Introduction
Cerebral ischemia usually occurs as a result of a
disruption of the blood supply, leading to a decrease in the supply
of glucose and oxygen to brain tissues. Following the restoration
of the supply, the return of blood flow can result in secondary
brain tissue damage (1). The
progression of cerebral ischemia/reperfusion (I/R) is dependent on
the interaction of several mechanisms, such as the production of
free radicals, glutamate over-release and calcium overload
(2,3). Early during this process, due to
the elevated oxygen supply, the lesioned area rapidly produces
large quantities of oxygen radicals, and these toxic products are
the main cause of DNA damage (4,5).
DNA double-stranded breaks (DSBs) are the primary form of DNA
damage and are one of the hallmarks of cell death (6,7).
It has been previously demonstrated that DNA repair is increased in
ischemic neurons, producing an endogenous repair effect (8,9).
The enhancement of the effective repair of damaged DNA and the
inhibition of cell death may therefore have a positive protective
effect against central nervous system-damaging diseases, such as
ischemic stroke.
Autophagy is a procedure that selectively degrades
cellular components to maintain the balance of the internal
environment (10). Being an
essential molecule in autophagosome formation, Beclin-1 mediates
the localization of other autophagic proteins to phagocytic
vesicles, thereby regulating the formation and maturation of
mammalian autophagosomes (11,12). Beclin-1 expression is typically
elevated and activates autophagy in stroke and cerebral I/R injury.
It has been shown that homocysteine accumulation in a brain model
of I/R can promote the release of Beclin-1 and the apoptosis of
neuronal cells, enhance DNA damage and induce neuronal death; this
indicates that, on the one hand, Beclin-1, apoptosis and DNA damage
are positively associated, and on the other hand, the high
expression level of Beclin-1 is positively linked to severe I/R
injury (13). On the contrary, a
protective role of Beclin-1 in I/R injury was suggested by another
study which demonstrated that increased levels of Beclin-1
activated autophagy and contributed to neuroprotection against
focal cerebral ischemia in rats (14). However, from the current
evidence, no definite conclusions have yet been reached as regards
the role of Beclin-1 in I/R, and autophagy and apoptosis may not be
sufficient to fully explain the contradictory role of Beclin-1 in
I/R injury. It is worth noting that there is a close association
between Beclin-1 and DNA damage repair. It has been demonstrated
that the knockdown of Beclin-1 significantly blocks the formation
of DNA-dependent protein kinase (DNA-PK) complexes, while the
Mre-11 (DSB repair protein), Nijmegen breakage syndrome 1 and
Rad-50 (DNA repair protein) (MNR) complex is slightly affected,
both of which are crucial for repairing damage to DSBs. This
suggests that Beclin-1 can influence DNA repair ability by
regulating the expression of several DSB repair proteins and the
formation of repair complexes (15,16).
At present, there remain unresolved issues and
controversies as regards the physiological and pathological roles
of the increase and decrease in Beclin-1 expression levels
following cerebral I/R injury. Considering the close connection
between Beclin-1 and DNA damage, as well as the close association
among DNA damage, apoptosis and I/R injury as aforementioned, the
present study thus aimed to further investigate the role and
regulatory mechanisms of Beclin-1 in DNA damage, autophagy and
apoptosis following cerebral I/R injury.
Materials and methods
Animal model of cerebral I/R
A total of 27 male Sprague-Dawley (SD) rats weighing
220-260 g and aged 7-8 weeks (Guangxi University Animal Experiment
Center, Guangxi, China) were used. All experiments were approved by
the Ethics Committee of the Affiliated Hospital of Youjiang Medical
University for Nationalities (approval no. YYFY-LL-2020-17), and
performed in accordance with the guidelines detailed in the
National Institutes of Health Guide for the Care and Use of
Laboratory Animals (National Institutes of Health) (17). All animals were housed at 22±2°C,
with 50-60% humidity, a 12-h light/dark cycle and were provided
with ad libitum access to food and water.
The rat model of cerebral I/R was established as
previously described (18). The
rats were anesthetized by an intraperitoneal injection of sodium
pentobarbital (30 mg/kg body weight; Sigma-Aldrich; Merck KGaA)
after 12 h of fasting. Following that, the dermatome was incised
slightly to the right along the middle of the neck, and the right
common carotid artery (CCA), right internal carotid artery (ICA)
and right external carotid artery (ECA) were bluntly dissected.
Surgical wires (5-0) were ligated proximal to the CCA and proximal
to the ECA. Upon clamping the ICA, a small incision was made at the
distal end of the ECA, and a wire plug (the diameter of the wire
plug was selected according to the weight of the rat) was inserted
towards the ICA at a depth of 16-20 mm, causing a flow block in the
middle cerebral artery and ligating the ICA. The removal of the
wire bolus caused reperfusion 90 min later. The sham-operated
(sham) group underwent the same surgery, but without cerebral
artery occlusion. The anal temperature of the rats was maintained
at 37±0.5°C throughout the procedure with an infrared heat lamp and
a heating pad. All rats were randomly divided into three groups
(n=9 per group): The sham group, model group and 3-methyladenine
(3-MA) group. Rats in the 3-MA group were injected with 10
µl 3-MA at 600 nmol, dissolved in normal saline
(MedChemExpress), a classical autophagy inhibitor, to the lateral
ventricles with a microinjector 60 min prior to modeling. Moreover,
the rats in the sham and model groups received an equivalent volume
of normal saline. After 24 h of modeling, the rats were euthanized
by an intraperitoneal injection of pentobarbital sodium (200 mg/kg
body weight) prior to collecting the brain tissues. Afterwards, 3
rats were used for 2,3,5-triphenyltetrazolium chloride (TTC)
staining. In the remaining 6 rats, the right brain tissues were
collected for use in western blot analysis, and the left brain
tissues were collected for analysis using staining: In total, three
left brain tissues were used for hematoxylin and eosin (H&E)
staining and immunohistochemistry (IHC), and the remaining three
left brain tissues were collected for immunofluorescence (IF) and
TUNEL assay.
In vitro model of oxygen-glucose
deprivation/reperfusion (OGD/R)
Mouse hippocampal cells (HT22; MilliporeSigma) were
used in all the in vitro experiments. The cells were
cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.),
100 U/ml penicillin and 100 mg/ml streptomycin (Gibco; Thermo
Fisher Scientific, Inc.), enclosed in a 5% CO2-95%
O2, 37°C constant temperature incubator.
The OGD/R model was used to simulate I/R injury
in vitro. Briefly, the cell culture medium was replaced with
glucose-free Earle's solution (Gibco; Thermo Fisher Scientific,
Inc.) and the cells were incubated in a hypoxic incubator (1%
O2, 5% CO2, 94% N2) at 37°C for 4
h. Subsequently, the medium was changed to normal medium and the
cells were incubated under normoxic atmospheric conditions (5%
CO2; 95% air) with reoxygenation for 12, 24 or 48 h. The
cells were divided into five groups as follows: The control, model,
3-MA, 3-MA + overexpression-negative control (OE-NC) and 3-MA +
Beclin-1 overexpression (OE-BECN1) groups. The cells in all groups,
apart from those in the control and model groups were treated with
3-MA (10 mM), which was added to the culture medium prior to
reoxygenation.
Cell transfection
Transfection was performed when the cells were 80%
confluent prior to the OGD/R experiments. OE-BECN1#1/2 and 0.8
µg OE-NC (Shanghai GenePharma, Co., Ltd.) vectors at a final
concentration of 50 nM were transfected into the cells using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) for 6 h at 37°C according to the manufacturer's
instructions. At 48 h post-transfection, the cells were used for
subsequent experiments.
Reverse transcription-quantitative PCR (RT-qPCR) was
used to assess the mRNA expression levels of BECN1 in the cells in
order to verify successful transfection. RNA extraction and
subsequent reverse transcription were performed according to the
manufacturer's instructions and as described below.
