Introduction
Ischemic heart disease (IHD) is a leading cause of
morbidity and mortality, affecting 112 million individuals
worldwide in 2015 (1). As one of
the main causes of IHD, coronary microvascular dysfunction (CMD)
accounts for approximately two-thirds of clinical conditions
presenting with symptoms and signs of myocardial ischemia without
obstructive coronary disease, including microvascular angina and
myocardial infarction with non-obstructive coronary artery disease
(2). Although CMD has a high
prevalence in IHD and is associated with adverse outcomes, the
diagnosis of CMD remains challenging and the underlying mechanisms
remain poorly understood (3). It
has been previously demonstrated that endothelial dysfunction,
oxidative stress, inflammation and other factors may be involved in
the development of CMD (4). At
present, treatment options for CMD are mainly focused on exploring
anti-inflammatory and anti-anginal pathways (5).
Higher serum free fatty acid (FFA) levels are often
observed in patients with cardiovascular disease and these are
associated with recurrence and poor prognosis (6-8).
Previous studies have demonstrated that a high level of serum FFAs
is an independent risk factor for cardiovascular disease (9,10).
This association is more pronounced in diabetes due to impaired
glucose utilization (11). An
excessive FFA oxidation rate causes abnormal energy metabolism and
myocardial dysfunction in diabetic patients (12,13). In addition, high FFAs levels have
been reported to promote lipid accumulation and lipotoxicity in
cardiomyocytes, which contributes to systemic inflammation,
oxidative stress and eventual cardiomyocyte apoptosis (14,15). Our previous study demonstrated
that a CMD mouse model could be successfully established via lipid
emulsion infusion, which resulted in an increase in the serum FFA
level (16). However, the
underlying mechanism remained unclear. AMP-activated protein kinase
(AMPK) is an essential component of the adaptive response to
cardiomyocyte stress that occurs during myocardial ischemia
(17). AMPK modulates fatty acid
metabolism in the ischemic heart, and activated AMPK appears to be
protective in reducing myocardial necrosis during
ischemia-reperfusion (18). Its
activation has also been revealed to improve exercise performance
and peripheral vascular insufficiency in mice fed on a high-fat
diet (19). AMPK also regulates
the downstream transcription factor, Krüppel-like factor 2 (KLF2),
which is expressed in endothelial cells and has the role of
preserving various endothelial functions (20).
The AMPK/KLF2 axis activates the expression of
endothelial nitric oxide (NO) synthase (eNOS) in endothelial cells,
subsequently promoting the release of vasodilator-NO (21). Endothelial cell-derived NO serves
key roles in vasodilation, inhibition of platelet aggregation and
reduction of leukocyte adhesion, and as an antioxidant (22). Activation of the AMPK/KLF2/eNOS
signaling pathway has been demonstrated to improve endothelial
dysfunction and slow down the progression of atherosclerosis,
pulmonary arterial hypertension and other diseases (23-25).
Cardiac microvascular endothelial cells (CMECs)
serve a key role in maintaining normal coronary circulation
(26). A previously published
study suggested that a high-fat diet could inhibit AMPK expression
(27), thus we hypothesized that
the AMPK pathway may be involved in the detrimental effects of FFAs
on CMECs.
Therefore, the aim of the present study was to
explore the AMPK/KLF2/eNOS signaling axis both in vivo (in a
CMD mouse model established by lipid infusion) and in vitro
[in CMECs treated with palmitic acid (PA)], and to investigate the
underlying mechanism.
Materials and methods
Animals and reagents
A total of 45 C57BL/6J mice (male; 6-8 weeks old;
weight, 20±2 g) were obtained from the Shanghai Jihui Laboratory
Animal Care Co., Ltd. All mice were housed individually and were
maintained at 22°C with 40-60% humidity and a 12-h light/dark cycle
starting at 6:00 am. The mice were fed with normal chow and had
free access to food and water throughout the experiment. Nicorandil
was purchased from Beijing Sihuan Kebao Pharmaceutical Co., Ltd.
FFAs and reactive oxygen species (ROS) assay kits were obtained
from Beijing Solarbio Science & Technology Co., Ltd. The
anti-GAPDH antibody was obtained from Abcam, whereas the anti-KLF2
antibody was purchased from BIOSS, anti-p-eNOS antibody was
purchased from Affinity Biosciences, and the anti-AMPK,
anti-phosphorylated (p-)AMPK and anti-eNOS antibodies were obtained
from Cell Signaling Technology, Inc. The lipid emulsion was
purchased from Fresenius Kabi Huarui Pharmaceutical Co., Ltd.
Anti-CD62L and anti-CD11b antibodies were purchased from BD
Biosciences. Mouse interferon-γ (IFN-γ) ELISA kit (cat. no.
F10660), mouse interleukin-6 (IL-6) ELISA kit (cat. no. F10830),
mouse NO ELISA kit (cat. no. F11321), mouse superoxide dismutase
(SOD) ELISA kit (cat. no. F11502) and mouse tumor necrosis factor-α
(TNF-α) ELISA kit (cat. no. F11630) were obtained from Shanghai
Xitang Biotechnology Co., Ltd. The AMPK activator,
5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), was
purchased from MedChemExpress. The Cell Counting Kit-8 (CCK-8)
assay kit was obtained from Shanghai DODGEN Chemical Technology
Co., Ltd. PA was obtained from Sigma-Aldrich; Merck KGaA. The KLF2
overexpression plasmid was obtained from Shanghai GeneChem Co.,
Ltd. Finally, Lipofectamine® 3000 transfection reagent
was obtained from Thermo Fisher Scientific, Inc.
The entire animal experimental protocol used in the
present study was carefully examined and approved by the Animal
Experiment Ethics Committee of the Second Affiliated Hospital of
Naval Medical University (approval no. 2020SL037; Shanghai, China).
All animal care procedures were carried out in accordance with the
Animal Research: Reporting of In Vivo Experiments guidelines
on animal research (28).
Establishment of the CMD model
A total of 30 male C57BL/6J mice were randomly
assigned to the control, model and nicorandil groups (n=10 mice in
each group). Normal saline solution (200 µl once per day)
was administered to the mice in the control and model groups via
intraperitoneal (i.p.) injection for 3 days, whereas the mice in
the nicorandil group were treated with an injection (i.p.) of
nicorandil (200 µl nicorandil at a concentration of 1.95
mg/kg once per day for 3 days). Nicorandil, a drug for CMD, is used
to stimulate the upregulation of eNOS (29) and was used in the present study to
investigate the AMPK/KLF2/eNOS signaling pathway. As shown in
Fig. 1A, the mice were
subsequently anaesthetized with sodium pentobarbital (40 mg/kg),
and the jugular vein was catheterized using a microcatheter.
Afterwards, the mice underwent a 6-h infusion with either normal
saline or lipid emulsion combined with heparin using a micro-pump
(the lipid component was 20% Intralipid®; Fresenius Kabi
Huarui Pharmaceutical Co., Ltd.; heparin was infused at a
concentration 20 U/ml, and at a rate of 0.1 ml/h).
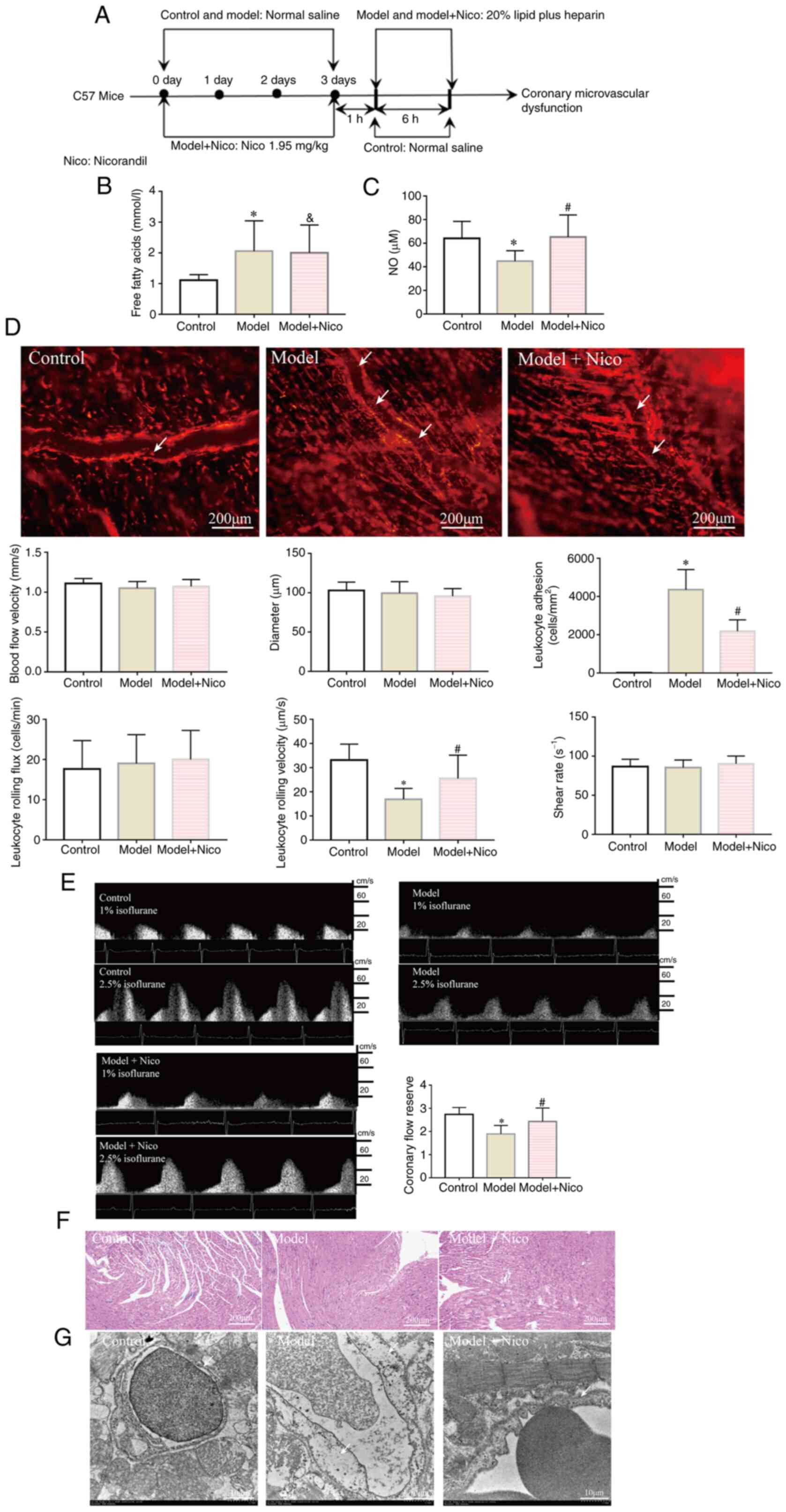 | Figure 1Lipid infusion induces CMD, whereas
treatment with nicorandil improves CMD. (A) Flow chart of the
protocol for the animal experiments. (B) Serum free fatty acid
levels were increased in the Model and Model + Nico groups. (C)
Serum NO levels were decreased in the Model group, whereas the NO
levels were increased in the Model + Nico group. (D) Lipid
infusion-activated leukocytes in cremaster microcirculation (white
arrows indicate the leukocytes that adhered to the cremaster
microvascular wall). Scale bar, 200 µm. (E) Coronary flow
reserve was decreased in the Model group. (F) Images captured under
a light microscope (magnification, ×200; scale bar, 200 µm).
