Introduction
Thrombotic diseases and events such as cerebral
infarction (CI) are among the most common causes of global
morbidity and mortality. Endothelial cell activation, particularly
in response to inflammation-associated damage, is thought to be a
key driver of these thrombotic processes. Antiphospholipid syndrome
(APS) is a rare form of autoimmunity that may increase the risk of
thrombotic events and eclampsia in affected individuals (1). Patients with APS exhibit persistent
upregulation of autoantibodies including anti-phospholipid
antibody, lupus anticoagulant, anti-cardiolipin and
anti-β2-glycoprotein I (anti-β2GPI).
Anti-β2GPI is thought to have a particularly critical
role in the context of thrombus formation (2,3),
yet its precise mechanistic pro-thrombotic function remains to be
fully elucidated.
β2GPI is a five-domain phospholipid-bound
protein and the primary target antigen for anti-β2GPI.
Circulating anti-β2GPI/β2GPI immune complexes
(ICs) in patients with APS are associated with thrombotic events
(4,5). A previous study by our group
indicated that these anti-β2GPI/β2GPI ICs may
activate platelets and thereby induce thrombosis (6), while also contributing to the
neutrophil-mediated release of pro-thrombotic neutrophil
extracellular traps (NETs) (7).
Neutrophils are the most common leukocytes in the circulation and
function as key mediators of innate immune and inflammatory
responses (8). In the present
study, it was hypothesized that neutrophil-related inflammation may
be involved in endothelial cell activation and thrombosis in
patients with APS.
Pyroptosis is a form of programmed cell death that
was first detected in Shigella flexneri-infected macrophages
in 1992 (9), before ultimately
being named by Cookson and Brennan (10) in 2001. Pyroptotic cell death is
characterized by nucleotide-binding oligomerization domain-like
receptor pyrin domain containing 3 (NLRP3) inflammasome activation,
cell membrane pore formation and the release of mature
interleukin-1β (IL-1β) through these pores. Caspase-1 is an
integral mediator of this process, functioning to directly cleave
the pro-form of IL-1β in order to facilitate cytokine maturation
while also promoting the activation of gasdermin D (GSDMD)
(11,12). GSDMD, in turn, forms pores in the
cell membrane that are 10-15 nm in diameter, thereby triggering
pyroptosis. The activation of the NLRP3 inflammasome is closely
associated with CI and other thrombotic diseases (13,14). Double-stranded RNA-dependent
protein kinase (PKR) is a serine/threonine protein kinase that
regulates inflammatory responses in mammalian cells. Upon
activation, PKR is able to bind the NLRP3 inflammasome, thus
triggering caspase-1 activation and IL-1β release (15). Yim et al (16) also reported that PKR-induced
eukaryotic initiation factor 2α (eIF2α) is able to suppress
inflammasome activation and associated inflammatory responses. PKR
may regulate inflammatory immune responses through the p38MAPK
pathway (17). In addition, PKR
may induce apoptotic cell death via p38MAPK (18).
Anti-β2GPI/β2GPI is able to activate p38MAPK
signaling via Toll-like receptor 4 (TLR4), thus inducing
neutrophil-derived NET release (7). The present study thus posits that
anti-β2GPI/β2GPI ICs may promote neutrophil
pyroptosis and associated IL-1β release in a TLR4-dependent
manner.
High mobility group box 1 protein (HMGB1) is a
eukaryotic non-histone chromosome-binding protein that is primarily
present within the nucleus. However, in response to certain stimuli
or stressors, HMGB1 may be released into the extracellular matrix,
wherein it may promote inflammatory immune responses (19). The hepatitis B virus X protein
promotes hepatocyte NLRP3 inflammasome activation, resulting in
IL-1β, IL-18 and HMGB1 release from these cells (20). Of note, both IL-1β and HMGB1 may
alter inflammation and cell growth, and stimulate leukocyte
activation by binding to specific cell surface receptors, thus
shaping immune responses (21,22).
In the present study, it was determined that serum
anti-β2GPI levels and neutrophil NLRP3 expression were
significantly increased in patients with CI compared to healthy
controls. Through a series of in vitro analyses, it was
further determined that
anti-β2GPI/β2GPI-induced neutrophil
pyroptosis may activate the NLRP3 inflammasome via the
TLR4/PKR/p38MAPK signaling axis, leading to the release of HMGB1
and IL-1β from these neutrophils. These inflammatory factors, in
turn, enhance intercellular cell adhesion molecule-1 (ICAM-1) and
IL-8 expression in endothelial cells.
Materials and methods
Patients
A total of 52 patients with acute CI (ACI; 33 males,
19 females) treated at The Second Affiliated Hospital of Harbin
Medical University (Harbin, China) between August 2017 and December
2020 were selected for the present study. Patients were excluded if
they had a history of autoimmunity, diabetes, malignant tumors,
prior CI, coronary heart disease, acute infections, severe renal
insufficiency (serum creatinine >3 mg/dl), nervous system
diseases and/or were undergoing immunosuppressive therapy. In
addition, 29 healthy individuals (18 males, 11 females) undergoing
physical examinations at The Second Affiliated Hospital of Harbin
Medical University (Harbin, China) during this same time period
were recruited as healthy controls. Control patients were free of
CI, acute or chronic infections and major comorbidities other than
autoimmune diseases. The patients' characteristics are detailed in
Table I. Samples were collected
from patients with the approval of the Institutional Ethics
Committee of Harbin Medical University (Harbin, China) and informed
consent was obtained from the patients in accordance with the
Declaration of Helsinki.
 | Table IPatient characteristics. |
Table I
Patient characteristics.
| Variable | Acute cerebral
infarction group (n=52) | Control group
(n=29) | Normal ranges | P-value |
|---|
| Sex | | | | 0.901 |
| Male | 33 (63.5) | 18 (62.1) | | |
| Female | 19 (36.5) | 11 (37.9) | | |
| Mean age,
years | 52.69±8.78 | 51.44±6.93 | | 0.513 |
| Glu, mmol/l | 5.15±0.61 | 5.29±0.62 | 3.90-6.10 | 0.348 |
| UA, mmol/l | 311.68±83.30 | 324.76±54.84 | 150.0-440.0 | 0.453 |
| TC, mmol/l | 4.85±1.00 | 4.67±0.89 | 1.8-5.17 | 0.424 |
| TG, mmol/l | 1.59
(1.17-2.47) | 1.41
(1.14-1.71) | 0.56-1.70 | 0.319 |
| HDL, mmol/l | 1.30
(1.14-1.57) | 1.42
(1.17-1.60) | 1.04-1.70 | 0.267 |
| LDL, mmol/l | 2.77
(2.31-3.36) | 2.43
(2.01-3.02) | 0.45-3.15 | 0.084 |
Cell isolation and culture
Neutrophils were isolated from healthy donor
peripheral blood at The Second Affiliated Hospital of Harbin
Medical University (Harbin, China) from individuals who had
provided informed consent. Blood was collected using EDTA as an
anticoagulant and was treated for 45 min at 4°C via Dextran T-500
(Beijing Solarbio Science & Technology Co., Ltd.) sedimentation
to remove erythrocytes. Neutrophil-containing supernatants were
then transferred to a lymphocyte separation solution (Tianjin Hao
Yang Biological Manufacture Co., Ltd.) and centrifuged for 15 min
at 500 × g at room temperature. Neutrophils were isolated from the
precipitate and treated with Red Blood Cell Lysis buffer (Beijing
Solarbio Science & Technology Co., Ltd.). After isolation,
these neutrophils were rinsed with PBS and resuspended in RPMI-1640
medium (Cellgro; Corning, Inc.) with or without 10% fetal bovine
serum (FBS; Biological Industries) (7).
Human umbilical vein endothelial cells (HUVECs) were
obtained from the Shanghai Institute of Biochemistry and Cell
Biology and cultured in RPMI-1640 containing 10% FBS in an
incubator containing a humidified atmosphere with 5% CO2
at 37°C. These cells were passaged using trypsin and replated at a
dilution of 1:2, with culture media being replaced every 1-2
days.
Reverse transcription-quantitative
(RT-q)PCR
Neutrophils or HUVECs were plated in 12-well plates
(1×106 cells/ml). Neutrophils were treated with PBS,
M-IgG (cat. no. sc-8432; 10 mg/ml; Santa Cruz Biotechnology,
Inc.)/BSA (100 mg/ml; MilliporeSigma), anti-β2GPI (cat.
no. 11221-MM06; 10 μg/ml; SinoBiological)/β2GPI
(100 μg/ml; MilliporeSigma) or lipopolysaccharide (LPS; 100
ng/ml; MilliporeSigma) as experimentally appropriate, while HUVECs
were treated for 24 h with recombinant HMGB1 (4 ng/ml; Prospec-Tany
TechnoGene, Ltd.) or recombinant IL-1β (2 ng/ml; SinoBiological).
In certain experiments, neutrophils were pretreated for 30 min with
1 μM of the TLR4 inhibitor TAK-242 [MedChemExpress (MCE)].
TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) was used to
extract RNA from these cells after treatment and cDNA was prepared
with a Transcriptor First Strand cDNA Synthesis Kit (Roche
Diagnostics). RT of RNA to cDNA required a 60-min incubation at
50°C, followed by 5 min of incubation at 85°C. Subsequently qPCR
analyses for NLRP3, caspase-1, pro-IL-1β, TLR4, HMGB1, IL-8 and
ICAM-1 mRNA levels were performed with SYBR Green I dye (Roche
Diagnostics) with double distilled water, cDNA template and
upstream/downstream primer using the following primers: NLRP3
forward, 5′-CTACACACGACTGCGTCTCATCAA-3′; and reverse,
5′-CGGGGTCAAACAGCAACTCCAT-3′; caspase-1 forward,
5′-TGGAAGAGCAGAAAGCGATAA-3′ and reverse,
5′-TTTGAAGGACAAACCGAAGGT-3′; pro-IL-1β forward,
5′-TCCAGGGACAGGATATGGAG-3′ and reverse, 5′-TCTTTCAACACGCAGGACAG-3′;
TLR4 forward, 5′-GCCCCTACTCAATCTCTCTT-3′ and reverse,
5′-GGACTTCTAAACCAGCCA-3′; HMGB1 forward, 5′-GATCCCAATGCACCCAAGAG-3′
and reverse, 5′-TCGCAACATCACCAATGGAC-3′; IL-8 forward,
5′-CAGCCTTCCTGATTTCTGC-3′ and reverse, 5′-GGGTGGAAAGGTTTGGAGTA-3′;
ICAM-1 forward, 5′-AGCTTCGTGTCCTGTATGGC-3′ and reverse,
5′-TTTTCTGGCCACGTCCAGTT-3′; β-actin forward,
5′-CTACCTCATGAAGATCCTCACCGA-3′ and reverse,
5′-TTCTCCTTAATGTCACGCACGATT-3′. Shanghai Generay Biotech
Corporation synthesized all primers for the present study.
Amplifications were performed on a CFX96 real-time PCR detection
system (Bio-Rad Laboratories, Inc.) for 40 cycles with a melting
temperature of 30 sec, an annealing temperature of 58°C for 30 sec
and an extension temperature of 72°C for 30 sec. Quantification
cycle (Cq) counts were determined for each sample and relative mRNA
expression was calculated using the 2−ΔΔCq method
(23).
Western blot analysis
Neutrophils (5×106 cells/ml) were treated
for 1 h with PBS, M-IgG (10 mg/ml)/BSA (100 mg/ml),
anti-β2GPI (10 μg/ml)/β2GPI (100
μg/ml) or LPS (100 ng/ml), while HUVECs were treated for 24
h with recombinant HMGB1 (4 ng/ml; Prospec-Tany TechnoGene, Ltd.)
or recombinant IL-1β (2 ng/ml; SinoBiological). In certain
experiments, neutrophils were pretreated for 30 min with 1
μM of the TLR4 inhibitor TAK-242, 10 μM of the PKR
inhibitor GC17925 (GLPBIO), 10 μM of the p38MAPK inhibitor
SB203580 (MCE) or 1 μM of the NLRP3 inhibitor MCC950 (MCE).
Cells were then lysed on ice for 30 min with RIPA buffer (Beyotime
Institute of Biotechnology) containing PMSF (1 mM) and a
phosphatase inhibitor cocktail (1 mM; Roche Diagnostics). Protein
levels in these lysates were measured with a BCA Protein Assay kit
(Beyotime Institute of Biotechnology), after which equal amounts
(30 μg) of protein per sample were separated via 10%
SDS-PAGE and transferred onto PVDF membranes (Cytiva). Blots were
then blocked [5% w/v nonfat dry milk in Tris-buffered saline
containing Tween-20) for 30 min at room temperature and stained at
4°C overnight with primary antibodies specific for NLRP3 (cat. no.
WL02635; 1:1,000 dilution), caspase-1 (cat. no. WL02996a; 1:500
dilution), pro-IL-1β (cat. no. WL02257; 1:1,000 dilution), IL-8
(cat. no. WL03074; 1:500 dilution), ICAM-1 (cat. no. WL02268; 1:500
dilution; all from Wanleibio Co., Ltd.), phosphorylated (p)-p38MAPK
(cat. no. 4511), p38MAPK (cat. no. 8690; both 1:1,000 dilution;
both from Cell Signaling Technology, Inc.), p-PKR (cat. no.
bs-3335R; 1:500 dilution), PKR (cat. no. bs-1493R; 1:1,000
dilution), HMGB1 (cat. no. bs-55098R; 1:1,000 dilution; all from
Bioss) or β-actin (cat. no. TA-09; 1:1,000 dilution; OriGene
Technologies, Inc.). After probing for 1 h at room temperature with
an HRP-conjugated secondary antibody (cat. no. ZB-2301/ZB-2305;
1:5,000 dilution; OriGene Technologies, Inc.), protein bands were
detected using a Fluorescence/Chemiluminescence Imaging System
(CLINX Science Instruments).
Immunofluorescence staining
GSDMD and HMGB1 expression in neutrophils and ICAM-1
and IL-8 expression in HUVECs was assessed via immunofluorescent
staining. In brief, cells were fixed for 20 min with 4%
paraformaldehyde and permeabilized for 30 min with 0.2% Triton
X-100 at room temperature, then blocked with 50% goat serum
(Beyotime Institute of Biotechnology) in PBS for 30 min at 37°C and
incubated overnight with anti-GSDMD (cat. no. 20770-1-AP; 1:100
dilution; Proteintech Group, Inc.), anti-HMGB1 (cat. no. bs-55098R;
1:50 dilution; Bioss) or anti-ICAM-1 (cat. no. WL02268) or
anti-IL-8 (cat. no. WL03074; both 1:50 dilution; both from
Wanleibio Co., Ltd.) at 4°C. Cells were then stained with an
AF488-conjugated secondary antibody (cat. no. R37118; 1:1,000
dilution; Invitrogen; Thermo Fisher Scientific, Inc.) for 2 h and
DAPI (1 μg/ml; Invitrogen; Thermo Fisher Scientific, Inc.)
was applied for nuclear staining for 10 min at room temperature.
Cells were rinsed with PBS and imaged via a fluorescence microscope
(ECLIPSE Ti; Nikon Corporation).
ELISA
Serum anti-β2GPI was detected using an
Anti-β2GPI antibody ELISA kit (cat. no. EA1632-9601P;
K-BIOanalytica Rayan Company). Neutrophils (5×106
cells/ml) were treated with PBS, anti-β2GPI (10
μg/ml)/β2GPI (100 μg/ml) or LPS (100
ng/ml) for 3 h following pretreatment for 30 min with different
inhibitors (1 μM TAK-242, 10 μM GC1725 or 10
μM SB203580). Supernatants were then collected and IL-1β
concentrations therein were assessed via a Human IL-1β ELISA Kit
(cat. no. JL13662; J&L Biological) according to the
manufacturer's instructions.
Statistical analysis
Count data (sex distribution) were assessed via
χ2 test and expressed as n (%). For continuous
variables, normally distributed data were tested via Student's
t-test and expressed as the mean ± standard error of the mean,
while non-normally distributed data were assessed via Mann-Whitney
U tests and represented as the median (interquartile range). Three
or more groups of data were tested via one-way ANOVA with Dunnet's
post-hoc test. These data were analyzed using GraphPad Prism 7
(GraphPad Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Patients with CI exhibit elevated serum
anti-β2GPI levels and increased neutrophil NLRP3
expression
The patients with ACI (age, 31-69 years) included in
the present study exhibited no significant differences in age or
sex compared with the healthy controls (age, 35-68 years)
(P>0.05; Table I), nor were
there any differences in serum glucose, uric acid, triglyceride,
total cholesterol or low/high-density lipoprotein levels between
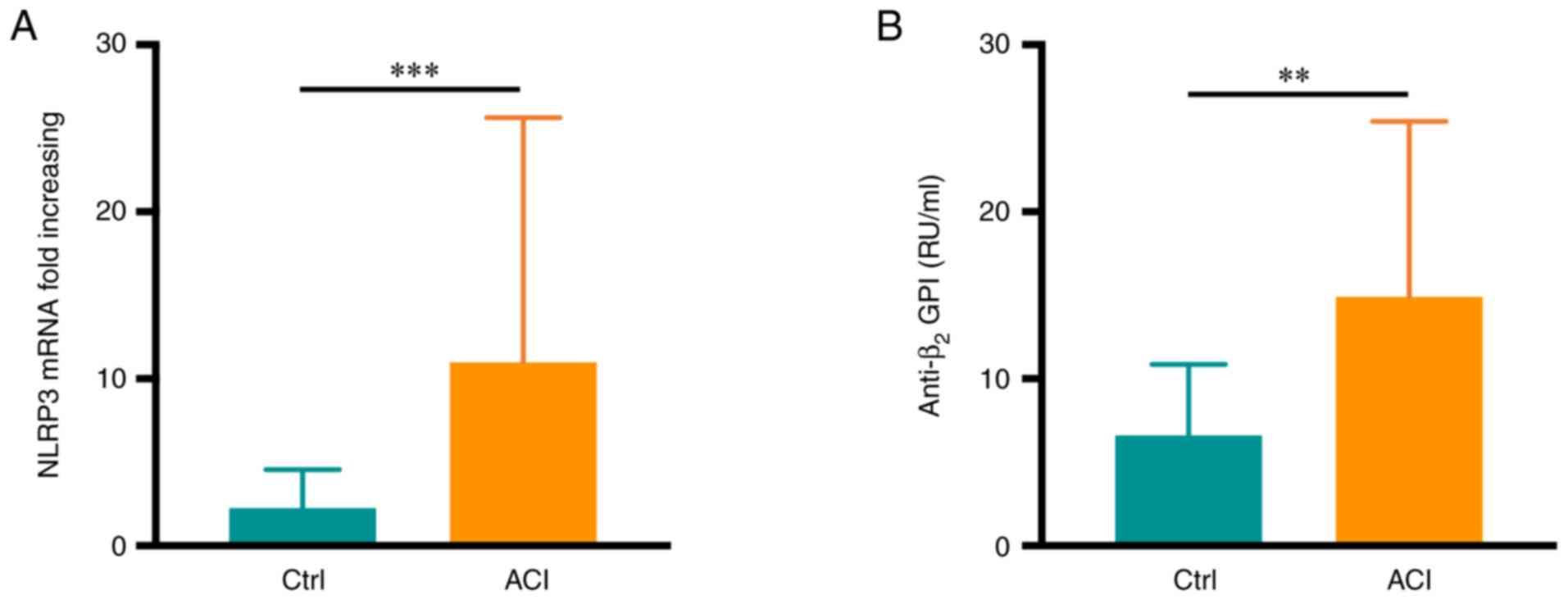
these groups (P>0.05). Of note, increased neutrophil NLRP3 mRNA
expression was observed in patients with ACI relative to healthy
controls (P<0.001; Fig. 1A).