RT-qPCR
The extraction of RNA from cells was performed using
TRIzol® reagent (Thermo Fisher Scientific, Inc.). The
reverse transcription of individual RNA samples was then performed
using total RNA and a RevertAid First Strand DNA Synthesis kit
(Fermentas; Thermo Fisher Scientific, Inc.) following the
manufacturer's protocol as follows: 25°C for 5 min, 42°C for 60 min
and 70°C for 5 min. The newly synthesized first-strand cDNA was
ready for immediate downstream applications, or for long-term
storage at -80°C. mRNA levels in all samples were measured using
qPCR with the DreamTaq Green PCR MasterMix kit (Thermo Fisher
Scientific, Inc.) on a CFX Connect fluorescent quantitative PCR
instrument (Bio-Rad Laboratories, Inc.). The RT-qPCR conditions
were as follows: Pre-denaturation at 95°C for 2 min; followed by 35
cycles of 95°C for 35 sec, 58°C for 45 sec and 72°C for 30 sec; and
72°C for 5 min. Relative expression levels were calculated using
the 2−ΔΔCq method with GAPDH as the housekeeping gene
(19). The primer sequences used
were as follows: Beclin-1 forward, 5′-GCT GTA GCC AGC CTC TGA AA-3′
and reverse, 5′-AAT GGC TCC TGT GAG TTC CTG-3′; and GAPDH forward,
5′-AAG AAG GTG GTG AAG CAG G-3′ and reverse, 5′-GAA GGT GGA AGA GTG
GGA GT-3′.
TCC staining
Following sacrifice, the rat brains were removed,
and six consecutive 2-mm-thick coronal sections were prepared to
examine the infarct volume in the model rats. The sections were
completely immersed in 2% TTC staining solution (Jiancheng
Bioengineering Institute) for 30 min at room temperature, protected
from light, and then fixed in 4% paraformaldehyde at room
temperature; the infarcted tissue appeared white. Images were
acquired using a digital camera (Canon, Inc.) and analyzed using
ImageJ version 4.6.2 (National Institutes of Health). The infarct
volume was calculated by multiplying the added infarct areas of
each slices by slice thickness, and the results were presented as a
ratio of (infarct volume/the whole brain volume) ×100%
H&E staining
At 24 h after modeling, the rats were euthanized by
an intraperitoneal injection of pentobarbital sodium (200 mg/kg
body weight) prior to harvesting the brain tissue. After fixing in
4% paraformaldehyde, the brain tissues were paraffin-embedded and
cut into sections (4-µm-thick). The H&E staining kit
(Beyotime Institute of Biotechnology) was then used and the
experimental procedure strictly adhered to the manufacturer's
instructions as follows: The sections were stained with hematoxylin
solution at 37°C for 5 min and stained with eosin solution at 37°C
for 3 min. The sections were finally observed under a light
microscope (Nikon Corporation); five fields of view were randomly
selected for analysis in each group.
TUNEL assay
All operations were performed according to the
instructions provided with the TUNEL Apoptosis Assay kit (Beyotime
Institute of Biotechnology). After returning the frozen brain
tissue sections to room temperature, they were fixed in 4%
paraformaldehyde for 30 min at room temperature, and permeabilized
with PBS containing 0.5% Triton X-100 for 5 min. The tissues were
then incubated with TUNEL reaction mixture for 60 min at 37°C,
protected from light. After blocking with mounting medium
containing 4,6-diamidino-2-phenylindole (DAPI; Beijing Solarbio
Science & Technology Co., Ltd.), five random fields of view
were observed under a fluorescence microscope (Nikon Corporation;
magnification, ×200).
Western blot analysis
Brain tissues or HT22 cells were homogenized using
RIPA lysis reagent (Beyotime Institute of Biotechnology) containing
protease inhibitors and phosphatase inhibitors to extract total
protein. The protein content was determined using a Bradford assay
(MilliporeSigma), according to the manufacturer's instructions.
Proteins (20 µg/lane) were immobilized onto PVDF
(MilliporeSigma) membranes using 8-12% SDS-PAGE (Beyotime Institute
of Biotechnology). The PVDF membranes were blocked with 5% BSA at
room temperature for 1 h and incubated with the corresponding
primary antibodies overnight at 4°C. Subsequently, the membranes
were incubated with the secondary antibody (1:2,000; cat. no.
ab96899, Abcam) at room temperature for 2 h. Signals were
visualized using ECL reagent (Thermo Fisher Scientific, Inc.) and
the signal intensity was measured using ImageJ software (version
1.48v; National Institutes of Health). The primary antibodies used
were the following: Bax (1:1,000; cat. no. ab32503, Abcam), Bcl-2
(1:1,000; cat. no. ab182858, Abcam), cleaved caspase-3 (1:1,000;
cat. no. 9661, Cell Signaling Technology, Inc.), Beclin-1 (1:500;
cat. no. ab62557, Abcam), autophagy related (ATG)5 (1:1,000; cat.
no. ab221604, Abcam), ATG7 (1:1,000; cat. no. ab133528, Abcam),
light chain 3 (LC3; 1:500; cat. no. 43566, Cell Signaling
Technology, Inc.), γ histone family member X (γH2AX; 1:500; cat.
no. ab81299, Abcam), DNA-PK (1:1,000; cat. no. ab32566, Abcam) and
GAPDH (1:1,000; cat. no. ab181602, Abcam).
IF assay
Cells or brain tissue sections were fixed with 4%
paraformaldehyde at room temperature and permeabilized with 1%
Triton X-100 at room temperature and then blocked with 5% BSA for 1
h. Subsequently, cells/tissues were incubated overnight at 4°C with
the corresponding primary antibodies (NeuN; 1:200; cat. no. 94403
or LC3; 1:200; cat. no. 43566; Cell Signaling Technology, Inc.).
The following day, goat anti-rabbit IgG H&L/HRP (1:200; BIOSS;
cat. no. bs-40295G-HRP) and rabbit anti-mouse IgG-Fc/FITC
antibodies (1:200; BIOSS; cat. no. bs-0377R-FITC) were co-incubated
with the cells/tissues at room temperature for 1 h. The nuclei were
stained with DAPI (Jiancheng Bioengineering Institute) for 15 min
at room temperature. The samples were washed with PBS between all
steps. The results were observed under a fluorescence microscope
(Olympus BX51; Olympus Corporation) with five random fields of view
per group.
IHC
An IHC detection system kit (BIOSS) was used in the
present study. The brains sections were deparaffinized and
rehydrated, followed by antigen retrieval and cooling to room
temperature. The sections were then immersed in 3%
H2O2-methanol for 10 min, blocked dropwise
with 10% goat serum, and incubated with the primary antibody
(γH2AX, 1:500; cat. no. ab124781; Abcam) at 4°C overnight.
According to the manufacturer's instructions, anti-rabbit lgG
HRP-conjugated secondary (1:2,000; cat. no. ab205718; Abcam) for 1
h at room temperature were added and 3,3′-diaminobenzidine
tetrahydrochloride (DAB; Beijing Solarbio Science & Technology
Co., Ltd.) was used for chromogenic development.
Cell Counting Kit-8 (CCK-8) assay
The activity of the OGD/R cells following treatment
with 3-MA and transfection to induce Beclin-1 overexpression was
investigated using a CCK-8 assay (Beyotime Institute of
Biotechnology) for 2 h of culture at 37°C, according to the
manufacturer's protocol, and the absorbance at 450 nm was measured
used using a microplate reader (Bio-Rad Laboratories, Inc.).
Flow cytometric analysis for
apoptosis
Apoptosis was detected using an Annexin V-FITC
Apoptosis Detection kit (eBioscience; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. The cells were
resuspended in binding buffer at a density of 5×105
cells/ml, mixed with 5 µl Annexin V-FITC per 195 µl
binding buffer, incubated for 10 min at room temperature, washed
with binding buffer and then resuspended with 190 µl binding
buffer, adding 10 µl propidium iodide (20 µg/ml). The
apoptosis of the cells was analyzed using a flow cytometer (FC500;
Beckman Coulter, Inc.).
