Tumor necrosis factor-α (TNF-α) is a complex
cytokine that impacts various physiological and pathological
conditions. It can function as an immune regulator, contributing to
the development and function regulation of B-cells, T-lymphocytes
and dendritic cells, as a pro-inflammatory mediator, modulating the
generation and preservation of inflammatory processes, or as an
apoptotic inducer, promoting cell death (1,2).
TNF-α is involved in the pathogenesis of numerous autoimmune
disorders, such as rheumatoid arthritis (RA) (3), inflammatory bowel disease (4,5),
psoriatic arthritis (6) and
multiple sclerosis (7,8); however, its role in systemic lupus
erythematosus (SLE) disease remains unclear. From the genetic point
of view, several investigations have demonstrated a link between
the TNF-α gene polymorphism and the susceptibility to SLE (9–11).
Furthermore, there is a strong connection between TNF-α gene
expression and clinical manifestations in patients with SLE
(12).
Furthermore, TNF-α is a growth factor for
B-lymphocytes, which can produce large quantities of TNF-α in an
autocrine loop (13–15). Serum levels of TNF-α have been
discovered to be high in patients with SLE and have been linked to
disease activity and several systemic manifestations, such as
SLE-related cardiovascular disease and lupus nephritis (12,16–20).
The dysregulation of TNF-α is clearly linked to tissue destruction
observed in lupus organ disease, to the death of lymphocytes and to
the impaired clearance of apoptotic cells, resulting in the
presentation of self-antigens and autoantibody formation (21). However, TNF-α has pro-apoptotic and
anti-apoptotic properties, depending on the underlying contextual
circumstances (22–24). Treatment with anti-TNF drugs or
recombinant TNF has been demonstrated to have conflicting results
in murine models of SLE. As previously demonstrated, TNF-competent
New Zealand Black (NZB) mice displayed an autoimmune phenotype,
whereas TNF-deficient New Zealand Black (NZB) mice developed severe
lupus-like disease (25,26).
Moreover, the disease was shown to be reversible by
the administration of recombinant TNF; indeed, the early
application of TNF in NZB/White (NZB/W) mice postponed the
development of autoantibodies and lupus nephritis (25,27).
In addition, TNF-blocking therapies have sometimes induced the
production of antinuclear antibodies and IgM antibodies to
double-stranded DNA in individuals with RA or Crohn's disease,
suggesting the potential propensity of anti-TNF agents to stimulate
pathogenic autoantibody production (28,29).
These patients rarely exhibited a reversible drug-induced
lupus-like syndrome (29). In this
context, a critical view of the relevance of TNF in SLE is
necessary. The present review article thus aimed to enhance the
understanding of the functions of TNF-α in the pathogenesis of SLE
and discuss the benefits associated with anti-TNF therapies in
patients with SLE.
It has been demonstrated that TNF-α functions as a
multifunctional cytokine which plays a vital role in controlling
inflammation, secondary and tertiary lymphoid tissue development,
and immune regulation (42). The
functional mechanisms of TNF-α are highly diverse and somewhat
complex. This protein plays conflicting roles: On the one hand, it
combats certain types of infections and, on the other hand, induces
pathological complications. This may be due to the stimulation of
various signaling pathways involved in diverse cellular reactions,
such as survival, differentiation, cell proliferation and cell
death (43). A recent study
indicated that TNF-α deregulation was directly associated with
chronic inflammation, autoimmune diseases and other pathologies,
such as neuroinflammation (44).
Therefore, understanding the exact mechanisms of action of the
TNF-α signaling pathways may lead to the development of effective
therapies for the treatment of immune diseases.
The TNFR1 gene (also known as p55, p60, CD120a or
TNFRSF1A), located on chromosome 12p13, has 10 exons and produces a
60-kDa protein (45). TNFR2 (also
known as p75, p80, CD120b or TNFRSF1B) encoded via the gene located
on chromosome 1p36.2, consists of 10 exons and gives rise to a
protein of 80 kDa (46,47). These receptors are membrane
glycoproteins and members of the TNF receptor superfamily (48). They are crucial to the development
and homeostasis of the immune and neurological systems, and
ectodermal organs (49,50). The extracellular domain is very
similar between these two receptors and consists of multiple
cysteine-rich domains involved in ligand binding; however, the
intracellular domains clearly differ; thus, they can activate
different signaling pathways by interacting with a variety of
cytosolic proteins (51).
Receptors are dependent on adapter proteins, including
TNFR-associated death domain protein (TRADD), Fas-associated
protein with death domain (FADD) and the TNFR-associated factor
(TRAF)-1 to activate intracellular signaling pathways and induce a
biological response. These proteins form a scaffold that allows
other proteins to be absorbed to trigger the signaling pathway
(52,53). TNFR1 expression has been observed
in a number of cell types; however, TNFR2 expression has been
observed in a small number of cells, such as T-cells and
endothelial cells (54). TNF-α
strongly binds to both receptors, and the differential engagement
of the receptors is associated with distinct functions. sTNF
interacts with both TNF receptors, while tmTNF mainly activates
TNFR2 (51). TNFR2 has a lower
affinity for TNF-α than for TNFR1, suggesting that TNFR2 can
momentarily bind and can subsequently be release, playing a role in
amplifying or synergizing TNFR1 signaling (55,56).
The stimulation of TNFR1 is responsible for several biological
effects of TNF-α, such as cytotoxicity and proliferation. The
activation of TNFR1 stimulates various cellular responses, such as
the induction of proliferation processes, apoptosis, or
necroptosis, depending on the cell type and environmental
conditions (56). TNFR1, in its
cytoplasmic part, has a death domain (DD) related to TNF-α-mediated
cytotoxicity, while TNFR2 lacks this domain (57). The engagement of TNF with TNFR1
leads to the successive formation of two different TNF receptor
signaling complexes (complex I and complex II) that are separated
both temporally and spatially. Complex I induces the expression of
anti-apoptotic genes, which inhibit cell death processes mainly by
activating transcription factors, such as NF-κB, whereas the second
signaling pathway (complex II) leads to apoptosis or necroptosis
(1). Compared to TNFR1, knowledge
of TNFR2 signaling pathways is limited. Since TNFR2 lacks the DD,
it cannot directly induce cell death. In contrast to the functions
of TNFR1, which is able to induce inflammation or apoptotic
responses, TNFR2 engagement significantly enhances cell
stimulation, migration and propagation (58). The binding of TRAF2 to TNFR2
activates the canonical and non-canonical NF-κB signaling pathways
(59). However, TNFR is able to
activate NF-κB slowly, although with a longer activation time
compared to TNFR (60). In
addition, it has been shown that TNFR2 can induce cell survival
(61). Other researchers have
indicated that TNFR2 is required for antigen-associated
differentiation and T-cell survival. TNFR2 regulates several
adhesion molecules, including intercellular adhesion molecule-1 and
selectin-E, which are central molecules in angiogenesis (62).
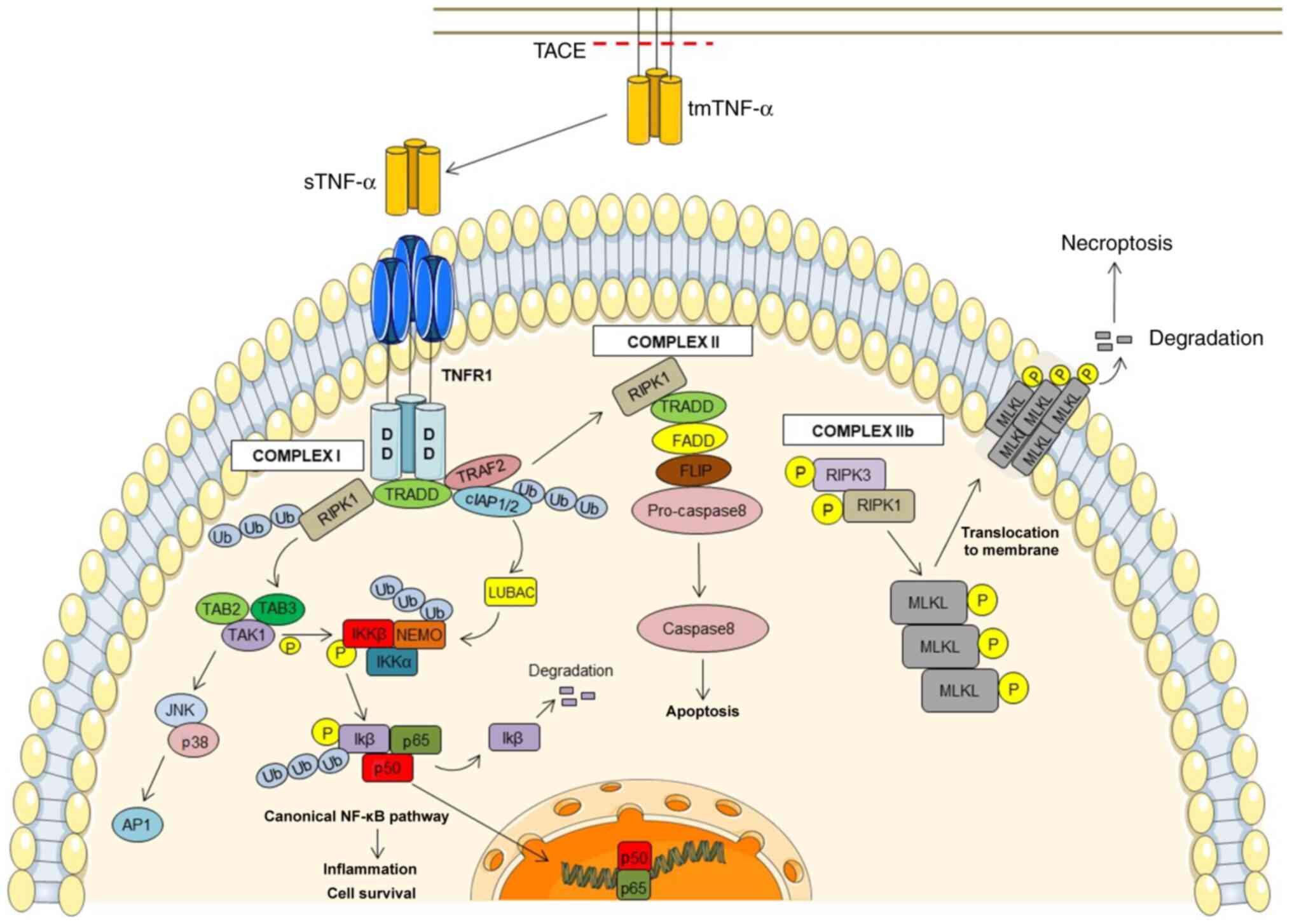
TNF signaling appears to be quite complex and can
cover various downstream signaling pathways. TNFR1 is triggered
through both membrane-bound and soluble TNF (51) (Fig.
