1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is the
excessive accumulation of lipids in hepatocytes and is a result of
a combination of circulating free fatty acids, de novo
adipogenesis and dietary fat intake. NAFLD is diagnosed following
the exclusion of other causes of hepatic steatosis, such as
excessive alcohol consumption or other diseases (1,2).
NAFLD is one of the most common chronic liver diseases worldwide
and the incidence of NAFLD in Europe and America is >20%
(3). According to the
pathological status of the liver, NAFLD can be divided into simple
fatty liver and non-alcoholic steatohepatitis (NASH), which will
eventually develop into liver fibrosis, cirrhosis and
hepatocellular carcinoma (HCC) (1,4).
Extrahepatic problems, including cardiovascular disease, obesity,
type 2 diabetes, dyslipidemia and neurodegenerative conditions, are
frequently associated with NAFLD. It has been reported that as the
incidence of obesity and diabetes rise the prevalence of NAFLD also
increases (5,6).
The pathophysiology of NAFLD is complex and involves
free fatty acid accumulation, liver inflammation, oxidative stress,
lipid peroxidation and hepatocyte damage (7). The double-hit model was previously
considered to be the most widely accepted and popular NAFLD theory.
In this model the first strike is insulin resistance (IR), which
causes fat to build up in the liver, and the second blow is
lipotoxicity, which causes mitochondrial dysfunction, oxidative
stress and endoplasmic reticulum (ER) stress, as a result of fat
accumulation. As a result, the vulnerability of the liver to
inflammatory necrosis and fibrosis increases and inflammatory
damage leads to NASH, liver fibrosis, cirrhosis and HCC, which
hastens NAFLD progression (8). It
is now widely accepted that multiple factors are involved in the
development of NAFLD (9,10). As there is presently no
satisfactory therapeutic approach for NAFLD due to its uncertain
pathophysiology, NAFLD therapeutics are still dependent on
improving the lifestyle and changing the eating habits of
individuals (11,12). Overall, the prevalence of NAFLD
has increased over the past few decades, which imposes an
increasingly severe economic burden (13). It has also been reported that
NAFLD will become the leading indication for liver transplantation
within a decade (14). Therefore,
it is important to understand in detail the regulatory mechanism of
lipid metabolism in hepatocytes and to develop novel therapeutic
strategies for the control of NAFLD.
Ion channels are polymeric proteins that generate
ion selective holes in the plasma or organelle membrane in response
to certain stimuli, such as membrane potential or ligand binding
(15). Calcium (Ca2+)
channels are highly selective for Ca2+ permeability and
mainly mediate Ca2+ flow, which thereby regulates
Ca2+ homeostasis inside and outside the cell and
therefore it serves an important role in specific physiological
functions. Ca2+ channels are mainly divided into the
following two types: i) Voltage-gated Ca2+ channels,
including T, L, N, P/Q and R types; and ii) non-voltage-gated
Ca2+ channels, including transient receptor potential
(TRP) and Orai calcium release-activated calcium modulator (Orai)
families and their partners, stromal interaction molecules (STIMs)
(16). Ca2+ channels
are distributed in the cell membranes and organelles of various
organs and regulate numerous physiological functions of the body
(Table I). Increasing evidence
has demonstrated that Ca2+ channels serve a key role in
the pathological process of liver diseases, including NAFLD. These
channels mediate pathophysiological changes via the upregulation or
downregulation of expression levels, such as those of diagnostic
markers of early liver cancer and drug treatment targets (17,18).
 | Table ILocalization and function of
Ca2+ channels. |
Table I
Localization and function of
Ca2+ channels.
| Name | Location | Function |
|---|
| TPC2 | Lysosome | Ca2+
release from the lysosome |
| TRPV1 | Plasma
membrane | Ca2+
entry from the extracellular space to the cytoplasm |
| TRPV4 | Plasma
membrane | Ca2+
entry from the extracellular space to the cytoplasm |
| TRPM2 | Plasma
membrane | Ca2+
entry from the extracellular space to the cytoplasm |
| IP3R1 | ER membrane | Ca2+
release from the ER to the cytoplasm |
| IP3R2 | ER membrane | Ca2+
release from the ER to the cytoplasm |
| VDAC1 | Mitochondrial outer
membrane | Entry of
Ca2+ and other metabolites |
| Orai | Plasma
membrane | Ca2+
entry from the extracellular space to the cytoplasm |
| P2X7R | Plasma
membrane | Ca2+ and
Na+ entry from the extracellular space to the cytoplasm,
and K+ outflow from the cytoplasm to the extracellular
space |
| KCa3.1 | Plasma
membrane | Ca2+
entry from the extracellular space to the cytoplasm, and
K+ outflow from the cytoplasm to the extracellular
space |
The present review aimed to summarize the regulatory
mechanism of Ca2+ channels and discuss its role in the
development and progression of NAFLD. Furthermore, the latest
research progress in drugs that target Ca2+ channels to
treat NAFLD was examined. Moreover, the present review aimed to
propose novel potential therapeutic strategies involving
Ca2+ channels for the treatment of NAFLD and to
highlight current issues and the direction of future research in
this field. The present review provided new opportunities for the
prevention and treatment of NAFLD.
The databases used in the present review included
PubMed, Web of Science and Embase. The key words used included
'non-alcoholic fatty liver disease', 'calcium channels',
'endoplasmic reticulum stress', 'mitochondrial dysfunction', and
'therapeutic strategies'. The references included studies performed
from 1998-2021.
2. Physiological role of Ca2+ in
the liver
The liver is a complex organ composed of a variety
of different types of cells, which serves a crucial role as the
metabolic center of the body, including in the metabolism of
lipids, carbohydrates, various drugs and toxins (19). Ca2+ is a ubiquitous
secondary messenger that regulates a variety of liver functions,
including lipid and carbohydrate metabolism as well as bile
secretion and cholestasis. Furthermore, the balance of
Ca2+ signals between the nucleus, cytoplasm and
mitochondria controls liver regeneration following injury and is
also related to Ca2+ channel expression and distribution
in different organelles (20,21). Changes in intracellular
Ca2+ concentration are affected by the influx of
extracellular Ca2+ and the release of Ca2+
from intracellular Ca2+ stores, such as the ER,
mitochondria and lysosomes. When cells are excited by external
stimuli, various Ca2+ channels are opened or closed,
which results in the dysregulation of Ca2+ homeostasis
(22,23). Previous studies have reported that
the Ca2+ signaling pathway is a key regulator of
nutrient uptake, metabolism and utilization (24,25). Therefore, if Ca2+
homeostasis in liver cells is disrupted, a series of different
metabolic damage and liver regeneration disorders will be caused,
including NAFLD.
3. Dysregulation of Ca2+ channel
expression and the disturbance of Ca2+ homeostasis in
NAFLD
Ca2+ maintains a dynamic balance between
internal and external cells and organelles and regulates normal
physiological functions (Fig. 1).
However, Ca2+ channels partially regulate
Ca2+ levels and once channel expression levels are
impaired, including the upregulation, downregulation and even
channel defects, a series of pathophysiological changes occur. In
NAFLD the imbalance of Ca2+ in the cytoplasm,
organelles, including the mitochondria and ER, and the nucleus is
one of the contributing factors that promotes the development of
hepatic steatosis (26). The ER
and mitochondria are important organelles for lipid synthesis and
glycolipid metabolism in the liver and therefore serve an important
role in controlling lipid synthesis and metabolism in the liver
(27). The ER is the main
Ca2+ reservoir that releases Ca2+ under the
stimulation of various hormones. Matrix interaction molecules,
STIM1 and STIM2, are Ca2+ sensors of the ER and detect
the reduction of ER Ca2+ (28). When ER Ca2+ is
depleted, STIMs rapidly connect the ER and the plasma membrane,
causing the plasma membrane to recruit and activate Orai
Ca2+ channels, which result in Ca2+ release
and activation of the Ca2+ release-activated channel.
Ca2+ flows from extracellular space via the Orai
channel, which results in increased cytoplasmic Ca2+
levels and downstream signaling. This process is called storage
operation Ca2+ entry (SOCE). The excess Ca2+
is subsequently pumped back into the ER to replenish the deficiency
and this therefore maintains a dynamic Ca2+ equilibrium
(29). Liver hyperlipidemia leads
to an imbalance between ER membrane lipids and
phosphatidylethanolamine, which impairs the sarco/ER
Ca2+ ATPase (SERCA) function of the ER Ca2+
uptake pump and decreases the opening of the inositol triphosphate
receptor (InsP3R) Ca2+ channel. These disturbances lead
to increased cytoplasmic Ca2+ levels and decreased ER
Ca2+ levels (30-32). Furthermore, in the ER certain
Ca2+-dependent molecular chaperones, including calin and
calreticulin, control the folding of secretory and membrane
proteins, such as insulin receptors. Therefore, low Ca2+
levels in the ER can lead to an excess of misfolded proteins and
this therefore results in ER stress. Furthermore, in response to ER
stress, hepatocytes activate the unfolded protein response (UPR)
that causes IR via different signaling pathways (24).