(G) Edema of endothelial cells (white arrow; scale bar, 10
µm). Data are presented as the mean ± SD.
*P<0.05, Model (n=10) vs. Control (n=10);
#P<0.05, Model + Nico (n=10) vs. Model (n=10);
&P<0.05, Model + Nico (n=10) vs. Control (n=10).
CMD, coronary microvascular dysfunction; Nico, nicorandil; NO,
nitric oxide. |
Coronary flow reserve (CFR)
measurement
Following lipid infusion, the mice were immediately
anaesthetized using isoflurane (3% for induction and 1-2.5% for
maintenance). Hyperemic flow was achieved by increasing the
percentage of isoflurane to 2.5%, whereas the baseline flow was
achieved by reducing the isoflurane percentage to 1% (30). Data were measured using a Doppler
signal processing workstation. The CFR was calculated according to
the following formula:
CFR=P/R=Vhigh/Vlow.(where P is the peak blood
flow, R is the resting blood flow, Vhigh is the
hyperemic blood flow velocity and Vlow is the baseline
blood flow velocity).
Cremaster microcirculation
assessment
Mice were anesthetized with sodium pentobarbital (40
mg/kg) immediately after the lipid infusion. Intravital microscopy
(Olympus BX51 WI upright microscope; Olympus Corporation) was used
to record the cremaster microvascular blood flow and leukocyte
adhesion in venules. Leukocyte flux and leukocyte endothelium
interactions (rolling and adhesion) analysis were conducted as
described previously (31).
Briefly, rolling leukocyte flux was measured at the indicated time
points by counting the number of rolling leukocytes per 20 sec that
passed a reference point in the microvessel and the number was then
presented as units of cells/min. Leukocyte rolling velocity was
calculated by the velocity of 10 leukocytes rolling along the
endothelial cell lining (mm/sec) and leukocyte adhesion (defined as
being stationary for 20 sec) was counted in 100-µm-long
vascular segments, and presented as the number of adherent
cells/mm2. Diameters (in mm) were measured
perpendicularly to the vessel path. Finally, the venular wall shear
rate was calculated based on the Newtonian definition, according to
the following formula: Wall shear rate (sec−1)=8 × [red
blood cell velocity (µm·sec−1)/venular diameter
(µm)] (32).
AICAR intervention
A total of 15 male mice were randomly assigned to
control (n=5), model (n=5) and AICAR (n=5) groups. Normal saline
solution (200 µl once per day) was administered to the mice
in the control and model groups via i.p. injection for 3 days. Mice
in the AICAR group were treated with 200 µl AICAR
(concentration, 0.5 mg/g) by i.p. injection for 3 days prior to
lipid infusion (once per day for 3 days). Subsequently,
microvascular dysfunction was induced in the model and AICAR groups
by lipid infusion as aforementioned. Cultured CMECs in vitro
were treated with AICAR (500 µM) for 3 h at 37°C, then 400
µM PA was added and cells were incubated at 37°C for 24
h.
Heart histological examination and
ultrastructural analysis
Mice were sacrificed by cervical dislocation while
under anesthesia induced by an i.p. injection of 40 mg/kg
pentobarbital, and respiratory arrest was used to confirm animal
death. Fresh heart sections were fixed in 4% paraformaldehyde at
room temperature for 24 h, embedded in paraffin and serial
5-µm-thick sections were prepared. Morphological analysis of
cardiomyocytes and microvessels was subsequently performed using
H&E staining. Then, the sections were stained with hematoxylin
for 5 min at room temperature, then washed with running water for 5
min and differentiated in 1% acid alcohol for 10 sec at room
temperature. The sections were then stained with 1% eosin for 1
min, washed with running water and dehydrated in increasing
concentrations of alcohol (70, 80, 90 and 100%) and cleared in
xylene. Finally, H&E staining was observed under a BX43 light
microscope (Olympus Corporation).
For the immunohistochemical analysis of KLF2, eNOS
and p-eNOS, fresh heart sections were fixed in 4% paraformaldehyde
at room temperature for 24 h, embedded in paraffin and serial
5-µm-thick sections were prepared. The sections were dewaxed
in xylene, then rehydrated in a descending alcohol series (hydrated
in 100, 95 and 80% ethanol and water for 5 min each). Antigen
retrieval was performed by pre-treatment of the slides in citrate
buffer (pH 6.0) (cat. no. G1202; Wuhan Servicebio Technology Co.,
Ltd.) in a microwave (720 W heating) oven for 12 min. Thereafter,
the slides were cooled to room temperature in deionized water for 5
min. The sections were incubated in 0.3%
H2O2-methanol solution for 10 min and then
washed with PBS three times. To block endogenous peroxidase
activity, 50 µl peroxidase blocking solution was added to
each section and these were incubated for 10 min at room
temperature. Each section was then incubated with 50 µl 10%
nonimmune goat serum (OriGene Technologies, Inc.) for 10 min at
room temperature, rinsed with PBS and incubated with 50 µl
primary antibody [KLF2 (dilution, 1:400; cat. no. bs-2772R; BIOSS),
eNOS (dilution, 1:1,000; cat. no. GB12086; Wuhan Servicebio
Technology Co., Ltd.) and p-eNOS (diluted 1:100; cat. no. AF3247;
Affinity Biosciences)] at 4°C overnight. The sections were then
incubated with 50 µl secondary antibody [HRP conjugated goat
anti-rabbit IgG (dilution, 1:200; cat. no. GB23303; Wuhan
Servicebio Technology Co., Ltd.) or HRP-conjugated goat anti-mouse
IgG (diluted 1:200; cat. no. GB23301; Wuhan Servicebio Technology
Co., Ltd.)] at room temperature for 10 min. The reaction was
developed with freshly prepared 3,3′-diaminobenzidine, and observed
under a Leica DM4000 microscope (Leica Microsystems GmbH) for 3-10
min, with brown indicating positive staining. The slides were then
counterstained with hematoxylin for 3 min at room temperature,
washed with running water, and dehydrated in increasing
concentrations of alcohol (75, 85 and 100%) and cleared in xylene.
The pathological and immunohistochemical changes were evaluated and
photographed under a BX43 light microscope (Olympus Corporation).
Data analysis was performed using Multiplex IHC v2.2.0 module
analysis software (Indica Labs, Inc.).
Heart samples were fixed in 2.5% glutaraldehyde in
0.1 mol/l cacodylate buffer at 4°C overnight, fixed in 1% osmium
tetroxide at 4°C for 2 h and then embedded in Epon using an
Embed-812 kit (Electron Microscopy Sciences) at room temperature
overnight. Ultrathin sections (80-100 nm) were obtained from
selected areas using an ultrathin microtome (Leica EM UC7; Leica
Microsystems GmbH). The ultrathin sections of the cubes were
stained with 3% uranyl acetate and lead citrate at 25°C for 30 min.
Then, the sections were examined using transmission electron
microscopy (TEM; Tecnai™ G2 20; FEI; Thermo Fisher Scientific,
Inc.).
ELISAs and measurements of plasma
FFAs
Blood samples (100 µl) were collected from
the tail-veins of the mice in heparin-containing tubes, and plasma
was prepared after centrifugation at 4,500 × g for 10 min at room
temperature. Subsequently, aliquots were stored at -80°C until use.
The levels of plasma biomarkers, including IFN-γ, IL-6, NO, SOD and
TNF-α, were determined using ELISA kits (Shanghai Xitang
Biotechnology Co., Ltd.). FFAs were measured using an FFA Content
Assay Kit (cat. no. BC0595; Beijing Solarbio Science &
Technology Co., Ltd.) was conducted according to the manufacturer's
user guide. The preparation of reagent1 was accomplished following
the ratio of n-heptane: absolute methanol: chloroform=24:1:25 at
room temperature. The physical state of diphenylcarbazide was
powder, absolute ethanol was added at the time of application and
the liquid volume was 13 ml. The standard solution was
chloroform-dissolved 5 µmol/ml palmitic acid solution. The
microplate reader was preheated for 30 min, the wavelength was
adjusted to 550 nm, and absolute ethanol was used as a blank (set
to 0). Copper reagent was water-bathed at 37°C for 30 min. Standard
solution was diluted to 0.05, 0.1, 0.2, 0.4, 0.6, 0.8 and 1
µmol/ml with chloroform. Each 1.5-ml Eppendorf tube was
first supplemented with 10 µl distilled water, sample,
chloroform or standard solution. Then 100 µl reagent1 and 40
µl copper reagent were added sequentially, followed by
shaking for 10 min at room temperature and finally centrifugation
at 850 g for 10 min at room temperature. A total of 50 µl of
the supernatant was transferred to a 1.5-ml Eppendorf tube and 200
µl diphenylcarbazide was added, followed by shaking for 2
min and standing for 15 min at room temperature. Absorbance at 550
nm of each well of a 96-well plate was measured using a microplate
reader (200 µl liquid volume was added per well). FFA
concentrations were calculated for each plasma sample according to
the standard curve.
Measurement of intracellular ROS in
leukocytes and CMECs
The intracellular levels of ROS were measured by a
fluorometric assay using the probe 2′-7′-dichlorodihydrofluorescin
diacetate (DCFH-DA). Cells were resuspended in 2 ml PBS
(106 cells/ml) and were subsequently incubated with 10
µM DCFH-DA at 37°C for 20 min with continuous shaking. Cells
were washed with PBS three times (2 min each at room temperature)
and were transferred from a tube to a microplate prior to
fluorescence being measured. To minimize the process of
photo-oxidation, samples were stored in the dark. Finally, DCF
fluorescence was measured using a microplate reader (488 nm
excitation and 525 nm emission wavelengths).
Flow cytometric analysis of
leukocytes
Blood (100 µl) was drawn from the great
saphenous vein of the mice after lipid infusion. Ammonium
chloride-potassium lysis buffer was used for erythrocyte removal.