Serum anti-β2GPI levels in these patients were detected
via ELISA, revealing significantly higher levels of this
autoantibody in samples from patients with ACI than in the healthy
controls (P<0.01; Fig. 1B).
Specifically, 9 patients in the ACI group (17.3%) were positive for
anti-β2GPI autoantibodies.
Anti-β2GPI/β2GPI
treatment triggers neutrophil pyroptosis
In previous studies, it was demonstrated that
β2GPI or anti-β2GPI alone cannot effectively
activate neutrophils (7). In
light of the above clinical findings, the potential mechanistic
role of β2GPI in the context of CI was then examined
in vitro. First, neutrophils were extracted from healthy
subjects and their purity and viability were confirmed (Fig. 2A). To evaluate neutrophil
pyroptosis, neutrophils were treated with PBS,
anti-β2GPI (10 μg/ml)/β2GPI (100
μg/ml) or LPS (100 ng/ml; positive control) for up to 3 h.
The NLRP3 inflammasome is an important mediator of pyroptosis
(24).
Anti-β2GPI/β2GPI IC but not IgG/BSA treatment
enhanced NLRP3 mRNA expression at 1 h of treatment relative to
control treatment or other incubation times (P<0.05; Fig. 2B-D). Caspase-1 and pro-IL-1β mRNA
expression was also enhanced by
anti-β2GPI/β2GPI treatment, with comparable
increases in NLRP3/caspase-1/pro-IL-1β protein levels (Fig. 2E and F). During pyroptosis, more
cell fragmentation was associated with the release of more
inflammatory factors into the cell supernatant. Expression of IL-1β
was present in the cell supernatant at 3 h. Supernatant IL-1β
levels also rose significantly following exposure to
anti-β2GPI/β2GPI (P<0.01; Fig. 2G). GSDMD mediates membrane pore
formation during pyroptosis (25). To more fully characterize
neutrophil pyroptosis, these cells were immunostained for GSDMD,
revealing significant increases in GSDMD-positive cell frequencies
among anti-β2GPI/β2GPI-treated neutrophils
(Fig. 2H). Together, these data
indicated that anti-β2GPI/β2GPI treatment was
sufficient to stimulate neutrophil pyroptosis and IL-1β
release.
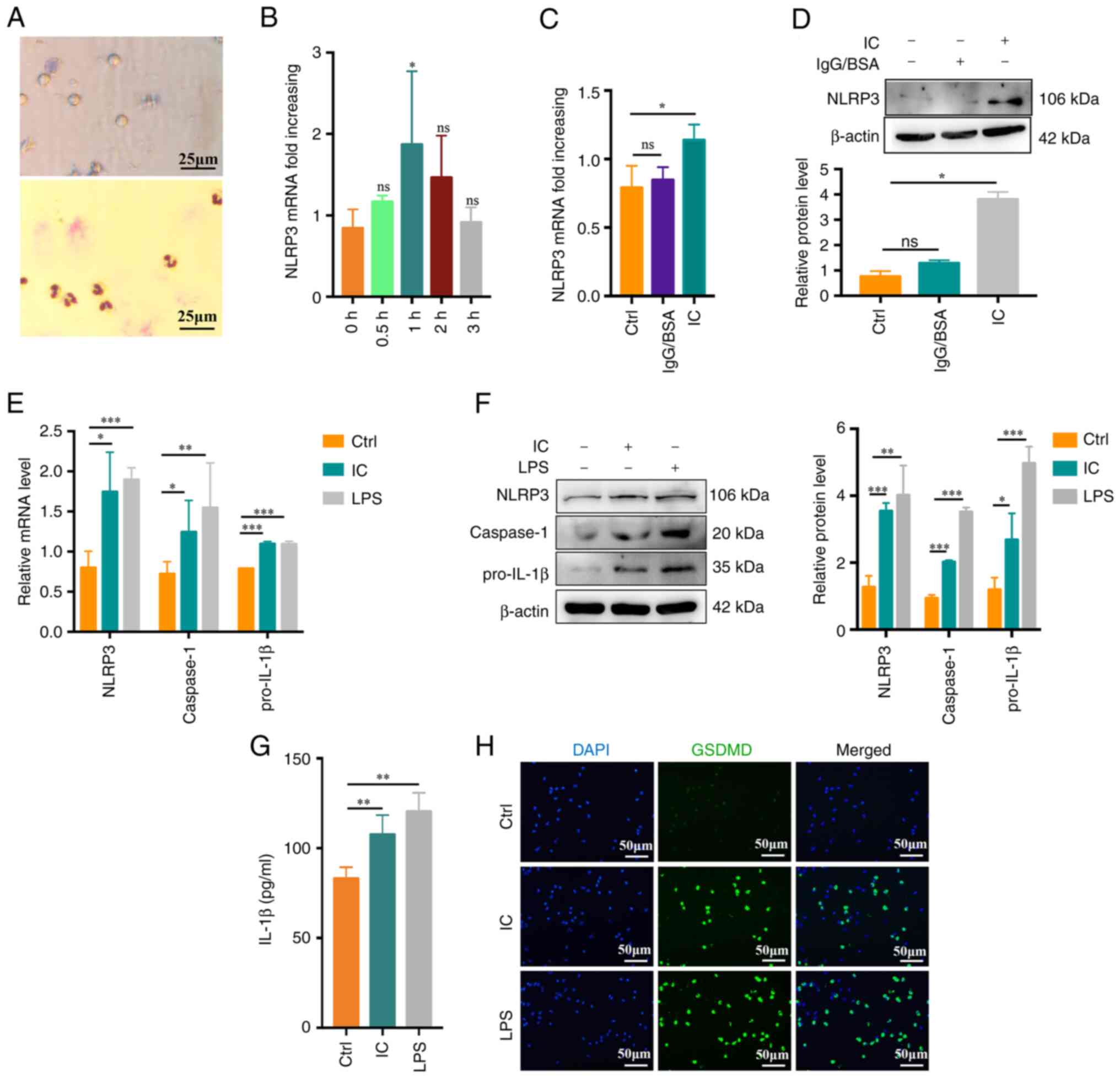 | Figure 2
Anti-β2GPI/β2GPI treatment triggers
neutrophil pyroptosis. (A) Representative images of neutrophil
viability (upper) and purity (lower) as assessed via trypan blue
dye exclusion assays and Giemsa staining, respectively (scale bar,
25 μm). (B) Following treatment with anti-β2GPI
(10 μg/ml)/β2GPI (100 μg/ml), neutrophils
exhibited significantly increased NLRP3 mRNA upregulation at 1 vs.
0 h. (C and D) Anti-β2GPI/β2GPI treatment
enhanced the expression of NLRP3 more efficiently than IgG/BSA
treatment as measured via (C) reverse transcription-quantitative
PCR and (D) western blot analysis. (E) Relative NLRP3, caspase-1
and pro-IL-1β mRNA levels rose in neutrophils following 1 h of
treatment with anti-β2GPI/β2GPI. (F) NLRP3,
caspase-1 and pro-IL-1β protein levels were increased in
neutrophils following 1 h of treatment with
anti-β2GPI/β2GPI, as measured via western
blot analysis. (G) IL-1β levels increased significantly in
neutrophils treated for 3 h with
anti-β2GPI/β2GPI as measured by ELISA. (H)
GSDMD-positive cells (green) following treatment with
anti-β2GPI/β2GPI (IC). DAPI (blue) was used
for nuclear staining (scale bar, 50 μm). Values are
expressed as the mean ± standard error of the mean (n=3 or n=4 in
G). *P<0.05, **P<0.01,
***P<0.001 as indicated or vs. 0 h. NLRP3, NLR family
pyrin domain containing 3; anti-β2GPI, anti-β2-glycoprotein I; LPS,
lipopolysaccharide; Ctrl, control; ns, no significance; IC,
anti-β2GPI/β2GPI immune complex; GSDMD,
gasdermin D. |
Anti-β2GPI/β2GPI IC
treatment induces neutrophil pyroptosis in a TLR4-dependent
manner
A previous study by our group suggested that TLR4
has a role in anti-β2GPI/β2GPI-induced NETs
formation (7). In the present
study, TLR4 activation was therefore assessed and its functional
importance in the context of anti-β2GPI/β2GPI
IC-induced neutrophil pyroptosis was explored.