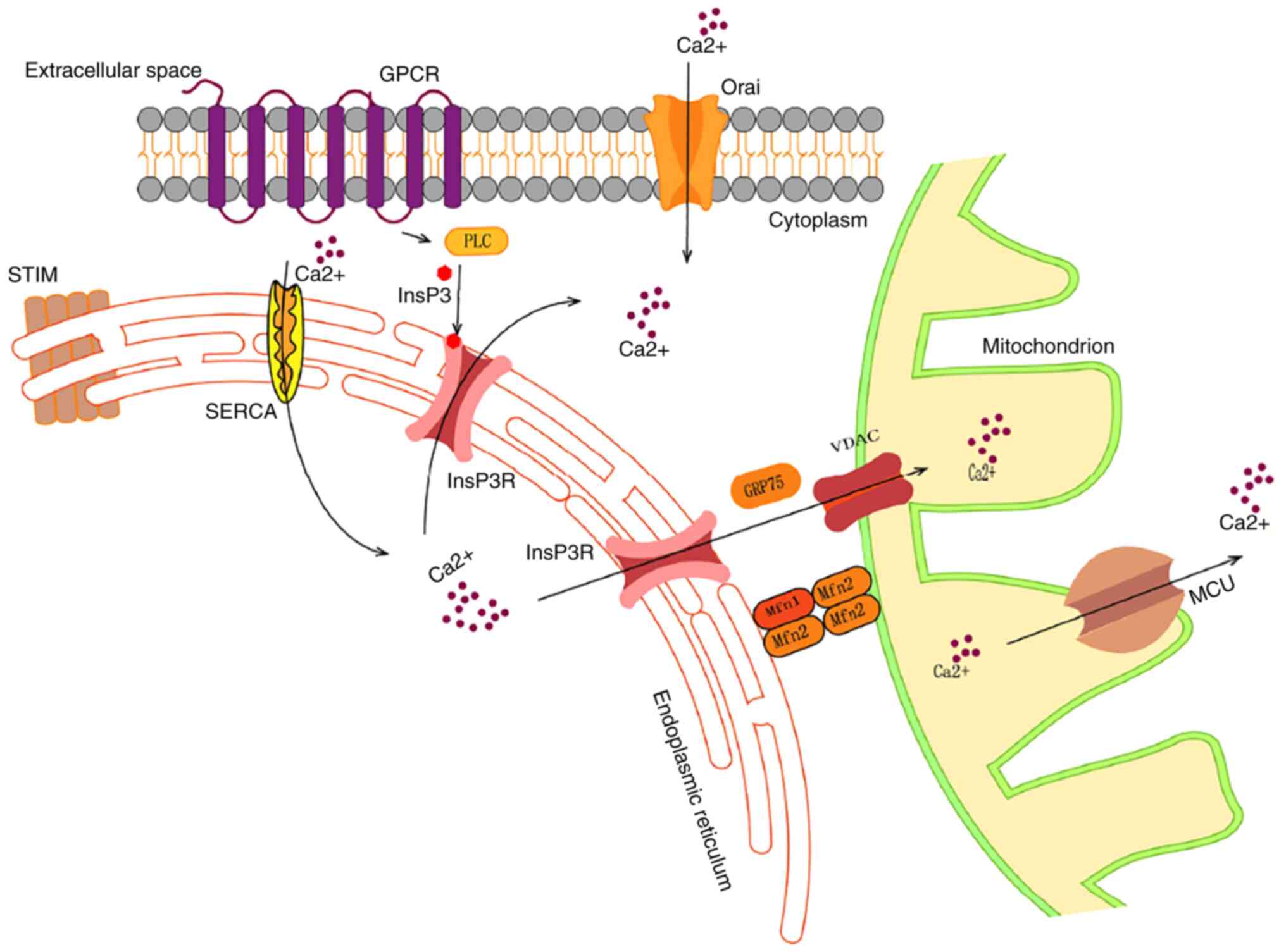 | Figure 1Schematic of intracellular
Ca2+ flow. When GPCRs are activated, the action of PLCs
leads to the formation of InsP3. Binding to the InsP3R triggers the
release of Ca2+ from the ER to the cytoplasm. Subsequent
isolation of Ca2+ from the STIM protein leads to its
interaction with Orai, which allows Ca2+ to enter the
cell from the extracellular space. Furthermore, cytoplasmic
Ca2+ is absorbed into the ER via SERCA activity. The ER
and mitochondria function via MAMs, whereby the chaperone molecule
GRP75 connects InsP3R and VDAC, which allows Ca2+ to be
transported from the ER to the mitochondria. Tethering proteins
Mfn1/2 regulate the stability of MAM. Ca2+ enters the
mitochondrial matrix via the MCU complex of the inner mitochondrial
membrane. Ca2+, calcium; GPCR, G-protein-coupled
receptor; PLC, phospholipase C; InsP3, inositol triphosphate;
InsP3R, InsP3 receptor; ER, endoplasmic reticulum; STIM, stromal
interaction molecule; Orai, Orai calcium release-activated calcium
modulator; SERCA, sarco/ER Ca2+ ATPase; MAM,
mitochondria-associated membrane; GRP75, glucose-regulated protein
75; VDAC, voltage-dependent anion channel; Mfn1/2, mitofusin 1/2;
MCU, mitochondrial Ca2+ uniporter. |
Moreover, the concentration of Ca2+ in
the mitochondria is equivalent to that in the cytoplasm under
normal physiological conditions. The close proximity between the ER
and mitochondria generates Ca2+ microdomains that allow
the rapid transport of Ca2+ to the mitochondria when
needed (33). Ca2+
absorption is achieved by a voltage-dependent anion channel (VDAC)
in the outer membrane of mitochondria that is connected with the ER
InsP3R1 via a molecular partner, glucose-regulated protein 75
(34). Mitochondrial
Ca2+ concentration is controlled via the coordinated
interaction between the mitochondrial Ca2+ uniporter
(MCU) complex and the sodium or hydrogen
(H+)/Ca2+ reverse transporters, solute
carrier family 8 member B1 and leucine zipper and EF-hand
containing transmembrane protein 1. The fine-regulation of
mitochondrial Ca2+ levels according to cellular needs
contributes to the regulation of numerous liver functions,
including lipid and carbohydrate metabolism, proliferation and
apoptosis (24). In compensatory
diseases with chronic elevated cytoplasmic Ca2+ levels,
Ca2+ uptake via the MCU increases and the exit via the
Ca2+ transporter becomes saturated. The overload of
mitochondrial Ca2+ buffer capacity leads to an increase
in mitochondrial Ca2+ levels, which can lead to
mitochondrial dysfunction and increase reactive oxygen species
levels. Oxidative stress promotes hepatocyte apoptosis via the
consumption of ATP, NAD and glutathione, as well as via the damage
of DNA, lipids and proteins (24,35).
Lysosomes are also important for intracellular
Ca2+ storage and H+-ATPase establishes a
H+ gradient on the lysosome membrane that provides
energy for Ca2+ entry via the
Ca2+/H+ exchanger (36). Lysosomal Ca2+ release
is triggered via numerous Ca2+ mobilization messengers,
including nicotinic acid adenine dinucleotide phosphate (NAADP) and
phosphatidylinositol 3,5-diphosphate, via Ca2+ permeable
channels, including two-pore channels (TPCs) and TRP mucilipids
(TRPMLs) (26). Furthermore,
lysosomal Ca2+ is a key regulator of autophagy and the
process of lipophagy decomposition of intracellular lipid drops is
named lipid phagocytosis (37).
Autophagy is important in NASH and it has previously been reported
that drugs enhancing autophagy have therapeutic potential for the
treatment of NASH (38).
Therefore, targeting Ca2+ channels on lysosomes to
regulate Ca2+ levels may also be a potential therapeutic
approach for the treatment of NAFLD.