Blood samples were centrifuged at room temperature for 10 min at
850 × g, and the plasma was subsequently removed. Leukocytes were
resuspended in 20 µl normal saline and then incubated with
the appropriate antibodies as previously described (33). Phycoerythrin rat anti-mouse CD11b
(cat. no. 557397) and allophycocyanin rat CD62L (cat. no. 5333152)
antibodies (BD Biosciences) were diluted at a ratio of 1:5. Flow
cytometry was performed using a Cytek DXP 8 Color upgraded BD FACS
Calibur™ flow cytometer (BD Biosciences) by staining with
appropriate anti-bodies, and the data were analyzed using FlowJo
10.0 software (Tree Star, Inc.).
Western blot analysis of heart tissue and
CMECs
Heart tissue (50 mg) and CMECs were collected for
quantitative analysis of the proteins. Proteins were extracted
using RIPA lysis buffer [Hangzhou Multi Sciences (Lianke) Biotech
Co., Ltd.]. Protein concentrations of heart tissues and CMECs were
subsequently determined using a Takara Bradford Protein Assay kit
(Takara Bio, Inc.). The proteins were diluted to a final protein
concentration of 2.5 µg/µl in 1X loading buffer
(Wuhan Servicebio Technology Co., Ltd.), and 25 µg protein
was loaded onto each lane of the western blot gel (10%). After the
proteins were separated, they were transferred to polyvinylidene
fluoride membranes (Bio-Rad Laboratories, Inc.), which were blocked
with 5% BSA (Shanghai Siding Biotechnology Co., Ltd.) for 2 h at
room temperature. The membrane was incubated with the corresponding
primary antibody overnight at 4°C. The following day, HRP-labeled
secondary antibodies (cat. no. 98164; Cell Signaling Technology,
Inc.) were incubated with the membranes at room temperature for 2
h. The dilution of rabbit anti-KLF2 polyclonal antibody (cat. no.
bs-2772R; BIOSS) was 1:300, and the dilution of rabbit anti GAPDH
monoclonal antibody (cat. no. ab181602; Abcam) was 1:10,000. The
dilution of rabbit anti-eNOS (cat. no. 32027; Cell Signaling
Technology, Inc.), anti-AMPK (cat. no. 5832; Cell Signaling
Technology, Inc.), anti-p-eNOS (Ser1177) (cat. no. AF3247; Affinity
Biosciences) and anti-p-AMPK (Thr172) (cat. no. 50081; Cell
Signaling Technology, Inc.) polyclonal antibody was 1:1,000.
HRP-labeled secondary antibodies were used at a dilution of
1:5,000. All antibodies were diluted using TBS with 0.1% Tween-20
(Wuhan Servicebio Technology Co., Ltd.). Enhanced chemiluminescence
western blotting substrate (Wuhan Servicebio Technology Co., Ltd.)
was used to detect the immuno-reactive signals. The protein gray
values were analyzed using ImageJ 1.6 software (National Institutes
of Health).
Isolation and identification of
CMECs
Primary mice CMECs were purchased from Saibaikang
(Shanghai) Biotechnology Co., Ltd. and cultured in Complete™
Endothelial Cell Medium (ECM; Sigma-Aldrich; Merck KGaA) containing
10% FBS (ScienCell Research Laboratories. Inc.), 1% endothelial
cell growth supplement and 1% penicillin/streptomycin at 37°C in an
atmosphere of 5% CO2. CMECs were identified by
immunofluorescence using an antibody against von Willebrand factor
VIII, which is constitutively expressed on the surface of CMECs
(34) (Fig. S1). In order to identify the
CMECs, cells were seeded in 24-well plates (2.0×104
cells per well). The cells were then washed twice with PBS and
fixed in ice-cold 100% methanol for 15 min at -20°C. After rinsing
with PBS three times for 5 min each, the samples were incubated
with 0.1% Triton X-100 at room temperature for 10 min, blocked with
1% BSA at room temperature for 60 min, and incubated with rabbit
anti-vWF polyclonal antibody (1:200; cat. no. 11778-1-AP;
Proteintech Group, Inc.) at 4°C overnight. The samples were then
washed three times with PBS, followed by incubation with Alexa
Fluor 488-conjugated secondary antibodies (1:500; cat. no.
SA00006-2; Proteintech Group, Inc.) at room temperature for 120 min
after 4′,6-diamidino-2-phenylindole counterstaining at room
temperature for 5 min. Images were captured with a fluorescence
microscope system (DS-Ri2; Nikon Corporation). The Image-Pro Plus
6.0 image analysis software (Media Cybernetics, Inc.) was used for
quantification analysis.
Determination of viability of CMECs
PA was dissolved in sodium hydroxide solution. Then,
10% BSA was added to the solution in order to form a PA and BSA
complex. The solution was filter-sterilized, and then diluted with
1% BSA to prepare different concentrations of PA solutions, which
were adjusted to pH 7.4. CMECs were seeded in 96-well plates (5,000
cells per well) and subjected to treatment with different
concentrations (0, 100, 200, 400 and 800 µM) of PA with or
without nicorandil (100 µM) at 37°C for 24 h. Subsequently,
10 µl CCK-8 solution was added to the cell culture and the
cells were incubated for 2 h at 37°C in a humidifier (containing 5%
CO2 and 95% O2). Finally, cell viability was
determined at an optical density of 450 nm using a microplate
reader.
Cell transfection
KLF2 overexpression and empty control pCMV plasmids
were purchased from Shanghai GeneChem Co., Ltd. CMECs were seeded
in 6-well plates (1.0×105 cells per well) for 24 h prior
to transfection. Cells were divided into the control group, control
overexpression group and KLF2 overexpression group. Transient
transfection was performed using Invitrogen
Lipofectamine® 3000 (Thermo Fisher Scientific, Inc.)
according to the standard protocol of the manufacturer. P3000
(Thermo Fisher Scientific, Inc.) combined with the
overexpression-DNA (2.5 µg) or the control
overexpression-DNA vectors (2.5 µg) was added to serum-free
medium and incubated with the cells at 25°C for 10 min, while the
control group was treated without vectors. Subsequently,
Lipofectamine 3000 was added to each group and cultured with the
cells in serum-free ECM. Following 6 h of culture (37°C; 5%
CO2), the medium was replaced with ECM containing 10%
FBS. After 48 h, subsequent experiments were performed.
Small interfering RNAs targeting KLF2 (siKLF2) and
the corresponding non-specific control (NC) siRNA were obtained
from Sigma-Aldrich; Merck KGaA. Cells were divided into the control
group, NC group and siKLF2 group. The control group was treated
without siRNAs. The transfection procedures were performed
according to the protocol described by Wang et al (35). siRNA (100 nM) was used for cell
transfection using Lipofectamine® 3000 (Invitrogen;
Thermo Fisher Scientific, Inc.). Cells were incubated at 37°C with
5% CO2. After a 6-h antibiotic-free medium incubation,
the transfection medium was removed, and the cells were incubated
in fresh medium for 24 h. Then, subsequent experiments were
performed. All siRNA sequences are shown in Table SI. The transfection of KLF2
overexpression vector and siKLF2 was successful and KLF2 expression
was increased and decreased, respectively (Fig. S2).
Statistical analysis
All experiments were performed at least three times,
and all data are presented as the mean ± standard deviation.
One-way analysis of variance was used to compare the differences
among groups, and Tukey's post hoc test was used for multiple
comparisons. All the statistical analyses were performed using IBM
SPSS 25.0 software (IBM Corp.). P<0.05 (two-sided) was
considered to indicate a statistically significant difference.
Results
Lipid infusion induces CMD
The CMD mouse model was successfully established by
lipid infusion, which was characterized by a reduction of the CFR
(model group vs. control group, 1.89±0.37 vs. 2.74±0.30; Fig. 1E). Serum tests revealed that the
levels of FFAs in model mice were significantly increased, whereas
the concentration of NO was decreased compared with those in
control mice (Fig. 1B and C).
Leukocyte activation in the cremaster microvascular wall was
subsequently investigated (Fig.
1D). Lipid infusion led to a significantly increased level of
leukocyte adhesion to the cremaster microvascular wall (model group
vs. control group, 4,350±1,057.5 vs. 11.8±5.4 cells/mm2)
and a decreased leukocyte rolling velocity. However, the
microvascular blood flow velocity, vascular diameter, leukocyte
rolling flux and shear stress were found not to be impacted by
lipid infusion.
To confirm whether lipid infusion caused structural
changes in the mouse heart, the cardiac tissue was examined using
H&E staining and TEM. No obvious structural changes were
observed under the light microscope (Fig. 1F). However, TEM revealed severely
swollen endothelial cells in the model group (Fig. 1G), demonstrating the damage of
coronary microvascular ultrastructure.
To further investigate the effects of FFAs on CMECs,
different concentrations of PA (0, 100, 200, 400 and 800 µM)
were used to treat cultured CMECs in vitro, which revealed
that 400 or 800 µM PA suppressed cell viability and
increased ROS production in a dose-dependent manner (Fig. S3). Treatment with PA at a
concentration of 400 µM for 24 h led to a reduction in cell
viability of nearly 40% (Fig.
S3A). Collectively, these results suggested that FFAs could
induce CMD by damaging CMECs.
Nicorandil was found to alleviate CMD induced by
lipid infusion, as demonstrated by an increase in the CFR and
leukocyte rolling velocity, a decrease in the level of leukocyte
adhesion and the improved endothelial damage (Fig. 1), the underlying mechanism of
which was hypothesized to be associated with the upregulation of
NO.
Lipid infusion induces an abnormal acute
inflammatory response
Subsequently, the effects of lipid infusion on the
inflammatory response were examined. Expression levels of the
leukocyte adhesion molecule CD11b (Fig. 2A and B) and intracellular ROS
levels (Fig. 2D) were found to be
increased in the model mice, while the expression levels of CD62L
on leukocytes were not altered (Fig.
2A and C). Subsequently, the levels of serum inflammatory
markers were examined using ELISA. Higher serum levels of TNF-α and
IL-6 were identified in the model group (Fig. 2F and G), whereas the serum SOD and
IFN-γ levels were not significantly different between the model and
control groups (Fig. 2E and H).
Experiments using the AMPK activator, AICAR, also revealed
anti-inflammatory effects with resultant reduced serum levels of
TNF-α and IL-6 identified in AICAR-treated model mice (Fig. S4).
