1. Introduction
Inflammatory bowel disease (IBD) is a chronic
inflammatory disorder that affects the gastrointestinal tract,
specifically the colon and/or the small intestine. IBD encompasses
two main types of conditions: Crohn's disease (CD) and ulcerative
colitis (UC). IBD can have a significant impact on a the quality of
life of affected individuals, and the condition requires lifelong
management with medications, lifestyle changes and sometimes even
surgery (1). Current medical
treatments for IBD focus on the use of drugs, such as
corticosteroids, aminosalicylates, immunomodulators and biologics
to manage symptoms, alongside other measures or surgery if deemed
necessary. However, a significant number of patients either do not
respond to these treatments or eventually, these lose their
effectiveness, indicating a need for alternative therapies
(2). Various new treatment
options are being explored, such as the use of small molecules,
apheresis therapy, enhancing gut microecology, cell therapy and
exosome therapy (3,4). The exact cause of IBD is not yet
fully understood, although it is considered to occur due to a
complex interplay between genetic, environmental and immune system
factors. These contribute to the activation of various regulated
cell death mechanisms, including ferroptosis, which has been
implicated in the pathogenesis of IBD (5,6).
Since its discovery in 2012 (7), ferroptosis has gained the attention
of researches worldwide and has been studied across several
diseases (8-10). In contrast to other types of cell
death, such as apoptosis, necroptosis and autophagy, ferroptosis
exhibits unique sets of biological processes and pathophysiological
characteristics and is mainly accompanied by iron accumulation and
lipid peroxidation (11,12). In other words, the execution
machinery of ferroptosis is comprised of the availability of
redox-active iron, peroxidation of polyunsaturated fatty
acid-(PUFA) containing phospholipids (PLs), and the inactivation of
the lipid peroxide repair system (13). Distinctively, the cytological
characteristic hallmarks of apoptotic cell death, including
chromatin condensation and plasma membrane blebbing, as well as the
morphological characteristics of necroptosis, i.e., the swelling of
cytoplasmic organelles, are rarely observed during ferroptotic cell
death (7,13). Approximately 10 years following
its discovery, substantial progress has been made concerning
studies on ferroptosis, including the characterization of core
molecular marks and regulatory components, such as glutathione
peroxidase (GPX)4, solute carrier family 7 member 11 (SLC7A11),
ferroptosis suppressor protein 1 (FSP1, also known as AIFM2),
nuclear factor erythroid 2-related factor 2 (Nrf2) and p53, the
identification of inducers, such as erastin, FINO2, RAS-selective
lethal 3 (RSL3) and FIN56, and specific inhibitors ferrostatin-1,
liproxstatin-1 and deferoxamine, which have together strengthened
the fundamental understanding of ferroptotic cell death (11,12).
Current evidence implicates ferroptosis in
gastrointestinal conditions, such as IBD and its associated
colorectal cancer (12,14), presenting it as a potential novel
therapeutic target. The pathological engagement of ferroptosis has
been demonstrated in both patients with IBD and experimental
models, including elevated iron deposition, glutathione (GSH)
exhaustion, GPX4 inactivation and lipid peroxidation, which
together contribute to intestinal cell death and sustained
inflammation (5,11,12,15). These factors further drive
inflammation and upregulate ferroptosis, exacerbating intestinal
tissue and barrier injury (16).
A number of studies have demonstrated that the inhibition of
ferroptosis significantly mitigates the characteristic clinical
features of IBD, including improved intestinal barrier function,
body weight, tissue repair, anti-inflammation, microbiota and a
decreased disease activity index (15,17-19). These findings present not only a
novel understanding of the pathogenesis of IBD, but may also aid in
the development of therapeutic approaches for IBD and other
intestinal diseases. The present review comprehensively summarizes
and discusses the role of ferroptosis in the pathology of IBD and
explores the currently available data on therapeutic targets of
ferroptosis in IBD. The mechanisms and key mediators of
ferroptosis, as well as pathogenic involvement and therapeutic
targets in other intestinal diseases, are also discussed.