Statistical analysis
All data were statistically analyzed using SPSS
software version 18.0 (SPSS, Inc.), and data are presented as the
mean ± standard deviation. Differences between multiple groups were
analyzed using one-way ANOVA, followed by Tukey's post hoc test.
GraphPad Prism version 8.0 (GraphPad Software, Inc.) was used for
plotting data. P<0.05 was considered to indicate a statistically
significant difference.
Results
Inhibition of autophagy partially
attenuates I/R-induced brain tissue lesions
A rat model of focal cerebral I/R was established
and the autophagy inhibitor, 3-MA, was administered for
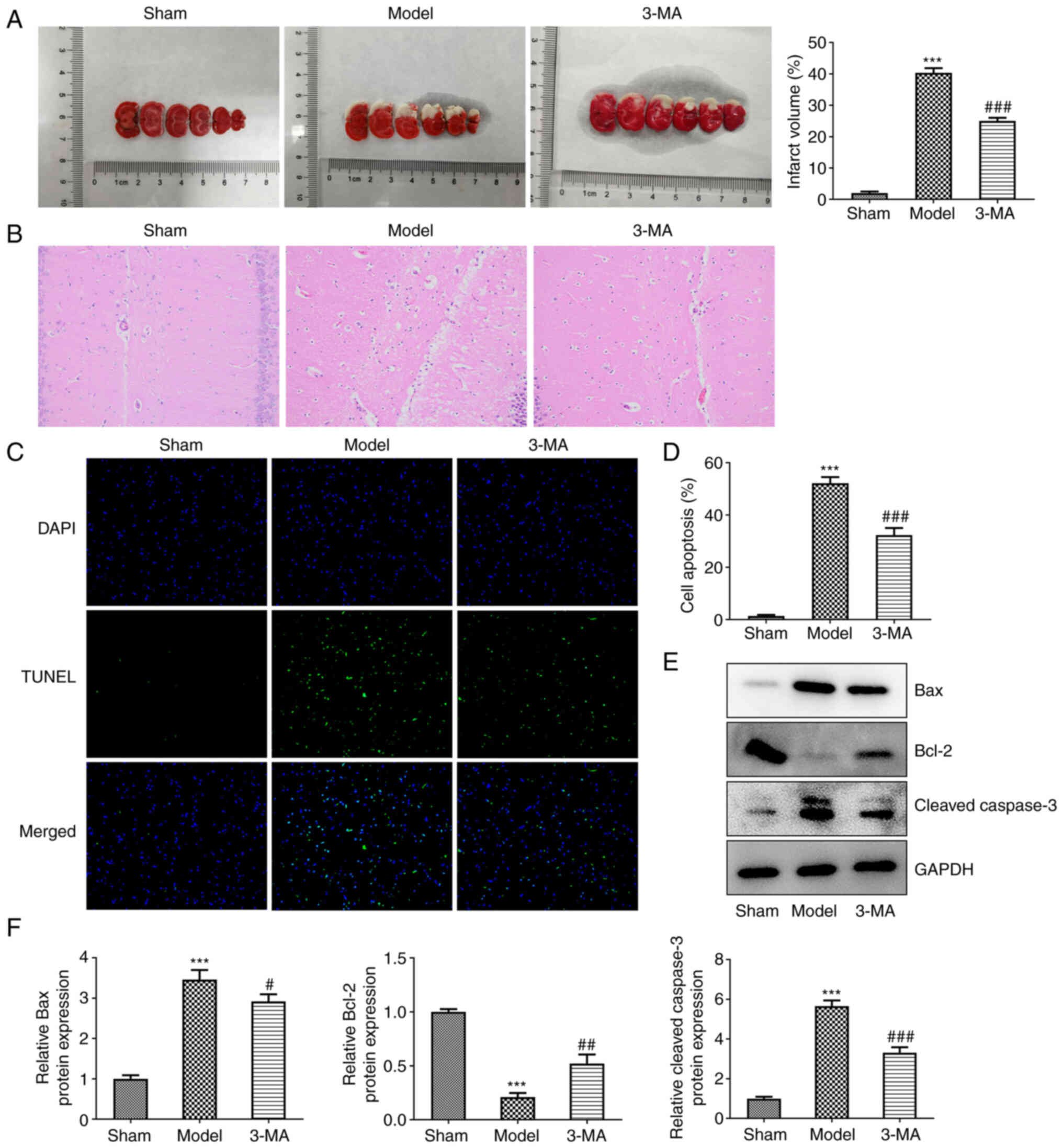
experimental validation. In comparison with the model group, the
results of TTC staining revealed that the area of cerebral
infarction (brain areas remaining white) was notably reduced in the
3-MA group (25.00±1.00 vs. 40.33±1.53, P<0.001) (Fig. 1A). As also revealed using H&E
staining, the cells in the sham group were uniformly colored,
densely distributed, neatly arranged, with a full cytosol, intact
cell structures and visible nucleoli (Fig. 1B). However, the cells in the
model group were disordered, with a reduced cytosolic volume,
densely stained cytoplasm, loose tissue and unclear nuclear
membranes. The 3-MA group exhibited an altered cellular state with
more ordered cell alignment, fewer tissue voids and fewer nuclear
alterations. The pathological results thus revealed that the
inhibition of autophagy significantly reduced cerebral I/R damage
to brain tissue, enhancing neuroprotection.
Apoptosis was analyzed using a TUNEL assay, as well
as western blot analysis. In the TUNEL assay, apoptotic cells
exhibited a fluorescent green color (Fig. 1C and D). The inhibition of
autophagy decreased the apoptosis of brain neuronal cells, compared
with the model group (32.33±2.72 vs. 52.17±2.40, P<0.001). This
was validated by the subsequent quantitative analysis of
apoptosis-related proteins. The results of western blot analysis
revealed that cerebral I/R injury led to an increase in the
expression of the the pro-apoptotic proteins, Bax (3.46±0.24 vs.
1.00±0.09, P<0.001) and cleaved-caspase-3 (5.66±0.28 vs.
1.00±0.09, P<0.001), with a decrease in the expression of the
anti-apoptotic protein, Bcl-2 (0.21±0.04 vs. 1.00±0.03, P<0.001)
(Fig. 1E and F). By contrast,
pre-treatment with 3-MA was able to partially reverse the
disorganized expression levels of both classes of apoptosis-related
proteins (2.92±0.18 vs. 3.46±0.24, P<0.05 for Bax; 3.32±0.27 vs.
5.66±0.28, P<0.01 for cleaved caspase-3; and 0.52±0.08 vs.
0.21±0.04, P<0.001 for Bcl-2).