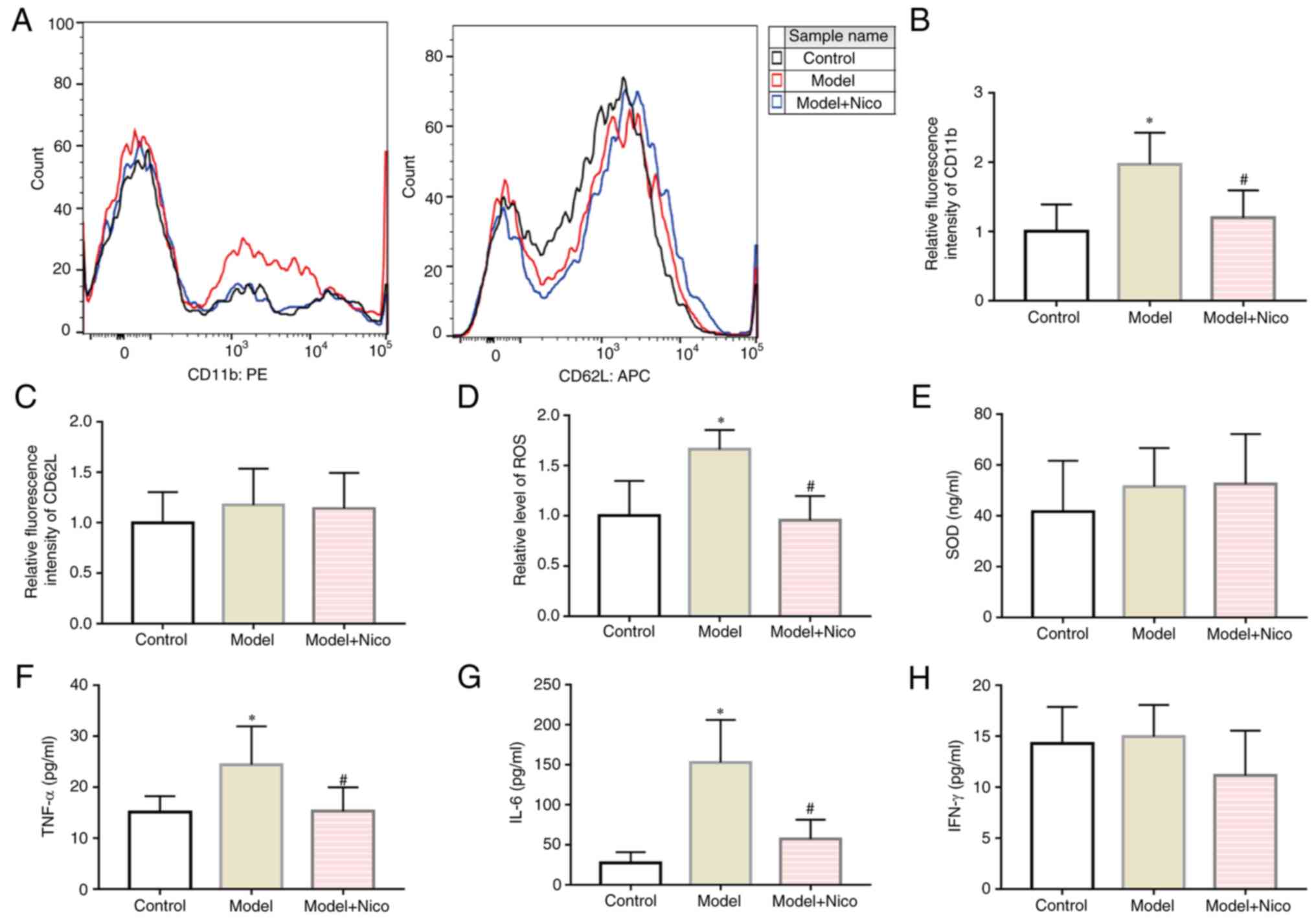 | Figure 2Lipid infusion upregulates CD11b
expression in leukocytes, and exacerbates inflammation, whereas
nicorandil treatment exerts anti-inflammatory effects. (A) Flow
cytometric analysis of CD11b and CD62L. (B) CD11b expression on
leukocytes relative to control group. (C) CD62L expression on
leukocytes relative to control group. (D) Intracellular ROS levels
of leukocytes relative to the control group. Serum (E) SOD, (F)
TNF-α, (G) IL-6 and (H) IFN-γ levels. Data are presented as the
mean ± SD. *P<0.05, Model (n=10) vs. Control (n=10);
#P<0.05, Model + Nico (n=10) vs. Model (n=10). PE,
phycoerythrin; APC, allophycocyanin; Nico, nicorandil; SOD,
superoxide dismutase; ROS, reactive oxygen species. |
Lipid infusion inhibits the
AMPK/KLF2/eNOS pathway
To assess the underlying mechanistic basis whereby
FFAs could induce CMD, the AMPK/KLF2/eNOS pathway was subsequently
analyzed via immunohistochemical and western blot analyses.
Activation of AMPK and eNOS was assessed by detecting the levels of
AMPK phosphorylation at Thr172 and eNOS phosphorylation at Ser117.
As shown in Fig. 3A and B, lipid
infusion led to downregulation of the expression level of KLF-2 and
a decrease of the p-AMPK/AMPK and p-eNOS/eNOS ratios in the
myocardium of model mice. Treatment of CMECs with 200, 400 and 800
µM PA in vitro caused significant suppression of the
expression level of KLF2 and a decrease of the p-AMPK/AMPK and
p-eNOS/eNOS ratios in a dose-dependent manner (Fig. 3C). Nicorandil increased the ratio
of p-eNOS/eNOS and did not influence the expression of KLF2 or the
ratio of p-AMPK/AMPK both in vivo (Fig. 3A and B) and in vitro
(Fig. 3C). Nicorandil also
improved CMEC viability and oxidative stress induced by PA
(Fig. S5).
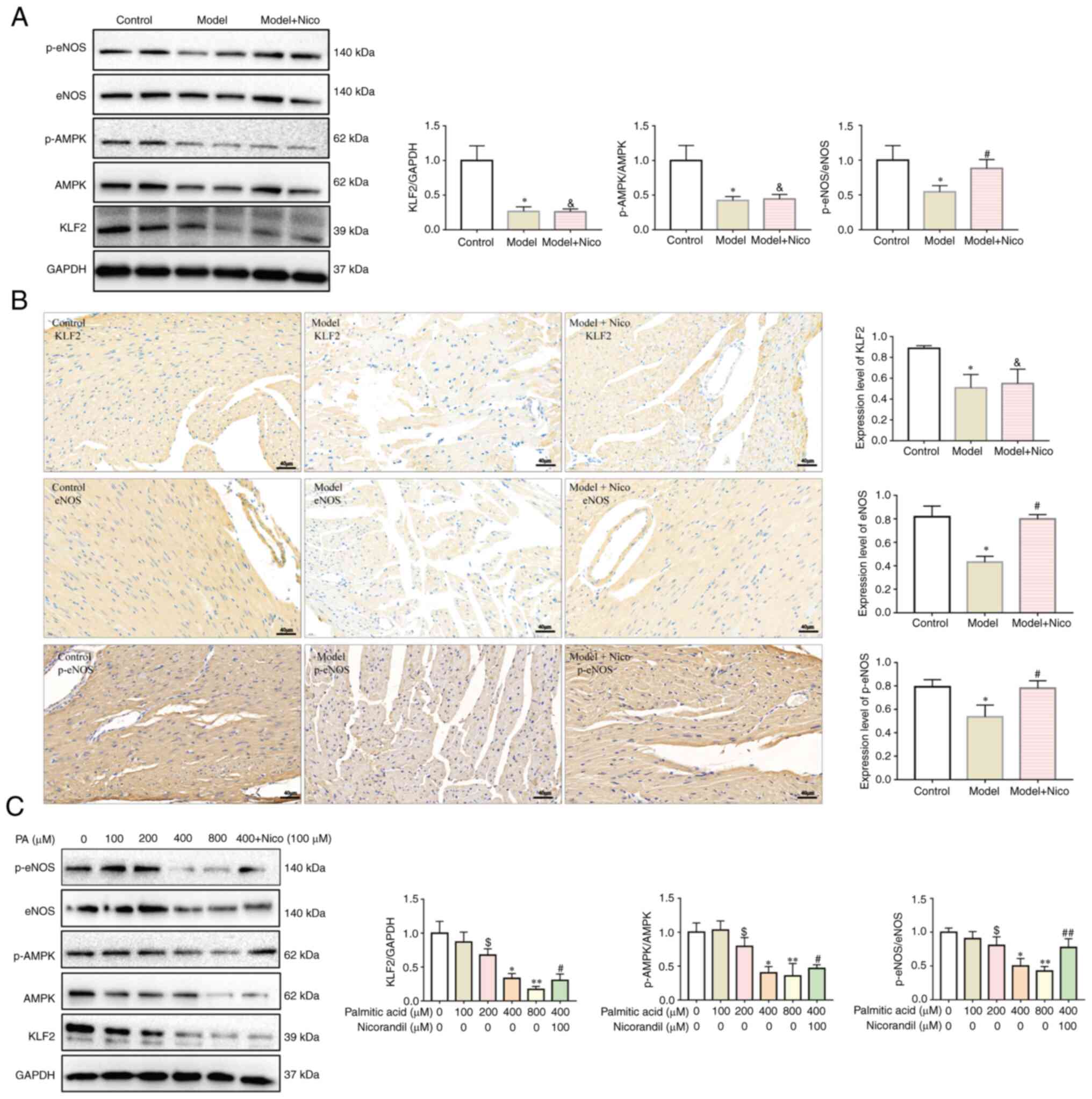 | Figure 3Lipid infusion inhibits the
AMPK/KLF2/eNOS signaling pathway, whereas nicorandil treatment
upregulates eNOS expression. (A) Expression levels of KLF2,
p-AMPK/AMPK and p-eNOS/eNOS were decreased in the heart tissues of
model mice, as assessed by western blotting. Data are presented as
the mean ± SD. *P<0.05, Model (n=6) vs. Control
(n=6). #P<0.05, Model + Nico (n=6) vs. Model (n=6).
&P<0.05, Model + Nico (n=6) vs. Control (n=6).
(B) Expression levels of KLF2, eNOS and p-eNOS were decreased in
heart tissues of model mice, as assessed by immunohistochemistry.
Scale bar, 40 µm. Data are presented as the mean ± SD.
*P<0.05, Model (n=5) vs. Control (n=5).
#P<0.05, Model + Nico (n=5) vs. Model (n=5).
&P<0.05, Model + Nico (n=5) vs. Control (n=5).
(C) Expression levels of KLF2, and ratios of p-AMPK/AMPK and
p-eNOS/eNOS were decreased in cardiac microvascular endothelial
cells treated with PA in a dose-dependent manner, as assessed by
western blotting. Data are presented as the mean ± SD.
$P<0.05, PA, 200 µM (n=6) vs. PA, 0 µM
(n=6). *P<0.05, PA, 400 µM (n=6) vs. PA, 0
µM (n=6). **P<0.05, PA, 800 µM (n=6)
vs. PA, 0 µM (n=6). #P<0.05, PA, 400 µM
+ Nicorandil, 100 µM (n=6) vs. PA, 0 µM (n=6).