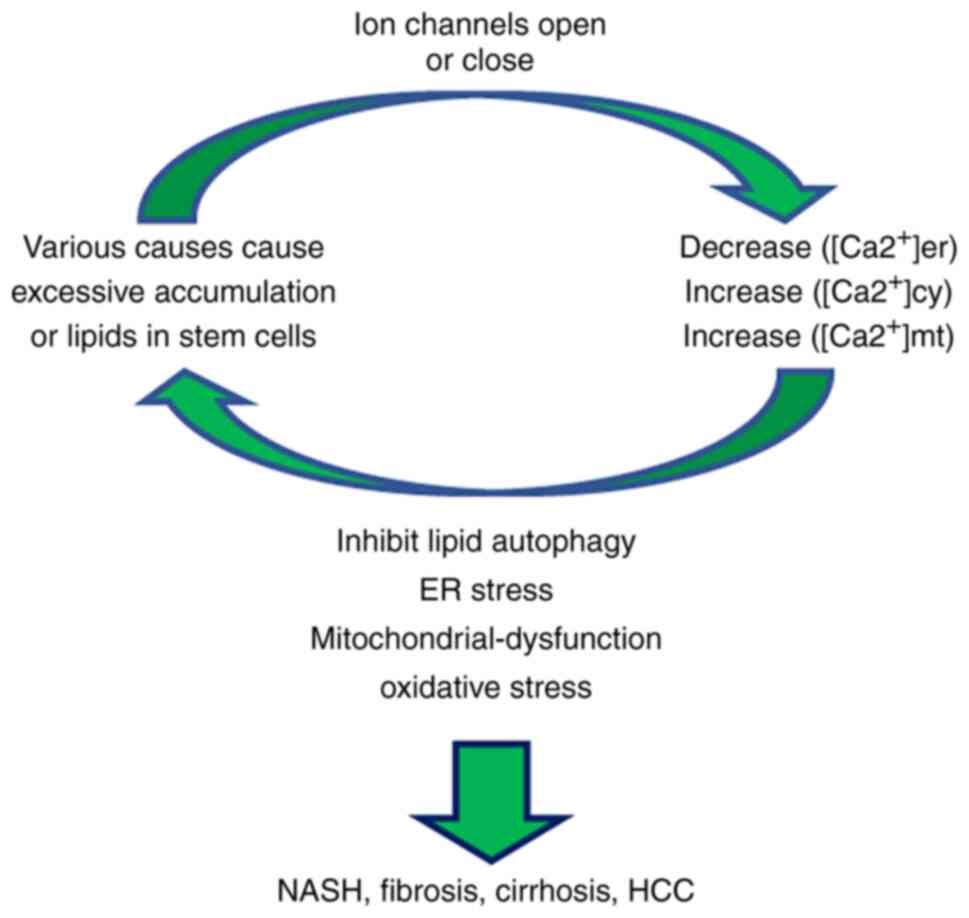
Lipid accumulation in liver cells induces changes in
Ca2+ signaling, mainly via the decreased concentration
of ER Ca2+ and the increased concentration of
Ca2+ in the cytoplasmic matrix [(Ca2+)cyt]
and mitochondrial matrix (39).
It can also lead to the inhibition of lipid autophagy (40), ER stress (41), mitochondrial dysfunction (2) and oxidative stress, which results in
increased liver fat accumulation, steatosis and the progression of
NASH (Fig. 2) (27,42). These aforementioned studies have
therefore indicated that Ca2+ signaling serves an
important role in NAFLD. By directly targeting Ca2+
channels that regulate Ca2+ levels, novel NAFLD
therapeutics may be developed to prevent steatosis and the
transformation of inflammation into fibrosis.
4. Ca2+ channels in the
development and progression of NAFLD and as an NAFLD therapeutic
target
Ca2+ channels serve an important role in
the development and progression of NAFLD, including via their
overexpression and low expression. Therefore, Ca2+
channels may be used as a potential target for the diagnosis,
prevention and treatment of NAFLD. The following is a summary of
the mechanism of action and/or treatment strategies of several
types of Ca2+ channels in NAFLD (Table II) (43-62).
| Table IIRole of Ca2+ channels in
the development of NAFLD. |
Table II
Role of Ca2+ channels in
the development of NAFLD.
| Name | Role and
therapeutic target | Therapeutic
strategy | (Refs.) |
|---|
| TPC2 | Downregulated
expression; promotes the progression | Inhibition | (43) |
| TRPV1 | After activation
inhibits the progression | Activation | (44,45) |
| TRPV4 | Upregulated
expression; promotes the progression | Inhibition | (46,47) |
| TRPM2 | Upregulated
expression; promotes the progression | Inhibition | (48,49) |
| IP3R1 | Upregulated
expression; promotes the progression | Inhibition | (50,51) |
| IP3R2 | Downregulated
expression; promotes the progression | Activation | (52) |
| VACD1 | Upregulated
expression; promotes the progression | Inhibition | (53,54) |
| SOCE | Downregulated
expression; promotes the progression | Activation | (55,56) |
| Orai1 | Upregulated
expression; promotes the progression | Inhibition | (57,58) |
| P2X7R | Upregulated
expression; promotes the progression | Inhibition | (59,60) |
| KCa3.1 | Upregulated
expression; promotes the progression | Inhibition | (61,62) |
TPC2 defects contribute to NAFLD
progression
TPCs are endolysomal ion channels. There are three
TPC subtypes. TPC1 and TPC2 are ubiquitous, whereas TPC3 is not
present in numerous animal genomes (63). TPCs are mainly located in acidic
organelles, such as lysosomes, with TPC2 expression being
predominantly lysosomal and TPC1 having a wider distribution within
the endolysomal system. TPC1 is found in lysosomes and early and
recycling endosomes (43,63). Previous studies have reported that
TPCs mediate the release of Ca2+ in lysosomes following
activation via NAADP, which regulates Ca2+ signal
stabilization to control a series of Ca2+-dependent
events (64). In NAFLD, the
opening of the TPC2 channel initiates the final fusion between late
endosomes and lysosomes, which mediates lipid degradation
transport. However, TPC2 deficiency leads to impaired transport
that promotes hepatic cholesterol accumulation and
hyperlipoproteinemia, which thereby significantly contributes to
the development of NASH (43).
Furthermore, in a previous study it was demonstrated that in the
liver cells of TPC2-knockout embryonic mice, substances and
receptors transported along the degradation pathway, such as
low-density lipoprotein or epidermal growth factor (EGF)/EGFR,
accumulate rapidly in intracellular vesicles. This resulted in
increased cholesterol accumulation and liver transaminase, which
suggested that the TPC2-deficient mice exhibited cholesterol
overload and liver damage consistent with NAFLD (43). Furthermore, high Ca2+
can cause lysosome damage, which leads to impaired lipid
degradation, liver cholesterol accumulation and hyperlipidemia,
which further progresses NAFLD into NASH (17,43). Therefore, it can be hypothesized
that regulating the high expression of TPC2 may prevent NAFLD
development.
TRP channels
TRP channels are a superfamily of univalent and
bivalent cation osmotic ion channels. More than 30 channels have
been identified, including TRP channel, TRP vanilloid (TRPV), TRP
melastatin (TRPM), TRP subfamily A, TRP polycystic, TRPML and TRP
no mechanoreceptor potential C (65). TRP channels mainly exist on the
plasma membrane of metabolically active tissues, including the
liver, gastrointestinal tract, brain, pancreas and adipose tissue
(66). They serve an important
role in sensory function (sight, hearing, taste, smell, pain,
mechanical and thermal sensations), homeostasis (absorption and
reabsorption of ions and fluid flow), as well as cell survival
(67,68). TRP channels have the following
three important characteristics: i) Ca2+ permeability;
ii) thermal sensation; and iii) mechanical sense. Moreover, TPC
channels have a high permeability for Ca2+ ions,
regulate the intracellular Ca2+ steady-state and affect
the energy intake, storage and consumption of the body (65,66). It has previously been reported
that the following TRP channels induce the development and
progression of NAFLD via the mediation of intracellular
Ca2+ homeostasis, which could help identify new
treatment strategies for NAFLD.
Following activation TRPV1 upregulates
the expression of uncoupling protein 2 (UCP2) and decreases
peroxisome proliferator activated receptor δ (PPARδ) and
autophagy
TRPV1 is found in mouse preadipocytes and
adipocytes, as well as in human and mouse visceral adipose tissue.