Anti-β2GPI/β2GPI treatment was associated
with an increase in TLR4 mRNA expression in treated neutrophils
(Fig. 3A). When these cells were
pretreated with the TLR4 inhibitor TAK-242, significant reductions
in anti-β2GPI/β2GPI-induced
NLRP3/caspase-1/pro-IL-1β protein expression we observed (Fig. 3B). Similarly, TAK-242 treatment
markedly suppressed neutrophil IL-1β secretion (P<0.01; Fig. 3C) and decreased the frequency of
GSDMD-positive cells, as determined via immunofluorescent staining,
relative to the control treatment (Fig. 3D).
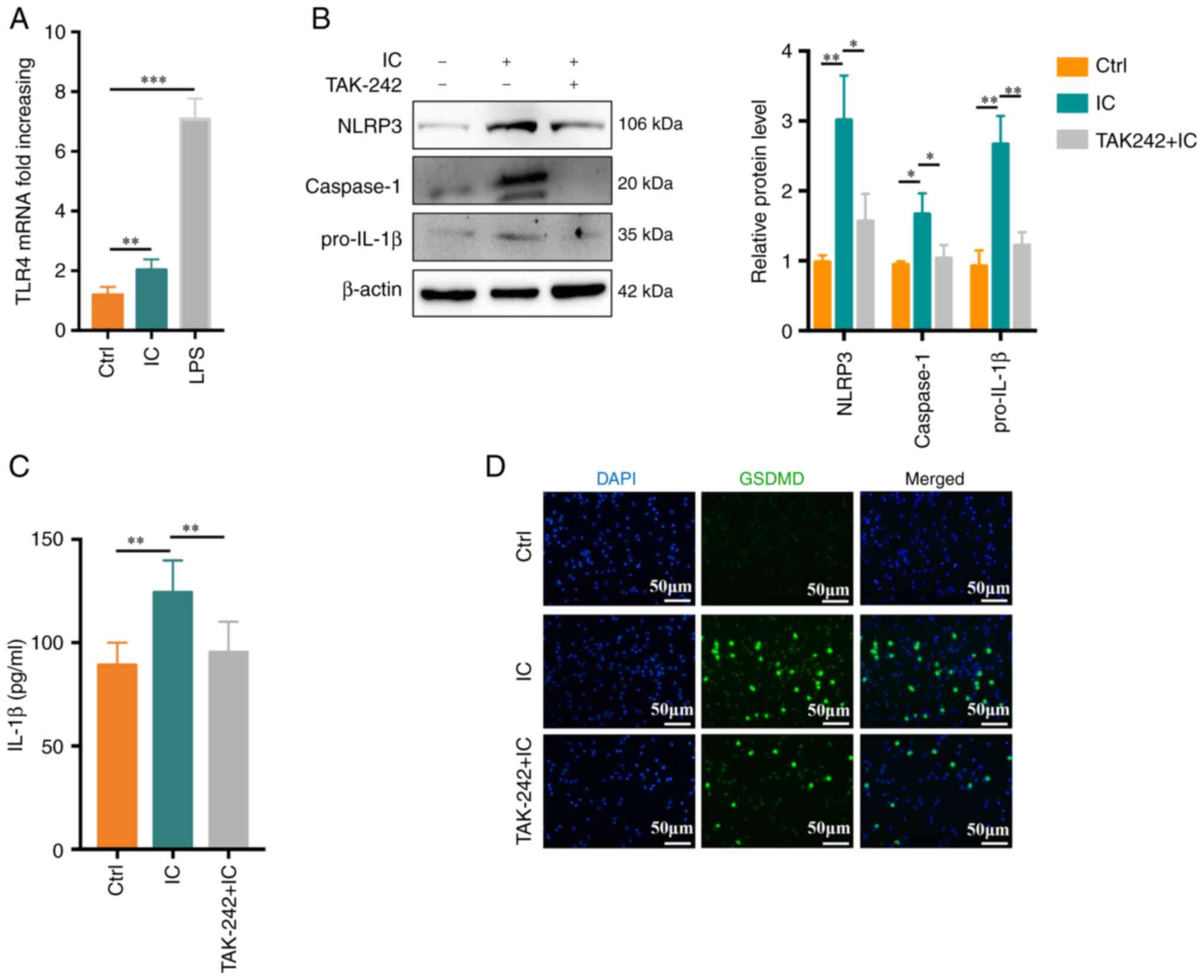 | Figure 3
Anti-β2GPI/β2GPI treatment induces neutrophil
pyroptosis in a TLR4-dependent manner. (A) Relative TLR4 mRNA
levels rose significantly in neutrophils following 1 h of treatment
with anti-β2GPI/β2GPI (n=5). (B) Following
pretreatment for 30 min with TAK-242 (1 μM), NLRP3,
caspase-1 and pro-IL-1β protein levels in neutrophils were reduced
(n=3). (C) IL-1β production was suppressed by TAK-242, as measured
via ELISA (n=6). (D) TAK-242 suppressed the number of
GSDMD-positive cells (scale bar, 50 μm). Values are
expressed as the mean ± standard error of the mean.
*P<0.05, **P<0.01,
***P<0.001. TLR4, Toll-like receptor 4; NLRP3, NLR
family pyrin domain containing 3; anti-β2GPI, anti-β2-glycoprotein
I; LPS, lipopolysaccharide; Ctrl, control; IC,
anti-β2GPI/β2GPI immune complex; GSDMD,
gasdermin D. |
Anti-β2GPI/β2GPI-induced neutrophil
pyroptosis is associated with the PKR/p38MAPK axis
PKR has previously been identified as a key
regulator of inflammasome activation, while PKR-induced eIF2α is
able to inhibit such inflammasome activity (16). A recent study suggested that
p38MAPK is involved in PKR activation and consequent human
chondrocyte apoptosis (18).
Thus, the role of PKR/p38MAPK in the context of
anti-β2GPI/β2GPI-induced neutrophil
pyroptosis was explored in the present study. After pretreatment
for 30 min with 1 μM TAK-242, 10 μM GC17925 (a PKR
inhibitor) or 10 μM SB203580 (a p38MAPK inhibitor),
neutrophils were stimulated for 1 h with anti-β2GPI (10
μg/ml)/β2GPI (100 μg/ml). It was indicated
that anti-β2GPI/β2GPI treatment was able to
stimulate PKR (Fig. 4A) and
p38MAPK (Fig. 4B)
phosphorylation, whereas TAK-242 and GC17925 suppressed the
activation of these proteins. GC17925 and SB203580 also suppressed
NLRP3/caspase-1/pro-IL-1β expression (Fig. 4C) and inhibited IL-1β release
(P<0.01; Fig. 4D) and GSDMD
activation (Fig. 4E) in
neutrophils. These results suggested that the PKR/p38MAPK signaling
pathway has an important role in
anti-β2GPI/β2GPI IC-induced neutrophil
pyroptosis.
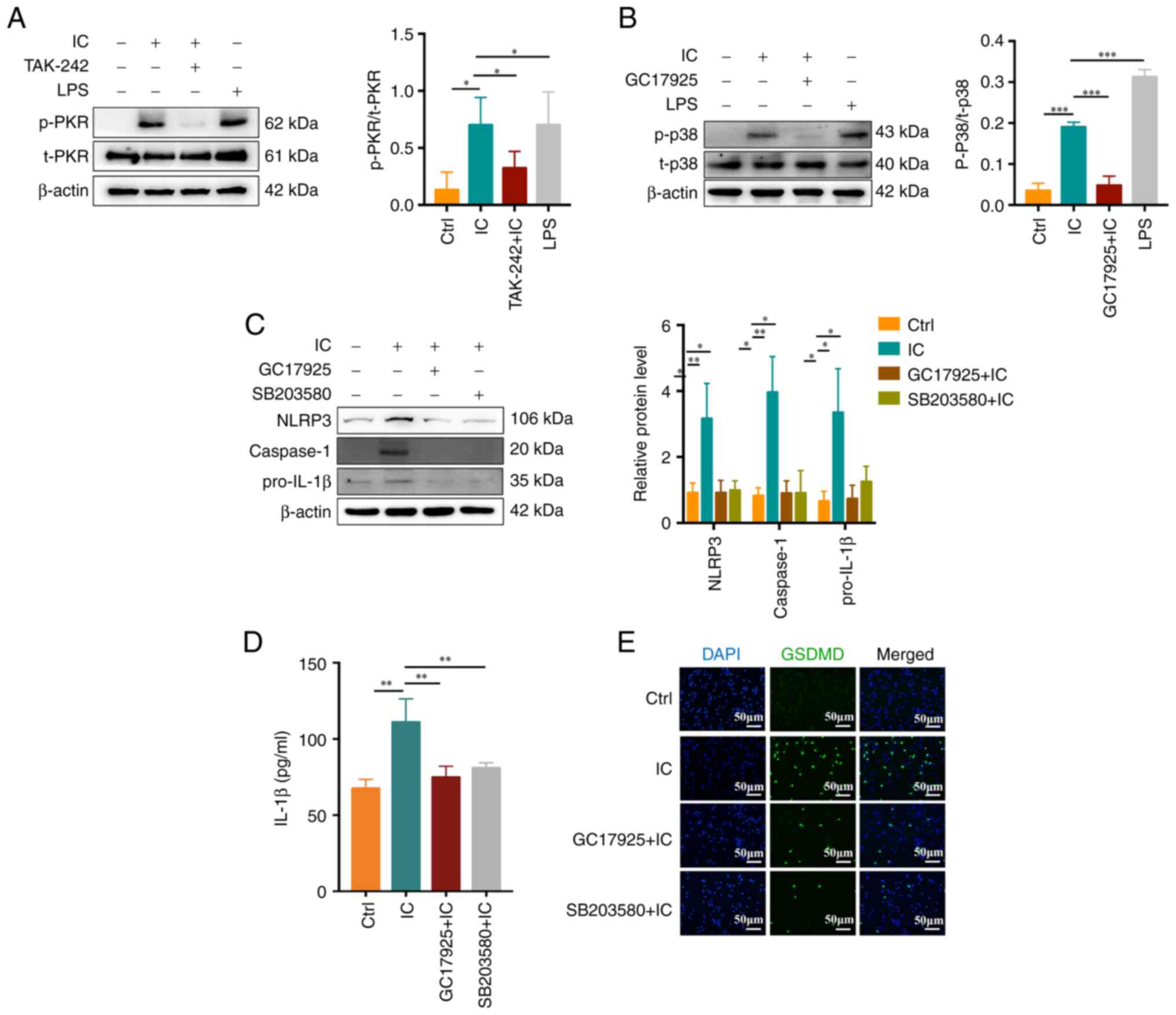 | Figure 4Association between the PKR/p38MAPK