##P<0.05, PA, 400 µM + Nicorandil, 100
µM (n=6) vs. PA, 400 µM (n=6). AMPK, AMP-activated
protein kinase; eNOS, endothelial nitric oxide synthase; KLF2,
Krüppel-like factor 2; Nico, nicorandil; p-, phosphorylated; PA,
palmitic acid. |
AMPK/KLF2/eNOS signaling pathway exerts
an important role in regulating microvascular function
To further demonstrate that inhibition of the
AMPK/KLF2/eNOS signaling pathway could account for CMD induced by
FFAs, AICAR was utilized to confirm whether activating AMPK could
improve the condition of CMD in model mice. As shown in Fig. 4G and H, AICAR intervention in
vivo did lead to an improvement in CFR and a decrease in
leukocyte adhesion compared with those in model mice. In
vitro, AICAR was found to enhance the viability of the CMECs
and to inhibit oxidative stress (Fig.
4B and C). AICAR also led to an increase in the expression of
both AMPK and the downstream proteins, KLF2 and eNOS, in CMECs
(Fig. 4A). Subsequently, KLF2
overexpression plasmid was used to evaluate the effect of KLF2 on
the phosphorylation of eNOS. As shown in Fig. S2A, KLF2 was found to be
overexpressed. KLF2 overexpression, in itself, led to significant
upregulation of eNOS in CMECs treated with PA, although it exerted
no significant effects on AMPK (Fig.
4D). Either upregulation of KLF2 with the overexpression
plasmid or increasing the concentration of NO with nicorandil led
to both an improvement in the viability of CMECs and inhibition of
oxidative stress (Figs. 4E and F,
and S5). However, when siKLF2
was used to suppress KLF2 expression in CMECs, AICAR did increase
the ratio of p-AMPK/AMPK but did not increase the expression of
KLF2 or the ratio of p-eNOS/eNOS (Fig. S6). Taken together, the underlying
mechanism through which FFAs induce CMD may involve disruption of
the AMPK/KLF2/eNOS signaling pathway.
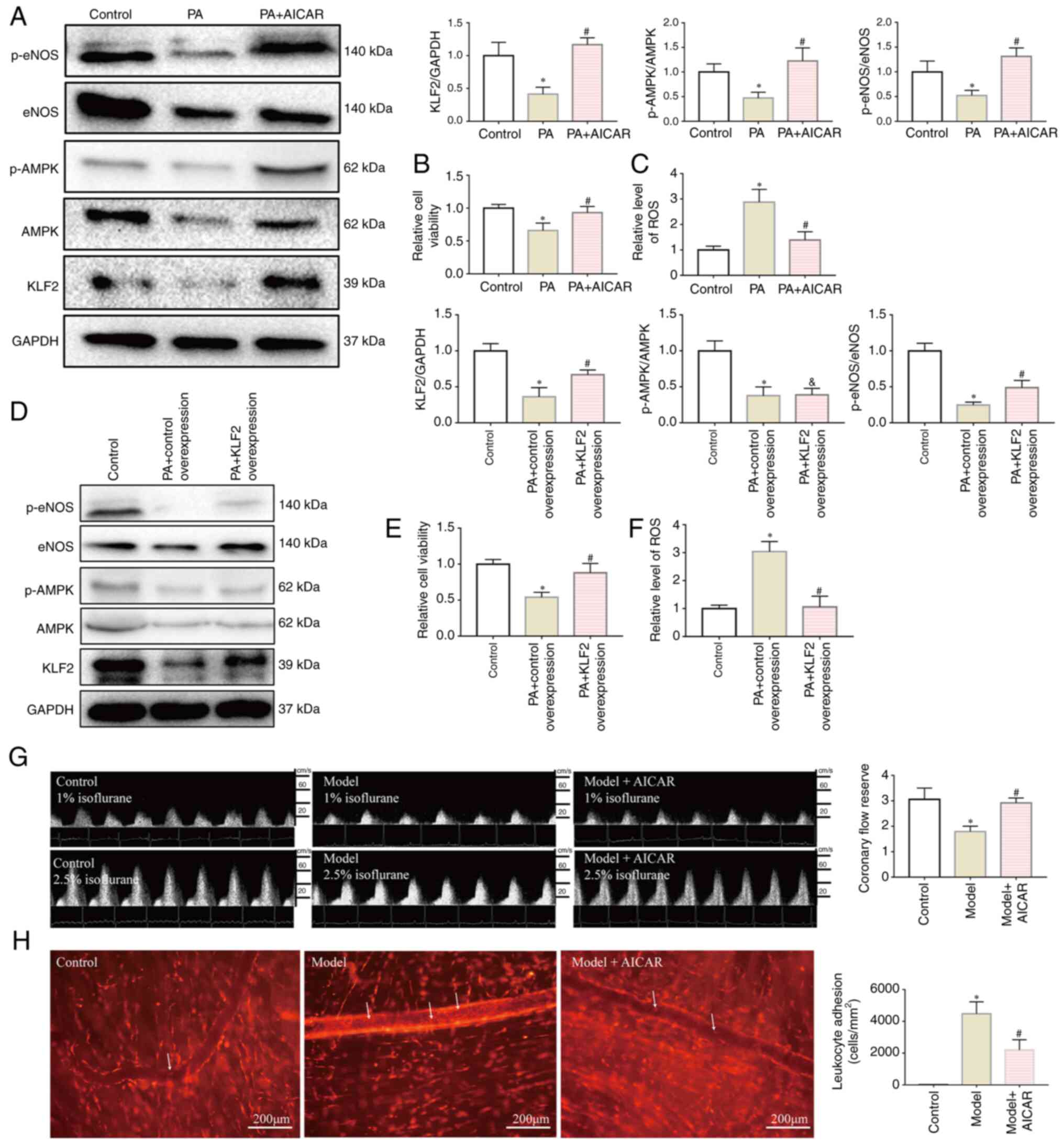 | Figure 4AMPK/KLF2/eNOS pathway serves an
important role in regulating microvascular function. (A) AICAR
activated the AMPK/KLF2/eNOS pathway in CMECs. AICAR treatment
upregulated the levels of KLF2, p-AMPK/AMPK and p-eNOS/eNOS. Data
are presented as the mean ± SD. *P<0.05, PA (n=6) vs.
Control (n=6). #P<0.05; PA + AICAR (n=6) vs. PA
(n=6). (B and C) AICAR restored the viability of CMECs and caused a
decrease in the levels of intracellular ROS. (B) Cell viability
relative to the control group. (C) ROS level relative to the
control group. Data are presented as the mean ± SD.
*P<0.05, PA (n=6) vs. Control (n=6);
#P<0.05, PA + AICAR (n=6) vs. PA (n=6). (D) KLF2
overexpression activated eNOS in CMECs and did not influence the
expression of AMPK. Data are presented as the mean ± SD.
*P<0.05, PA + Control Overexpression (n=6) vs.
Control (n=6); #P<0.05, PA + KLF2 Overexpression
(n=6) vs. PA + Control Overexpression (n=6);
&P<0.05, PA + KLF2 Overexpression (n=6) vs.
Control (n=6). (E and F) KLF2 overexpression restored the viability
of the CMECs and led to a decrease in intracellular ROS. (E) Cell
viability relative to the control group. (F) ROS level relative to
the control group. Data are presented as the mean ± SD.
*P<0.05, PA + Control Overexpression (n=6) vs.
Control (n=6); #P<0.05, PA + KLF2 Overexpression
(n=6) vs. PA + Control Overexpression (n=6). (G) AICAR treatment
increased the coronary flow reserve of model mice. (H) AICAR
treatment decreased the levels of leukocyte adhesion (white arrows
indicate the leukocytes that adhered to the cremaster microvascular
wall). Data are presented as the mean ± SD. *P<0.05,
Model (n=5) vs. Control (n=5); #P<0.05, Model + AICAR
(n=5) vs. Model (n=5). Scale bar, 200 µm. AICAR,
5-aminoimidazole-4-carboxamide ribonucleotide; AMPK, AMP-activated
protein kinase; CMD, coronary microvascular dysfunction; CMECs,
cardiac microvascular endothelial cells; eNOS, endothelial nitric
oxide synthase; KLF2, Krüppel-like factor 2; p-, phosphorylated;
PA, palmitic acid; ROS, reactive oxygen species. |
Discussion
A high level of FFAs is a risk factor for
cardiovascular disease due to their detrimental effects on vascular
endothelial cells, which is common in patients with myocardial
ischemia (36). This may be due
to the increased sympathetic drive under myocardial ischemia
promoting the release of FFAs into the blood through the activation
of lipolysis in adipose tissue (37). However, elevation of the level of
FFAs in myocardial ischemia has been demonstrated to increase
ischemic damage of the myocardium (38). Availability of FFAs in the
circulation reciprocally reduces cardiac glucose utilization,
whereas oxidation of fatty acids requires more oxygen per adenosine
triphosphate produced than glucose (39). Therefore, increased cardiac
utilization of fatty acids will lead to increased oxygen
consumption and make the heart more susceptible to ischemic events
(40).
An acute increase in the serum FFA level triggers
inflammation and oxidative stress in the endothelium (41). Ko et al (42) reported that FFAs, as nutrient
stress factors, could activate cardiac inflammation and suppress
myocardial glucose metabolism via inhibition of AMPK in the heart.
Han et al (43) also
revealed that FFAs could induce cardiac dysfunction and alter
insulin-signaling pathways. High serum concentrations of FFAs can
lead to both microvascular and metabolic insulin resistance
(44). Endothelial dysfunction is
a major mechanism associated with CMD (2) and, based on these findings, our
hypothesis was that abnormal FFA metabolism may serve an important
role in the pathogenesis of CMD by influencing endothelial
function.
A common approach for experimentally elevating
plasma FFA levels is to simultaneously infuse heparin and a lipid
emulsion (45-47), as was accomplished in our previous
study (16), and in the present
study. Heparin can activate lipoprotein lipase and catalyzes
triglyceride hydrolysis, which increases the serum concentration of
FFAs (48). The elevation of FFAs
achieved by the infusion of heparin and lipids leads to rapid
induction of endothelial dysfunction (49,50). To further understand the
deleterious effects of FFAs on CMD, a mouse model of systemic
microvascular dysfunction was established via infusion of lipid
emulsion with heparin to increase the concentration of serum FFAs.
After lipid emulsion infusion, the serum concentration of FFAs was
found to increase to more than twice the normal value.
In the present study, the model group exhibited a
reduction in CFR of >30%, indicating that CMD had been
established. A large number of leukocytes were found to adhere to
the walls of microvessels, indicating that leukocyte activation and
the impairment of microvascular endothelial function had occurred.
In addition, the use of TEM revealed the presence of swelling of
microvascular endothelial cells in the heart tissues and palmitic
acid were demonstrated to cause a decrease in the viability of
CMECs in a dose-dependent manner. Both phenomena indicated the
damage to CMECs that had been caused by FFAs. Endothelial
dysfunction and leukocyte activation may be the key processes of
CMD induced by FFAs. Following intravenous infusion of the lipid
emulsion, the production of ROS in the model group was found to be
increased. In the present study, high levels of FFAs were able to
stimulate the production of ROS in both leukocytes and CMECs.