2. The mechanisms of ferroptosis
Ferroptosis is primarily triggered by small
molecules, such as erastin and the inhibition of glutathione
biosynthesis or GPX4, in the presence of iron-dependent
accumulation of lipid reactive oxygen species (ROS) and the
depletion of plasma membrane PUFA (20). Ferroptosis can occur through two
major pathways, the extrinsic or transporter-dependent pathway that
includes increased iron uptake and decreased cysteine/glutamine
uptake, and the intrinsic or enzyme-regulated pathway that includes
the inhibition of GPX4 (21). In
addition to the initial discovery of the inhibitory role of the
cystine-import-GSH-GPX4 machinery in ferroptosis, the role of PL
hydroperoxides (PLOOHs) as the executioners of ferroptosis has also
been established. Studies on the mechanism of PLOOH synthesis,
particularly, the synthesis and activation of PUFAs, the precursor
of PLOOHs, have been extensively reported (22). In the inhibition of ferroptosis,
GPX4 serves as the key PLOOH-neutralizing enzyme. Thus, in the
canonical GPX4-regulated ferroptotic pathway, the inhibition of two
cellular components, GPX4 and system XC−
cystine/glutamate antiporter, by RSL3 and erastin, respectively,
triggers ferroptotic cell death (7,23).
In the exploration of the general mechanism underlying
erastin/RSL3-induced ferroptosis, it has been noted that both
compounds inactivate GPX4, as RSL3 directly inactivates GPX4, while
erastin indirectly inactivates GPX4 by preventing the import of
cystine, leading to the deprivation of cysteine, a crucial building
block of GSH and cellular antioxidants. As a result, PLOOHs
accumulate, leading to rapid and unrepairable plasma membrane
damage, and consequently cell death (24).
A key hallmark of ferroptosis is uncontrolled lipid
peroxidation. In this process, a bis-allylic hydrogen atom is
abstracted from the polyunsaturated fatty acyl moieties found on
PLs (PUFA-PLs) located in lipid bilayers, causing the formation of
a carbon-centered radical, and subsequently reacting with molecular
oxygen to produce peroxyl radicals (25). The free radicals mediate further
reactions that result in the formation of a myriad of secondary
products, which subsequently cause the breakdown of cell membrane
integrity and ultimately rupturing of organelle and cell membranes.
Therefore, the membrane-repairing enzymes, acyl-CoA synthetase
long-chain family member 4 (ACSL4) and lysophosphatidylcholine
acyltransferase (LPCAT)3 are crucial in ferroptosis. ACSL4 has the
ability to ligate long-chain PUFAs with coenzyme A, whereas LPCAT
enzymes re-esterify the ligated long-chain PUFAs in phospholipids
(26,27). Lipoxygenases (LOXs), a group of
non-heme iron-dependent dioxygenases, participate in ferroptosis by
directly oxygenating PUFAs of cellular membranes; thus, their
inhibition prevents ferroptosis (28). While the ferroptotic cell death
processes can be reversible, the cell recovery phenomena could be
mediated by different mechanisms yet to be fully clarified.
Currently, there is no direct lipid peroxidation rescue experiment
using ACSL4. Instead, studies have shown that the application of
GSH or ferrostatin-1 can promote the reversal of ferroptosis,
possibly because GSH enhances the GPX4 activity to arrest ROS
accumulation (29), while
ferrostatin-1 is a ROS scavenger that can remove the excessive
cytosolic and lipid ROS (30).