1). The TNFR1 cytoplasmic DD allows interactions with other
DD-containing proteins, including TRADD, E3 ubiquitin ligases,
cellular inhibitor of apoptosis protein (cIAP)1/2, the
receptor-interacting serine/threonine-protein kinase (RIPK)1 and
TRAF2, resulting in complex I signaling (52,53).
In turn, polyubiquitinated RIPK1 and cIAP1/2 proteins have a
crucial function in the uptake of other proteins, such as the
TGF-β-activated kinase 1 (TAK1) in the TAK1-binding protein
(TAB)2/3 complex and the linear ubiquitin chain assembly complex
(LUBAC), respectively (63,64).
LUBAC can polyubiquitinate numerous molecules, such as LUBAC itself
and NF-κB essential modulator (NEMO) in the IκB kinase (IKK)
complex comprised of IKK1/IKKα, IKK2/IKKβ and NEMO/IKKγ (64,65).
Furthermore, TAK1 phosphorylates IκB, a prerequisite for its
ubiquitylation and proteasome degradation. NF-κB then translocates
to the nucleus and prompts the transcription of target genes
involved in inflammation and cell survival (64). In addition, TAK1 in complex with
TAB2/TAB3 can also induce the triggering of AP-1 transcription
factor through the phosphorylation of MAP kinases, such as cJun
NH2-terminal kinase (JNK) and p38 (66,67).
This signaling pathway activates the transcription of various
pro-inflammatory genes. Moreover, TNF-TNFR1 interaction can
activate other signaling pathways involved in programmed cell
death, such as apoptosis and necroptosis through complex II and IIb
signaling, respectively (68,69).
In this case, the separation of RIPK1 and TRADD from complex I
leads to the instability of complex I and in the formation of
complex II, which includes FADD, cellular FLICE-inhibitory protein
and pro-caspase-8 molecules, thus inducing apoptosis (70). In addition, when caspase is
inhibited, the interaction of TNF with TNFR1 induces the formation
of complex IIb, leading to the activation of the cell death pathway
known as necroptosis. Complex IIb consists of the phosphorylated
molecules, RIPK1 and RIPK3, and mixed lineage kinase domain-like
pseudokinase (MLKL). Thereafter, MLKL oligomerization occurs, and
phosphorylated MLKL is translocated to the plasma membrane, which
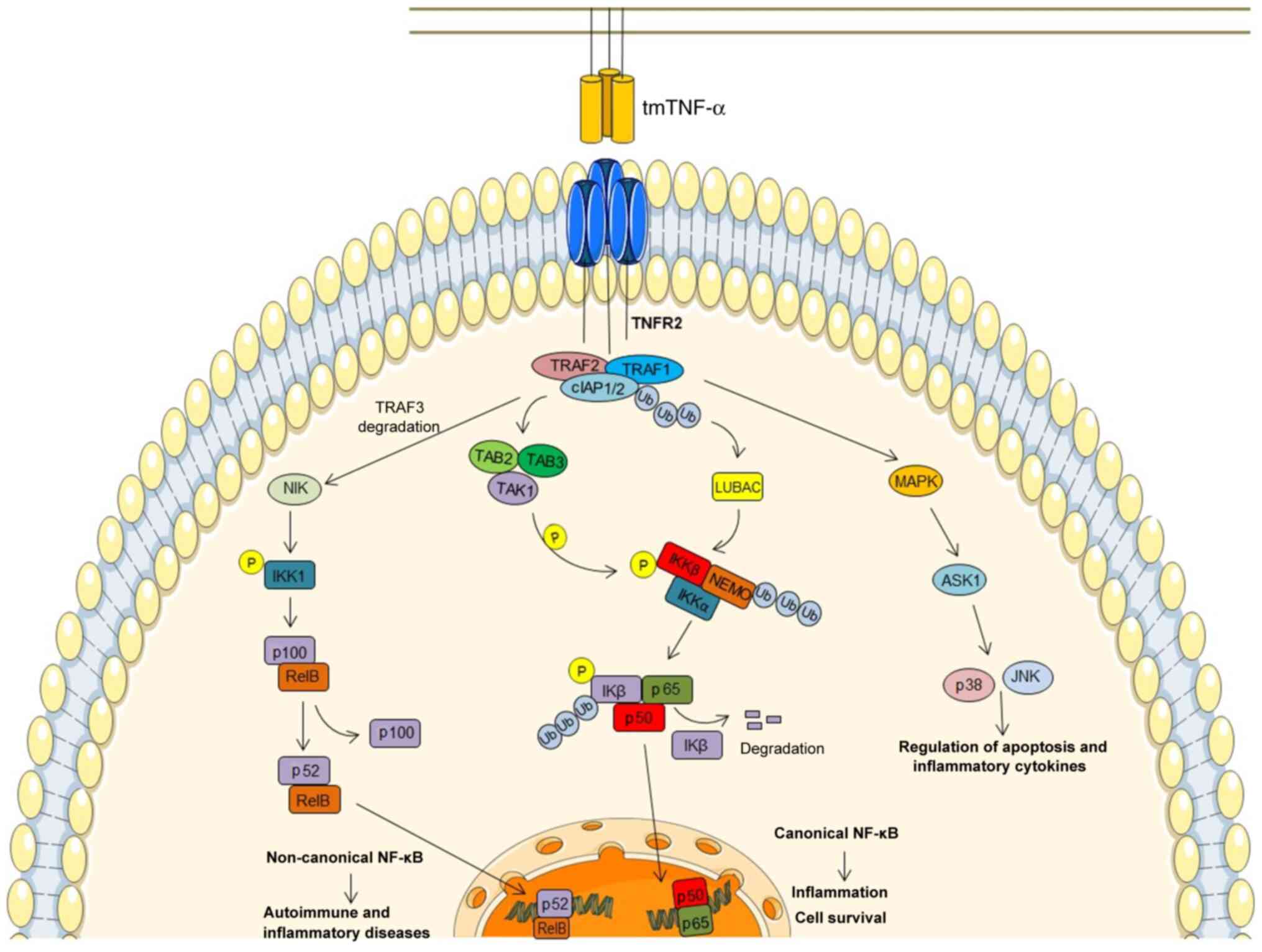
is disrupted to stimulate necroptosis (71). In contrast to TNFR1, the
interaction of TNFR2 with TNF (Fig.
2) causes the direct recruitment of TRAF1 or TRAF2 together
with cIAP1/2 and LUBAC molecules (72,73).
Subsequently, this signaling pathway may, similar to the TNFR1
signaling pathway, recruit the TAK1/TAB2/TAB3 and NEMO/IKKα/β
complexes, resulting in the downstream stimulation of the canonical
NF-κB pathway (60).
Alternatively, the only membrane-bound TNF and non-soluble TNF
trimers can activate, via TNFR2, the non-canonical NF-κB pathway
via the TRAF2/cIAP1⁄2 complex interaction, resulting in the
accumulation of NF-κB-inducing kinase (NIK). NIK phosphorylates the
NF-κB precursor protein p100, thus eliciting its proteasomal
proteolysis to p52, which results in the transcription of
p52/RelB-containing NF-κB heterodimer (74,75).
Under normal conditions, the basal level of NIK is maintained at a
low level by TRAF3, which induces NIK ubiquitination and
constitutive degradation by the proteasome. In response to tmTNF,
NIK becomes stabilized due to TRAF3 degradation, and its
accumulation activates non-canonical NF-κB signaling, resulting in
autoimmune and inflammatory diseases (75).
In contrast to the common belief that TNFR1
signaling triggers apoptosis and TNFR2 signaling promotes
pro-survival, there is increasing evidence to indicate that
exclusive TNFR2 stimulation can induce apoptosis (despite the fact
that TNFR2 does not contain a DD) through crosstalk among the two
receptors and TRAFs, which are involved in initiating TNFR1 and
TNFR2 signaling (60). Moreover,
TNFR2 can enhance TNFR1-mediated apoptosis, despite the enhanced
NF-κB activation (22,76–80).
The upregulation of TNFR2 induces proteasomal degradation and the
consequent depletion of TRAF2 (77,78).
Upon the activation of TNF, a member of the MAPK kinase family
termed apoptosis signal-regulating kinase-1 is activated by TRAF2,
and induces p38 and JNK activation (81). This pathway is triggered by stress
stimuli and results in the regulation of apoptosis and inflammatory
cytokine expression (66).