It mediates the elevation of Ca2+ in fat cells and
therefore significantly inhibits adipogenesis and reduces the
accumulation of triglycerides (69). Capsaicin, the main irritant in
pepper, is an effective agonist of TRPV1 (70) that blocks adipogenesis and obesity
in anterior fat cells in vivo and in vitro via the
activation of TRPV1 channels (71). Previous studies have reported that
NAFLD is often associated with obesity, which is specifically
reflected in autophagy disorders during obesity that contribute to
the pathological progression of fatty liver disease, such as
steatohepatitis and HCC (71-73). In an in vitro model of
NAFLD induced via a high-fat diet (HFD), capsaicin treatment was
demonstrated to reduce liver steatosis and inflammation, but not in
TRPV1-knockout mice (44,45). In an in vitro model of
NAFLD induced by HFD, UCP2 expression was upregulated in the liver
of wild-type mice following TRPV1 activation, but not in
TRPV1-knockout mice. UCP2 promotes liver β-oxidation and lipid
metabolism (74). These
aforementioned results suggested that the activation of TRPV1
prevents the development of fatty liver and may be associated with
increased hepatic β-oxidation, mediated via UCP2 (45,75). Another study reported that a
long-term HFD significantly reduced the expression of liver PPARδ
and autophagy molecules in wild-type mouse liver tissue (44). Autophagy regulates intracellular
lipid levels by removing lipid droplets, which consequently
enhances autophagy, liver steatosis and IR, and leads to
improvements in HFD-treated mice (76). It was also demonstrated that TRPV1
activation via capsaicin treatment promoted lipolysis and enhanced
autophagy, whereas dietary capsaicin did not have this effect in
TRPV1-deficient mice. Therefore, capsaicin also inhibits NAFLD by
increasing the expression levels of PPARδ, which causes autophagy
enhancement (44). In conclusion,
the activation of TRPV1 potentially has clinical value in the
treatment of the development and progression of NAFLD.
TRPV4 inhibits the action of cytochrome
P450 family 2 subfamily E member 1 (CYP2E1) via the activation of
Kupffer cell nitric oxide (NO) synthase 3 (NOS3) and NO release via
the paracrine system
TRPV4 is a Ca2+ channel that is widely
distributed in the kidneys, lungs, brain, bladder, fat, liver,
heart, skin, airway smooth muscle cells, vascular endothelial
cells, chondrocytes, osteoclasts and other tissues. TRPV4 is
activated by numerous physical and chemical stimuli, including
heat, mechanical stress, arachidonic acid and its derivatives
(77). Certain studies have
reported that TRPV4 has a pro-inflammatory effect on adipose
tissue. Its inhibition by drugs will lead to an increase in the
thermogenic gene program and a decrease in adipose tissue
inflammation. This indicates that TRPV4 has therapeutic benefits
for obesity and metabolic diseases (78). TRPV4 can also increase autophagy
and inhibit stellate cell apoptosis (79). Furthermore, TRPV4 expression is
significantly increased in hepatic fibrosis tissues (80). CYP2E1 is a cytochrome P450 enzyme,
which can lead to lipid peroxidation following its activation and
serves a pro-inflammatory role in NAFLD and enhances the
progression of NAFLD (46). In
progressive NAFLD, the promoter region of the TRPV4 gene is
methylated, which inhibits TRPV4 expression and therefore reduces
the levels of channel proteins associated with the progression of
NAFLD (47). In a previous study,
the NAFLD model was established in TRPV4-knockout mice and the
deterioration of NAFLD was observed 1 week following CYP2E1
activation. This was exhibited via necrosis, liver cell swelling,
the upregulation of inflammatory factor expression levels, Kupffer
cell activation and an increase in IL-1β levels. Furthermore, the
mRNA and protein expression levels of the damage-related nuclear
DNA binding protein, high mobility group box 1 (HMGB1), were also
increased, which promoted the further development of NAFLD. This
aforementioned study therefore concluded that TRPV4 serves a
protective role in the liver (47). This protective effect of TRPV4 may
block the role of CYP2E1 via the activation of Kupffer cell NOS3
and the release of NO via the paracrine system (47). Therefore, as an endogenous defense
molecule, TRPV4 has the potential to treat NAFLD.
Salidroside prevents the further
development of NAFLD via inhibition of the
TRPM2/Ca2+/Ca2+/calmodulin-dependent protein
kinase II (CaMKII) signaling pathway
TRPM2 is a non-selective cationic channel that
mediates Ca2+ transmission and is expressed in the skin,
brain, pancreas, spleen, kidney and immune cells (65). However, its presence in the liver
has only been confirmed at the mRNA expression level (81). TRPM2 is activated via temperature,
oxidative stress and intracellular endogenous ligands, of which
oxidative stress is the most important (48,65). It also regulates Ca2+
signal transduction in tissues and mediates various
pathophysiological processes. TRPM2-mediated Ca2+ entry
has been reported to contribute to drug-induced hepatotoxicity and
the progression of NAFLD into cirrhosis, fibrosis and HCC (49). In a previous study, following the
establishment of NAFLD models in vitro with palmitic acid
(PA), the expression of TRPM2 was increased and cytoplasmic
Ca2+ levels were significantly increased. This
subsequently resulted in the downstream phosphorylation of CaMKII.
Furthermore, treatment with salidroside reduced the accumulation of
fat droplets in liver L02 cells of PA-treated mice in a
dose-dependent manner, which was potentially a result of the
inhibition of TRPM2 channel activation in NAFLD liver cells.
Moreover, this reduced the progression of NAFLD-related disease
symptoms via reducing Ca2+ influx-induced
phosphorylated-CaMKII downregulation. These results suggested that
the TRPM2/Ca2+/CaMKII signaling pathway further leads to
lipid accumulation, mitochondrial damage and ER stress, which may
exacerbate the progression of NAFLD. This can lead to NASH and
progression to cirrhosis and HCC (48). Therefore, TRPM2 inhibitors may be
useful therapeutic targets for NAFLD treatment (48,49,82).
1,4,5-Trisphosphate receptor (IP3R)1
deletion inhibits the development of NAFLD, whereas IP3R2 deletion
can promote the development of NAFLD
The IP3R is a type of important intracellular
Ca2+ release channel that mediates a series of
pathophysiological processes by participating in the regulation of
intracellular Ca2+ levels (83). There are three main types, named
IP3R1, IP3R2 and IP3R3. Moreover, IP3R1 and IP3R2 are both
expressed in liver cells, whereas IP3R3 is physiologically
deficient. IP3R1 is mainly distributed in the ER, which controls
mitochondrial Ca2+ signaling and lipid metabolism via
connecting with VDAC on the outer membrane of the mitochondria.
IP3R2 is concentrated near the apical membrane and regulates the
bile solute secretion of liver cells (50). Therefore, IP3R1 and IP3R2 serve
different roles in the progression of NAFLD. It has previously been
reported that IP3R1 expression is increased in the
mitochondria-bound ER of HFD mice, which leads to mitochondrial
dysfunction and the disruption of metabolic homeostasis (84). Furthermore, the results of this
previous study suggested that IP3R1 may promote the development of
NAFLD. A HFD diet was used to induce fatty liver in mice. When
compared with the control group, IP3R1-knockout mice exhibited
reduced triglycerides in the serum and liver and lipid droplet
accumulation. Moreover, liver histological examinations
demonstrated that IP3R1-knockout mice were free from steatosis
(51). Further investigations
also detected a significant decrease in Ca2+ signaling
in the mitochondria of IP3R1-deficient mice (51). Therefore, this aforementioned
study concluded that IP3R1-knockout mice have reduced mitochondrial
Ca2+ signal impairment, reduced hepatic triglycerides
and reduced lipid droplet formation and are resistant to the
development of fatty liver disease (51). In a study, hepatocytes were
treated with PA to establish an NAFLD model in vitro. The
increased expression of IP3R1, mitochondrial dysfunction and
apoptosis were detected. The results also demonstrated that the
inhibition of IP3R1 expression alleviated the decrease in
mitochondrial membrane potential and of mitochondrial superoxide
accumulation in hepatocytes following PA treatment. This may have
been due to the increased phosphorylation of Tyr353 in IP3R1 via
the tyrosine kinase signaling pathway that led to the increased
stability of the IP3R1 protein. Therefore, mitochondrial
Ca2+ overload may induce mitochondrial dysfunction in
hepatocytes (50). In summary,
the specific inhibition of IP3R1 channels to reduce mitochondrial
dysfunction in hepatocytes may be a potential novel clinical
strategy for the treatment of NAFLD and the prevention of its
further development.
In a previous study, liver cell models of NAFLD were
constructed using a HFD. c-Jun expression was reported to be
elevated, whereas IP3R2 expression was significantly decreased
(52). Moreover, liver biopsies
from patients with simple steatosis and NASH were also analyzed and
the results were consistent with the in vitro models
(52). Compared with normal
cells, IP3R2-deficient cells displayed a significantly reduced
nuclear Ca2+ signal amplitude, which suggested that
nuclear Ca2+ signaling may be dependent on inositol
1,4,5-trisphosphate receptor type 2 channel activation (52). [Ca2+]cyt is known to be
essential for cell proliferation (85). Further research has reported
significant diffuse steatosis and impaired hepatocyte regeneration
in IP3R2-deficient livers following a hepatectomy (52). These aforementioned results
suggested that increased c-Jun expression levels may potentially
negatively regulate IP3R2 expression in the livers of patients with
NAFLD. This may therefore affect nuclear Ca2+ and lead
to impaired liver regeneration and cell proliferation, which
eventually causes the progression of NAFLD (52). Although the specific underlying
mechanism remains to be studied further, it can be hypothesized
that the factors regulating IP3R2 expression have potential
clinical significance in the treatment of NAFLD.