signaling and anti-β2GPI/β2GPI
complex-induced neutrophil pyroptosis. (A and B)
Anti-β2GPI/β2GPI induced (A) PKR and (B)
p38MAPK phosphorylation, which was respectively inhibited by
TAK-242 and GC17925 treatment. (C) NLRP3, caspase-1 and pro-IL-1β
protein levels were reduced by GC17925 or SB203580 treatment. (D)
IL-1β secretion was decreased by GC17925 or SB203580 treatment. (E)
GSDMD fluorescent staining demonstrated inhibition of
anti-β2GPI/β2GPI-induced neutrophil
pyroptosis mediated by GC17925 or SB203580 (scale bar, 50
μm). Values are expressed as the mean ± standard error of
the mean (n=3 and n=4 in D). *P<0.05,
**P<0.01, ***P<0.001. NLRP3, NLR family
pyrin domain containing 3; anti-β2GPI, anti-β2-glycoprotein I; LPS,
lipopolysaccharide; Ctrl, control; IC,
anti-β2GPI/β2GPI immune complex; GSDMD,
gasdermin D; p-/t-PKR, phosphorylated/total double-stranded
RNA-dependent protein kinase. |
Anti-β2GPI/β2GPI
ICs induce neutrophil pyroptosis-mediated HMGB1 release
HMGB1 is an inflammatory protein that is present in
most cell types and that is released extracellularly in response to
specific stimuli or stress conditions. To explore its relevance in
the experimental system of the present study, neutrophils were
treated with PBS, anti-β2GPI (10
μg/ml)/β2GPI (100 μg/ml) or LPS (100
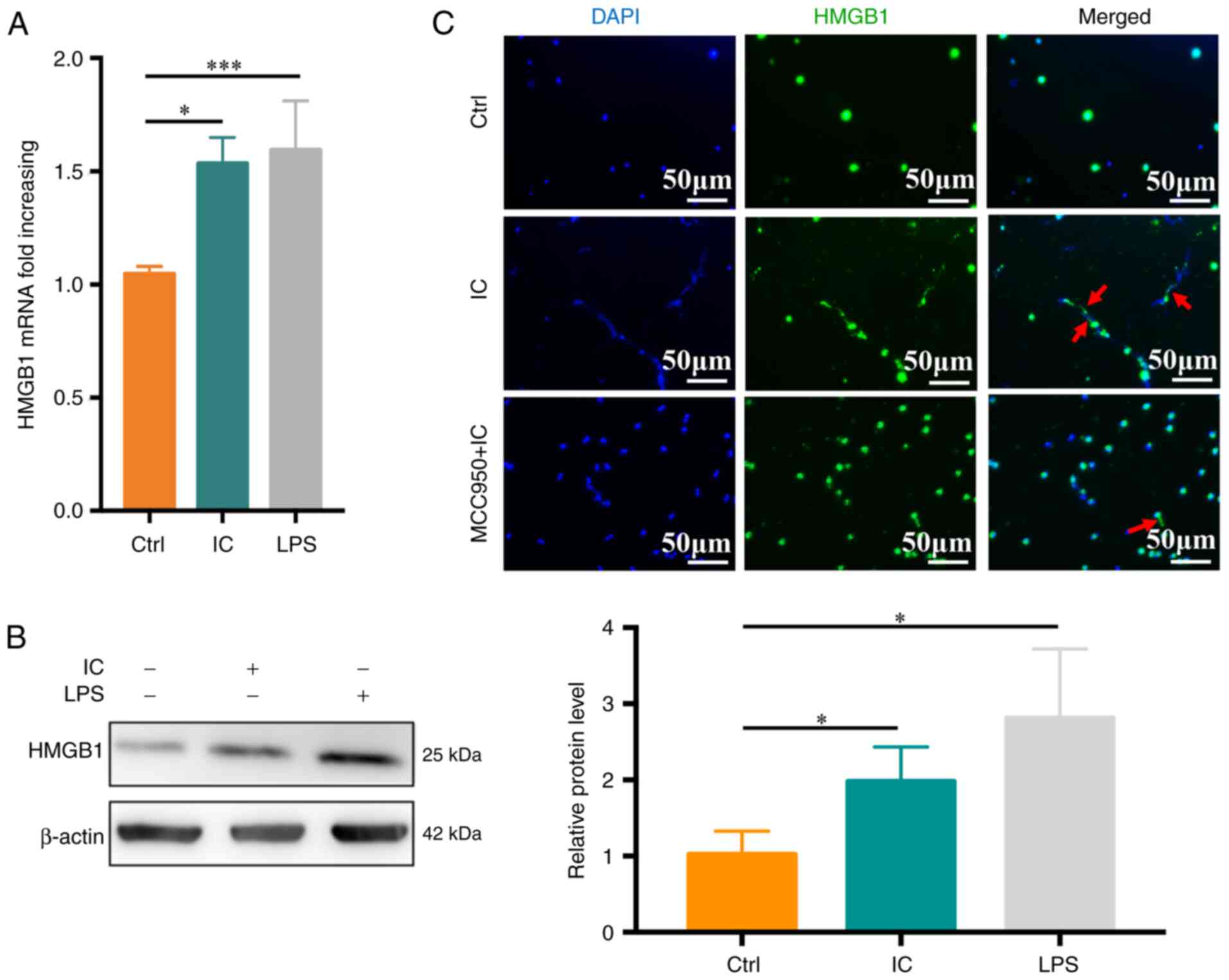
ng/ml), and HMGB1 expression was then monitored.
Anti-β2GPI/β2GP treatment was sufficient to
significantly enhance HMGB1 mRNA levels relative to the control
treatment (P<0.05; Fig. 5A),
with similar changes being observed at the protein level (Fig. 5B). To assess whether
anti-β2GPI/β2GP-stimulated HMGB1 release was
dependent on pyroptosis, immunofluorescent staining was performed,
which revealed that HMGB1 release was significantly reduced in the
anti-β2GPI/β2GP-MCC950-treated group relative
to the anti-β2GPI/β2GP-treated group
(Fig. 5C).
HMGB1 and IL-1β promote the activation of
endothelial cells
To determine the optimal stimulation time,
microscopy was used to confirm that the endothelial cells were in
good condition (Fig. 6A). The
cells were then treated with recombinant human HMGB1 (4 ng/ml) for
0-24 h. Maximal ICAM-1 upregulation relative to the baseline was
observed at 24 h via RT-qPCR (P<0.05; Fig. 6B). HUVECs were then cultured for
24 h with recombinant human HMGB1 (4 ng/ml) or recombinant human
IL-1β (2 ng/ml) and when using this incubation time, both IL-8 and
ICAM-1 were upregulated at the mRNA level relative to control cells
(Fig. 6C and D), with consistent
increases in IL-8 and ICAM-1 protein levels following stimulation
(Fig. 6E and F). Fluorescence
microscopy further confirmed these results (Fig. 6G and H).