Excessive ROS production leads to the oxidative damage of numerous
biological macromolecules, including nucleic acids, lipids,
proteins and carbohydrates, which subsequently affects multiple
physiological metabolic pathways (51). Postprandial hyperlipidemia has
been reported to be associated with acute endothelial dysfunction
(52). In the present study, it
was also found that CD11b expression was markedly increased in the
model group. CD11b is directly involved in cell adhesion and
leukocyte activation (53).
Endothelial cell adhesion molecules bind to leukocytes, thereby
activating them and leading to their infiltration into the
underlying tissues (54).
Activated leukocytes not only block microvessels, but also cause an
increase in the vascular permeability (55). Compared with those in control
mice, the levels of the plasma pro-inflammatory factors, IL-6 and
TNF-α, were found to be significantly increased following lipid
emulsion infusion. An increase in the level of IL-6 is able to
initiate the acute inflammatory reaction, and inhibition of IL-6
has previously been reported to reduce the risk of atherosclerotic
thrombotic events (56). As far
as TNF-α is concerned, an increase in its level has been
demonstrated to induce endothelial cells to express adhesion
factors, accelerate the adhesion and penetration of leukocytes into
the vascular endothelium, and cause local inflammatory reactions
and pannus formation (57).
Microvascular endothelial dysfunction and abnormal activation of
leukocytes are processes that interact with and mutually enhance
each other (58), thereby
fulfilling an important role in the pathophysiological mechanism of
FFA-induced CMD.
Extensive studies have demonstrated that AMPK is
activated by an energy deficiency but suppressed by overnutrition
(59,60). In the present study, the increased
serum concentration of FFAs led to a significant inhibition of the
activation of AMPK. Dysregulation of the AMPK/KLF2/eNOS signaling
pathway may be responsible for the mechanism that accounts for the
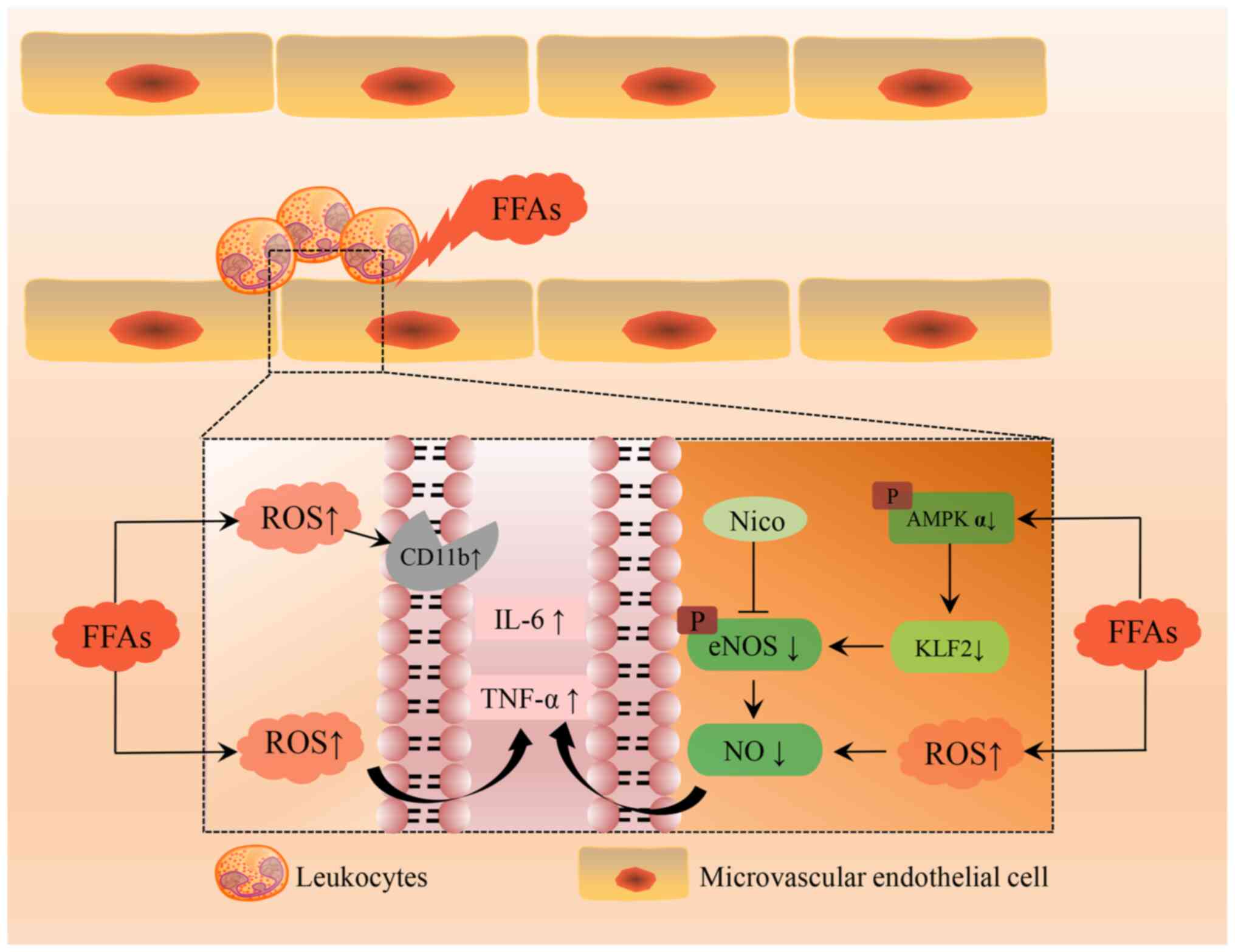
induction of CMD mediated by FFAs (Fig. 5). On one side, elevated FFAs can
activate leukocytes and increase ROS, promoting the production of
CD11b, IL-6 and TNF-α. On the other side, FFAs caused NO reduction
in CMECs by blocking the AMPK/KLF2/eNOS pathway. This resulted in
decreased cell viability and impaired endothelial function, which
eventually induces the development of CMD. In the model group, the
expression levels of AMPK, KLF2 and eNOS in myocardial tissue were
downregulated, and the serum NO level was decreased. In
vitro experiments subsequently demonstrated that treatment with
PA caused a decrease in the expression levels of AMPK, KLF2 and
eNOS in a dose-dependent manner. These results suggested that FFAs
may suppress the AMPK/KLF2 signaling pathway in CMECs, thereby
inhibiting endothelial function and reducing NO production.
Increased serum levels of FFAs can cause endoplasmic reticulum
stress and mitochondrial dysfunction, which lead to a disordering
of energy metabolism and biosynthesis (13). It is well established that the
AMPK signaling pathway is closely associated with energy metabolism
(18). Wang et al
(61) found that PA reduced AMPK
phosphorylation, and activation of the AMPK signaling pathway could
improve mitochondrial function by inhibiting lipid aggregation.
KLF2 is an important regulator of endothelial function, which is
able to activate eNOS (24).
Decreased KLF2 expression levels are associated with endothelial
dysfunction induced by advanced glycation end products (62). KLF2 is widely regarded as the most
potent inducer of eNOS, especially under laminar shear stress
(63). Furthermore, the AMPK/KLF2
signaling pathway has been demonstrated to participate in
neovascular development following cerebral ischemia via the
regulation of eNOS protein expression (25). In order to further verify that the
decreased AMPK activity was associated with the downstream proteins
KLF2 and eNOS, the AMPK activator AICAR was used to treat the CMECs
and these experiments revealed that activation of AMPK could
significantly improve PA-induced inhibition of KLF2 and eNOS. AICAR
is an AMPK agonist, which can promote AMPK phosphorylation
(64). Viglino et al
(65) reported that chronic
treatment with AICAR induces a metabolic shift in FFA-exposed
cardiomyocytes, characterized by improved glucose transport and
glycolysis and redirection of fatty acids towards neutral storage.
Hu et al (66) found that
AICAR treatment led to an improvement in liver fibrosis in rats
with bile duct ligation by increasing the level of NO. Acute and
chronic use of AICAR have been demonstrated to relieve portal vein
pressure without changing systemic hemodynamics (66). AICAR can also alleviate
endothelial dysfunction and promote vasodilation by improving eNOS
activity and increasing NO production (67,68). The present study also demonstrated
that an injection of AICAR relieved FFA-induced CMD and enhanced
CFR. In addition, a KLF2 overexpression plasmid was transfected
into CMECs, and KLF2 overexpression was found to activate eNOS.
However, when combing AICAR and siKLF2 to treat CMECs, KLF2 or eNOS
expression was not increased. Therefore, the aforementioned results
further suggested that FFAs may serve a role in coronary
microcirculation damage by interfering with the AMPK/KLF2/eNOS
signaling pathway.
Nicorandil is commonly used to relieve the
microvascular angina that is called cardiac syndrome X (69). A recent meta-analysis of
randomized controlled trials confirmed that nicorandil has
potential in terms of improving angina symptoms in cardiac syndrome
X (69). Nicorandil is also one
of the few drugs available that have been evidentially demonstrated
to effectively treat CMD (70).
The present study demonstrated that nicorandil could increase the
CFR and reduce the level of leukocyte adhesion in FFA-induced CMD
model mice. Nicorandil also alleviated swelling of the
microvascular endothelium caused by FFAs, increased the viability
of CMECs, and caused a reduction in ROS production. Increased
production of NO was likely to account for the effects of
nicorandil. Nicorandil can not only function as a NO donor, but
also modulate eNOS (71,72). The present study demonstrated that
nicorandil could directly activate eNOS without the participation
of AMPK. In addition, nicorandil fulfilled an anti-inflammatory
role, leading to reduced levels of IL-6 and TNF-α in the plasma. A
clinical trial has suggested that nicorandil treatment may result
in cardiovascular protection by inhibiting systemic inflammation
(73). In the present study,
nicorandil could stimulate eNOS expression and facilitate NO
production in CMECs. Nicorandil also exerted anti-inflammatory and
antioxidant effects. Previous studies have concluded that
nicorandil could attenuate organ injury by increasing eNOS
expression and via its antiapoptotic, anti-inflammatory and
antioxidant properties (71,74).
Reducing chronic serum FFA levels may reduce the
cardiovascular disease burden, and thus, efforts have been made to
develop pharmaceuticals to reduce the level of serum FFAs (75). Oral phytosterol supplementation
has been demonstrated to cause a reduction in serum FFAs (76). Lifestyle interventions are also
useful; for example, a previous meta-analysis of randomized
controlled trials revealed a reduction of FFAs with chronic
exercise training (77). Research
in this field is ongoing, although, at present, it has not yet
yielded any approved drugs aimed at suppressing the serum FFA
concentration (78).