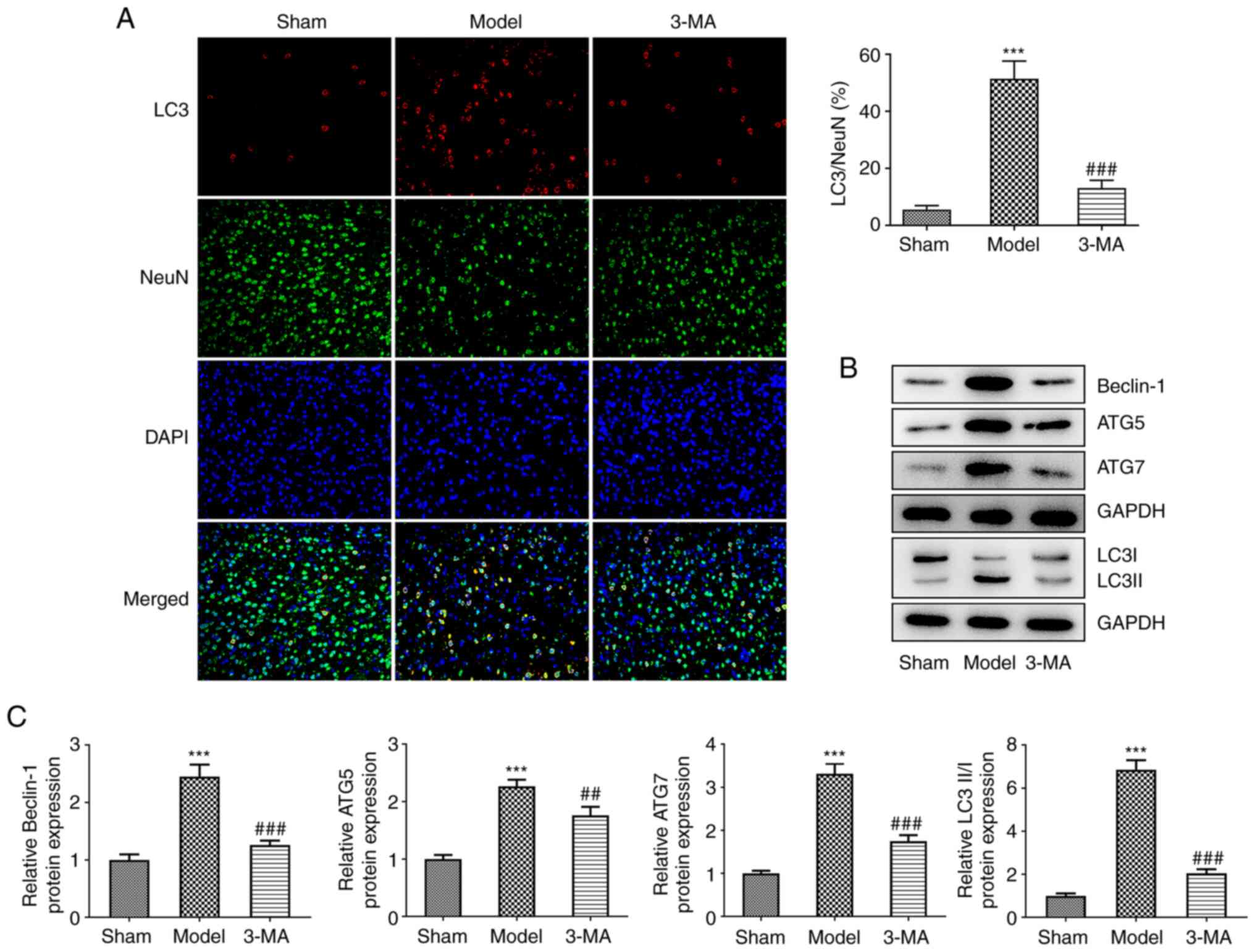
The brain sections following I/R injury were
specifically stained for the key autophagy protein, LC3, as well as
the neuronal marker, NeuN, which revealed that I/R injury resulted
in an increase in LC3 and NeuN fluorescent double-positive cells,
and the ratio of LC3 to NeuN-positive cells was markedly
upregulated compared to the sham group, indicating that LC3 was
primarily expressed in the cytoplasm of neurons, and autophagy
induced by I/R occurred primarily in neurons (Fig. 2A). Following pre-treatment with
3-MA, a decrease in neuronal autophagy levels was observed,
characterized by a significant decrease in LC3 and NeuN
fluorescence double-positive staining. In addition, the results of
IF assay demonstrated that 3-MA upregulated the number of
NeuN-positive cells, suggesting that the inhibition of autophagy
reduced neuronal death induced by I/R. Additionally, using western
blot analysis of autophagy-related proteins (Fig. 2B and C), it was found that the
levels of Beclin-1 (2.45±0.21 vs. 1.00±0.10, P<0.001), ATG5
(2.27±0.11 vs. 1.00±0.07, P<0.001), ATG7 (3.31±0.24 vs.
1.00±0.06, P<0.001) and LC3II/LC3I (6.86±0.44 vs. 1.00±0.11,
P<0.001) were increased in the brain tissue of rats following
I/R injury compared with the sham group; I/R injury also
significantly promoted the development of autophagy in the brain.
This process was greatly antagonized by 3-MA. Taken together, these
results suggested that I/R significantly enhanced autophagy in the
brain and contributed to neuronal cell death, whereas 3-MA
administration prior to modeling inhibited autophagy and thus
attenuated the damage induced by cerebral ischemia-reperfusion.
Such protective effects were however, limited and the damage
observed was still greater than that in the control group.
Inhibition of autophagy may exacerbate
DNA damage induced by cerebral I/R
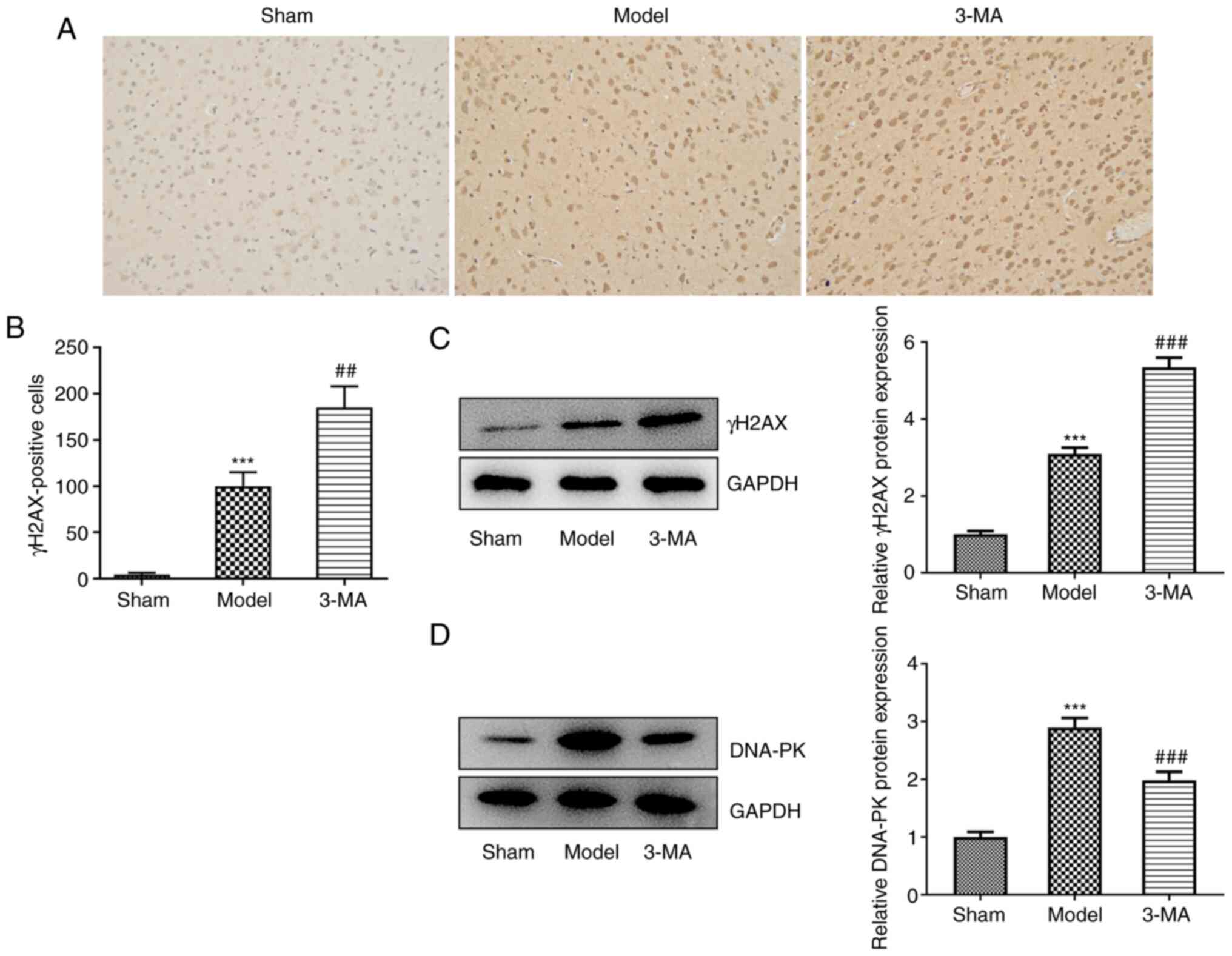
γH2AX is considered a biological marker for the
detection of DSBs (20). In the
present study. IHC staining of the rat brain sections was performed
to assess the protein expression of γH2AX in each group as a
reflection of cellular DSBs (Fig. 3A
and B). It was found that the number of cells specifically
immunostained for γH2AX was significantly increased compared with
the sham group, suggesting that I/R induced a substantial increase
in the degree of cellular DSBs, which may have been responsible for
neuronal cell death. However, the addition of 3-MA, instead of
ameliorating the severe cellular DNA damage observed, further
increased the degree of DSBs. Furthermore, western blot analysis
clearly demonstrated an increase in the protein expression levels
of γH2AX in the 3-MA group compared with the model group (5.35±0.25
vs. 3.10±0.16, P<0.001) (Fig.