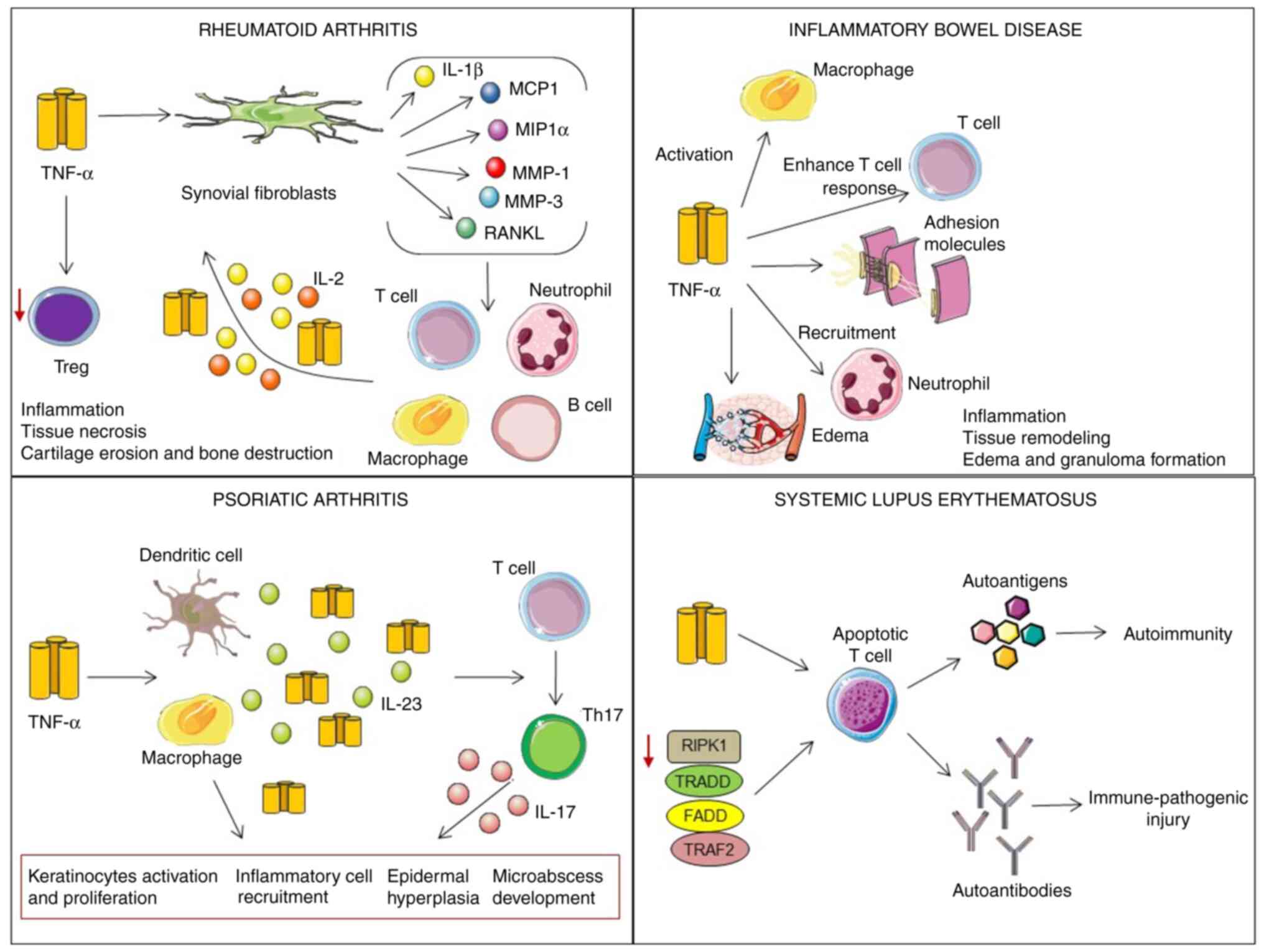
A similar role is played by TNF-α in the
gastrointestinal tract in patients with ulcerative colitis and
Crohn's. TNF-α activates macrophages, enhances the T-cell response,
induces the expression of adhesion molecules by the vascular
endothelium, and the recruitment of neutrophils to local sites of
inflammation, promotes tissue remodeling, edema and granuloma
formation (4,5). TNF-α-dependent inflammation is
extended through triggered NF-κβ-dependent pathways, which provide
the release of MMPs with the consequent degradation of the mucosa
and ulceration (91).
In patients with psoriatic arthritis, elevated
levels of TNF-α stimulate DCs and macrophages to secrete high
amounts of TNF-α and IL-23, promoting the differentiation of naive
T-cells into Th17 cells, with the consequent overproduction of the
pro-inflammatory cytokine, IL-17. IL-17 and TNF-α trigger the NF-κB
signaling pathway, leading to keratinocyte activation and
proliferation, the recruitment of inflammatory cells, epidermal
hyperplasia and microabscess development (92).
There is also emerging evidence to support the
involvement of TNF-α in the pathogenesis of SLE, which is discussed
in the following section (Fig.
3).
SLE is a systemic autoimmune disease featured by
heterogeneous clinical manifestations and immunological
abnormalities. Its pathogenesis remains poorly understood, and even
though the etiology of SLE is undetermined, multiple elements are
associated with disease development, including genetic (93–95),
epigenetic (96), immunoregulatory
(97), ethnic (98), hormonal (99) and environmental factors (100–103). The role TNF-α in the pathogenesis
of SLE is controversial; some investigators have found that TNF-α
confers SLE susceptibility (10,11,18,104), while others have described a
protective role of TNF-α in patients with SLE (105,106). Multiple have studies indicated
that TNF-α, along with other cytokines, such as IFN-α, IL-12, IL-4,
IL-10, IL-6, A proliferation-inducing ligand (APRIL) and B
cell-activating factor, IL-17 and IL-21 are the main SLE-related
cytokines (107–111). In particular, Svenungsson et
al (19,20) emphasized the high triglyceride and
low HDL levels as disease activity markers, and the elevated levels
of TNF-α/TNFR in patients with SLE, as well as the link between
inflammation, dyslipoproteinemia and cardiovascular disease in
patients with SLE. Furthermore, an increased TNF-α concentration
has been observed in the blood and in the inflamed kidneys of
patients with SLE (112–116). Further studies have also
demonstrated a significant genetic relation between TNF-α promoter
polymorphism and SLE susceptibility (9–11,117–121). Increased levels of TNF-α have
been found to be associated with disease severity in patients with
SLE (18,107,122). Higher serum levels of TNF-α and
its soluble receptors have been observed in patients with SLE with
active disease compared with SLE patients with inactive disease
(122). Moreover, patients with
SLE with high TNF-α levels present T-lymphocytes which are more
susceptible to apoptosis than T-cells from healthy controls
(104). This enhanced
TNF-α-induced apoptosis increases the autoantigen load, promoting
autoimmune responses in patients with SLE (104). This enhanced TNF-α-induced
apoptosis also increases the load of autoantigens, promoting
autoimmune responses in patients with SLE (104).
Moreover, genetic variation at TNF alpha induced
protein 3 (TNFAIP3) and TNF superfamily member 4 (TNFSF4) have been
associated with lymphocyte dysregulation and different SLE ethnic
groups (123–126). Polymorphisms in TNFR2 may also
play a role in the genetic susceptibility to SLE. A previous
genotype analysis manifested that the existence of one 196R allele
was sufficient for delivering SLE susceptibility in the Japanese
population (127). Both TNFR1 and
TNFR2 expression levels are highly enhanced in active the serum of
patients with SLE (122,128,129), and sTNFRs are crucial modulators
of the inflammatory responses in lupus nephritis (130,131). In Japanese patients, a mutation
in exon 3 in position 61 of the tumor necrosis factor receptor
superfamily 1A gene (TNFRSF1A) was shown to be associated with SLE.
These patients were characterized by a high concentration of serum
TNF, sTNFRSF1B and a low concentration of sTNFRSF1A (132).
On the contrary, some researchers have observed
decreased levels of TNF-α in patients with SLE, particularly in
patients with severe disease (105). Zhu et al (106) indicated that the expression
levels of TNF-α adapter proteins TRADD, FADD, TRAF-2 and RIPK-1 in
peripheral blood mononuclear cells were markedly diminished in
patients with SLE and were negatively associated with the SLE
activity index. Reduced levels of TNF-α adapter proteins have been
shown to be related to advanced lymphocyte apoptosis and enormous
autoantibody secretion, resulting in immune-pathogenic injury in
patients with SLE (106).
Moreover, several studies did not demonstrate any association
between polymorphisms in the TNFR2 gene and SLE (133–135). Sullivan et al (135) analyzed the frequency of genetic
polymorphisms in the 3′ untranslated region of the TNFR2 gene in
patients with SLE and did not find an association, although the
study examined only Caucasian patients. Furthermore, Chadha et
al (134) did not find any
association between TNFRSF14, TNFRSF8, TNFRSF1B locus and SLE in
European-Caucasian families. In line with this, Al-Ansari et
al did not find any connection between the TNFRII 196R allele
and SLE neither in Spanish or in UK populations (133).
Murine disease models are genetically homogeneous
populations used to research disease initiation and progression
(136). There are different mouse
models for lupus; some of them develop lupus spontaneously [e.g.,
NZB/W F1hybrid mice, medical research laboratory
lymphoproliferation (MRL/lpr) mice, BXSB/Yaa mice], and in others,
lupus is induced in the animals (e.g., pristane-induced lupus)
(137). Due to the dual function
of TNF-α (mediator of inflammation and regulator of autoimmunity),
the efficacy of TNF-based therapies in SLE is controversial and can
vary, depending on the subsets of patients (138). TNF-α is well observed in NZB/W F1
hybrid mice, MRL/lpr and C3H.SW lupus-prone mouse models. The NZB/W
F1 lupus model denotes an F1 cross between the NZB and NZW strains
(139). In 1988, Jacob and
McDevitt (25) demonstrated that,
unlike NZW mice (healthy mouse strains), NZB/W mice were defective
in TNF-α production and developed severe lupus-like phenotypes.
They also noted that the early application of recombinant TNF-α to
NZB/W mice attenuated the progression of lupus nephritis (25). In 1989, Gordon et al
(27) continued research on NZB/W
mice, demonstrating that the use of TNF-α, even following the onset
of renal symptoms, increased survival, reduced the progression of
kidney damage and delayed the emergence of lupus in these mice. In
2000, the study by Kontoyiannis and Kollias (26) demonstrated that NZB mice with an
engineered heterozygous TNF deficit developed lupus nephritis and
autoimmunity due to a lower production of TNF. Contrary to these
findings, Brennan et al (140) found high steady-state levels of
TNF-α and IL-1 β in the renal cortices of NZB/W mice with lupus
nephritis. They also noted that the administration of a lower dose
of TNF-α increased kidney injury (140). Furthermore, in MRL/lpr mice, an
elevation in TNF-α expression was previously detected, which was
linked to the degree of inflammation and organ dysfunction
(141–143). The upregulation of TNF mRNA was
discovered in the lungs of MRL/lpr mice in the study by Deguchi and
Kishimoto (144). Overall these
findings suggest that TNF-α may have both beneficial and harmful
effects in experimental lupus models, based on its concentration
and ability to play both immune-regulatory and pro-inflammatory
functions (116). Thus, this
cytokine can be considered as a therapeutic target in SLE. Rabbit
anti-mouse TNF-α immunoglobulin (Ig)G antibody therapy has been
shown to reduce autoimmune pulmonary inflammation in lupus-prone
mice (144). It has been
demonstrated that therapies directed at blocking TNF/TNFR
interactions, such as soluble, dimericTNFR I (sTNFRI), which binds
to TNF-α with high affinity, thus neutralizing it, reduce the
infiltration of mononuclear cells into joints, lungs and skin in
NZB/W mice, improving the symptoms of the disease and extending the
lifespan (145). Bethunaickan
et al (146) used a NZB/W
murine model of IFN-induced lupus nephritis and treated mice with
recombinant fusion proteins, such as TNFR2-Ig. They revealed that
TNFR2-Ig treatment reduced the renal inflammatory response to
immune complex deposition, stabilizing nephritis, thus prolonging
survival (146).