The present study still has several limitations to
be disclosed. Firstly, there are endothelia-dependent and
endothelia-independent mechanisms operative in the pathogenesis of
CMD (4). In the present study,
only the effects on endothelial factors were explored and further
research is required in this regard. Secondly, pan-endothelial cell
markers were not tested by immunohistochemistry analysis. Thirdly,
the long-term efficacy of nicorandil remains to be validated.
In conclusion, the present study demonstrated that
FFAs could induce CMD via inhibition of the AMPK/KLF2/eNOS
signaling pathway, whereas activation of this pathway alleviated
FFA-induced CMD, which could be a therapeutic option for CMD.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ, JZ, ZH and CL designed the experiments. YZ, JZ,
CR and BH performed the experiments. YZ, JZ and RD analyzed the
data. YZ, JZ, ZH and CL wrote and revised the manuscript. JZ and CL
acquired the funding. YZ and CL confirmed the authenticity of all
the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The entire animal experimental protocol used in the
present study was carefully checked and approved by the Animal
Experiment Ethics Committee of the Second Affiliated Hospital of
Naval Medical University (no. 2020SL037; Shanghai, China). All
animal care procedures were carried out in accordance with the
Animal Research: Reporting of In Vivo Experiments guidelines
on animal research.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (grant nos. 82120108004 and 82104588), the
Shanghai Science and Technology Innovation Action Plan (grant no.
22DZ2304500), and the Pyramid Talent Program of Changzheng Hospital
(grant no. YQ662).
References
|
1
|
GBD 2015 Mortality and Causes of Death
Collaborators: Global, regional, and national life expectancy,
all-cause mortality, and cause-specific mortality for 249 causes of
death, 1980-2015: A systematic analysis for the global burden of
disease study 2015. Lancet. 388:1459–1544. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vancheri F, Longo G, Vancheri S and Henein
M: Coronary microvascular dysfunction. J Clin Med. 9:28802020.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Murthy VL, Naya M, Taqueti VR, Foster CR,
Gaber M, Hainer J, Dorbala S, Blankstein R, Rimoldi O, Camici PG
and Di Carli MF: Effects of sex on coronary microvascular
dysfunction and cardiac outcomes. Circulation. 129:2518–2527. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Del BM, Montone RA, Camilli M, Carbone S,
Narula J, Lavie CJ, Niccoli G and Crea F: Coronary microvascular
dysfunction across the spectrum of cardiovascular diseases: JACC
state-of-the-art review. J Am Coll Cardiol. 78:1352–1371. 2021.
View Article : Google Scholar
|
|
5
|
Thakker RA, Rodriguez Lozano J, Rodriguez
Lozano P, Motiwala A, Rangasetty U, Khalife W and Chatila K:
Coronary microvascular disease. Cardiol Ther. 11:23–31. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Niu Z, Hu H and Tang F: High free fatty
acid levels are associated with stroke recurrence and poor
functional outcome in Chinese patients with ischemic stroke. J Nutr
Health Aging. 21:1102–1106. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jung Y, Cho Y, Kim N, Oh IY, Kang SW, Choi
EK and Hwang GS: Lipidomic profiling reveals free fatty acid
alterations in plasma from patients with atrial fibrillation. PLoS
One. 13:e1967092018. View Article : Google Scholar
|
|
8
|
Skidmore PML, Woodside JV, Mc Master C,
Bingham A, Mercer C, Evans A, Young IS and Yarnell JW: Plasma free
fatty acid patterns and their relationship with CVD risk in a male
middle-aged population. Eur J Clin Nutr. 64:239–244. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khawaja O, Maziarz M, Biggs ML, Longstreth
WT Jr, Ix JH, Kizer JR, Zieman S, Tracy RP, Mozaffarian D, Mukamal
KJ, et al: Plasma free fatty acids and risk of stroke in the
cardiovascular health study. Int J Stroke. 9:917–920. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pilz S and März W: Free fatty acids as a
cardiovascular risk factor. Clin Chem Lab Med. 46:429–434. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hu T, Zhang W, Han F, Zhao R, Liu L and An
Z: Plasma fingerprint of free fatty acids and their correlations
with the traditional cardiac biomarkers in patients with type 2
diabetes complicated by coronary heart disease. Front Cardiovasc
Med. 9:9034122022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lopaschuk GD, Ussher JR, Folmes CD, Jaswal
JS and Stanley WC: Myocardial fatty acid metabolism in health and
disease. Physiol Rev. 90:207–258. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fillmore N, Mori J and Lopaschuk GD:
Mitochondrial fatty acid oxidation alterations in heart failure,
ischaemic heart disease and diabetic cardiomyopathy. Br J
Pharmacol. 171:2080–2090. 2014. View Article : Google Scholar :
|
|
14
|
Goldberg IJ, Trent CM and Schulze PC:
Lipid metabolism and toxicity in the heart. Cell Metab. 15:805–812.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wende AR and Abel ED: Lipotoxicity in the
heart. Biochim Biophys Acta. 1801:311–319. 2010. View Article : Google Scholar :
|
|
16
|
Zhang Y, Zhao J, Ding R, Niu W, He Z and
Liang C: Pre-treatment with compound Danshen dripping pills
prevents lipid infusion-induced microvascular dysfunction in mice.
Pharm Biol. 58:701–706. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Niu Y, Li S, Na L, Feng R, Liu L, Li Y and
Sun C: Mangiferin decreases plasma free fatty acids through
promoting its catabolism in liver by activation of AMPK. PLoS One.
7:e307822012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qi D and Young LH: AMPK: Energy sensor and
survival mechanism in the ischemic heart. Trends Endocrinol Metab.
26:422–429. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baltgalvis KA, White K, Li W, Claypool MD,
Lang W, Alcantara R, Singh BK, Friera AM, McLaughlin J, Hansen D,
et al: Exercise performance and peripheral vascular insufficiency
improve with AMPK activation in high-fat diet-fed mice. Am J
Physiol Heart Circ Physiol. 306:H1128–H1145. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Atkins GB and Jain MK: Role of
Krüppel-like transcription factors in endothelial biology. Circ
Res. 100:1686–1695. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chatauret N, Coudroy R, Delpech PO,
Vandebrouck C, Hosni S, Scepi M and Hauet T: Mechanistic analysis
of nonoxygenated hypothermic machine perfusion's protection on warm
ischemic kidney uncovers greater eNOS phosphorylation and
vasodilation. Am J Transplant. 14:2500–2514. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hong FF, Liang XY, Liu W, Lv S, He SJ,
Kuang HB and Yang SL: Roles of eNOS in atherosclerosis treatment.
Inflamm Res. 68:429–441. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yan J, Wang A, Cao J and Chen L:
Apelin/APJ system: An emerging therapeutic target for respiratory
diseases. Cell Mol Life Sci. 77:2919–2930. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee GH, Park JS, Jin SW, Pham TH, Thai TN,
Kim JY, Kim CY, Choi JH, Han EH and Jeong HG: Betulinic acid
induces eNOS expression via the AMPK-dependent KLF2 signaling
pathway. J Agric Food Chem. 68:14523–14530. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen GH, Li XL, Deng YQ, Zhou FM, Zou WQ,
Jiang WX, Shangguan SQ and Lu ZN: The molecular mechanism of EPO
regulates the angiogenesis after cerebral ischemia through
AMPK-KLF2 signaling pathway. Crit Rev Eukaryot Gene Expr.
29:105–112. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Segers VFM, Brutsaert DL and De Keulenaer
GW: Cardiac remodeling: endothelial cells have more to say than
just NO. Front Physiol. 9:3822018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang P, Ren L, Zhi L, Yu Z, Lv F, Xu F,
Peng W, Bai X, Cheng K, Quan L, et al: Negative regulation of AMPK
signaling by high glucose via E3 ubiquitin ligase MG53. Mol Cell.
81:629–637.e5. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Percie du Sert N, Hurst V, Ahluwalia A,
Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl
U, et al: The ARRIVE guidelines 2.0: Updated guidelines for
reporting animal research. Br J Pharmacol. 177:3617–3624. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhan B, Xu Z, Zhang Y, Wan K, Deng H, Wang
D, Bao H, Wu Q, Hu X, Wang H, et al: Nicorandil reversed
homocysteine-induced coronary microvascular dysfunction via
regulating PI3K/Akt/eNOS pathway. Biomed Pharmacother.
127:1101212020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chang WT, Fisch S, Chen M, Qiu Y, Cheng S
and Liao R: Ultrasound based assessment of coronary artery flow and
coronary flow reserve using the pressure overload model in mice. J
Vis Exp. e525982015.PubMed/NCBI
|
|
31
|
Wang Y, Du F, Hawez A, Mörgelin M and
Thorlacius H: Neutrophil extracellular trap-microparticle complexes
trigger neutrophil recruitment via high-mobility group protein 1
(HMGB1)-toll-like receptors (TLR2)/TLR4 signalling. Br J Pharmacol.
176:3350–3363. 2019.PubMed/NCBI
|
|
32
|
House SD and Lipowsky HH:
Leukocyte-endothelium adhesion: Microhemodynamics in mesentery of
the cat. Microvasc Res. 34:363–379. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
McDonald CA, Payne NL, Sun G, Moussa L,
Siatskas C, Lim R, Wallace EM, Jenkin G and Bernard CC:
Immunosuppressive potential of human amnion epithelial cells in the
treatment of experimental autoimmune encephalomyelitis. J
Neuroinflammation. 12:1122015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Johnson EK, Schelling ME, Quitadamo IJ,
Andrew S and Johnson EC: Cultivation and characterization of
coronary microvascular endothelial cells: A novel porcine model
using micropigs. Microvasc Res. 64:278–288. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang C, He H, Liu G, Ma H, Li L, Jiang M,
Lu Q, Li P and Qi H: DT-13 induced apoptosis and promoted
differentiation of acute myeloid leukemia cells by activating
AMPK-KLF2 pathway. Pharmacol Res. 158:1048642020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Roy VK, Kumar A, Joshi P, Arora J and
Ahanger AM: Plasma free fatty acid concentrations as a marker for
acute myocardial infarction. J Clin Diagn Res. 7:2432–2434.
2013.
|
|
37
|
Hendrickson SC, St Louis JD, Lowe JE and
Abdel-aleem S: Free fatty acid metabolism during myocardial
ischemia and reperfusion. Mol Cell Biochem. 166:85–94. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Manzella D, Barbieri M, Rizzo MR, Ragno E,
Passariello N, Gambardella A, Marfella R, Giugliano D and Paolisso
G: Role of free fatty acids on cardiac autonomic nervous system in
noninsulin-dependent diabetic patients: Effects of metabolic
control. J Clin Endocrinol Metab. 86:2769–2774. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Knuuti MJ, Mäki M, Yki-Järvinen H,
Voipio-Pulkki LM, Härkönen R, Haaparanta M and Nuutila P: The
effect of insulin and FFA on myocardial glucose uptake. J Mol Cell
Cardiol. 27:1359–1367. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lopaschuk GD: Metabolic modulators in
heart disease: Past, present, and future. Can J Cardiol.