3C). This may suggest that the inhibition of autophagy, whilst
possibly reducing brain tissue damage to a certain extent, also
impeded DNA damage repair.
The results of the analysis of DNA-PK expression
also supported this view. DNA-PK belongs to the
phosphatidylinositol 3-kinase-related kinase family, and is most
well-known for its role in the repair of DSBs (20). The quantitative protein analysis
of DNA-PK in each group revealed that brain injury induced an
endogenous repair mechanism, as indicated by a significant increase
in DNA-PK protein expression in the model group compared with the
sham group (2.90±0.17 vs. 1.00±0.09, P<0.001); however, this
endogenous repair mechanism was diminished by pre-treatment with
3-MA (1.98±0.14 vs. 2.90±0.17, P<0.001) (Fig. 3D).
Combining brain tissue damage and DNA damage
studies, it was found that while the inhibition of autophagy could
lead to a certain degree of reduction in tissue damage, DNA damage
was not reduced, but instead increased significantly. Thus,
although Beclin-1 plays a central role in autophagy, it may also
affect the repair of DSBs by influencing the formation of DNA-PK.
Combined with this background, it was hypothesized that Beclin-1
itself had mechanisms related to the induction of repair of DSBs
resulting in the aforementioned experimental results, and that
although 3-MA inhibited autophagy, it also inhibited the expression
of Beclin-1, and thus its induction of DSB repair; thus, the tissue
damage remained at a higher level, not decreasing as markedly as
the autophagy level itself.
Overexpression of Beclin-1 attenuates
OGD/R-induced cell damage independently of autophagy, and this is
further augmented by 3-MA treatment
In vitro cell experiments were performed to
determine the effects of Beclin-1 overexpression on OGD/R-induced
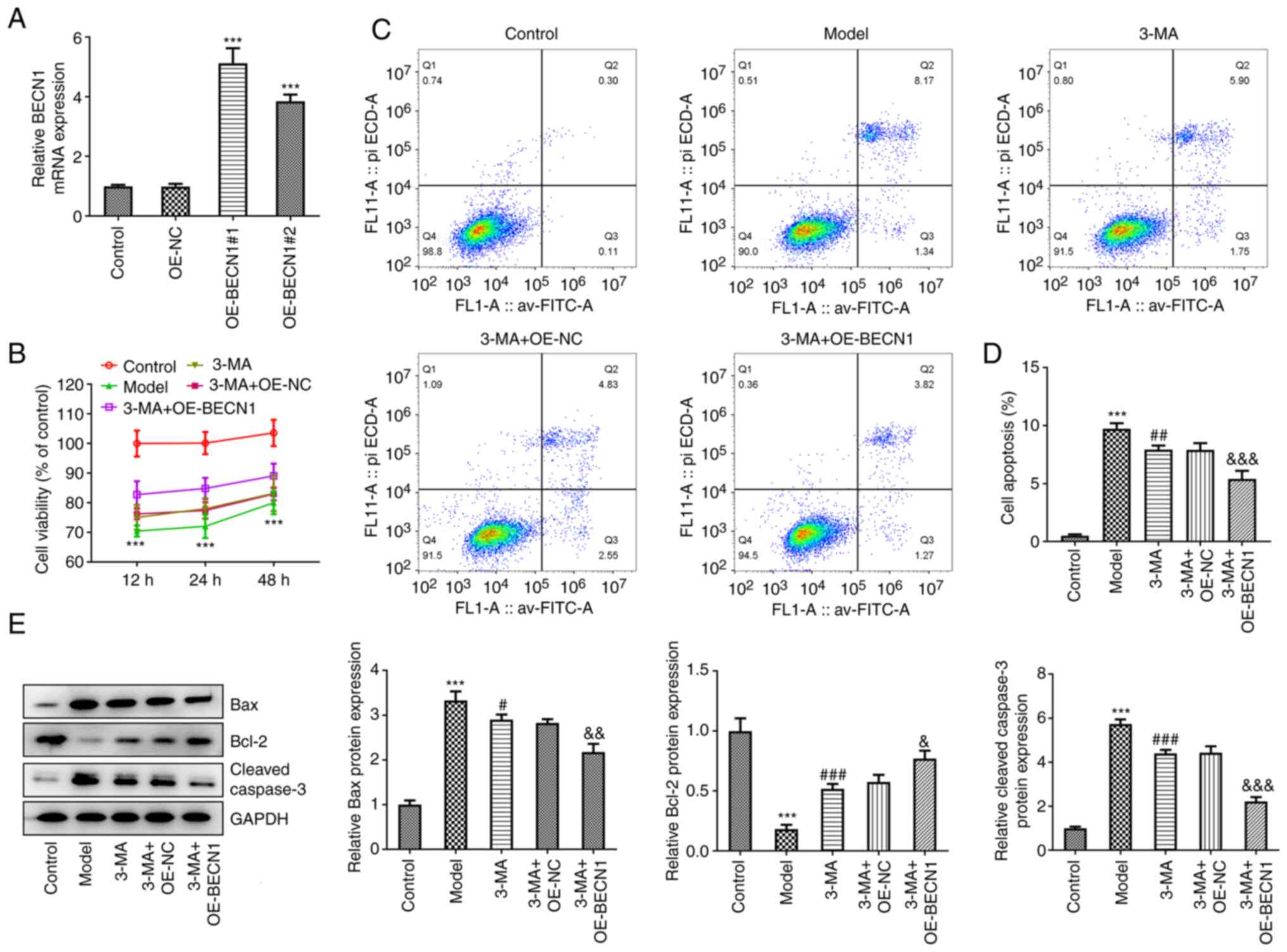
cell damage. Initially, a Beclin-1 overexpression vector was
constructed and transfected into HT22 cells. RT-qPCR revealed that
the overexpression was successful (5.12±0.51 and 3.84±0.23 vs.
0.99±0.09, P<0.001) (Fig.
4A). The more efficient OE-BECN1#1 was selected for use in
subsequent experiments. An OGD/R cell model was prepared for
subsequent experiments. The results of CCK-8 assay revealed that,
consistent with the results of the in vivo experiments,
pre-treatment with 3-MA enhanced the activity of cells exposed to
OGD/R injury to a modest level; transfection with the blank plasmid
did not alter the effects of 3-MA (Fig. 4B). The 3-MA + OE-BECN1 group
exhibited a pronounced elevation in cellular activity compared with
the 3-MA + OE-NC group. Moreover, the cells in the model group were
in the lowest activity state following 12 h of reoxygenation; thus
the time duration of 12 h of reoxygenation was selected for use in
subsequent experiments.