Given the promising results of TNF blockade in SLE
mouse models, the inhibition of this cytokine was previously
investigated in patients with SLE. Clinically authorized TNF-α
suppressors have been revealed to be effective in several
autoimmune disorders, and novel TNF-α signaling blockers are
currently being investigated in clinical trials. Infliximab
(Remicade), adalimumab (Humira), certolizumab pegol (Cimzia),
golimumab (Simponi) and etanercept (Enbrel) are the five anti-TNF
drugs approved by the US Food and Drug Administration (FDA) for the
treatment of rheumatic inflammatory diseases, such as RA,
psoriasis, psoriatic arthritis and Crohn's disease (Table I) are currently being studied in
patients with SLE (44,147).
However, these agents may induce autoimmunity,
leading to the production of antinuclear antibodies and/or
anti-double-stranded DNA antibodies, and may occasionally trigger
the anti-TNF-α-induced lupus-like syndrome (ATIL) defined by
clinical features suggestive of SLE (148). The majority of cases occur in
patients with RA, inflammatory bowel disease and ankylosing
spondylitis (29,149–152). Previous studies have demonstrated
that nephritis may occur following the administration of anti-TNF-α
drugs (153,154).
Infliximab is a chimeric genetically modified
monoclonal antibody that includes a murine variable region and a
human IgG1 constant region. It is particular for all types of TNF
in humans and effectively prevents TNF from attaching to both
transmembrane and soluble receptors (147). Due to its chimeric structure,
infliximab is the anti-TNF-α molecule with a larger degree of
immunogenicity (152).
Nevertheless, open-label studies and case reports have reported the
effectiveness, acceptable safety and tolerability profile of
infliximab in patients with SLE. Aringer et al (155–157) observed that short-term induction
therapy with infliximab along with azathioprine or methotrexate
elicited long-term improvement in individuals with lupus nephritis.
The majority of patients with SLE exhibited a transient elevation
in autoantibodies against phospholipids and nuclear antigens, which
was not associated with disease flares (NCT00368264) (155–157). Other studies have confirmed the
safety and efficiency of infliximab in patients with
difficult-to-treat lupus nephritis (158,159). Hayat and Uppal (159) also demonstrated the efficacy of
infliximab in a patient with difficult-to-treat active non-renal
SLE. In a pilot study, Uppal et al (160) demonstrated that infliximab
significantly decreased the SLE disease activity index (SLEDAI)
without raising any safety concerns.
Etanercept is a full human monoclonal antibody with
reduced immunogenicity. It is a fusion protein consisting of two
equal extracellular regions of TNFR2 linked to the Fc fragment of
human IgG1 and strongly binds to sTNF-α or tmTNF-α (161,162). The FDA has approved the
therapeutic application of this drug for the treatment of RA,
polyarticular juvenile idiopathic arthritis (JIA), psoriatic
arthritis, ankylosing spondylitis and plaque psoriasis (44). Although the FDA has not yet
approved etanercept for the treatment of SLE, it has been used in
several clinical studies, including a randomized, double-blind,
phase II, multi-center study for the treatment of lupus nephritis
(NCT00447265), and in two phase II open-label trials for the
treatment of discoid lupus erythematosus (NCT02656082 and
NCT00797784). In an observational study, long-term treatment with
etanercept was revealed to be relatively safe and efficacious in
refractory lupus arthritis (163). In a previous case report study,
an enhancement of clinical symptoms and the quality of life were
described in subacute cutaneous lupus erythematosus individuals by
etanercept treatment (164). The
efficacy and the acceptable safety profile of etanercept were also
shown to treat rhupus, a disease with characteristics of both RA
and SLE (165,166). Micheloud et al (167) described a pregnant woman with SLE
with a severe diffuse proliferative nephritis who was successfully
treated with etanercept, plasmapheresis and high-dose intravenous
gammaglobulin.
Using molecular docking approach, a recent study
investigated the potential of selected anti-inflammatory peptides
from plant and animal sources as novel inhibitors for the treatment
of SLE. Protein-ligand and peptide-protein docking of twenty
anti-inflammatory peptides targeting IFN-γ, IL-3 and TNF-α were
developed to reduce inflammatory events which lead to autoantibody
production. The study represents an initial step for employment of
these peptides in the treatment of autoimmune disorders (168).
The present review article has noted the paradoxical
involvement of TNF-α in lupus and explained the advantages and
disadvantages of blocking this cytokine in preclinical and clinical
studies. In addition to inducing ATIL, and autoantibodies to dsDNA
and phospholipids, TNF-α inhibitors can increase the risk of
infections, malignancies (169),
central nervous system demyelinating disorders, and other
autoimmune diseases, such as type I diabetes, psoriasis and
multiple sclerosis (53,170–172). A probable cause of these
side-effects is that prevailing TNF-α suppressors prevent the
engagement between TNF-α and the receptors TNFR1 (with
pro-inflammatory and pro-apoptotic role) and TNFR2 (with a
regulatory function), leading to a loss of TNFR2 signaling
regulatory function (173). Van
Hauwermeiren et al (174)
noted that TNFR1+/− mice, which express 50% of TNFR1 on
cells, were highly resistant to lethal TNF-induced inflammation.
Moreover, the decrease in p55TNFR mitigated TNF toxicity without
compromising effectiveness (174), suggesting that TNFR can be
considered as a therapeutic target (175). In SLE, the significance of
TNF-α-TNFR1 interaction has been emphasized (176). Wu et al (177) demonstrated that the TNFR1 levels
in the urine of mice and individuals with lupus nephritis
increased; sTNFR1 and sTNFR2 levels have also been shown to be
higher in patients with lupus nephritis (131). According to Deng et al
(178) TNFR1 is abundantly
expressed in skin lesions of MRL/lpr mice, unlike TNFR2, and the
inhibition of TNFR1 signaling relieved skin lesions. On the other
hand, thye acceleration of the disease course occurred in NZB/F1
mice defective in both TNFR1 and TNFR2 (179). However, the lack of the p55TNFR
has been shown to lead to significantly increased
lymphoproliferation and autoimmune disorder in the Fas deficient
MRL-lpr/lpr mouse (180). Aderka
et al (128) suggested
that elevated serum sTNFR levels may be a valuable marker for
assessing the progression of SLE. The effect of Brentuximab Vedotin
targeting TNFR was investigated in adults with active SLE in a
phase II, multi-center, randomized, double-blinded,
multiple-ascending-dose study (NCT02533570) (Table II).
TNF-α is a potent pleiotropic cytokine with
multiple cellular activities, also involved in developing
autoimmune disorders. The impact of TNF-α on these diseases is not
yet completely understood. On the one hand, TNF-α can play a
pro-inflammatory and pro-apoptotic role, and on the other hand, it
has a regulatory function. Currently, therapeutic strategies that
target TNF-α are clinically utilized for the treatment of
inflammatory and autoimmune diseases, such as RA, inflammatory
bowel disease and psoriasis. However, notwithstanding their
clinical achievement, the application of anti-TNF drugs is
restricted due to severe side-effects and ATIL development.
Alternative therapeutic strategies that selectively target TNFRs
have exhibited immense therapeutic potential. Thus, the majority of
available evidence suggests that the usability of anti-TNF drugs
could be broadened. Understanding the dual role of TNF-α in
autoimmunity is difficult, particularly in a complex disease, such
as SLE. The use of drugs targeting TNF-α and TNFRs in SLE remains
controversial. Further investigations are thus required to
establish the favorable therapeutics benefits/risk ratio associated
with the use of anti-TNF-α drugs, as well as to determine the
treatment's effectiveness and side-effects in patients with
SLE.
The authors would like to thank Dr Afshin
Derakhshani, Immunology Research Center, Tabriz University of
Medical Sciences, Tabriz 5165665811, Iran, for critically reading
and commenting on the manuscript.
Funding: No funding was received.
Not applicable.
FG, PL, HA, BN, NSN, MP, EM, HS, NJT, VR and BB
contributed to the conceptualization, methodology, data curation,
investigation, visualization, and the drafting and editing of the
manuscript. VR and BB critically reviewed the manuscript. All
authors have read and approved the final version of the manuscript.
Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Micheau O and Tschopp J: Induction of TNF
receptor I-mediated apoptosis via two sequential signaling
complexes. Cell. 114:181–190. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Graninger WB, Steiner CW, Graninger MT,
Aringer M and Smolen JS: Cytokine regulation of apoptosis and Bcl-2
expression in lymphocytes of patients with systemic lupus
erythematosus. Cell Death Differ. 7:966–972. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Choy EH and Panayi GS: Cytokine pathways
and joint inflammation in rheumatoid arthritis. N Engl J Med.
344:907–916. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sands BE and Kaplan GG: The role of
TNFalpha in ulcerative colitis. J Clin Pharmacol. 47:930–941. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Adegbola SO, Sahnan K, Warusavitarne J,
Hart A and Tozer P: Anti-TNF therapy in Crohn's disease. Int J Mol
Sci. 19:22442018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Celis R, Cuervo A, Ramirez J and Cañete
JD: Psoriatic synovitis: Singularity and potential clinical
implications. Front Med (Lausanne). 6:142019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Derakhshani A, Asadzadeh Z, Safarpour H,
Leone P, Shadbad MA, Heydari A, Baradaran B and Racanelli V:
Regulation of CTLA-4 and PD-L1 expression in relapsing-remitting
multiple sclerosis patients after treatment with fingolimod,
IFNbeta-1α, glatiramer acetate, and dimethyl fumarate drugs. J Pers
Med. 11:7212021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pegoretti V, Baron W, Laman JD and Eisel
ULM: Selective modulation of TNF-TNFRs signaling: Insights for
multiple sclerosis treatment. Front Immunol. 9:9252018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen L, Huang Z, Liao Y, Yang B and Zhang
J: Association between tumor necrosis factor polymorphisms and
rheumatoid arthritis as well as systemic lupus erythematosus: A
meta-analysis. Braz J Med Biol Res. 52:e79272019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mahto H, Tripathy R, Meher BR, Prusty BK,
Sharma M, Deogharia D, Saha AK, Panda AK and Das BK: TNF-α promoter
polymorphisms (G-238A and G-308A) are associated with
susceptibility to Systemic Lupus Erythematosus (SLE) and P.
falciparum malaria: A study in malaria endemic area. Sci Rep.