Iron is involved in the mechanism of ferroptosis
through the implication of the metabolic enzymes of phospholipid
peroxidation (LOXs and POR) which require iron for catalysis, and
through the iron-dependent Fenton chain reaction (a non-enzymatic
reaction). In the Fenton reaction, ferrous (Fe2+) or
ferric (Fe3+) ions react with hydrogen peroxide
(H2O2) to generate hydroxyl radicals, a form
of ROS that triggers the peroxidation of PUFAs in membrane lipids,
leading to ferroptosis (31,32). Metabolism plays a central role in
ferroptosis, as several cellular metabolic reactions can result in
the production of PLOOHs and autophagy (33). For example, the influence of
autophagy in cystine-deprivation-induced ferroptosis is mediated
through the autophagic degradation of the iron-storage protein
ferritin (ferritinophagy), which leads to increased cellular labile
iron content, and consequently to sensitization to ferroptosis
(8). Multiple autophagy-related
genes have been identified as positive regulators of ferroptosis,
where the induction of ferroptosis led to autophagy activation and
consequent degradation of ferritin and ferritinophagy cargo
receptor nuclear receptor coactivator 4 (NCOA4). The inhibition of
ferritinophagy by the knockdown of NCOA4 or the blockage of
autophagy prevents the accumulation of ferroptosis-associated
cellular labile iron and ROS, as well as eventual ferroptotic cell
death; thus, ferroptosis is an autophagic cell death process
(34). Moreover, the
participation of the mitochondria in ferroptosis underscores its
metabolic nature, as one of the hallmarks of ferroptosis is a
specific morphological phenotype characterized by extensive
ultrastructural changes of mitochondria (8). The critical role of the mitochondria
in lipid peroxidation-driven ferroptotic cell death has been
observed in several conditions, including excess free-iron
accumulation in the mitochondria, impaired mitochondrial
metabolism, resulting in the extensive production of ROS, and
mitochondrial cysteine deprivation (9,35).
As a major fuel of the mitochondrial TCA cycle, glucose regulates
ferroptosis, since its deprivation inhibits ferroptosis via
5'AMP-activated protein kinase (AMPK) signaling (36). On the mitochondrial membrane,
cytoplasmic iron activates mitoferrin 2, which in turn increases