The degree of apoptosis in each group was
subsequently analyzed. As demonstrated by the results of flow
cytometric analysis, 3-MA inhibited autophagy and was able to
reduce apoptosis following injury (7.93±0.36 vs. 9.74±0.47,
P<0.01). In the cells overexpressing Beclin-1 and treated with
3-MA, the degree of inhibition of apoptosis was more potent than
that observed in the cells treated with 3-MA and transfected with
the blank vector (5.40±0.70 vs. 7.90±0.58, P<0.001) (Fig. 4C and D). In the quantitative
analysis of proteins in each group, the 3-MA + OE-BECN1 group
exhibited a significant decrease in the expression levels of Bax
(2.17±0.19 vs. 2.83±0.08, P<0.01) and cleaved caspase-3
(2.23±0.19 vs. 4.44±0.29, P<0.001), and a significant increase
in the levels of Bcl-2 compared with the 3-MA + OE-NC group
(0.77±0.07±0.58±0.06, P<0.05) (Fig. 4E).
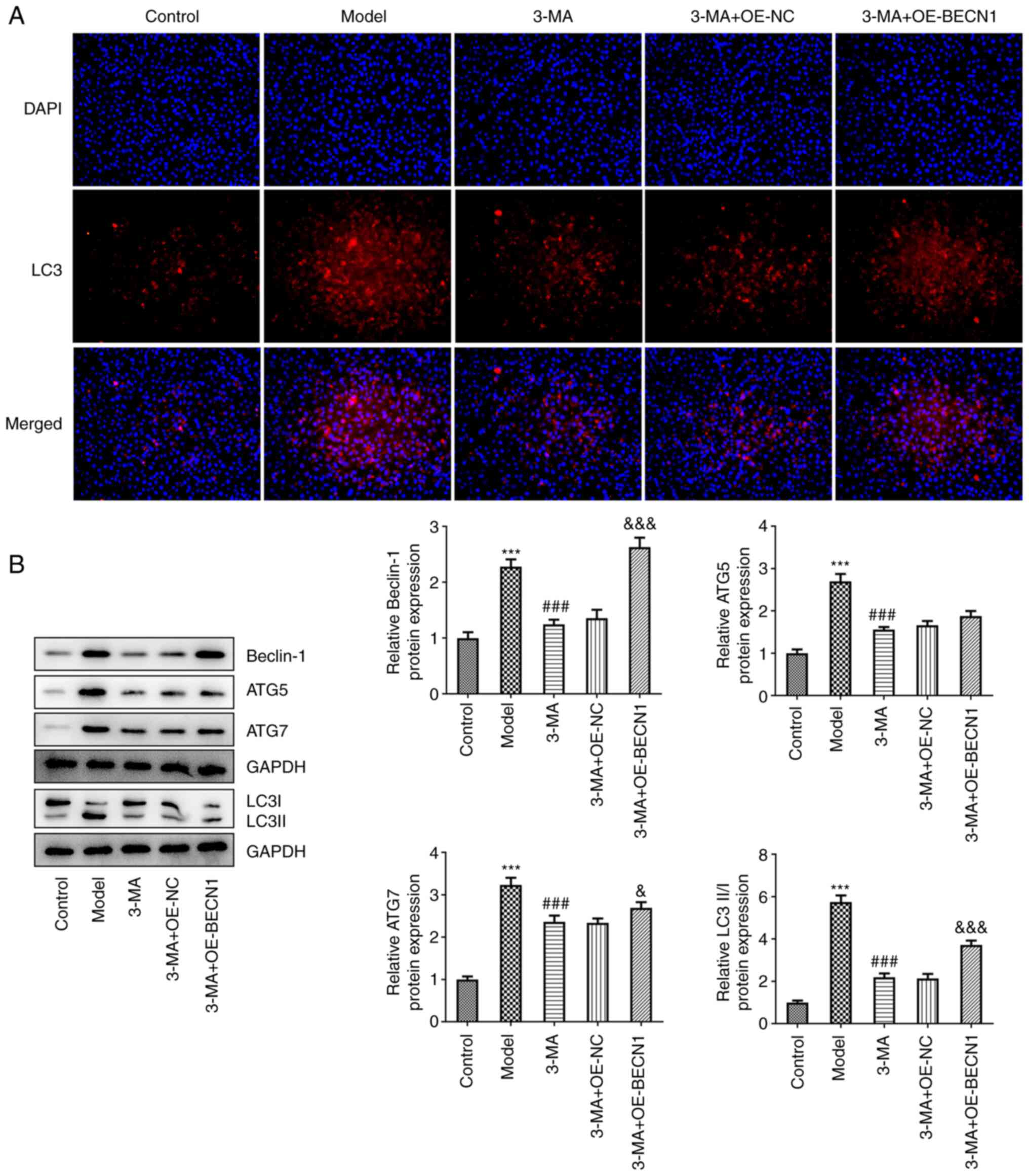
The levels of autophagy in each group were also
measured. The results of IF assay revealed that there was a notable
enhancement in the fluorescence intensity of LC3 in the 3-MA +
OE-BECN1 group (Fig. 5A). The
results of western blot analysis also revealed that Beclin-1
expression in the 3-MA + OE-BECN1 group was significantly increased
compared with the group transfected with the blank vector
(2.63±0.17 vs. 1.36±0.15, P<0.001); in addition, the expression
of ATG7 (2.69±0.13 vs. 2.34±0.11, P<0.05) and the LC3II/LC3I
ratio (3.72±0.20 vs. 2.14±0.21, P<0.001) in the 3-MA + OE-BECN1
group also moderately increased; the levels of ATG5 were also
increased, alhtough this increase was not significant (1.88±0.12
vs. 1.66±0.10, P=0.225) (Fig.
5B). In the 3-MA group, the overexpression of BECN1 limited the
expression of autophagy-related proteins.
Thus, it could be concluded that despite the modest
increase in autophagy, the overexpression of Beclin-1 exerted a
pronounced cytoprotective effect, suggesting that this effect may
not be dependent on the cellular autophagic pathway.
Beclin-1 augments DNA repair
independently of autophagy
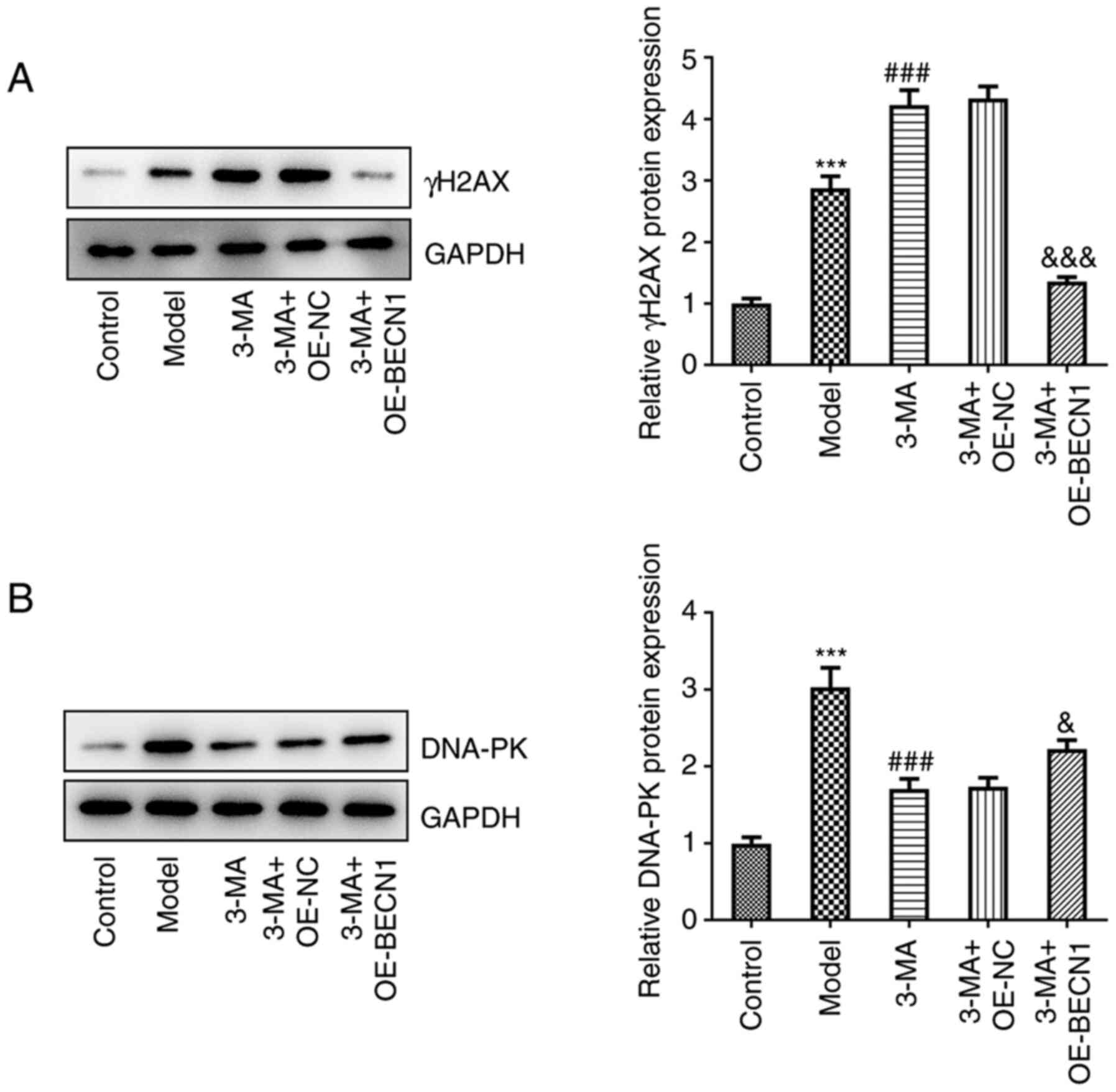
To investigate whether Beclin-1 itself can affect
DNA DSB repair, and whether this effect is dependent on the
regulation of cellular autophagy, the γH2AX and DNA-PK levels were
quantified in each group of cells (Fig. 6A and B). In the cells treated
with 3-MA, the high expression levels of Beclin-1 potently
suppressed the levels of γH2AX compared with the group transfected
with the blank vector (1.35±0.08 vs. 4.33±0.20, P<0.001), and
there was a rebound in the weakened DNA-PK formation (2.22±0.12 vs.