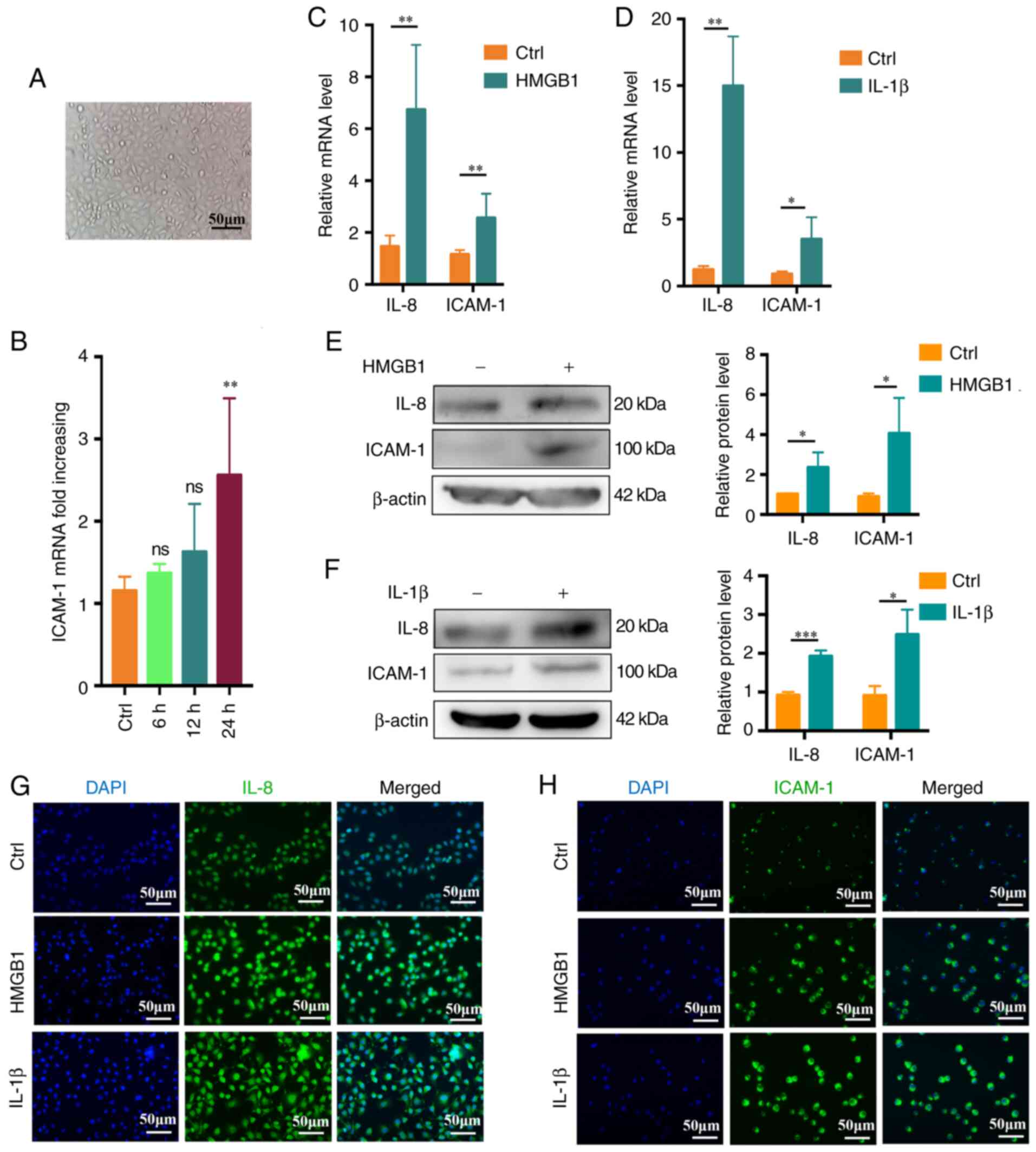 | Figure 6HMGB1 and IL-1β promote in
vitro endothelial cell activation. (A) Representative
microscopic image of endothelial cell morphology (scale bar, 50
μm). (B) Changes in endothelial cell ICAM-1 expression over
time (0-24 h) (n=3). (C) IL-8 and ICAM-1 expression levels in
endothelial cells following treatment for 24 h with HMGB1 (4
ng/ml), with β-actin being used for normalization (n=4). (D) IL-8
and ICAM-1 expression levels in endothelial cells stimulated for 24
h with IL-1β (2 ng/ml), with β-actin being used for normalization
(n=4). (E and F) IL-8 and ICAM-1 protein levels following (E) HMGB1
or (F) IL-1β stimulation (n=3). (G) IL-8 expression as assessed via
fluorescence microscopy following a 24 h stimulation with HMGB1 (4
ng/ml) or IL-1β (2 ng/ml). (H) ICAM-1 expression was assessed via
fluorescence microscopy following a 24 h stimulation with HMGB1 (4
ng/ml) or IL-1β (2 ng/ml) (scale bar, 50 μm). Values are
expressed as the mean ± standard error of the mean.
*P<0.05, **P<0.01,
***P<0.001 as indicated or vs. Ctrl. Ctrl, control;
ns, no significance; HMGB1, high mobility group box protein 1;
ICAM-1, intercellular adhesion molecule-1. |
Discussion
Anti-β2GPI antibodies are closely linked
to increased rates of coagulation and thrombosis in patients with
APS (3). β2GPI is a
single-stranded 50-kDa glycoprotein produced by liver cells that
forms a complex with phospholipids and thereby adopts an open
conformation that facilitates anti-β2GPI binding, such
that a single antibody is able to bind to two β2GPI
molecules (26). The resultant
anti-β2GPI/β2GPI complexes are associated with an increased risk of
thrombotic events (5). Zhou
et al (27) determined
that anti-β2GPI/β2GPI ICs, rather than
IgG/β2GPI or anti-β2GPI/BSA complexes, were responsible for in
vitro THP-1 cell activation. A previous study by our group also
indicated that an anti-β2GPI to β2GPI ratio of 1:10 is able to
facilitate efficient IC formation in vitro (6). The resultant complexes may promote
neutrophil-mediated NETs release, thereby driving thrombosis
(7). In the present study,
increased anti-β2GPI levels and neutrophil NLRP3
expression were observed in patients with CI.
Neutrophils function as critical mediators of innate
immune responses against a wide array of pathogens, releasing IL-1β
in an NLRP3-dependent manner in the context of bacterial infection
or inflammation (28).
Anti-β2GPI may induce trophoblast-derived IL-1β release
via promoting NLRP3 activation, thereby contributing to the
incidence of morbid pregnancy (29). There is robust evidence that
NLRP3 participates in thrombosis via the regulation of inflammatory
responses (30). In the present
study, it was determined that
anti-β2GPI/β2GPI treatment resulted in
increased NLRP3 and caspase-1 expression in neutrophils. During
pyroptosis, activated NLRP3 recruits apoptosis-associated
speck-like protein containing CARD and subsequently activates
caspase-1, thereby forming the multi-protein NLRP3 inflammasome
complex (24). Activated
caspase-1 additionally cleaves pro-IL-1β to yield activated IL-1β,
which may be secreted from cells (31). However, data reported by Karmakar
et al (32) suggest that,
while neutrophils release IL-1β in an NLRP3-dependent manner in
certain inflammatory contexts with concomitant caspase-1
activation, this is not indicative of ongoing neutrophil
pyroptosis. Indeed, pyroptosis is characterized by the presence of
small pores in the cell membrane that are detectable via electron
microscopy, ultimately leading to cellular swelling, rupture and
the release of the contents therein. GSDMD has been indicated to
bind to and form pores in the cell membrane following caspase-1
activation in the context of pyroptosis. While the loss of GSDMD
has no impact on caspase-1-mediated IL-1β processing, it does
nonetheless suppress the secretion of this cytokine (33). In the present study, it was
determined that anti-β2GPI/β2GPI ICss are able to trigger
neutrophil pyroptosis and consequent IL-1β via an NLRP3-caspase-1
pathway with concomitantly increased GSDMD expression, and in
addition, control for the potential interference of IgG/BSA was
provided. The above results confirmed that non-pathogenic immune
complexes did not have any effect on neutrophil pyroptosis. MCC950
is a selective NLRP3 inhibitor that also inhibits inflammatory
responses (34). MCC950 or
related inhibitors may thus be effective tools for preventing
APS-related thrombosis.
Anti-β2GPI/β2GPI ICs have
previously been indicated to bind to TLR4 on the surface of
neutrophils, thereby triggering NET release (7). TLR4 is an important inducer of both
innate and adaptive immunity (35). Tumor-derived autophagosomes may
activate TLRs, thereby promoting NLRP3 inflammasome-dependent
innate immune responses (36).
The present study suggested that anti-β2GPI/β2GPI IC-induced
neutrophil pyroptosis was dependent upon TLR4 activation, as such
activity was blunted by treatment with the TLR4 inhibitor TAK-242.
Consistently, TAK-242 was recently reported to inhibit
GSDMD-mediated pyroptosis in tubular cells in the context of
diabetic kidney disease (37).
PKR controls translation and inflammatory signaling in the context
of antiviral immune responses. Bat-Erdene et al (38) posited that breast cancer cell
glycosaminoglycans may induce a proliferation-inducing ligand
secretion through a mechanism dependent on neutrophil TLR4 and PKR.
In the present study, the ability of anti-β2GPI/β2GPI IC treatment
to induce PKR phosphorylation in neutrophils was established, and
TAK-242 treatment inhibited this process. Previous studies have
indicated that PKR is able to regulate inflammatory responses
through the JNK, p38MAPK, interferon regulatory factor 3 and NF-κB
pathways (17,39,40). Consistently, the present study
suggested that anti-β2GPI/β2GPI ICs were able
to induce p38MAPK phosphorylation, while treatment with the PKR
inhibitor GC17925 prevented this process. Yim et al
(16) determined that PKR is
able to suppress inflammasome activity. Ma et al (18) further indicated that PKR promotes
apoptosis via p38MAPK signaling. However, Lu et al (15) proposed that PKR promotes
inflammasome activation. The present study confirmed that
inhibitors of PKR (GC17925) or p38MAPK (SB203580) were able to
suppress NLRP3 inflammasome activation and associated IL-1β
secretion, while also inhibiting GSDMD expression in treated
neutrophils. It was thus demonstrated that the PKR/p38MAPK
signaling pathway has an essential role in
anti-β2GPI/β2GPI-induced neutrophil
pyroptosis.
In addition to IL-1β, pyroptosis also induces the
release of the nuclear protein HMGB1 (15). Barlan et al (41) confirmed that macrophages are able
to recognize adenovirus 5 upon infection via a TLR9-dependent
mechanism that induces NLRP3 and caspase-1 expression and
consequent reactive oxygen species production, leading to NLRP3
inflammasome activation and the release of IL-1β and HMGB1. MCC950
is able to selectively suppress NLRP3 and thereby inhibit
inflammatory responses (35).