VDAC1 expression is upregulated in
NAFLD-associated HCC
VDAC1 is an anionic channel that is located on the
outer membrane of the mitochondria and can form hydrophilic
channels to regulate the passage of anions, cations, ATP and other
metabolites into and out of the mitochondria. It also interacts
with numerous proteins that are also involved in lipid metabolism
and cholesterol transport. VDAC1 serves an important role in
regulating cell metabolism, maintaining intracellular
Ca2+ homeostasis, apoptosis and necrosis (86,87). Cardiolipin (CL), the signature
phospholipid of the inner mitochondrial membrane (IMM), serves an
important role in maintaining the structure and function of
mitochondria (88). CL deletion
occurs in various metabolic disorders, including NAFLD. Certain
studies have analyzed patients with NAFLD-driven HCC, and have
demonstrated that VDAC1 is dysregulated in NAFLD-driven HCC.
Furthermore, immunohistochemical analysis has previously
demonstrated that compared with non-tumor tissues, VDAC1 expression
levels in HCC tissues are upregulated and gradually increase with
the progression of the tumor (53,54). Furthermore, high levels of VDAC1
expression are associated with poor clinical outcomes, possibly
because VDAC1 is significantly positively correlated with protein
tyrosine phosphatase mitochondrial 1 and negatively correlated with
the CL remodeling enzyme tafazzin. This results in changes to CL
composition and damages the stability of the IMM structure, which
ultimately leads to mitochondrial dysfunction. VDAC1 therefore
promotes the development of NAFLD to HCC (54). Although the specific underlying
mechanism remains unclear, the targeted inhibition of VDAC1 may be
a potential therapeutic approach to prevent the progression of
NAFLD to HCC.
Inhibition of SOCE and SERCA2b leads to
the development of NAFLD, whereas inhibition of Orai1 inhibits this
effect
SOCE, composed of STIM1, is located on the ER and
the Orai1 protein is located on the cell membrane and is an
important channel that mediates the entry of extracellular
Ca2+ into cells (89).
As a Ca2+ sensor, when STIM1 senses a decrease in ER
Ca2+ concentration it will undergo rapid activation
reactions, such as translocation and polymerization, and will
subsequently combine with Orai1 on the plasma membrane, leading to
the activation of the Orai1 pore, which results in the entry of
intracellular Ca2+ (89,90). SOCE signals control a variety of
functions, including gene expression, migration, proliferation and
apoptosis. Dysregulation of STIM and Orai1 protein expression
levels can lead to SOCE dysfunction and consequently induce
pathophysiological changes (90).
Ca2+ entry via the SOCE signaling pathway induces cAMP
production, protein kinase C (PKC) activation, hormone-sensitive
lipase activation and enhanced lipolysis (39). The accumulation of cholesterol
during steatosis is associated with ER Ca2+ depletion,
which leads to the induction of UPR and apoptosis (91) and participates in the regulation
of lipid metabolism (92).
Previous studies have reported that lipid accumulation induces the
inhibition of SOCE and SERCA2b, the major isoform of SERCA protein
in the liver, and reduces the entry of Ca2+ via the
activation of PKC-mediated phosphorylation of the Orai1 protein. As
the ER stress response is related to Ca2+ homeostasis, a
decrease of Ca2+ concentration in the ER induces the ER
stress response and leads to a decrease in protein folding, as well
as protein chaperone activity and glucose tolerance. This therefore
stimulates the synthesis of diacylglycerol and triacylglycerol
(55,56). This process ultimately intensifies
lipid accumulation and leads to IR, which promotes NAFLD
progression to NASH, liver fibrosis and even HCC. Furthermore, the
inhibition of SOCE and subsequent increase in intracellular lipid
accumulation are associated with increased lipid autophagy
(4,55,56). Previous research on the Orai1
protein has reported that Orai1 expression regulates de novo
fat formation (57). Hepatic
BRL-3A cells were previously treated with high concentrations of
nesfatins to construct a NAFLD cell model. The expression levels of
Orai1 and NF-κB p65 were significantly upregulated, which promoted
ER and oxidative stress and led to the inflammatory response of
hepatic lipid deposition. Treatment with Orai1 inhibitor
2-aminoethoxydiphenyl borate and NF-κB inhibitor wogonin reduced
these effects (57,58). Therefore, it can be hypothesized
that the activation of SOCE or the reduction of SOCE inhibition
increases Ca2+ in the ER, which may reduce ER stress and
decrease lipid accumulation. However, lipid deposition may also be
reduced via the regulation of Orai1 protein expression. The
aforementioned strategies may serve as potential therapeutic
approaches to prevent NAFLD progression.
Deletion of the purinergic receptor X7
(P2X7R) against fatty liver injury and fibrosis
P2X7R, a member of the purinergic receptor family,
is an ion-type ligand-gated ion channel that is expressed on the
membrane of hepatic cells and is activated via ATP to regulate
liver metabolic processes, such as the insulin response, glycogen
and lipid metabolism and bile secretion (93). P2X7R has been reported to increase
NADPH oxidase activity, increase the expression of major
histocompatibility complex II in Kupffer cells and participate in
the regulation of inflammation (59). Previous studies have demonstrated
that P2X7R expression was increased in hepatocytes, Kupffer cells
and hepatic sinusoidal endothelial cells via the construction of an
in vitro NASH model (60,94). P2X7R is associated with autophagy,
disruption of normal lysosomal function, as well as autophagosome
release into the extracellular matrix of microglia, which
ultimately leads to inflammation (95). Autophagy is one of the key
characteristics of NASH and drugs that enhance autophagy have been
reported to have therapeutic potential for the treatment of NASH
(38). Moreover, P2X7R is a key
regulator of autophagy in NASH (96). LC3B depletion, lysosome-associated
membrane protein 2 and heat shock cognate protein 70 increasing,
which act as autophagy markers, have been reported in mice with the
P2X7R gene deletion. Furthermore, LI-1, IFN-α and HMGB1 release was
demonstrated to be subsequently reduced, which resulted in enhanced
autophagy and reduced inflammation (60). Other studies have reported that
liver cells treated with a HFD and carbon tetrachloride (CCL4)
exhibited improved apoptosis, as well as reduced inflammation and
fibrosis in mice with a P2X7R deletion (59,60). Furthermore, P2X7R activated
Kupffer cells and increased the production of TNF-α and monocyte
chemotactic protein-2 in HFD mice treated with CCL4 (59). In summary, P2X7R deletion mice
were protected from the damage of steatosis and fibrosis. However,
the exact molecular underlying mechanism of P2X7R is unclear. It
can be hypothesized that enhancing lipid autophagy via the
inhibition of P2X7R may be a novel therapeutic approach for the
treatment of NASH.
Senicapoc inhibits the KCa3.1 signaling
pathway and serves anti-inflammatory and antisteatotic roles
Medium conductance Ca2+-activated
potassium (K+) channels (KCa3.1) are widely expressed
throughout the body, mainly in hematopoietic cells, the
gastrointestinal tract, lungs, endocrine glands, exocrine glands,
vascular endothelial cells, fibroblasts and proliferating
neointimal vascular smooth muscle cells (97). Via the promotion of K+
outflow from hyperpolarized cell membranes, Ca2+ entry
is promoted, which affects cell proliferation, migration and
vascular resistance (61).
Triarylmethane-34, an inhibitor of KCa3.1, can reduce the
proliferation of stellate cells via the induction of cell cycle
arrest and reduces the gene expression of TGF-β1-induced collagen,
α-smooth muscle actin and TGF-β1 to serve an anti-fibrotic role
(98). Moreover, in a previous
study, to further confirm the role of KCa3.1 in NAFLD-related liver
fibrosis, rat liver fibrosis models were induced using
thioacetamide and HFD. Furthermore, the pharmacodynamic effects of
senicapoc were determined via detecting biomarkers of apoptosis,
inflammation, steatosis and fibrosis (62). The upregulation of the KCa3.1
signaling pathway has previously been observed in three different
pathological models. However, in the aforementioned previous study
senicapoc-induced inhibition of the KCa3.1 signaling pathway
reduced hepatic triglyceride content and served an
anti-inflammatory and anti-steatotic role (62). Therefore, drug inhibition of the
KCa3.1 channel may serve as a potential therapeutic target for
liver fibrosis induced via NAFLD.