1.74±0.12, P<0.05). This indicated that the high expression of
Beclin-1, although causing a minimal decrease in intracellular
autophagy, exerted a potent repairing effect on DSBs under 3-MA
treatment; this repairing effect which occurred asynchronously with
the autophagy levels may be attributed to the fact that Beclin-1
itself plays a role in repairing DSBs that may be independent of
the autophagic pathway.
Discussion
In the present study, a rat brain I/R model and
OGD/R neuronal model were established to simulate cerebral I/R
injury in vitro and in vivo. The findings revealed
that cerebral I/R significantly induced high levels of autophagy
and neuronal injury, whereas the autophagy inhibitor, 3-MA, was
able to partially prevent brain injury, but also aggravated DSBs.
The overexpression of Beclin-1 resulted in an increase in the
expression levels of anti-apoptotic proteins, whilst promoting the
synthesis of DNA repair proteins, and these roles were not
associated with its ability to regulate autophagy. It is clearly
evident from these results that Beclin-1 protects neurological
function in the brain following I/R injury by improving the DNA
damage repair capacity of cells, independent of the regulation of
autophagy.
The accumulation of sudden bursts of oxides in I/R
can cause DNA breaks and lead to the necrosis of ischemic neurons
(6,21,22). In fact, it has been found in
previous studies that the levels of DNA repair increase following
transient ischemia (6,9). In the present study, it was
likewise confirmed that the γH2AX levels, which are characteristic
of DSBs, were significantly elevated in response to stimuli; the
increased synthesis of DNA-PK also demonstrated that the organism
itself initiated a repair mechanism. In this view, the effective
improvement of the organism's DNA repair capacity enhanced
neuroprotection. It appears that the effective enhancement of
endogenous DNA repair enhances neuroprotection.
Autophagy is the process through which cells are
targeted by lysosomes to recycle functionally damaged organelles,
as well as misfolded proteins (23). Moderate autophagy facilitates
endogenous repair, and it has been shown that I/R can ameliorate
neuronal damage through mitochondrial clearance via the activation
of autophagy (24). However, the
excessive activation of autophagy may lead to cell death. For
example, it has been reported that the inhibition of autophagy can
shrink the infarct lesion volume and exerts a neuroprotective
effect in rats with cerebral I/R injury (25). Additionally, sevoflurane has been
reported to protect the brain against injury by inhibiting
autophagy in rats with cerebral I/R (26). On the contrary, eugenol has been
shown to attenuate cerebral I/R injury by enhancing autophagy
(27). Herein, it was found that
the lesioned areas exhibited a high level of autophagy in affected
neurons, accompanied by more severe tissue and cellular damages,
which may be responsible for neurodegeneration; 3-MA inhibited
autophagy, reduced the infarct volume and neuronal apoptosis, and
protected the nerve cells. However, although the addition of 3-MA
reduced brain tissue and neuronal damage, the amplitude of the
reduction did not correspond with the degree of the decrease in
autophagy, and the amount of damage remained relatively high. In
addition, it was also observed that 3-MA resulted in more severe
DSBs than the model group, as evidenced by an increase in γH2AX
levels and a reduction in DNA-PK levels, which may explain why the
inhibition of autophagy alone did not improve neurological function
following I/R in the brain.
Beclin-1 is an essential autophagy regulatory gene.
Although studies on the functional regulation of Beclin-1 through
autophagy have dominated prior efforts, increased attention is
currently being paid to its role in non-autophagy-dependent
pathways (15). It has been
shown that the non-autophagy-dependent effects of Beclin-1 manifest
in the antagonism of growth factors (28,29), the repair of cellular DNA
(15) and the aggregation of
cellular chromosomes (30). In
this regard, of great interest is the association between Beclin-1
and DNA damage repair. In the study by Xu et al (15), it was found that cells undergo
severe DNA damage during phase-responsive radiation, Beclin-1
nuclear localization is augmented and intra-nuclear Beclin-1 is
able to interact with DNA topoisomerase IIβ to recruit it toward
DNA break sites; the overexpression of Beclin-1 in wild-type cells
with physiologically normal autophagy enhances autophagy and
improves DNA repair, and in the absence of both, Beclin-1 will not
function. Of interest, in the present study, it was demonstrated
that 3-MA reduced the autophagy levels, and inhibited Beclin-1
expression in brain tissue, as well as in cells, affecting its
regulation of DNA repair, thus causing more severe DNA damage than
in the model group. The overexpression of Beclin-1 in cells with
3-MA-restricted autophagy and exposure to ODG/R resulted in a
decrease in Bax and in cleaved caspase-3 expression levels. Bax
enhanced the caspase-mediated cleavage of Beclin-1, thereby
inducing apoptosis. Additionally, the overexpression of Beclin-1
induces a significant increase in endogenous Bcl-2 content, as
demonstrated by several studies assessing the protective effects of
Bcl-2 on neurons from ischemic challenge and the enhanced
neuroprotection imbued (24,31,32). In the present study, the level of
autophagy was slightly recovered by the overexpression of Beclin-1,
and a significant decrease in γH2AX levels, as well as a notable
increase in the DNA-PK complex were observed during the same
period. This indicated that the overexpression of Beclin-1 could
correct the repair defects of DSBs under 3-MA pre-treatment, and
the degree of increase in repair was not associated with the degree
of recovery of autophagy. This highlights the potential involvement
of Beclin-1 in regulating DNA repair independently, rather than via
autophagy.