Adamiak et al (42)
determined that murine hematopoietic stem/progenitor cells
exhibited reductions in peripheral blood ATP stimulation-induced
IL-1β expression when MCC950 was applied to inhibit the NLRP3
inflammasome. In the present study, it was determined that
neutrophils were able to release IL-1β via pyroptosis following
anti-β2GPI/β2GPI stimulation. Such IC exposure also induced
neutrophil-mediated HMGB1 release, whereas MCC950 inhibited this
process. MCC950 or related inhibitors may thus be effective tools
for preventing APS-related thrombosis. In addition, PKR controls
the release of IL-1β and the HMGB1, and may thus be a relevant
target in this pathological context (15).
Circulating IL-1β and HMGB1 have previously been
indicated to participate in a range of pathological processes,
including inflammation, thrombosis and tumor metastasis (43,44). For instance, Schulze et al
(45) found plasma HMGB1 levels
to be correlated with stroke severity, while Yoshida et al
(46) identified IL-1β as a
mediator of extra-intestinal thrombosis in a model of experimental
colitis. Endothelial-cell activation has a central role in
thrombosis, with endothelial cell upregulation of the inflammatory
marker proteins ICAM-1 and IL-8 being closely linked to the
induction of thrombotic diseases (47,48). The present data revealed that
HUVECs stimulated with recombinant IL-1β and HMGB1 upregulated both
IL-8 and ICAM-1 in vitro. However, a limitation of the
present study is the lack of in vivo experiments to verify
neutrophil pyroptosis and rule out other cellular interference.
TLR4 is also expressed on vascular endothelial cells and the
anti-β2GPI/β2GPI IC may also directly
activate endothelial cells (49). Xia et al (50) indicated that
anti-β2GPI/β2GPI ICs induced thrombosis by
TLR4-induced monocyte activation.
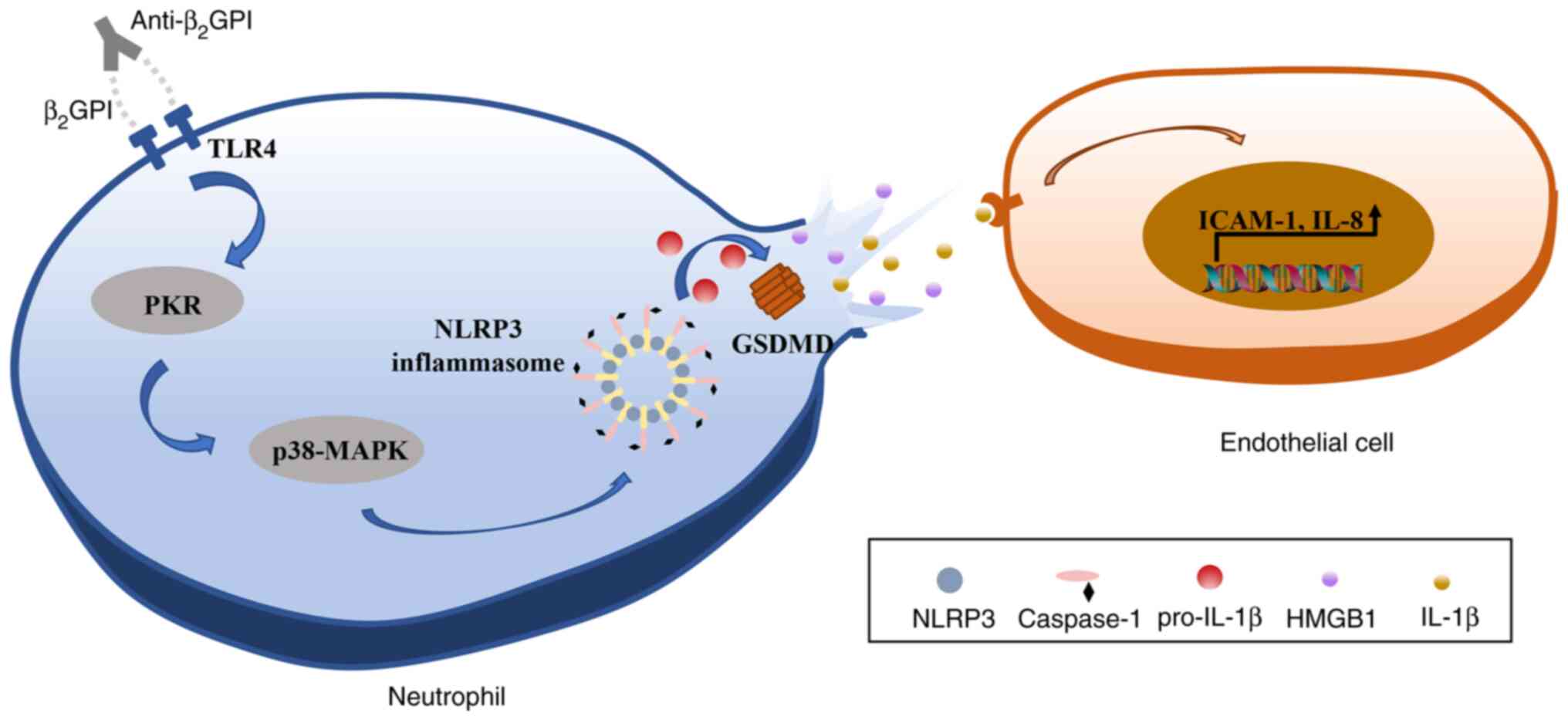
In conclusion, the results of the present study
indicated that anti-β2GPI/β2GPI is able to induce pyroptotic
neutrophil-mediated IL-1β and HMGB1 release, in turn activating
endothelial cells and promoting their upregulation of IL-8 and
ICAM-1 expression in vitro. It was further established that
these effects were mediated via TLR4 through an intracellular
PKR/p38MAPK/NLRP3 signaling pathway (Fig. 7). Together, these data suggest
that neutrophil pyroptosis may represent a key mechanism driving
thrombosis, thereby offering new insight into the potential
pathogenic etiology underlying CI and related thrombotic diseases
in patients with APS.
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
JL, MZ and YL designed this study; JL, MZ and ZW
performed the experiments; ZW and LY were involved in data curation
and formal analyses; JL and MZ wrote the manuscript; YL critically
revised the manuscript for important intellectual content, approved
the final manuscript version to be published and agreed to be
accountable for all aspects of the work. JL and MZ checked and
approved the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Samples were collected from patients with the
approval of the Institutional Ethics Committee of Harbin Medical
University (Harbin, China) and informed consent was obtained from
the patients in accordance with the Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (grant no. 81974108 to YL).
References
|
1
|
Tektonidou MG: Antiphospholipid syndrome
nephropathy: From pathogenesis to treatment. Front Immunol.
9:11812018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Giannakopoulos B, Passam F, Rahgozar S and
Krilis SA: Current concepts on the pathogenesis of the
antiphospholipid syndrome. Blood. 109:422–430. 2007. View Article : Google Scholar
|
|
3
|
Pierangeli SS, Chen PP, Raschi E, Scurati
S, Grossi C, Borghi MO, Palomo I, Harris EN and Meroni PL:
Antiphospholipid antibodies and the antiphospholipid syndrome:
Pathogenic mechanisms. Semin Thromb Hemost. 34:236–250. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McDonnell T, Wincup C, Buchholz I,
Pericleous C, Giles I, Ripoll V, Cohen H, Delcea M and Rahman A:
The role of beta-2-glycoprotein I in health and disease associating
structure with function: More than just APS. Blood Rev.
39:1006102020. View Article : Google Scholar :
|
|
5
|
Martínez-Flores JA, Serrano M, Pérez D,
Cámara A G, Lora D, Morillas L, Ayala R, Paz-Artal E, Morales JM
and Serrano A: Circulating immune complexes of IgA bound to beta 2
glycoprotein are strongly associated with the occurrence of acute
thrombotic events. J Atheroscler Thromb. 23:1242–1253. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang W, Gao F, Lu D, Sun N, Yin X, Jin M
and Liu Y: Anti-β2 glycoprotein I antibodies in complex with β2
glycoprotein I induce platelet activation via two receptors:
Apolipoprotein E receptor 2' and glycoprotein I bα. Front Med.
10:76–84. 2016. View Article : Google Scholar
|
|
7
|
Zha C, Zhang W, Gao F, Xu J, Jia R, Cai J
and Liu Y: Anti-β2GPI/β2GPI induces
neutrophil extracellular traps formation to promote thrombogenesis
via the TLR4/MyD88/MAPKs axis activation. Neuropharmacology.
138:140–150. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Segel GB, Halterman MW and Lichtman MA:
The paradox of the neutrophil's role in tissue injury. J Leukoc
Biol. 89:359–372. 2011. View Article : Google Scholar
|
|
9
|
Zychlinsky A, Prevost MC and Sansonetti
PJ: Shigella flexneri induces apoptosis in infected macrophages.
Nature. 358:167–169. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cookson BT and Brennan MA:
Pro-inflammatory programmed cell death. Trends Microbiol.
9:113–114. 2011. View Article : Google Scholar
|
|
11
|
Miao EA, Rajan JV and Aderem A:
Caspase-1-induced pyroptotic cell death. Immunol Rev. 243:206–214.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli
VG, Wu H and Lieberman J: Inflammasome-activated gasdermin D causes
pyroptosis by forming membrane pores. Nature. 535:153–158. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mezzaroma E, Toldo S, Farkas D, Seropian
IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF and
Abbate A: The inflammasome promotes adverse cardiac remodeling
following acute myocardial infarction in the mouse. Proc Natl Acad
Sci USA. 108:19725–19730. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Inoue Y, Shirasuna K, Kimura H, Usui F,
Kawashima A, Karasawa T, Tago K, Dezaki K, Nishimura S, Sagara J,
et al: NLRP3 regulates neutrophil functions and contributes to
hepatic ischemia-reperfusion injury independently of inflammasomes.