33:838–849. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
I S Sobczak A, A Blindauer C and J Stewart
A: Changes in plasma free fatty acids associated with type-2
diabetes. Nutrients. 11:20222019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ko HJ, Zhang Z, Jung DY, Jun JY, Ma Z,
Jones KE, Chan SY and Kim JK: Nutrient stress activates
inflammation and reduces glucose metabolism by suppressing
AMP-activated protein kinase in the heart. Diabetes. 58:2536–2546.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Han L, Liu J, Zhu L, Tan F, Qin Y, Huang H
and Yu Y: Free fatty acid can induce cardiac dysfunction and alter
insulin signaling pathways in the heart. Lipids Health Dis.
17:1852018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chai W, Liu J, Jahn LA, Fowler DE, Barrett
EJ and Liu Z: Salsalate attenuates free fatty acid-induced
microvascular and metabolic insulin resistance in humans. Diabetes
Care. 34:1634–1638. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tripathy D, Mohanty P, Dhindsa S, Syed T,
Ghanim H, Aljada A and Dandona P: Elevation of free fatty acids
induces inflammation and impairs vascular reactivity in healthy
subjects. Diabetes. 52:2882–2887. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Turpin SM, Ryall JG, Southgate R, Darby I,
Hevener AL, Febbraio MA, Kemp BE, Lynch GS and Watt MJ: Examination
of 'lipotoxicity' in skeletal muscle of high-fat fed and ob/ob
mice. J Physiol. 587:1593–1605. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Boden G, She P, Mozzoli M, Cheung P,
Gumireddy K, Reddy P, Xiang X, Luo Z and Ruderman N: Free fatty
acids produce insulin resistance and activate the proinflammatory
nuclear factor-kappaB pathway in rat liver. Diabetes. 54:3458–3465.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yasu T, Mutoh A, Wada H, Kobayashi M,
Kikuchi Y, Momomura S and Ueda S: Renin-angiotensin system
inhibitors can prevent intravenous lipid infusion-induced
myocardial microvascular dysfunction and leukocyte activation. Circ
J. 82:494–501. 2018. View Article : Google Scholar
|
|
49
|
Umpierrez GE, Smiley D, Robalino G, Peng
L, Kitabchi AE, Khan B, Le A, Quyyumi A, Brown V and Phillips LS:
Intravenous intralipid-induced blood pressure elevation and
endothelial dysfunction in obese African-Americans with type 2
diabetes. J Clin Endocrinol Metab. 94:609–614. 2009. View Article : Google Scholar :
|
|
50
|
Wang H, Li H, Hou Z, Pan L, Shen X and Li
G: Role of oxidative stress in elevated blood pressure induced by
high free fatty acids. Hypertens Res. 32:152–158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Guerra BA and Otton R: Impact of the
carotenoid astaxanthin on phagocytic capacity and ROS/RNS
production of human neutrophils treated with free fatty acids and
high glucose. Int Immunopharmacol. 11:2220–2226. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mah E, Noh SK, Ballard KD, Matos ME, Volek
JS and Bruno RS: Postprandial hyperglycemia impairs vascular
endothelial function in healthy men by inducing lipid peroxidation
and increasing asymmetric dimethylarginine:arginine. J Nutr.
141:1961–1968. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Dong G, Song L, Tian C, Wang Y, Miao F,
Zheng J, Lu C, Alsadun S and Graves DT: FOXO1 regulates
bacteria-induced neutrophil activity. Front Immunol. 8:10882017.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gomes NE, Brunialti MK, Mendes ME,
Freudenberg M, Galanos C and Salomão R: Lipopolysaccharide-induced
expression of cell surface receptors and cell activation of
neutrophils and monocytes in whole human blood. Braz J Med Biol
Res. 43:853–858. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang H, Wang Y, Qu M, Li W, Wu D, Cata JP
and Miao C: Neutrophil, neutrophil extracellular traps and
endothelial cell dysfunction in sepsis. Clin Transl Med.
13:e11702023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ridker PM, Libby P, MacFadyen JG, Thuren
T, Ballantyne C, Fonseca F, Koenig W, Shimokawa H, Everett BM and
Glynn RJ: Modulation of the interleukin-6 signalling pathway and
incidence rates of atherosclerotic events and all-cause mortality:
Analyses from the canakinumab anti-inflammatory thrombosis outcomes
study (CANTOS). Eur Heart J. 39:3499–3507. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jiang T, Zhang W and Wang Z: Laquinimod
protects Against TNF-α-induced attachment of monocytes to human
aortic endothelial cells (HAECs) by increasing the expression of
KLF2. Drug Des Devel Ther. 14:1683–1691. 2020. View Article : Google Scholar :
|
|
58
|
Wang Y, Zhang J, Wang Z, Wang C and Ma D:
Endothelial-cell-mediated mechanism of coronary microvascular
dysfunction leading to heart failure with preserved ejection
fraction. Heart Fail Rev. 28:169–178. 2023. View Article : Google Scholar :
|
|
59
|
Hardie DG and Carling D: The AMP-activated
protein kinase-fuel gauge of the mammalian cell? Eur J Biochem.
246:259–273. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Coughlan KA, Valentine RJ, Ruderman NB and
Saha AK: Nutrient excess in AMPK downregulation and insulin
resistance. J Endocrinol Diabetes Obes. 1:10082013.PubMed/NCBI
|
|
61
|
Wang Q, Wang Z, Xu M, Tu W, Hsin IF,
Stotland A, Kim JH, Liu P, Naiki M, Gottlieb RA and Seki E:
Neurotropin inhibits lipid accumulation by maintaining
mitochondrial function in hepatocytes via AMPK activation. Front
Physiol. 11:9502020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Saum K, Campos B, Celdran-Bonafonte D,
Nayak L, Sangwung P, Thakar C, Roy-Chaudhury P and Owens AP: Iii
PhD: Uremic advanced glycation end products and protein-bound
solutes induce endothelial dysfunction through suppression of
Krüppel-like factor 2. J Am Heart Assoc. 7:e0075662018. View Article : Google Scholar
|
|
63
|
SenBanerjee S, Lin Z, Atkins GB, Greif DM,
Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW, et
al: KLF2 is a novel transcriptional regulator of endothelial
proinflammatory activation. J Exp Med. 199:1305–1315. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Guo D, Hildebrandt IJ, Prins RM, Soto H,
Mazzotta MM, Dang J, Czernin J, Shyy JY, Watson AD, Phelps M, et
al: The AMPK agonist AICAR inhibits the growth of
EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc
Natl Acad Sci USA. 106:12932–12937. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Viglino C, Foglia B and Montessuit C:
Chronic AICAR treatment prevents metabolic changes in
cardiomyocytes exposed to free fatty acids. Pflugers Arch.
471:1219–1234. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hu L, Su L, Dong Z, Wu Y, Lv Y, George J
and Wang J: AMPK agonist AICAR ameliorates portal hypertension and
liver cirrhosis via NO pathway in the BDL rat model. J Mol Med
(Berl). 97:423–434. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yu H, Liu Q, Chen G, Huang L, Luo M, Lv D
and Luo S: SIRT3-AMPK signaling pathway as a protective target in
endothelial dysfunction of early sepsis. Int Immunopharmacol.
106:1086002022. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Li JM, Lu W, Ye J, Han Y, Chen H and Wang
LS: Association between expression of AMPK pathway and adiponectin,
leptin, and vascular endothelial function in rats with coronary
heart disease. Eur Rev Med Pharmacol Sci. 24:905–914.
2020.PubMed/NCBI
|
|
69
|
Jia Q, Shi S, Yuan G, Shi J, Shi S, Wei Y
and Hu Y: The effect of nicorandil in patients with cardiac
syndrome X: A meta-analysis of randomized controlled trials.
Medicine (Baltimore). 99:e221672020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhu H, Xu X, Fang X, Zheng J, Zhao Q, Chen
T and Huang J: Effects of the antianginal drugs ranolazine,
nicorandil, and ivabradine on coronary microvascular function in
patients with nonobstructive coronary artery disease: A
meta-analysis of randomized controlled trials. Clin Ther.
41:2137–2152.e12. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Abdelzaher WY, Khalaf HM, El-Hussieny M,
Bayoumi A, Shehata S and Refaie M: Role of nitric oxide donor in
methotrexate-induced testicular injury via modulation of
pro-inflammatory mediators, eNOS and P-glycoprotein. Hum Exp
Toxicol. 39:1700–1709. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Harb IA, Ashour H, Sabry D, El-Yasergy DF,
Hamza WM and Mostafa A: Nicorandil prevents the nephrotoxic effect
of cyclosporine-A in albino rats through modulation of
HIF-1α/VEGF/eNOS signaling. Can J Physiol Pharmacol. 99:411–417.
2021. View Article : Google Scholar
|
|
73
|
Ishibashi Y, Takahashi N, Tokumaru A,
Karino K, Sugamori T, Sakane T, Yoshitomi H, Sato H, Oyake N,
Murakami Y and Shimada T: Effects of long-term nicorandil
administration on endothelial function, inflammation, and oxidative
stress in patients without coronary artery disease. J Cardiovasc
Pharmacol. 51:311–316. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Refaie MMM, Shehata S, El-Hussieny M,
Abdelraheem WM and Bayoumi AMA: Role of ATP-sensitive potassium
channel (KATP) and eNOS in mediating the protective
effect of nicorandil in cyclophosphamide-induced cardiotoxicity.
Cardiovasc Toxicol. 20:71–81. 2020. View Article : Google Scholar
|
|
75
|
Sun X, Pan H, Tan H and Yu Y: High free
fatty acids level related with cardiac dysfunction in obese rats.
Diabetes Res Clin Pract. 95:251–259. 2012. View Article : Google Scholar
|
|
76
|
Fatahi S, Kord-Varkaneh H, Talaei S,
Mardali F, Rahmani J, Ghaedi E, Tan SC and Shidfar F: Impact of
phytosterol supplementation on plasma lipoprotein(a) and free fatty
acid (FFA) concentrations: A systematic review and meta-analysis of
randomized controlled trials. Nutr Metab Cardiovasc Dis.
29:1168–1175. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Smart NA, King N, McFarlane JR, Graham PL
and Dieberg G: Effect of exercise training on liver function in
adults who are overweight or exhibit fatty liver disease: A
systematic review and meta-analysis. Br J Sports Med. 52:834–843.
2018. View Article : Google Scholar
|
|
78
|
Henderson GC: Plasma free fatty acid
concentration as a modifiable risk factor for metabolic disease.
Nutrients. 13:25902021. View Article : Google Scholar : PubMed/NCBI
|