramirez-Carrozzi VR, Yamamoto M,
Mizutani T, Kuwata H, Iba H, Matsumoto M, Honda K, Smale ST and
Takeda K: Class-Specific regulation of pro-inflammatory genes by
MyD88 pathways and IκBζ. J Biol Chem. 290:48152015. View Article : Google Scholar
|
|
26
|
Hazeki K, Nigorikawa K and Hazeki O: Role
of phosphoinositide 3-kinase in innate immunity. Biol Pharm Bull.
30:1617–1623. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Androulidaki A, Iliopoulos D, Arranz A,
Doxaki C, Schworer S, Zacharioudaki V, Margioris AN, Tsichlis PN
and Tsatsanis C: The kinase akt1 controls macrophage response to
lipopolysaccharide by regulating microRNAs. Immunity. 21:220–231.
2009. View Article : Google Scholar
|
|
28
|
Ruse M and Knaus UG: New players in
TLR-mediated innate immunity: PI3K and small rho GTPases. Immunol
Res. 34:33–48. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rose MS, Smith LL and Wyatt I: Evidence
for energy-dependent accumulation of paraquat into rat lung.
Nature. 22:314–315. 1974. View
Article : Google Scholar
|
|
30
|
Wang X, Jiao W, Lin M, Lu C, Liu C, Wang
Y, Ma D, Wang X, Yin P, Feng J, et al: Resolution of inflammation
in neuromyelitis optica spectrum disorders. Mult Scler Relat
Disord. 27:34–41. 2019. View Article : Google Scholar
|
|
31
|
Börgeson E, Johnson AM, Lee YS, Till A,
Syed GH, Ali-Shah ST, Guiry PJ, Dalli J, Colas RA, Serhan CN, et
al: Lipoxin A4 attenuates obesity-induced adipose inflammation and
associated liver and kidney disease. Cell Metab. 7:125–137. 2015.
View Article : Google Scholar
|
|
32
|
Liu L, Zhang P, Zhang Z, Hu Q, He J, Liu
H, Zhao J, Liang Y, He Z, Li X, et al: LXA4 ameliorates
cerebrovascular endothelial dysfunction by reducing acute
inflammation after subarachnoid hemorrhage in rats. Neuroscience.
1:105–114. 2019. View Article : Google Scholar
|
|
33
|
Lu H, Zhen J, Wu T, Peng A, Ye T, Wang T,
Yu X, Vaziri ND, Mohan C and Zhou XJ: Superoxide dismutase mimetic
drug tempol aggravates anti-GBM antibody-induced glomerulonephritis
in mice. Am J Physiol Renal Physiol. 299:F445–F452. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Y, Liu Y, Peng X, Liu W, Zhao F, Feng
D, Han J, Huang Y, Luo S, Li L, et al: NMDA receptor antagonist
attenuates bleomycin-induced acute lung injury. PLoS One.
5:e01258732015. View Article : Google Scholar
|
|
35
|
Kovach MA and Standiford TJ: Toll like
receptors in diseases of the lung. Int Immunopharmacol.
11:1399–1406. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Oakes JL, O'Connor BP, Warg LA, Burton R,
Hock A, Loader J, Laflamme D, Jing J, Hui L, Schwartz DA and Yang
IV: Ozone enhances pulmonary innate immune response to a toll-like
receptor-2 agonist. Am J Respir Cell Mol Biol. 48:27–34. 2013.
View Article : Google Scholar :
|
|
37
|
Qin C, Zhang B, Zhang L, Zhang Z, Wang L,
Tang L, Li S, Yang Y, Yang F, Zhang P and Yang B: MyD88-Dependent
toll-like receptor 4 signal pathway in intervertebral disc
degeneration. Exp Ther Med. 12:611–618. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu W, Shan LP, Dong XS and Liu Z:
Toll-Like receptor 4 implicated in acute lung injury induced by
paraquat poisoning in mice. Int J Clin Exp Med. 5:3392–3397.
2014.
|
|
39
|
Dong XS, Xu XY, Sun YQ, Wei-Liu, Jiang ZH
and Liu Z: Toll-Like receptor 4 is involved in myocardial damage
following paraquat poisoning in mice. Toxicology. 312:115–122.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hu R, Xu H, Jiang H, Zhang Y and Sun Y:
The role of TLR4 in the pathogenesis of indirect acute lung injury.
Front Biosci (Landmark Ed). 2013 Jun 1;18:1244–55. View Article : Google Scholar
|
|
41
|
Li TT, Ogino S and Qian ZR: Toll-Like
receptor signaling in colorectal cancer: Carcinogenesis to cancer
therapy. World J Gastroenterol. 1:17699–17708. 2014. View Article : Google Scholar
|
|
42
|
Lawrence T: The nuclear factor NF-kappaB
pathway in inflammation. Cold Spring Harb Perspect Biol.
1:a0016512009. View Article : Google Scholar
|
|
43
|
Liu P, Zhou YS, Qin YL, Li L, Liu Y, Xu B,
Huang K, Ji CC, Lin F, Wang YG, et al: Mechanism of action for
oligomeric proanthocyaniclins in pava qnat-induced acute lung
injury. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 20:818–822.
2017.In Chinese.
|
|
44
|
Hu X, Shen H, Wang Y and Zhao M: Liver X
receptor agonist TO901317 attenuates paraquat-induced acute lung
injury through inhibition of NF-κB and JNK/p38 MAPK signal
pathways. Biomed Res Int. 2017:46526952017. View Article : Google Scholar
|
|
45
|
Liu Mw, Su Mx, Zhang W, Wang Yq, Chen M,
Wang L and Qian Cy: Protective effect of Xuebijing injection on
paraquat-induced pulmonary injury via down-regulating the
expression of p38 MAPK in rats. BMC Complement Altern Med.
16:4982014. View Article : Google Scholar
|
|
46
|
Troutman TD, Hu W, Fulenchek S, Yamazaki
T, Kurosaki T, Bazan JF and Pasare C: Role for B-cell adapter for
PI3K (BCAP) as a signaling adapter linking Toll-like receptors
(TLRs) to serine/threonine kinases PI3K/Akt. Proc Natl Acad Sci
USA. 3:273–278. 2012. View Article : Google Scholar
|
|
47
|
Chu AJ: Antagonism by bioactive
polyphenols against inflammation: A systematic view. Inflamm
Allergy Drug Targets. 13:34–64. 2014. View Article : Google Scholar
|
|
48
|
Lo JY, Kamarudin MN, Hamdi OA, Awang K and
Kadir HA: Curcumenol isolated from curcuma zedoaria suppresses
akt-mediated NF-κB activation and p38 MAPK signaling pathway in
LPS-stimulated BV-2 microglial cells. Food Funct. 6:3550–3559.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Madrid LV, Mayo MW, Reuther JY and Baldwin
AS Jr: Akt stimulates the transactivation potential of the RelA/p65
Subunit of NF-kappa B through utilization of the Ikappa B kinase
and activation of the mitogen-activated protein kinase p38. J Biol
Chem. 1:18934–18940. 2001. View Article : Google Scholar
|