9:117522019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ramirez-Bello J, Cadena-Sandoval D,
Mendoza-Rincon JF, Barbosa-Cobos RE, Sánchez-Muñoz F,
Amezcua-Guerra LM, Sierra-Martínez M and Jiménez-Morales S: Tumor
necrosis factor gene polymorphisms are associated with systemic
lupus erythematosus susceptibility or lupus nephritis in Mexican
patients. Immunol Res. 66:348–354. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Idborg H, Eketjall S, Pettersson S,
Gustafsson JT, Zickert A, Kvarnström M, Oke V, Jakobsson PJ,
Gunnarsson I and Svenungsson E: TNF-α and plasma albumin as
biomarkers of disease activity in systemic lupus erythematosus.
Lupus Sci Med. 5:e0002602018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kehrl JH, Miller A and Fauci AS: Effect of
tumor necrosis factor alpha on mitogen-activated human B cells. J
Exp Med. 166:786–791. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Boussiotis VA, Nadler LM, Strominger JL
and Goldfeld AE: Tumor necrosis factor alpha is an autocrine growth
factor for normal human B cells. Proc Natl Acad Sci USA.
91:7007–7011. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rieckmann P, Tuscano JM and Kehrl JH:
Tumor necrosis factor-alpha (TNF-alpha) and interleukin-6 (IL-6) in
B-lymphocyte function. Methods. 11:128–132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aringer M, Stummvoll GH, Steiner G, Köller
M, Steiner CW, Höfler E, Hiesberger H, Smolen JS and Graninger WB:
Serum interleukin-15 is elevated in systemic lupus erythematosus.
Rheumatology (Oxford). 40:876–881. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gabay C, Cakir N, Moral F, Roux-Lombard P,
Meyer O, Dayer JM, Vischer T, Yazici H and Guerne PA: Circulating
levels of tumor necrosis factor soluble receptors in systemic lupus
erythematosus are significantly higher than in other rheumatic
diseases and correlate with disease activity. J Rheumatol.
24:303–308. 1997.PubMed/NCBI
|
|
18
|
Studnicka-Benke A, Steiner G, Petera P and
Smolen JS: Tumour necrosis factor alpha and its soluble receptors
parallel clinical disease and autoimmune activity in systemic lupus
erythematosus. Br J Rheumatol. 35:1067–1074. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Svenungsson E, Fei GZ, Jensen-Urstad K, de
Faire U, Hamsten A and Frostegard J: TNF-alpha: A link between
hypertriglyceridaemia and inflammation in SLE patients with
cardiovascular disease. Lupus. 12:454–461. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Svenungsson E, Gunnarsson I, Fei GZ,
Lundberg IE, Klareskog L and Frostegård J: Elevated triglycerides
and low levels of high-density lipoprotein as markers of disease
activity in association with up-regulation of the tumor necrosis
factor alpha/tumor necrosis factor receptor system in systemic
lupus erythematosus. Arthritis Rheum. 48:2533–2540. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gordon C and Salmon M: Update on systemic
lupus erythematosus: Autoantibodies and apoptosis. Clin Med (Lond).
1:10–14. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang CY, Mayo MW, Korneluk RG, Goeddel DV
and Baldwin AS Jr: NF-kappaB antiapoptosis: Induction of TRAF1 and
TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation.
Science. 281:1680–1683. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Micheau O, Lens S, Gaide O, Alevizopoulos
K and Tschopp J: NF-kappaB signals induce the expression of c-FLIP.
Mol Cell Biol. 21:5299–5305. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yan X, Xiao CW, Sun M, Tsang BK and Gibb
W: Nuclear factor kappa B activation and regulation of
cyclooxygenase type-2 expression in human amnion mesenchymal cells
by interleukin-1beta. Biol Reprod. 66:1667–1671. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jacob CO and McDevitt HO: Tumour necrosis
factor-alpha in murine autoimmune ‘lupus’ nephritis. Nature.
331:356–358. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kontoyiannis D and Kollias G: Accelerated
autoimmunity and lupus nephritis in NZB mice with an engineered
heterozygous deficiency in tumor necrosis factor. Eur J Immunol.
30:2038–2047. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gordon C, Ranges GE, Greenspan JS and
Wofsy D: Chronic therapy with recombinant tumor necrosis
factor-alpha in autoimmune NZB/NZW F1 mice. Clin Immunol
Immunopathol. 52:421–434. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mohan AK, Edwards ET, Coté TR, Siegel JN
and Braun MM: Drug-induced systemic lupus erythematosus and
TNF-alpha blockers. Lancet. 360:6462002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Charles PJ, Smeenk RJ, Jong JD, Feldmann M
and Maini RN: Assessment of antibodies to double-stranded DNA
induced in rheumatoid arthritis patients following treatment with
infliximab, a monoclonal antibody to tumor necrosis factor alpha:
Findings in open-label and randomized placebo-controlled trials.
Arthritis Rheum. 43:2383–2390. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Carswell EA, Old LJ, Kassel RL, Green S,
Fiore N and Williamson B: An endotoxin-induced serum factor that
causes necrosis of tumors. Proc Natl Acad Sci USA. 72:3666–3670.
1975. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Spriggs DR, Deutsch S and Kufe DW: Genomic
structure, induction, and production of TNF-alpha. Immunol Ser.
56:3–34. 1992.PubMed/NCBI
|
|
32
|
Shakhov AN, Collart MA, Vassalli P,
Nedospasov SA and Jongeneel CV: Kappa B-type enhancers are involved
in lipopolysaccharide-mediated transcriptional activation of the
tumor necrosis factor alpha gene in primary macrophages. J Exp Med.
171:35–47. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Smith RA and Baglioni C: The active form
of tumor necrosis factor is a trimer. J Biol Chem. 262:6951–6954.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Eck MJ and Sprang SR: The structure of
tumor necrosis factor-alpha at 2.6 A resolution. Implications for
receptor binding. J Biol Chem. 264:17595–17605. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Watts AD, Hunt NH, Wanigasekara Y,
Bloomfield G, Wallach D, Roufogalis BD and Chaudhri G: A casein
kinase I motif present in the cytoplasmic domain of members of the
tumour necrosis factor ligand family is implicated in ‘reverse
signalling’. EMBO J. 18:2119–2126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vilcek J and Lee TH: Tumor necrosis
factor. New insights into the molecular mechanisms of its multiple
actions. J Biol Chem. 266:7313–7316. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cairns CB, Panacek EA, Harken AH and
Banerjee A: Bench to bedside: Tumor necrosis factor-alpha: From
inflammation to resuscitation. Acad Emerg Med. 7:930–941. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Camussi G, Albano E, Tetta C and Bussolino
F: The molecular action of tumor necrosis factor-alpha. Eur J
Biochem. 202:3–14. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang L, Lindholm K, Konishi Y, Li R and
Shen Y: Target depletion of distinct tumor necrosis factor receptor
subtypes reveals hippocampal neuron death and survival through
different signal transduction pathways. J Neurosci. 22:3025–3032.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Beyaert R and Fiers W: Molecular
mechanisms of tumor necrosis factor-induced cytotoxicity. What we
do understand and what we do not. FEBS Lett. 340:9–16. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Darnay BG and Aggarwal BB: Signal
transduction by tumour necrosis factor and tumour necrosis factor
related ligands and their receptors. Ann Rheum Dis. 58 (Suppl
1):I2–I13. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kalliolias GD and Ivashkiv LB: TNF
biology, pathogenic mechanisms and emerging therapeutic strategies.
Nat Rev Rheumatol. 12:49–62. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fiers W: Tumor necrosis factor.
Characterization at the molecular, cellular and in vivo level. FEBS
Lett. 285:199–212. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jang DI, Lee AH, Shin HY, Song HR, Park
JH, Kang TB, Lee SR and Yang SH: The role of tumor necrosis factor
alpha (TNF-α) in autoimmune disease and current TNF-α inhibitors in
therapeutics. Int J Mol Sci. 22:27192021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fuchs P, Strehl S, Dworzak M, Himmler A
and Ambros PF: Structure of the human TNF receptor 1 (p60) gene
(TNFR1) and localization to chromosome 12p13 [corrected]. Genomics.
13:219–224. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang XY, Kafka M, Dvilansky A and Nathan
I: The roles of protein phosphorylation/dephosphorylation in tumor
necrosis factor antitumor effects. J Interferon Cytokine Res.
16:1021–1025. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kemper O and Wallach D: Cloning and
partial characterization of the promoter for the human p55 tumor
necrosis factor (TNF) receptor. Gene. 134:209–216. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Desplat-Jego S, Burkly L and Putterman C:
Targeting TNF and its family members in autoimmune/inflammatory
disease. Mediators Inflamm. 2014:6287482014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bodmer JL, Schneider P and Tschopp J: The
molecular architecture of the TNF superfamily. Trends Biochem Sci.
27:19–26. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Locksley RM, Killeen N and Lenardo MJ: The
TNF and TNF receptor superfamilies: Integrating mammalian biology.
Cell. 104:487–501. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Grell M: Tumor necrosis factor (TNF)
receptors in cellular signaling of soluble and membrane-expressed
TNF. J Inflamm. 47:8–17. 1995.PubMed/NCBI
|
|
52
|
Hsu H, Xiong J and Goeddel DV: The TNF
receptor 1-associated protein TRADD signals cell death and NF-kappa
B activation. Cell. 81:495–504. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sedger LM and McDermott MF: TNF and
TNF-receptors: From mediators of cell death and inflammation to
therapeutic giants-past, present and future. Cytokine Growth Factor
Rev. 25:453–472. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Faustman D and Davis M: TNF receptor 2
pathway: Drug target for autoimmune diseases. Nat Rev Drug Discov.
9:482–493. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Grell M, Wajant H, Zimmermann G and
Scheurich P: The type 1 receptor (CD120a) is the high-affinity
receptor for soluble tumor necrosis factor. Proc Natl Acad Sci USA.
95:570–575. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wajant H, Pfizenmaier K and Scheurich P:
Tumor necrosis factor signaling. Cell Death Differ. 10:45–65. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Tartaglia LA, Ayres TM, Wong GH and
Goeddel DV: A novel domain within the 55 kd TNF receptor signals
cell death. Cell. 74:845–853. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bradley JR: TNF-mediated inflammatory
disease. J Pathol. 214:149–160. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Fischer R, Maier O, Naumer M,
Krippner-Heidenreich A, Scheurich P and Pfizenmaier K:
Ligand-induced internalization of TNF receptor 2 mediated by a
di-leucin motif is dispensable for activation of the NFκB pathway.