5. Conclusion and future perspectives
NAFLD is one of the most common causes of cirrhosis
and HCC, however to date, there are no effective treatments
(1). Therefore, improving the
quality of human life with respect to NAFLD is a challenge. In
conclusion, NAFLD involves increased hepatic fatty acid synthesis,
decreased fatty acid catabolism and cellular stress, including
oxidative stress, the inflammatory response and ER stress (57). Dysregulation of Ca2+
channel expression leads to the disruption of Ca2+
signal homeostasis, which results in the aforementioned changes
discussed in the present review. This suggests that ion channels
serve an important role in the occurrence and development of NAFLD.
The inhibition or activation of these channel proteins may be a
potential therapeutic approach to prevent NAFLD progression to
NASH, fibrosis, cirrhosis and HCC. Furthermore, it is unclear if
Ca2+ channels serve a role in all stages of NAFLD,
including simple steatosis, chronic inflammation, fibrosis or
cirrhosis. Therefore, further research is required in this area of
research. At present, there are relatively few studies concerning
NAFLD and most of these are still at the laboratory stage. The
clinical feasibility and effectiveness of NAFLD Ca2+
channel-targeted therapeutics need to be studied. Therefore, more
systematic studies are needed in the future.
Availability of data and materials
Not applicable.
Authors' contributions
XC made substantial contributions to the conception
design of the study as well as wrote the manuscript and performed
the literature search. LZha, LZhe and BT were involved in revising
the manuscript critically for important intellectual content. Data
authentication is not applicable. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
We thank Dr Hai Jin (Department of
Gastroenterology, Digestive Disease Hospital, Affiliated Hospital
of Zunyi Medical University, Zunyi, Guizhou) for providing
suggestions for the review.
Funding
The present study was supported by a grant from the National
Natural Science Foundation of China (grant no. 82073087), the
National Natural Science Foundation of China (grant no. 81960507)
and the Science and Technology Bureau fund of Zunyi city [grant no.
ZUN SHI KE HE HZ ZI(2019)93-HAO] Collaborative Innovation Center of
Chinese Ministry of Education (2020-39).
References
|
1
|
Brunt EM, Wong VW, Nobili V, Day CP,
Sookoian S, Maher JJ, Bugianesi E, Sirlin CB, Neuschwander-Tetri BA
and Rinella ME: Nonalcoholic fatty liver disease. Nat Rev Dis
Primers. 1:150802015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gusdon AM, Song KX and Qu S: Nonalcoholic
fatty liver disease: Pathogenesis and therapeutics from a
mitochondria-centric perspective. Oxid Med Cell Longev.
2014:6370272014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhu JZ, Dai YN, Wang YM, Zhou QY, Yu CH
and Li YM: Prevalence of nonalcoholic fatty liver disease and
economy. Dig Dis Sci 2015. Nov;60:3194–3202. 2015. View Article : Google Scholar
|
|
4
|
Neuschwander-Tetri BA: Non-alcoholic fatty
liver disease. BMC Med. 15:452017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stefan N, Häring H and Cusi K:
Non-alcoholic fatty liver disease: Causes, diagnosis,
cardiometabolic consequences, and treatment strategies. Lancet
Diabetes Endocrinol. 7:313–324. 2019. View Article : Google Scholar
|
|
6
|
Manne V, Handa P and Kowdley KV:
Pathophysiology of nonalcoholic fatty liver disease/nonalcoholic
steatohepatitis. Clin Liver Dis. 22:23–37. 2018. View Article : Google Scholar
|
|
7
|
Varela-Rey M, Embade N, Ariz U, Lu SC,
Mato JM and Martínez-Chantar ML: Non-alcoholic steatohepatitis and
animal models: Understanding the human disease. Int J Biochem Cell
Biol. 41:969–976. 2009. View Article : Google Scholar
|
|
8
|
Day CP and James OF: Steatohepatitis: A
tale of two 'hits'? Gastroenterology. 114:842–845. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Friedman SL, Neuschwander-Tetri BA,
Rinella M and Sanyal AJ: Mechanisms of NAFLD development and
therapeutic strategies. Nat Med. 24:908–922. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Buzzetti E, Pinzani M and Tsochatzis E:
The multiple-hit pathogenesis of non-alcoholic fatty liver disease
(NAFLD). Metabolism. 65:1038–1048. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schuppan D and Schattenberg JM:
Non-alcoholic steatohepatitis: Pathogenesis and novel therapeutic
approaches. J Gastroenterol Hepatol. 28(Suppl 1): S68–S76. 2013.
View Article : Google Scholar
|
|
12
|
Cortez-Pinto H, de Moura MC and Day CP:
Non-alcoholic steatohepatitis: From cell biology to clinical
practice. J Hepatol. 44:197–208. 2006. View Article : Google Scholar
|
|
13
|
Younossi ZM, Blissett D, Blissett R, Henry
L, Stepanova M, Younossi Y, Racila A, Hunt S and Beckerman R: The
economic and clinical burden of nonalcoholic fatty liver disease in
the United States and Europe. Hepatology. 64:1577–1586. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wong RJ, Aguilar M, Cheung R, Perumpail
RB, Harrison SA, Younossi ZM and Ahmed A: Nonalcoholic
steatohepatitis is the second leading etiology of liver disease
among adults awaiting liver transplantation in the United States.
Gastroenterology. 148:547–555. 2015. View Article : Google Scholar
|
|
15
|
Kiselyov K and Muallem S: ROS and
intracellular ion channels. Cell Calcium. 60:108–114. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guéguinou M, Chantôme A, Fromont G,
Bougnoux P, Vandier C and Potier-Cartereau M: KCa and Ca(2+)
channels: The complex thought. Biochim Biophys Acta.
1843:2322–2333. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ramírez A, Vázquez-Sánchez AY,
Carrión-Robalino N and Camacho J: Ion channels and oxidative stress
as a potential link for the diagnosis or treatment of liver
diseases. Oxid Med Cell Longev. 2016:39287142016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ali ES, Rychkov GY and Barritt GJ:
Targeting Ca2+ signaling in the initiation, promotion
and progression of hepatocellular carcinoma. Cancers (Basel).
12:27552020. View Article : Google Scholar
|
|
19
|
Ben-Moshe S and Itzkovitz S: Spatial
heterogeneity in the mammalian liver. Nat Rev Gastroenterol
Hepatol. 16:395–410. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Garcin I and Tordjmann T: Calcium
signalling and liver regeneration. Int J Hepatol. 2012:6306702012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Taira Z, Ueda Y, Monmasu H, Yamase D,
Miyake S and Shiraishi M: Characteristics of intracellular
Ca2+ signals consisting of two successive peaks in
hepatocytes during liver regeneration after 70% partial hepatectomy
in rats. J Exp Pharmacol. 8:21–33. 2016. View Article : Google Scholar :
|
|
22
|
Berridge MJ, Bootman MD and Roderick HL:
Calcium signalling: Dynamics, homeostasis and remodelling. Nat Rev
Mol Cell Biol. 4:517–529. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu L, Lian W and Zhao L: Calcium signaling
in cancer progression and therapy. FEBS J. 288:6187–6205. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Oliva-Vilarnau N, Hankeova S, Vorrink SU,
Mkrtchian S, Andersson ER and Lauschke VM: Calcium signaling in
liver injury and regeneration. Front Med (Lausanne). 5:1922018.
View Article : Google Scholar
|
|
25
|
Bartlett P, Gaspers L, Pierobon N and
Thomas A: Calcium-dependent regulation of glucose homeostasis in
the liver. Cell Calcium. 55:306–316. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen CC, Hsu LW, Chen KD, Chiu KW, Chen CL
and Huang KT: Emerging roles of calcium signaling in the
development of non-alcoholic fatty liver disease. Int J Mol Sci.
23:2562021. View Article : Google Scholar
|
|
27
|
Wang J, He W, Tsai PJ, Chen PH, Ye M, Guo
J and Su Z: Mutual interaction between endoplasmic reticulum and
mitochondria in nonalcoholic fatty liver disease. Lipids Health
Dis. 19:722020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Arruda A, Pers B, Parlakgul G, Güney E,
Goh T, Cagampan E, Lee GY, Goncalves RL and Hotamisligil GS:
Defective STIM-mediated store operated Ca2+ entry in
hepatocytes leads to metabolic dysfunction in obesity. Elife.
6:e299682017. View Article : Google Scholar
|
|
29
|
Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck
TJ, Ellisman MH, Stauderman KA and Cahalan MD: STIM1 is a Ca2+
sensor that activates CRAC channels and migrates from the Ca2+
store to the plasma membrane. Nature. 437:902–905. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park SW, Zhou Y, Lee J, Lee J and Ozcan U:
Sarco(endo)plasmic reticulum Ca2+-ATPase 2b is a major regulator of
endoplasmic reticulum stress and glucose homeostasis in obesity.