Overall, to the best of our knowledge, the present
study is the first to examine the non-autophagic role of Beciln-1
in cerebral I/R, and Beclin-1 was shown to enhance neuronal DNA
repair to resist I/R damage through a non-autophagy-dependent
regulatory mechanism. Additionally, the present study demonstrated
that the inhibition of the excessive activation of autophagy
combined with Beclin-1 upregulation was more effective in
mitigating I/R injury than autophagy inhibition alone. The findings
of the present study may contribute to the investigation of the
mechanisms of I/R injury, and may assist in improving the
therapeutic effects, providing a novel perspective for studying the
role of Beclin-1 and autophagy. Nevertheless, the present study was
preliminary in its nature, and did not validate the effects of the
overexpression of Beclin-1 on DNA repair under the complete
inhibition of autophagy, which will thus form the basis of future
studies. In addition, the use of primary neurons as experimental
subjects and in vivo verification can further validate the
results, which needs to be taken into consideration in future
studies.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HL, WC and ZQ designed the study. HL and DH analyzed
the experimental data and wrote the manuscript. XT and YL assisted
in the revision of the manuscript. HL, DH, XT, YL, QL, CL and HH
performed the experiments. WC and ZQ designed the study,
coordinated technical support and project funding. HL and DH
confirm the authenticity of all the raw data. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
The animal experiments and the protocols in the
present study were approved by the Ethics Committee of Affiliated
Hospital of Youjiang Medical University for Nationalities (Approval
no. YYFY-LL-2020-17).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Key R&D
Program of China (grant no. 2018YFA0108300) and the Natural Science
Foundation of Guangxi Zhuang Autonomous Region (grant nos.
2019GXNSFBA245015 and 2020GXNSFAA297108).
References
|
1
|
Fann DY, Lee SY, Manzanero S, Chunduri P,
Sobey CG and Arumugam TV: Pathogenesis of acute stroke and the role
of inflammasomes. Ageing Res Rev. 12:941–966. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bai J and Lyden PD: Revisiting cerebral
postischemic reperfusion injury: New insights in understanding
reperfusion failure, hemorrhage, and edema. Int J Stroke.
10:143–152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Graham SH and Chen J: Programmed cell
death in cerebral ischemia. J Cereb Blood Flow Metab. 21:99–109.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen J, Jin K, Chen M, Pei W, Kawaguchi K,
Greenberg DA and Simon RP: Early detection of DNA strand breaks in
the brain after transient focal ischemia: Implications for the role
of DNA damage in apoptosis and neuronal cell death. J Neurochem.
69:232–245. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barber PA, Demchuk AM, Hirt L and Buchan
AM: Biochemistry of ischemic stroke. Adv Neurol. 92:151–164.
2003.PubMed/NCBI
|
|
6
|
Sun FY, Lin X, Mao LZ, Ge WH, Zhang LM,
Huang YL and Gu J: Neuroprotection by melatonin against ischemic
neuronal injury associated with modulation of DNA damage and repair
in the rat following a transient cerebral ischemia. J Pineal Res.
33:48–56. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nagata S: Apoptotic DNA fragmentation. Exp
Cell Res. 256:12–18. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu J, Li J, Yang Y, Wang X, Zhang Z and
Zhang L: Neuronal apoptosis in cerebral ischemia/reperfusion area
following electrical stimulation of fastigial nucleus. Neural Regen
Res. 9:727–734. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yin KJ and Sun FY: Effect of
dextromethorphan, a NMDA antagonist, on DNA repair in rat
photochemical thrombotic cerebral ischemia. Brain Res. 815:29–35.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Y, Zhang Y, Jin XF, Zhou XH, Dong
XH, Yu WT and Gao WJ: The role of astragaloside IV against cerebral
ischemia/reperfusion injury: Suppression of apoptosis via promotion
of P62-LC3-Autophagy. Molecules. 24:18382019. View Article : Google Scholar :
|
|
11
|
Yue Z, Jin S, Yang C, Levine AJ and Heintz
N: Beclin 1, an autophagy gene essential for early embryonic
development, is a haploinsufficient tumor suppressor. Proc Natl
Acad Sci USA. 100:15077–15082. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao Y, Huang G, Chen S, Gou Y, Dong Z and
Zhang X: Homocysteine aggravates cortical neural cell injury
through neuronal autophagy overactivation following rat cerebral
ischemia-reperfusion. Int J Mol Sci. 17:11962016. View Article : Google Scholar :
|
|
14
|
Su J, Zhang T, Wang K, Zhu T and Li X:
Autophagy activation contributes to the neuroprotection of remote
ischemic perconditioning against focal cerebral ischemia in rats.
Neurochem Res. 39:2068–2077. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu F, Fang Y, Yan L, Xu L, Zhang S, Cao Y,
Xu L, Zhang X, Xie J, Jiang G, et al: Nuclear localization of
Beclin 1 promotes radiation-induced DNA damage repair independent
of autophagy. Sci Rep. 7:453852017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee JH and Paull TT: Direct activation of
the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science.
304:93–96. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guide for the Care and Use of Laboratory
Animals. 8th edition. Washington, DC: 2011
|
|
18
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
20
|
Damia G: Targeting DNA-PK in cancer. Mutat
Res. 821:1116922020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schmidley JW: Free radicals in central
nervous system ischemia. Stroke. 21:1086–1090. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thiyagarajan M and Sharma SS:
Neuroprotective effect of curcumin in middle cerebral artery
occlusion induced focal cerebral ischemia in rats. Life Sci.
74:969–985. 2004. View Article : Google Scholar
|
|
23
|
Zhao G, Zhang W, Li L, Wu S and Du G:
Pinocembrin protects the brain against ischemia-reperfusion injury
and reverses the autophagy dysfunction in the penumbra area.
Molecules. 19:15786–15798. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang X, Yan H, Yuan Y, Gao J, Shen Z,
Cheng Y, Shen Y, Wang RR, Wang X, Hu WW, et al: Cerebral
ischemia-reperfusion-induced autophagy protects against neuronal
injury by mitochondrial clearance. Autophagy. 9:1321–1333. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xing S, Zhang Y, Li J, Zhang J, Li Y, Dang
C, Li C, Fan Y, Yu J, Pei Z and Zeng J: Beclin 1 knockdown inhibits
autophagic activation and prevents the secondary neurodegenerative
damage in the ipsilateral thalamus following focal cerebral
infarction. Autophagy. 8:63–76. 2012. View Article : Google Scholar
|
|
26
|
Shi CX, Jin J, Wang XQ, Song T, Li GH, Li
KZ and Ma JH: Sevoflurane attenuates brain damage through
inhibiting autophagy and apoptosis in cerebral ischemia-reperfusion
rats. Mol Med Rep. 21:123–130. 2020.
|
|
27
|
Sun X, Wang D, Zhang T, Lu X, Duan F, Ju
L, Zhuang X and Jiang X: Eugenol attenuates cerebral
ischemia-reperfusion injury by enhancing autophagy via
AMPK-mTOR-P70S6K pathway. Front Pharmacol. 11:842020. View Article : Google Scholar :
|
|
28
|
Rohatgi RA, Janusis J, Leonard D, Bellvé
KD, Fogarty KE, Baehrecke EH, Corvera S and Shaw LM: Beclin 1
regulates growth factor receptor signaling in breast cancer.
Oncogene. 34:5352–5362. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tian M, Chen Y, Tian D, Qiao X, Ma Z and
Li J: Beclin1 antagonizes LAPTM4B-mediated EGFR overactivation in
gastric cancer cells. Gene. 626:48–53. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fremont S, Gerard A, Galloux M, Janvier K,
Karess RE and Berlioz-Torrent C: Beclin-1 is required for
chromosome congression and proper outer kinetochore assembly. EMBO
Rep. 14:364–372. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun K, Fan J and Han J: Ameliorating
effects of traditional Chinese medicine preparation, Chinese
materia medica and active compounds on ischemia/reperfusion-induced
cerebral microcirculatory disturbances and neuron damage. Acta
Pharm Sin B. 5:8–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ji B, Cheng B, Pan Y, Wang C, Chen J and
Bai B: Neuroprotection of bradykinin/bradykinin B2 receptor system
in cerebral ischemia. Biomed Pharmacother. 94:1057–1063. 2017.
View Article : Google Scholar : PubMed/NCBI
|