Cell Signal. 23:161–170. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Naude PJ, den Boer JA, Luiten PG and Eisel
UL: Tumor necrosis factor receptor cross-talk. FEBS J. 278:888–898.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Marchetti L, Klein M, Schlett K,
Pfizenmaier K and Eisel ULM: Tumor necrosis factor (TNF)-mediated
neuroprotection against glutamate-induced excitotoxicity is
enhanced by N-methyl-D-aspartate receptor activation. Essential
role of a TNF receptor 2-mediated phosphatidylinositol
3-kinase-dependent NF-kappa B pathway. J Biol Chem.
279:32869–32881. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Goto N, Tsurumi H, Takemura M, Hara T,
Sawada M, Kasahara S, Kanemura N, Yamada T, Shimizu M, Takahashi T,
et al: Serum-soluble tumor necrosis factor receptor 2 (sTNF-R2)
level determines clinical outcome in patients with aggressive
non-Hodgkin's lymphoma. Eur J Haematol. 77:217–225. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kanayama A, Seth RB, Sun L, Ea CK, Hong M,
Shaito A, Chiu YH, Deng L and Chen ZJ: TAB2 and TAB3 activate the
NF-kappaB pathway through binding to polyubiquitin chains. Mol
Cell. 15:535–548. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Haas TL, Emmerich CH, Gerlach B, Schmukle
AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T,
et al: Recruitment of the linear ubiquitin chain assembly complex
stabilizes the TNF-R1 signaling complex and is required for
TNF-mediated gene induction. Mol Cell. 36:831–844. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Tokunaga F, Sakata S, Saeki Y, Satomi Y,
Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, et
al: Involvement of linear polyubiquitylation of NEMO in NF-kappaB
activation. Nat Cell Biol. 11:123–132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Sabio G and Davis RJ: TNF and MAP kinase
signalling pathways. Semin Immunol. 26:237–245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zeke A, Misheva M, Reményi A and
Bogoyevitch MA: JNK signaling: Regulation and functions based on
complex protein-protein partnerships. Microbiol Mol Biol Rev.
80:793–835. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Itoh N and Nagata S: A novel protein
domain required for apoptosis. Mutational analysis of human fas
antigen. J Biol Chem. 268:10932–10937. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Vercammen D, Beyaert R, Denecker G,
Goossens V, Loo GV, Declercq W, Grooten J, Fiers W and Vandenabeele
P: Inhibition of caspases increases the sensitivity of L929 cells
to necrosis mediated by tumor necrosis factor. J Exp Med.
187:1477–1485. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Shalini S, Dorstyn L, Dawar S and Kumar S:
Old, new and emerging functions of caspases. Cell Death Differ.
22:526–539. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Samson AL, Zhang Y, Geoghegan ND, Gavin
XJ, Davies KA, Mlodzianoski MJ, Whitehead LW, Frank D, Garnish SE,
Fitzgibbon C, et al: MLKL trafficking and accumulation at the
plasma membrane control the kinetics and threshold for necroptosis.
Nat Commun. 11:31512020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Rothe M, Wong SC, Henzel WJ and Goeddel
DV: A novel family of putative signal transducers associated with
the cytoplasmic domain of the 75 kDa tumor necrosis factor
receptor. Cell. 78:681–692. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Borghi A, Haegman M, Fischer R, Carpentier
I, Bertrand MJM, Libert C, Afonina IS and Beyaert R: The E3
ubiquitin ligases HOIP and cIAP1 are recruited to the TNFR2
signaling complex and mediate TNFR2-induced canonical NF-kappaB
signaling. Biochem Pharmacol. 153:292–298. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Rauert H, Wicovsky A, Muller N, Siegmund
D, Spindler V, Waschke J, Kneitz C and Wajant H: Membrane tumor
necrosis factor (TNF) induces p100 processing via TNF receptor-2
(TNFR2). J Biol Chem. 285:7394–7404. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Sun SC: The non-canonical NF-κB pathway in
immunity and inflammation. Nat Rev Immunol. 17:545–558. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Devin A, Cook A, Lin Y, Rodriguez Y,
Kelliher M and Liu Z: The distinct roles of TRAF2 and RIP in IKK
activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP
mediates IKK activation. Immunity. 12:419–429. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Fotin-Mleczek M, Henkler F, Samel D,
Reichwein M, Hausser A, Parmryd I, Scheurich P, Schmid JA and
Wajant H: Apoptotic crosstalk of TNF receptors: TNF-R2-induces
depletion of TRAF2 and IAP proteins and accelerates
TNF-R1-dependent activation of caspase-8. J Cell Sci.
115:2757–2770. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Li X, Yang Y and Ashwell JD: TNF-RII and
c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature.
416:345–347. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Tada K, Okazaki T, Sakon S, Kobarai T,
Kurosawa K, Yamaoka S, Hashimoto H, Mak TW, Yagita H, Okumura K, et
al: Critical roles of TRAF2 and TRAF5 in tumor necrosis
factor-induced NF-kappa B activation and protection from cell
death. J Biol Chem. 276:36530–36534. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Vince JE, Pantaki D, Feltham R, Mace PD,
Cordier SM, Schmukle AC, Davidson AJ, Callus BA, Wong WWL, Gentle
IE, et al: TRAF2 must bind to cellular inhibitors of apoptosis for
tumor necrosis factor (tnf) to efficiently activate nf-{kappa}b and
to prevent tnf-induced apoptosis. J Biol Chem. 284:35906–35915.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Liu ZG, Hsu H, Goeddel DV and Karin M:
Dissection of TNF receptor 1 effector functions: JNK activation is
not linked to apoptosis while NF-kappaB activation prevents cell
death. Cell. 87:565–576. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ridgley LA, Anderson AE and Pratt AG: What
are the dominant cytokines in early rheumatoid arthritis? Curr Opin
Rheumatol. 30:207–214. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Kobayashi M, Squires GR, Mousa A, Tanzer
M, Zukor DJ, Antoniou J, Feige U and Poole AR: Role of
interleukin-1 and tumor necrosis factor alpha in matrix degradation
of human osteoarthritic cartilage. Arthritis Rheum. 52:128–135.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Mirza F, Lorenzo J, Drissi H, Lee FY and
Soung DY: Dried plum alleviates symptoms of inflammatory arthritis
in TNF transgenic mice. J Nutr Biochem. 52:54–61. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Chen W, Li Z, Wang Z, Gao H, Ding J and He
Z: Intraarticular injection of infliximab-loaded thermosensitive
hydrogel alleviates pain and protects cartilage in rheumatoid
arthritis. J Pain Res. 13:3315–3329. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Rioja I, Bush KA, Buckton JB, Dickson MC
and Life PF: Joint cytokine quantification in two rodent arthritis
models: Kinetics of expression, correlation of mRNA and protein
levels and response to prednisolone treatment. Clin Exp Immunol.
137:65–73. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Williams RO, Marinova-Mutafchieva L,
Feldmann M and Maini RN: Evaluation of TNF-alpha and IL-1 blockade
in collagen-induced arthritis and comparison with combined
anti-TNF-alpha/anti-CD4 therapy. J Immunol. 165:7240–7245. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yu D, Ye X, Che R, Wu Q, Qi J, Song L, Guo
X, Zhang S, Wu H, Ren G and Li D: FGF21 exerts comparable
pharmacological efficacy with Adalimumab in ameliorating
collagen-induced rheumatoid arthritis by regulating systematic
inflammatory response. Biomed Pharmacother. 89:751–760. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wu AJ, Hua H, Munson SH and McDevitt HO:
Tumor necrosis factor-alpha regulation of
CD4+CD25+ T cell levels in NOD mice. Proc
Natl Acad Sci USA. 99:12287–12292. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ehrenstein MR, Evans JG, Singh A, Moore S,
Warnes G, Isenberg DA and Mauri C: Compromised function of
regulatory T cells in rheumatoid arthritis and reversal by
anti-TNFalpha therapy. J Exp Med. 200:277–285. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Louis E: The immuno-inflammatory reaction
in Crohn's disease and ulcerative colitis: Characterisation,
genetics and clinical application. Focus on TNF alpha. Acta
Gastroenterol Belg. 64:1–5. 2001.PubMed/NCBI
|
|
92
|
Gottlieb AB, Chamian F, Masud S, Cardinale
I, Abello MV, Lowes MA, Chen F, Magliocco M and Krueger JG: TNF
inhibition rapidly down-regulates multiple proinflammatory pathways
in psoriasis plaques. J Immunol. 175:2721–2729. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Catalina MD, Owen KA, Labonte AC, Grammer
AC and Lipsky PE: The pathogenesis of systemic lupus erythematosus:
Harnessing big data to understand the molecular basis of lupus. J
Autoimmun. 110:1023592020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kwon YC, Chun S, Kim K and Mak A: Update
on the genetics of systemic lupus erythematosus: Genome-wide
association studies and beyond. Cells. 8:11802019. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Marion TN and Postlethwaite AE: Chance,
genetics, and the heterogeneity of disease and pathogenesis in
systemic lupus erythematosus. Semin Immunopathol. 36:495–517. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Hedrich CM: Epigenetics in SLE. Curr
Rheumatol Rep. 19:582017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Pan L, Lu MP, Wang JH, Xu M and Yang SR:
Immunological pathogenesis and treatment of systemic lupus
erythematosus. World J Pediatr. 16:19–30. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Maningding E, Dall'Era M, Trupin L, Murphy
LB and Yazdany J: Racial and ethnic differences in the prevalence
and time to onset of manifestations of systemic lupus
erythematosus: The California lupus surveillance project. Arthritis
Care Res (Hoboken). 72:622–629. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Christou EAA, Banos A, Kosmara D, Bertsias
GK and Boumpas DT: Sexual dimorphism in SLE: Above and beyond sex
hormones. Lupus. 28:3–10. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Barbhaiya M and Costenbader KH:
Environmental exposures and the development of systemic lupus
erythematosus. Curr Opin Rheumatol. 28:497–505. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Constantin MM, Nita IE, Olteanu R,
Constantin T, Bucur S, Matei C and Raducan A: Significance and
impact of dietary factors on systemic lupus erythematosus
pathogenesis. Exp Ther Med. 17:1085–1090. 2019.PubMed/NCBI
|
|
102
|
Luo S, Long H and Lu Q: Recent advances in
understanding pathogenesis and therapeutic strategies of systemic
lupus erythematosus. Int Immunopharmacol. 89:1070282020. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Parks CG, de Souza Espindola Santos A,
Barbhaiya M and Costenbader KH: Understanding the role of
environmental factors in the development of systemic lupus
erythematosus. Best Pract Res Clin Rheumatol. 31:306–320. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Habib HM, Taher TE, Isenberg DA and Mageed
RA: Enhanced propensity of T lymphocytes in patients with systemic
lupus erythematosus to apoptosis in the presence of tumour necrosis
factor alpha. Scand J Rheumatol. 38:112–120. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Gómez D, Correa PA, Gómez LM, Cadena J,
Molina JF and Anaya JM: Th1/Th2 cytokines in patients with systemic
lupus erythematosus: Is tumor necrosis factor alpha protective?