Proc Natl Acad Sci USA. 107:19320–19325. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Egnatchik RA, Leamy AK, Jacobson DA,
Shiota M and Young JD: ER calcium release promotes mitochondrial
dysfunction and hepatic cell lipotoxicity in response to palmitate
overload. Mol Metab. 3:544–553. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fu S, Yang L, Li P, Hofmann O, Dicker L,
Hide W, Lin X, Watkins SM, Ivanov AR and Hotamisligil GS: Aberrant
lipid metabolism disrupts calcium homeostasis causing liver
endoplasmic reticulum stress in obesity. Nature. 473:528–531. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Amaya M and Nathanson M: Calcium signaling
in the liver. Compr Physiol. 3:515–539. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Szabadkai G, Bianchi K, Várnai P, De
Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T and Rizzuto
R: Chaperone-mediated coupling of endoplasmic reticulum and
mitochondrial Ca2+ channels. J Cell Biol. 175:901–911. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Masarone M, Rosato V, Dallio M, Gravina
AG, Aglitti A, Loguercio C, Federico A and Persico M: Role of
oxidative stress in pathophysiology of nonalcoholic fatty liver
disease. Oxid Med Cell Longev. 2018:95476132018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Luzio JP, Hackmann Y, Dieckmann NM and
Griffiths GM: The biogenesis of lysosomes and lysosome-related
organelles. Cold Spring Harb Perspect Biol. 6:a0168402014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Singh R, Kaushik S, Wang Y, Xiang Y, Novak
I, Komatsu M, Tanaka K, Cuervo AM and Czaja MJ: Autophagy regulates
lipid metabolism. Nature. 458:1131–1135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lin CW, Zhang H, Li M, Xiong X, Chen X,
Chen X, Dong XC and Yin XM: Pharmacological promotion of autophagy
alleviates steatosis and injury in alcoholic and non-alcoholic
fatty liver conditions in mice. J Hepatol. 58:993–999. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ali ES, Rychkov GY and Barritt GJ:
Deranged hepatocyte intracellular Ca2+ homeostasis and
the progression of non-alcoholic fatty liver disease to
hepatocellular carcinoma. Cell Calcium. 82:1020572019. View Article : Google Scholar
|
|
40
|
Miyagawa K, Oe S, Honma Y, Izumi H, Baba R
and Harada M: Lipid-induced endoplasmic reticulum stress impairs
selective autophagy at the step of autophagosome-lysosome fusion in
hepatocytes. Am J Pathol. 186:1861–1873. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Koo JH and Han CY: Signaling nodes
associated with endoplasmic reticulum stress during NAFLD
progression. Biomolecules. 11:2422021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ali ES and Petrovsky N: Calcium signaling
as a therapeutic target for liver steatosis. Trends Endocrinol
Metab. 30:270–281. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Grimm C, Holdt LM, Chen CC, Hassan S,
Müller C, Jörs S, Cuny H, Kissing S, Schröder B, Butz E, et al:
High susceptibility to fatty liver disease in two-pore channel
2-deficient mice. Nat Commun. 5:46992014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li Q, Li L, Wang F, Chen J, Zhao Y, Wang
P, Nilius B, Liu D and Zhu Z: Dietary capsaicin prevents
nonalcoholic fatty liver disease through transient receptor
potential vanilloid 1-mediated peroxisome proliferator-activate
receptor delta activation. Pflugers Arch. 465:1303–1316. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li L, Chen J, Ni Y, Feng X, Zhao Z, Wang
P, Sun J, Yu H, Yan Z, Liu D, et al: TRPV1 activation prevents
nonalcoholic fatty liver through UCP2 upregulation in mice.
Pflugers Arch. 463:727–732. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang K, Tan W, Liu X, Deng L, Huang L,
Wang X and Gao X: New insight and potential therapy for NAFLD:
CYP2E1 and flavonoids. Biomed Pharmacother. 137:1113262021.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Seth RK, Das S, Dattaroy D,
Chandrashekaran V, Alhasson F, Michelotti G, Nagarkatti M,
Nagarkatti P, Diehl AM, Bell PD, et al: TRPV4 activation of
endothelial nitric oxide synthase resists nonalcoholic fatty liver
disease by blocking CYP2E1-mediated redox toxicity. Free Radic Biol
Med. 102:260–273. 2017. View Article : Google Scholar
|
|
48
|
Feng Q, Liu C, Gao W, Geng XL and Dai N:
Salidroside-mitigated inflammatory injury of hepatocytes with
non-alcoholic fatty liver disease via inhibition TRPM2 ion channel
activation. Diabetes Metab Syndr Obes. 12:2755–2763. 2019.
View Article : Google Scholar
|
|
49
|
Ali ES, Rychkov GY and Barritt GJ: TRPM2
non-selective cation channels in liver injury mediated by reactive
oxygen species. Antioxidants (Basel). 10:12432021. View Article : Google Scholar
|
|
50
|
Yu T, Zheng E, Li Y, Li Y, Xia J, Ding Q,
Hou Z, Ruan XZ, Zhao L and Chen Y: Src-mediated Tyr353
phosphorylation of IP3R1 promotes its stability and causes
apoptosis in palmitic acid-treated hepatocytes. Exp Cell Res.
399:1124382021. View Article : Google Scholar
|
|
51
|
Feriod CN, Oliveira AG, Guerra MT, Nguyen
L, Richards KM, Jurczak MJ, Ruan HB, Camporez JP, Yang X, Shulman
GI, et al: Hepatic inositol 1,4,5 trisphosphate receptor type 1
mediates fatty liver. Hepatol Commun. 1:23–35. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Khamphaya T, Chukijrungroat N,
Saengsirisuwan V, Mitchell-Richards KA, Robert ME, Mennone A,
Ananthanarayanan M, Nathanson MH and Weerachayaphorn J:
Nonalcoholic fatty liver disease impairs expression of the type II
inositol 1,4,5-trisphosphate receptor. Hepatology. 67:560–574.
2018. View Article : Google Scholar
|
|
53
|
Smedlund K, Dube P and Vazquez G: Early
steatohepatitis in hyperlipidemic mice with endothelial-specific
gain of TRPC3 function precedes changes in aortic atherosclerosis.
Physiol Genomics. 48:644–649. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhu Y, Zhang C, Xu F, Zhao M, Bergquist J,
Yang C, Liu X, Tan Y, Wang X, Li S, et al: System biology analysis
reveals the role of voltage-dependent anion channel in
mitochondrial dysfunction during non-alcoholic fatty liver disease
progression into hepatocellular carcinoma. Cancer Sci.
111:4288–4302. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ali ES, Rychkov GY and Barritt GJ:
Metabolic disorders and cancer: Hepatocyte store-operated
Ca2+ channels in nonalcoholic fatty liver disease. Adv
Exp Med Biol. 993:595–621. 2017. View Article : Google Scholar
|
|
56
|
Wilson CH, Ali ES, Scrimgeour N, Martin
AM, Hua J, Tallis GA, Rychkov GY and Barritt GJ: Steatosis inhibits
liver cell store-operated Ca2+ entry and reduces ER
Ca2+ through a protein kinase C-dependent mechanism.
Biochem J. 466:379–390. 2015. View Article : Google Scholar
|
|
57
|
Zhang B, Yang W, Zou Y, Li M, Guo H, Zhang
H, Xia C and Xu C: NEFA-sensitive Orai1 expression in regulation of
de novo lipogenesis. Cell Physiol Biochem. 47:1310–1317. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang B, Li M, Zou Y, Guo H, Zhang B, Xia
C, Zhang H, Yang W and Xu C: NFκB/Orai1 facilitates endoplasmic
reticulum stress by oxidative stress in the pathogenesis of
non-alcoholic fatty liver disease. Front Cell Dev Biol. 7:2022019.
View Article : Google Scholar
|
|
59
|
Chatterjee S, Rana R, Corbett J, Kadiiska
MB, Goldstein J and Mason RP: P2X7 receptor-NADPH oxidase axis
mediates protein radical formation and Kupffer cell activation in
carbon tetrachloride-mediated steatohepatitis in obese mice. Free
Radic Biol Med. 52:1666–1679. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Das S, Seth RK, Kumar A, Kadiiska MB,
Michelotti G, Diehl AM and Chatterjee S: Purinergic receptor X7 is
a key modulator of metabolic oxidative stress-mediated autophagy
and inflammation in experimental nonalcoholic steatohepatitis. Am J
Physiol Gastrointest Liver Physiol. 305:G950–G963. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Freise C, Heldwein S, Erben U, Hoyer J,
Köhler R, Jöhrens K, Patsenker E, Ruehl M, Seehofer D, Stickel F
and Somasundaram R: K+-channel inhibition reduces portal
perfusion pressure in fibrotic rats and fibrosis associated
characteristics of hepatic stellate cells. Liver Int. 35:1244–1252.