J Immunol. 192:4342–4351. 2014. View Article : Google Scholar
|
|
15
|
Lu B, Nakamura T, Inouye K, Li J, Tang Y,
Lundbäck P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, et al:
Novel role of PKR in inflammasome activation and HMGB1 release.
Nature. 488:670–674. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yim HC, Wang D, Yu L, White CL, Faber PW,
Williams BR and Sadler AJ: The kinase activity of PKR represses
inflammasome activity. Cell Res. 26:367–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Goh KC, deVeer MJ and Williams BR: The
protein kinase PKR is required for p38 MAPK activation and the
innate immune response to bacterial endotoxin. EMBO J.
19:4292–4297. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ma CH, Wu CH, Jou IM, Tu YK, Hung CH, Chou
WC, Chang YC, Hsieh PL and Tsai KL: PKR promotes oxidative stress
and apoptosis of human articular chondrocytes by causing
mitochondrial dysfunction through p38 MAPK activation-PKR
activation causes apoptosis in human chondrocytes. Antioxidants
(Basel). 8:3702019. View Article : Google Scholar
|
|
19
|
Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao
L, Huang J, Yu Y, Fan XG, Yan Z, et al: HMGB1 in health and
disease. Mol Aspects Med. 40:1–116. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xie WH, Ding J, Xie XX, Yang XH, Wu XF,
Chen ZX, Guo QL, Gao WY, Wang XZ and Li D: Hepatitis B virus X
protein promotes liver cell pyroptosis under oxidative stress
through NLRP3 inflammasome activation. Inflamm Res. 69:683–696.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee PH, Yamamoto TN, Gurusamy D, Sukumar
M, Yu Z, Hu-Li J, Kawabe T, Gangaplara A, Kishton RJ, Henning AN,
et al: Host conditioning with IL-1β improves the antitumor function
of adoptively transferred T cells. J Exp Med. 216:2619–2634. 2019.
View Article : Google Scholar :
|
|
22
|
Huebener P, Pradere JP, Hernandez C, Gwak
GY, Caviglia JM, Mu X, Loike JD and Schwabe RF: The HMGB1/RAGE axis
triggers neutrophil-mediated injury amplification following
necrosis. J Clin Invest. 125:539–550. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu X, Zhang H, Qi W, Zhang Y, Li J, Li Z,
Lin Y, Bai X, Liu X, Chen X, et al: Nicotine promotes
atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis.
Cell Death Dis. 9:1712018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takahashi M: NLRP3 inflammasome as a novel
player in myocardial infarction. Int Heart J. 55:101–105. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kovacs SB and Miao EA: Gasdermins:
Effectors of pyroptosis. Trends Cell Biol. 27:673–684. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Agar C, van Os GM, Mörgelin M, Sprenger
RR, Marquart JA, Urbanus RT, Derksen RH, Meijers JC and de Groot
PG: Beta2-glycoprotein I can exist in 2 conformations: Implications
for our understanding of the antiphospholipid syndrome. Blood.
116:1336–1343. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou H, Sheng L, Wang H, Xie H, Mu Y, Wang
T and Yan J: Anti-β2GPI/β2GPI stimulates activation of THP-1 cells
through TLR4/MD-2/MyD88 and NF-κB signaling pathways. Thromb Res.
132:742–749. 2013. View Article : Google Scholar
|
|
28
|
Cho JS, Guo Y, Ramos RI, Hebroni F,
Plaisier SB, Xuan C, Granick JL, Matsushima H, Takashima A, Iwakura
Y, et al: Neutrophil-derived IL-1β is sufficient for abscess
formation in immunity against staphylococcus aureus in mice. PLoS
Pathog. 8:e10030472012. View Article : Google Scholar
|
|
29
|
Mulla MJ, Salmon JE, Chamley LW, Brosens
JJ, Boeras CM, Kavathas PB and Abrahams VM: A role for uric acid
and the Nalp3 inflammasome in antiphospholipid antibody-induced
IL-1β production by human first trimester trophoblast. PLoS One.
8:e652372013. View Article : Google Scholar
|
|
30
|
Guo Z, Yu S, Chen X, Ye R, Zhu W and Liu
X: NLRP3 is involved in ischemia/reperfusion injury. CNS Neurol
Disord Drug Targets. 15:699–712. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang W and Zhang T: Caspase-1-mediated
pyroptosis of the predominance for driving CD4[Formula: See text] T
cells death: A nonlocal spatial mathematical model. Bull Math Biol.
80:540–582. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Karmakar M, Katsnelson M, Malak HA, Greene
NG, Howell SJ, Hise AG, Camilli A, Kadioglu A, Dubyak GR and
Pearlman E: Neutrophil IL-1β processing induced by pneumolysin is
mediated by the NLRP3/ASC inflammasome and caspase-1 activation and
is dependent on K+ efflux. J Immunol. 194:1763–1775. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shi J, Zhao Y, Wang K, Shi X, Wang Y,
Huang H, Zhuang Y, Cai T, Wang F and Shao F: Cleavage of GSDMD by
inflammatory caspases determines pyroptotic cell death. Nature.
526:660–665. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ismael S, Zhao L, Nasoohi S and Ishrat T:
Inhibition of the NLRP3-inflammasome as a potential approach for
neuroprotection after stroke. Sci Rep. 8:59712018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
An H, Qian C and Cao X: Regulation of
Toll-like receptor signaling in the innate immunity. Sci China Life
Sci. 53:34–43. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xing Y, Cao R and Hu HM: TLR and NLRP3
inflammasome-dependent innate immune responses to tumor-derived
autophagosomes (DRibbles). Cell Death Dis. 7:e23222016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Y, Zhu X, Yuan S, Wen S, Liu X, Wang
C, Qu Z, Li J, Liu H, Sun L and Liu F: TLR4/NF-κB signaling induces
GSDMD-Related pyroptosis in tubular cells in diabetic kidney
disease. Front Endocrinol (Lausanne). 10:6032019. View Article : Google Scholar
|
|
38
|
Bat-Erdene U, Quan E, Chan K, Lee BM,
Matook W, Lee KY and Rosales JL: Neutrophil TLR4 and PKR are
targets of breast cancer cell glycosaminoglycans and effectors of
glycosaminoglycan-induced APRIL secretion. Oncogenesis. 7:452018.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bonnet MC, Weil R, Dam E, Hovanessian AG
and Meurs EF: PKR stimulates NF-kappaB irrespective of its kinase
function by interacting with the IkappaB kinase complex. Mol Cell
Biol. 20:4532–4542. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang P and Samuel CE: Induction of
protein kinase PKR-dependent activation of interferon regulatory
factor 3 by vaccinia virus occurs through adapter IPS-1 signaling.
J Biol Chem. 283:34580–34587. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Barlan AU, Griffin TM, McGuire KA and
Wiethoff CM: Adenovirus membrane penetration activates the NLRP3
inflammasome. J Virol. 85:146–155. 2011. View Article : Google Scholar :
|
|
42
|
Adamiak M, Ciechanowicz A, Skoda M, Cymer
M, Tracz M, Xu B and Ratajczak MZ: Novel Evidence that purinergic
signaling-Nlrp3 inflammasome axis regulates circadian rhythm of
hematopoietic stem/progenitor cells circulation in peripheral
blood. Stem Cell Rev Rep. 16:335–343. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dyer MR, Chen Q, Haldeman S, Yazdani H,
Hoffman R, Loughran P, Tsung A, Zuckerbraun BS, Simmons RL and Neal
MD: Deep vein thrombosis in mice is regulated by platelet HMGB1
through release of neutrophil-extracellular traps and DNA. Sci Rep.
8:20682018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tulotta C and Ottewell P: The role of
IL-1B in breast cancer bone metastasis. Endocr Relat Cancer.
25:R421–R434. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Schulze J, Zierath D, Tanzi P, Cain K,
Shibata D, Dressel A and Becker K: Severe stroke induces
long-lasting alterations of high-mobility group box 1. Stroke.
44:246–248. 2013. View Article : Google Scholar
|
|
46
|
Yoshida H, Russell J, Senchenkova EY,
Almeida Paula LD and Granger DN: Interleukin-1beta mediates the
extra-intestinal thrombosis associated with experimental colitis.
Am J Pathol. 177:2774–2781. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Khodabandehlou K, Masehi-Lano JJ, Poon C,
Wang J and Chung EJ: Targeting cell adhesion molecules with
nanoparticles using in vivo and flow-based in vitro models of
atherosclerosis. Exp Biol Med (Maywood). 242:799–812. 2017.
View Article : Google Scholar
|
|
48
|
Hosseinkhani B, Kuypers S, van den Akker
NMS, Molin DGM and Michiels L: Extracellular vesicles work as a
functional inflammatory mediator between vascular endothelial cells
and immune cells. Front Immunol. 9:17892018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Raschi E, Chighizola CB, Grossi C, Ronda
N, Gatti R, Meroni PL and Borghi MO: β2-glycoprotein I,
lipopolysaccharide and endothelial TLR4: Three players in the two
hit theory for anti-phospholipid-mediated thrombosis. J Autoimmun.
55:42–50. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xia L, Zhou H, Wang T, Xie Y, Wang T, Wang
X and Yan J: Activation of mTOR is involved in
anti-β2GPI/β2GPI-induced expression of tissue
factor and IL-8 in monocytes. Thromb Res. 157:103–110. 2017.
View Article : Google Scholar : PubMed/NCBI
|