Semin Arthritis Rheum. 33:404–413. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhu L, Yang X, Chen W, Li X, Ji Y, Mao H,
Nie J and Yu X: Decreased expressions of the TNF-alpha signaling
adapters in peripheral blood mononuclear cells (PBMCs) are
correlated with disease activity in patients with systemic lupus
erythematosus. Clin Rheumatol. 26:1481–1489. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
McCarthy EM, Smith S, Lee RZ, Cunnane G,
Doran MF, Donnelly S, Howard D, O'Connell P, Kearns G, Gabhann JN
and Jefferies CA: The association of cytokines with disease
activity and damage scores in systemic lupus erythematosus
patients. Rheumatology (Oxford). 53:1586–1594. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Prete M, Leone P, Frassanito MA, Desantis
V, Marasco C, Cicco S, Dammacco F, Vacca A and Racanelli V:
Belimumab restores Treg/Th17 balance in patients with refractory
systemic lupus erythematosus. Lupus. 27:1926–1935. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Su DL, Lu ZM, Shen MN, Li X and Sun LY:
Roles of pro- and anti-inflammatory cytokines in the pathogenesis
of SLE. J Biomed Biotechnol. 2012:3471412012. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Tahernia L, Alimadadi H, Tahghighi F,
Amini Z and Ziaee V: Frequency and type of hepatic and
gastrointestinal involvement in juvenile systemic lupus
erythematosus. Autoimmune Dis. 2017:80972732017.PubMed/NCBI
|
|
111
|
Yap DY and Lai KN: The role of cytokines
in the pathogenesis of systemic lupus erythematosus-from bench to
bedside. Nephrology (Carlton). 18:243–255. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Takemura T, Yoshioka K, Murakami K, Akano
N, Okada M, Aya N and Maki S: Cellular localization of inflammatory
cytokines in human glomerulonephritis. Virchows Arch. 424:459–464.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Malide D, Russo P and Bendayan M: Presence
of tumor necrosis factor alpha and interleukin-6 in renal mesangial
cells of lupus nephritis patients. Hum Pathol. 26:558–564. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Neale TJ, Ruger BM, Macaulay H, Dunbar PR,
Hasan Q, Bourke A, Murray-McIntosh RP and Kitching AR: Tumor
necrosis factor-alpha is expressed by glomerular visceral
epithelial cells in human membranous nephropathy. Am J Pathol.
146:1444–1454. 1995.PubMed/NCBI
|
|
115
|
Herrera-Esparza R, Barbosa-Cisneros O,
Villalobos-Hurtado R and Avalos-Díaz E: Renal expression of IL-6
and TNFalpha genes in lupus nephritis. Lupus. 7:154–158. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Aringer M and Smolen JS: The role of tumor
necrosis factor-alpha in systemic lupus erythematosus. Arthritis
Res Ther. 10:2022008. View
Article : Google Scholar : PubMed/NCBI
|
|
117
|
D'Alfonso S, Colombo G, Bella SD, Scorza R
and Momigliano-Richiardi P: Association between polymorphisms in
the TNF region and systemic lupus erythematosus in the Italian
population. Tissue Antigens. 47:551–555. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Dourmishev L, Kamenarska Z, Hristova M,
Dodova R, Kaneva R and Mitev V: Association of TNF-α polymorphisms
with adult dermatomyositis and systemic lupus erythematosus in
Bulgarian patients. Int J Dermatol. 51:1467–1473. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Lee YH, Harley JB and Nath SK:
Meta-analysis of TNF-alpha promoter-308 A/G polymorphism and SLE
susceptibility. Eur J Hum Genet. 14:364–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Lin YJ, Chen RH, Wan L, Sheu JC, Huang CM,
Lin CW, Chen SY, Lai CH, Lan YC, Hsueh KC, et al: Association of
TNF-alpha gene polymorphisms with systemic lupus erythematosus in
Taiwanese patients. Lupus. 18:974–979. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Zúñiga J, Vargas-Alarcón G,
Hernández-Pacheco G, Portal-Celhay C, Yamamoto-Furusho JK and
Granados J: Tumor necrosis factor-alpha promoter polymorphisms in
Mexican patients with systemic lupus erythematosus (SLE). Genes
Immun. 2:363–366. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Davas EM, Tsirogianni A, Kappou I,
Karamitsos D, Economidou I and Dantis PC: Serum IL-6, TNFalpha, p55
srTNFalpha, p75srTNFalpha, srIL-2alpha levels and disease activity
in systemic lupus erythematosus. Clin Rheumatol. 18:17–22. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Adrianto I, Wen F, Templeton A, Wiley G,
King JB, Lessard CJ, Bates JS, Hu Y, Kelly JA, Kaufman KM, et al:
Association of a functional variant downstream of TNFAIP3 with
systemic lupus erythematosus. Nat Genet. 43:253–258. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Bates JS, Lessard CJ, Leon JM, Nguyen T,
Battiest LJ, Rodgers J, Kaufman KM, James JA, Gilkeson GS, Kelly
JA, et al: Meta-analysis and imputation identifies a 109 kb risk
haplotype spanning TNFAIP3 associated with lupus nephritis and
hematologic manifestations. Genes Immun. 10:470–477. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Goulielmos GN, Zervou MI, Vazgiourakis VM,
Ghodke-Puranik Y, Garyfallos A and Niewold TB: The genetics and
molecular pathogenesis of systemic lupus erythematosus (SLE) in
populations of different ancestry. Gene. 668:59–72. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Manku H, Langefeld CD, Guerra SG, Malik
TH, Alarcon-Riquelme M, Anaya JM, Bae SC, Boackle SA, Brown EE,
Criswell LA, et al: Trans-ancestral studies fine map the
SLE-susceptibility locus TNFSF4. PLoS Genet. 9:e10035542013.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Komata T, Tsuchiya N, Matsushita M,
Hagiwara K and Tokunaga K: Association of tumor necrosis factor
receptor 2 (TNFR2) polymorphism with susceptibility to systemic
lupus erythematosus. Tissue Antigens. 53:527–533. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Aderka D, Wysenbeek A, Engelmann H, Cope
AP, Brennan F, Molad Y, Hornik V, Levo Y, Maini RN and Feldmann M:
Correlation between serum levels of soluble tumor necrosis factor
receptor and disease activity in systemic lupus erythematosus.
Arthritis Rheum. 36:1111–1120. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Munroe ME, Vista ES, Guthridge JM,
Thompson LF, Merrill JT and James JA: Proinflammatory adaptive
cytokine and shed tumor necrosis factor receptor levels are
elevated preceding systemic lupus erythematosus disease flare.
Arthritis Rheumatol. 66:1888–1899. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Horiuchi T, Mitoma H, Harashima S,
Tsukamoto H and Shimoda T: Transmembrane TNF-alpha: Structure,
function and interaction with anti-TNF agents. Rheumatology
(Oxford). 49:1215–1228. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Patel M, Oni L, Midgley A, Smith E, Tullus
K, Marks SD, Jones CA, Pilkington C and Beresford MW: Increased
concentration of plasma TNFR1 and TNFR2 in paediatric lupus
nephritis. Lupus. 25:1040–1044. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Ida H, Kawasaki E, Miyashita T, Tanaka F,
Kamachi M, Izumi Y, Huang M, Tamai M, Origuchi T, Kawakami A, et
al: A novel mutation (T61I) in the gene encoding tumour necrosis
factor receptor superfamily 1A (TNFRSF1A) in a Japanese patient
with tumour necrosis factor receptor-associated periodic syndrome
(TRAPS) associated with systemic lupus erythematosus. Rheumatology
(Oxford). 43:1292–1299. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Al-Ansari AS, Ollier WE, Villarreal J,
Ordi J, The LS and Hajeer AH: Tumor necrosis factor receptor II
(TNFRII) exon 6 polymorphism in systemic lupus erythematosus.
Tissue Antigens. 55:97–99. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Chadha S, Miller K, Farwell L, Sacks S,
Daly MJ, Rioux JD and Vyse TJ: Haplotype analysis of tumour
necrosis factor receptor genes in 1p36: No evidence for association
with systemic lupus erythematosus. Eur J Hum Genet. 14:69–78. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Sullivan KE, Piliero LM, Goldman D and
Petri MA: A TNFR2 3′ flanking region polymorphism in systemic lupus
erythematosus. Genes Immun. 1:225–227. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Fairhurst AM, Wandstrat AE and Wakeland
EK: Systemic lupus erythematosus: Multiple immunological phenotypes
in a complex genetic disease. Adv Immunol. 92:1–69. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Li W, Titov AA and Morel L: An update on
lupus animal models. Curr Opin Rheumatol. 29:434–441. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Aringer M and Smolen JS: Therapeutic
blockade of TNF in patients with SLE-promising or crazy? Autoimmun
Rev. 11:321–325. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Helyer BJ and Howie JB: Renal disease
associated with positive lupus erythematosus tests in a cross-bred
strain of mice. Nature. 197:1971963. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Brennan DC, Yui MA, Wuthrich RP and Kelley
VE: Tumor necrosis factor and IL-1 in New Zealand black/white mice.
Enhanced gene expression and acceleration of renal injury. J
Immunol. 143:3470–3475. 1989.PubMed/NCBI
|
|
141
|
Boswell JM, Yui MA, Burt DW and Kelley VE:
Increased tumor necrosis factor and IL-1 beta gene expression in
the kidneys of mice with lupus nephritis. J Immunol. 141:3050–3054.