2015. View Article : Google Scholar
|
|
62
|
Paka L, Smith DE, Jung D, McCormack S,
Zhou P, Duan B, Li JS, Shi J, Hao YJ, Jiang K, et al:
Anti-steatotic and anti-fibrotic effects of the KCa3.1 channel
inhibitor, senicapoc, in non-alcoholic liver disease. World J
Gastroenterol. 23:4181–4190. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Morgan AJ and Galione A: Two-pore channels
(TPCs): Current controversies. Bioessays. 36:173–183. 2014.
View Article : Google Scholar
|
|
64
|
Patel S and Kilpatrick BS: Two-pore
channels and disease. Biochim Biophys Acta Mol Cell Res.
1865:1678–1686. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Bishnoi M, Khare P, Brown L and Panchal
SK: Transient receptor potential (TRP) channels: A metabolic TR(i)P
to obesity prevention and therapy. Obes Rev. 19:1269–1292. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhu Z, Luo Z, Ma S and Liu D: TRP channels
and their implications in metabolic diseases. Pflugers Arch.
461:211–223. 2011. View Article : Google Scholar
|
|
67
|
Nilius B and Owsianik G: The transient
receptor potential family of ion channels. Genome Biol. 12:2182011.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Venkatachalam K and Montell C: TRP
channels. Annu Rev Biochem. 76:387–417. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhang LL, Yan Liu D, Ma LQ, Luo ZD, Cao
TB, Zhong J, Yan ZC, Wang LJ, Zhao ZG, Zhu SJ, et al: Activation of
transient receptor potential vanilloid type-1 channel prevents
adipogenesis and obesity. Circ Res. 100:1063–1070. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Uchida K, Dezaki K, Yoneshiro T, Watanabe
T, Yamazaki J, Saito M, Yada T, Tominaga M and Iwasaki Y:
Involvement of thermosensitive TRP channels in energy metabolism. J
Physiol Sci. 67:549–560. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Park HW and Lee JH: Calcium channel
blockers as potential therapeutics for obesity-associated autophagy
defects and fatty liver pathologies. Autophagy. 10:2385–2386. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Polyzos SA, Kountouras J and Mantzoros CS:
Obesity and nonalcoholic fatty liver disease: From pathophysiology
to therapeutics. Metabolism. 92:82–97. 2019. View Article : Google Scholar
|
|
73
|
Li J, Li X, Liu D, Zhang S, Tan N, Yokota
H and Zhang P: Phosphorylation of eIF2α signaling pathway
attenuates obesity-induced non-alcoholic fatty liver disease in an
ER stress and autophagy-dependent manner. Cell Death Dis.
11:10692020. View Article : Google Scholar
|
|
74
|
Baffy G: Uncoupling protein-2 and
non-alcoholic fatty liver disease. Front Biosci. 10:2082–2096.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Panchal SK, Bliss E and Brown L: Capsaicin
in metabolic syndrome. Nutrients. 10:6302018. View Article : Google Scholar :
|
|
76
|
Yang L, Li P, Fu S, Calay E and
Hotamisligil GS: Defective hepatic autophagy in obesity promotes ER
stress and causes insulin resistance. Cell Metab. 11:467–478. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Everaerts W, Nilius B and Owsianik G: The
vanilloid transient receptor potential channel TRPV4: From
structure to disease. Prog Biophys Mol Biol. 103:2–17. 2010.
View Article : Google Scholar
|
|
78
|
Ye L, Kleiner S, Wu J, Sah R, Gupta RK,
Banks AS, Cohen P, Khandekar MJ, Boström P, Mepani RJ, et al: TRPV4
is a regulator of adipose oxidative metabolism, inflammation, and
energy homeostasis. Cell. 151:96–110. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhan L, Yang Y, Ma TT, Huang C, Meng XM,
Zhang L and Li J: Transient receptor potential vanilloid 4 inhibits
rat HSC-T6 apoptosis through induction of autophagy. Mol Cell
Biochem. 402:9–22. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Song Y, Zhan L, Yu M, Huang C, Meng X, Ma
T, Zhang L and Li J: TRPV4 channel inhibits TGF-β1-induced
proliferation of hepatic stellate cells. PLoS One. 9:e1011792014.
View Article : Google Scholar
|
|
81
|
Fonfria E, Murdock PR, Cusdin FS, Benham
CD, Kelsell RE and McNulty S: Tissue distribution profiles of the
human TRPM cation channel family. J Recept Signal Transduct Res.
26:159–178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kheradpezhouh E, Ma L, Morphett A, Barritt
GJ and Rychkov GY: TRPM2 channels mediate acetaminophen-induced
liver damage. Proc Natl Acad Sci USA. 111:3176–3181. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Vanderheyden V, Devogelaere B, Missiaen L,
De Smedt H, Bultynck G and Parys JB: Regulation of inositol
1,4,5-trisphosphate-induced Ca2+ release by reversible
phosphorylation and dephosphorylation. Biochim Biophys Acta.
1793:959–970. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Arruda AP, Pers BM, Parlakgül G, Güney E,
Inouye K and Hotamisligil GS: Chronic enrichment of hepatic
endoplasmic reticulum-mitochondria contact leads to mitochondrial
dysfunction in obesity. Nat Med. 20:1427–1435. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Rodrigues MA, Gomes DA, Leite MF, Grant W,
Zhang L, Lam W, Cheng YC, Bennett AM and Nathanson MH:
Nucleoplasmic calcium is required for cell proliferation. J Biol
Chem. 282:17061–17068. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Lemasters JJ and Holmuhamedov E:
Voltage-dependent anion channel (VDAC) as mitochondrial
governator-thinking outside the box. Biochim Biophys Acta.
1762:181–190. 2006. View Article : Google Scholar
|
|
87
|
Shoshan-Barmatz V, De Pinto V,
Zweckstetter M, Raviv Z, Keinan N and Arbel N: VDAC, a
multi-functional mitochondrial protein regulating cell life and
death. Mol Aspects Med. 31:227–285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Pittala S, Krelin Y, Kuperman Y and
Shoshan-Barmatz V: A mitochondrial VDAC1-based peptide greatly
suppresses steatosis and NASH-associated pathologies in a mouse
model. Mol Ther. 27:1848–1862. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Prakriya M and Lewis RS: Store-operated
calcium channels. Physiol Rev. 95:1383–1436. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kappel S, Borgström A, Stoklosa P, Dörr K
and Peinelt C: Store-operated calcium entry in disease: Beyond
STIM/Orai expression levels. Semin Cell Dev Biol. 94:66–73. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Li Y, Ge M, Ciani L, Kuriakose G, Westover
EJ, Dura M, Covey DF, Freed JH, Maxfield FR, Lytton J and Tabas I:
Enrichment of endoplasmic reticulum with cholesterol inhibits
sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in
parallel with increased order of membrane lipids: Implications for
depletion of endoplasmic reticulum calcium stores and apoptosis in
cholesterol-loaded macrophages. J Biol Chem. 279:37030–37039. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Maus M, Cuk M, Patel B, Lian J, Ouimet M,
Kaufmann U, Yang J, Horvath R, Hornig-Do HT,
Chrzanowska-Lightowlers ZM, et al: Store-operated Ca2+
entry controls induction of lipolysis and the transcriptional
reprogramming to lipid metabolism. Cell Metab. 25:698–712. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Jain S and Jacobson KA: Purinergic
signaling in liver pathophysiology. Front Endocrinol (Lausanne).
12:7184292021. View Article : Google Scholar
|
|
94
|
Jiang M, Cui BW, Wu YL, Zhang Y, Shang Y,
Liu J, Yang HX, Qiao CY, Zhan ZY, Ye H, et al: P2X7R orchestrates
the progression of murine hepatic fibrosis by making a feedback
loop from macrophage to hepatic stellate cells. Toxicol Lett.
333:22–32. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Takenouchi T, Nakai M, Iwamaru Y, Sugama
S, Tsukimoto M, Fujita M, Wei J, Sekigawa A, Sato M, Kojima S, et
al: The activation of P2X7 receptor impairs lysosomal functions and
stimulates the release of autophagolysosomes in microglial cells. J
Immunol. 182:2051–2062. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Chatterjee S and Das S: P2X7 receptor as a
key player in oxidative stress-driven cell fate in nonalcoholic
steatohepatitis. Oxid Med Cell Longev. 2015:1724932015. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Wulff H and Castle NA: Therapeutic
potential of KCa3.1 blockers: Recent advances and promising trends.
Expert Rev Clin Pharmacol. 3:385–396. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Alkhouri N, Dixon LJ and Feldstein AE:
Lipotoxicity in nonalcoholic fatty liver disease: Not all lipids
are created equal. Expert Rev Gastroenterol Hepatol. 3:445–451.
2009. View Article : Google Scholar : PubMed/NCBI
|