1988.PubMed/NCBI
|
|
142
|
Yokoyama H, Kreft B and Kelley VR:
Biphasic increase in circulating and renal TNF-alpha in MRL-lpr
mice with differing regulatory mechanisms. Kidney Int. 47:122–130.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Tsai CY, Wu TH, Huang SF, Sun KH, Hsieh
SC, Han SH, Yu HS and Yu CL: Abnormal splenic and thymic IL-4 and
TNF-alpha expression in MRL-lpr/lpr mice. Scand J Immunol.
41:157–163. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Deguchi Y and Kishimoto S: Tumour necrosis
factor/cachectin plays a key role in autoimmune pulmonary
inflammation in lupus-prone mice. Clin Exp Immunol. 85:392–395.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Su X, Zhou T, Yang P, Edwards CK III and
Mountz JD: Reduction of arthritis and pneumonitis in motheaten mice
by soluble tumor necrosis factor receptor. Arthritis Rheum.
41:139–149. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Bethunaickan R, Sahu R, Liu Z, Tang YT,
Huang W, Edegbe O, Tao H, Ramanujam M, Madaio MP and Davidson A:
Anti-tumor necrosis factor alpha treatment of interferon-α-induced
murine lupus nephritis reduces the renal macrophage response but
does not alter glomerular immune complex formation. Arthritis
Rheum. 64:3399–3408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Monaco C, Nanchahal J, Taylor P and
Feldmann M: Anti-TNF therapy: Past, present and future. Int
Immunol. 27:55–62. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Williams EL, Gadola S and Edwards CJ:
Anti-TNF-induced lupus. Rheumatology (Oxford). 48:716–720. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Aghdashi MA, Khadir M and Dinparasti-Saleh
R: Antinuclear antibodies and lupus-like manifestations in
rheumatoid arthritis and ankylosing spondylitis patients at 4
months' follow-up after treatment with infliximab and etanercept.
Curr Rheumatol Rev. 16:61–66. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Gonnet-Gracia C, Barnetche T, Richez C,
Blanco P, Dehais J and Schaeverbeke T: Anti-nuclear antibodies,
anti-DNA and C4 complement evolution in rheumatoid arthritis and
ankylosing spondylitis treated with TNF-alpha blockers. Clin Exp
Rheumatol. 26:401–407. 2008.PubMed/NCBI
|
|
151
|
Ramos-Casals M, Brito-Zeron P, Munoz S,
Soria N, Galiana D, Bertolaccini L, Cuadrado MJ and Khamashta MA:
Autoimmune diseases induced by TNF-targeted therapies: Analysis of
233 cases. Medicine (Baltimore). 86:242–251. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Santos CS, Castro CA, Morales CM and
Álvarez ED: Anti-TNF-α-induced lupus syndrome: Two case reports and
review of current literature. Z Rheumatol. 80:481–486. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Stokes MB, Foster K, Markowitz GS,
Ebrahimi F, Hines W, Kaufman D, Moore B, Wolde D and D'Agati VD:
Development of glomerulonephritis during anti-TNF-alpha therapy for
rheumatoid arthritis. Nephrol Dial Transplant. 20:1400–1406. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Mor A, Bingham CO III, Barisoni L, Lydon E
and Belmont HM: Proliferative lupus nephritis and leukocytoclastic
vasculitis during treatment with etanercept. J Rheumatol.
32:740–743. 2005.PubMed/NCBI
|
|
155
|
Aringer M, Graninger WB, Steiner G and
Smolen JS: Safety and efficacy of tumor necrosis factor alpha
blockade in systemic lupus erythematosus: An open-label study.
Arthritis Rheum. 50:3161–3169. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Aringer M, Steiner G, Graninger WB, Höfler
E, Steiner CW and Smolen JS: Effects of short-term infliximab
therapy on autoantibodies in systemic lupus erythematosus.
Arthritis Rheum. 56:274–279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Aringer M, Houssiau F, Gordon C, Graninger
WB, Voll RE, Rath E, Steiner G and Smolen JS: Adverse events and
efficacy of TNF-alpha blockade with infliximab in patients with
systemic lupus erythematosus: long-term follow-up of 13 patients.
Rheumatology (Oxford). 48:1451–1454. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Matsumura R, Umemiya K, Sugiyama T,
Sueishi M, Umibe T, Ichikawa K and Yoshimura M; Study Group on
Nephrology at the National Hospital Organization of Japan, :
Anti-tumor necrosis factor therapy in patients with
difficult-to-treat lupus nephritis: A prospective series of nine
patients. Clin Exp Rheumatol. 27:416–421. 2009.PubMed/NCBI
|
|
159
|
Hayat SJ and Uppal SS: Therapeutic
efficacy and safety profile of infliximab in active systemic lupus
erythematosus. Mod Rheumatol. 17:174–177. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Uppal SS, Hayat SJ and Raghupathy R:
Efficacy and safety of infliximab in active SLE: A pilot study.
Lupus. 18:690–697. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Moreland LW, Baumgartner SW, Schiff MH,
Tindall EA, Fleischmann RM, Weaver AL, Ettlinger RE, Cohen S,
Koopman WJ, Mohler K, et al: Treatment of rheumatoid arthritis with
a recombinant human tumor necrosis factor receptor (p75)-Fc fusion
protein. N Engl J Med. 337:141–147. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Anderson PJ: Tumor necrosis factor
inhibitors: Clinical implications of their different immunogenicity
profiles. Semin Arthritis Rheum. 34 5 Suppl 1:S19–S22. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Cortes-Hernandez J, Egri N,
Vilardell-Tarres M and Ordi-Ros J: Etanercept in refractory lupus
arthritis: An observational study. Semin Arthritis Rheum.
44:672–679. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Norman R, Greenberg RG and Jackson JM:
Case reports of etanercept in inflammatory dermatoses. J Am Acad
Dermatol. 54 3 Suppl 2:S139–S142. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Yang BB, Xiao H, Li XJ and Zheng M: Safety
and efficacy of etanercept-methotrexate combination therapy in
patients with rhupus: An observational study of non-glucocorticoid
treatment for rheumatic diseases. Discov Med. 25:14–20.
2018.PubMed/NCBI
|
|
166
|
Danion F, Sparsa L, Arnaud L, Alsaleh G,
Lefebvre F, Gies V, Martin T, Lukas C, Durckel J, Ardizzone M, et
al: Long-term efficacy and safety of antitumour necrosis factor
alpha treatment in rhupus: An open-label study of 15 patients. RMD
Open. 3:e0005552017. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Micheloud D, Nuno L, Rodriguez-Mahou M,
Sánchez-Ramón S, Ortega MC, Aguarón A, Junco E, Carbone J,
Fernández-Cruzl E, Carreño L and López-Longo FJ: Efficacy and
safety of Etanercept, high-dose intravenous gammaglobulin and
plasmapheresis combined therapy for lupus diffuse proliferative
nephritis complicating pregnancy. Lupus. 15:881–885. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Mustafa G, Mahrosh HS, Salman M, Sharif S,
Jabeen R, Majeed T and Tahir H: Identification of peptides as novel
inhibitors to target IFN-γ, IL-3, and TNF-α in systemic lupus
erythematosus. Biomed Res Int. 2021:11240552021. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Bongartz T, Sutton AJ, Sweeting MJ, Buchan
I, Matteson EL and Montori V: Anti-TNF antibody therapy in
rheumatoid arthritis and the risk of serious infections and
malignancies: Systematic review and meta-analysis of rare harmful
effects in randomized controlled trials. JAMA. 295:2275–2285. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Tack CJ, Kleijwegt FS, Van Riel PL and
Roep BO: Development of type 1 diabetes in a patient treated with
anti-TNF-alpha therapy for active rheumatoid arthritis.
Diabetologia. 52:1442–1444. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Bloom BJ: Development of diabetes mellitus
during etanercept therapy in a child with systemic-onset juvenile
rheumatoid arthritis. Arthritis Rheum. 43:2606–2608. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Kollias G and Kontoyiannis D: Role of
TNF/TNFR in autoimmunity: Specific TNF receptor blockade may be
advantageous to anti-TNF treatments. Cytokine Growth Factor Rev.
13:315–321. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Zhang N, Wang Z and Zhao Y: Selective
inhibition of Tumor necrosis factor receptor-1 (TNFR1) for the
treatment of autoimmune diseases. Cytokine Growth Factor Rev.
55:80–85. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Van Hauwermeiren F, Armaka M, Karagianni
N, Kranidioti K, Vandenbroucke RE, Loges S, Roy MV, Staelens J,
Puimège L, Palagani A, et al: Safe TNF-based antitumor therapy
following p55TNFR reduction in intestinal epithelium. J Clin
Invest. 123:2590–2603. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Puimege L, Libert C and Van Hauwermeiren
F: Regulation and dysregulation of tumor necrosis factor
receptor-1. Cytokine Growth Factor Rev. 25:285–300. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Vinay DS and Kwon BS: The tumour necrosis
factor/TNF receptor superfamily: Therapeutic targets in autoimmune
diseases. Clin Exp Immunol. 164:145–157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Wu T, Xie C, Wang HW, Zhou XJ, Schwartz N,
Calixto S, Mackay M, Aranow C, Putterman C and Mohan C: Elevated
urinary VCAM-1, P-selectin, soluble TNF receptor-1, and CXC
chemokine ligand 16 in multiple murine lupus strains and human
lupus nephritis. J Immunol. 179:7166–7175. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Deng GM, Liu L and Tsokos GC: Targeted
tumor necrosis factor receptor I preligand assembly domain improves
skin lesions in MRL/lpr mice. Arthritis Rheum. 62:2424–2431. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Jacob N, Yang H, Pricop L, Liu Y, Gao X,
Zheng SG, Wang J, Gao HX, Putterman C, Koss MN, et al: Accelerated
pathological and clinical nephritis in systemic lupus
erythematosus-prone New Zealand mixed 2328 mice doubly deficient in
TNF receptor 1 and TNF receptor 2 via a Th17-associated pathway. J
Immunol. 182:2532–2541. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Zhou T, Edwards CK III, Yang P, Wang Z,
Bluethmann H and Mountz JD: Greatly accelerated lymphadenopathy and
autoimmune disease in lpr mice lacking tumor necrosis factor
receptor I. J Immunol. 156:2661–2665. 1996.PubMed/NCBI
|