iron transport into the mitochondria, leading to elevated ROS
production and ferroptosis. As previously demonstrated, mice fed an
iron-deficient diet for 3 weeks exhibited a significant reversal in
intestinal injury induced by abdominal ionizing radiation exposure
(37). The mechanisms involved in
ferroptosis and their interconnections are summarized in Fig. 1.
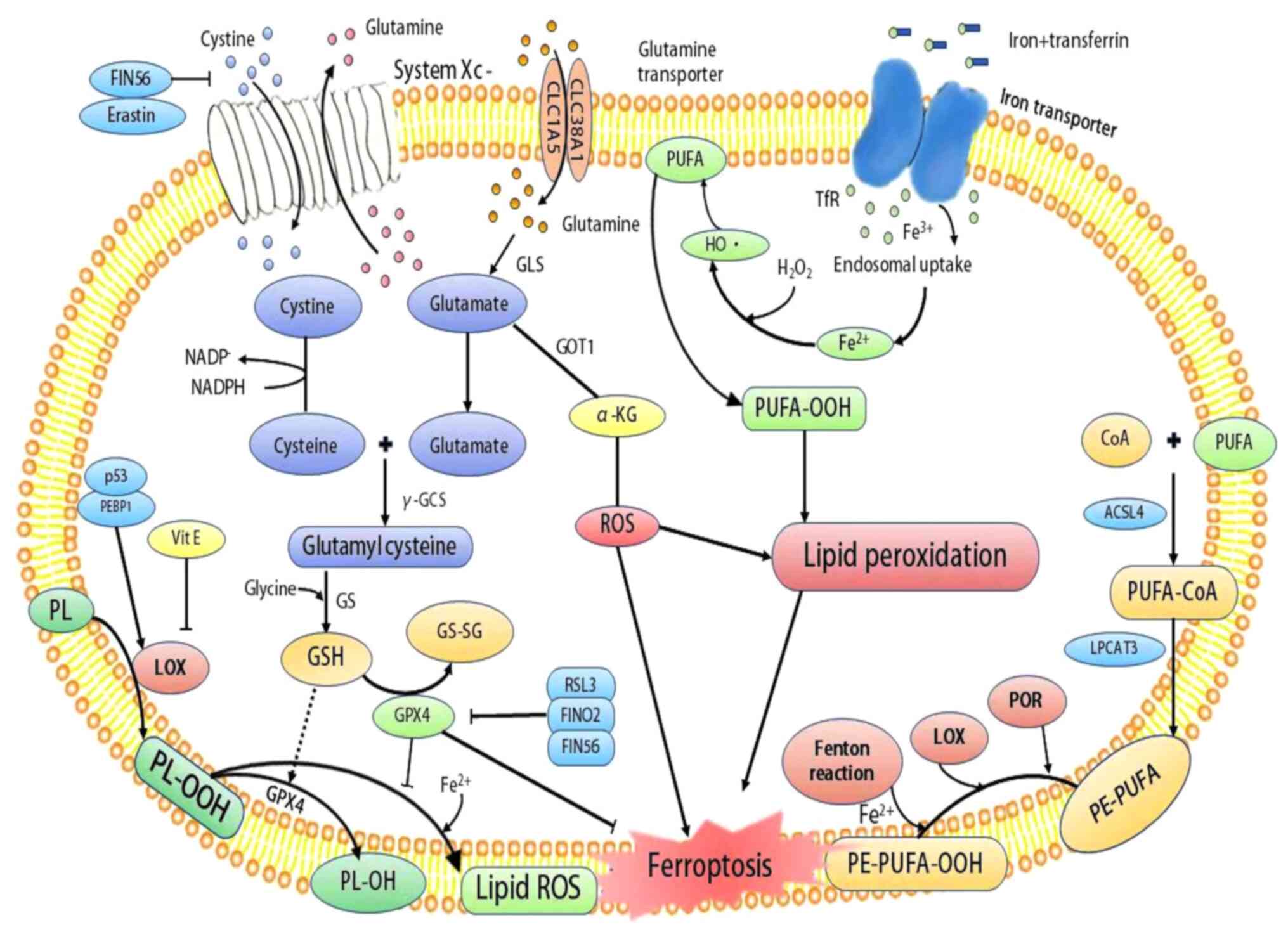 | Figure 1Mechanisms of ferroptosis. System
XC− and glutamine transporter allow the
influx of cystine and glutamine, respectively, which are converted
into cysteine and glutamate. Cysteine combines with glutamate to
form glutamylcysteine by γ-GCS, followed by conversion to GSH under
the action of GS. GSH and GPX4 serve as scavengers of ROS to
prevent oxidative stress and lipid peroxidation. However, the
inhibition of system Xc- by agents, such as erastin and FIN56
depletes GSH, leading to increased ROS and lipid peroxidation.
Moreover, while RSL3, FINO2 and FIN56 can directly inhibit GPX4 to
promote lipid peroxidation, p53 and PEBP1 enhance the activity of
LOX to promote lipid peroxidation, resulting in ferroptosis. In
addition, POR, LOX and the Fenton reaction chain promote the
conversion of PE-PUFA to PE-PUFA-OOH, leading to ferroptosis. GSH,
glutathione; GS, GSH synthetase; γ-GCS, γ-glutamylcysteine
synthetase; GPX4, glutathione peroxidase 4; PUFA, polyunsaturated
fatty acid; GOT1, glutamate oxaloacetate transaminase 1; GSSG,
oxidized glutathione; ROS, reactive oxygen species; LOX,
lipoxygenase; POR, cytochrome P450 oxidoreductase, ACSL4, acyl-CoA
synthetase long-chain family member 4; LPCAT3,
lysophosphatidylcholine acyltransferase 3; PEBP1,
phosphatidylethanolamine-binding protein-1; PL, phospholipid;
PLOOH, phospholipid hydroperoxides; PE, phosphatidylethanolamine;
RSL3, RAS-selective lethal 3. |
3. Key mediators of ferroptosis
PUFAs
The peroxidation of PUFAs by lipoxygenases is a key
driver of ferroptosis. It has been demonstrated that the mechanism
of lipid peroxidation during ferroptosis involves the phosphorylase
kinase G2 (PHKG2) regulation of iron availability to LOX enzymes,
which in turn drives ferroptosis through the peroxidation of PUFAs
at the bis-allylic position. The pretreatment of cells with PUFAs
that contain the heavy hydrogen isotope deuterium at the site of
peroxidation (D-PUFA) inhibits PUFA oxidation and blocks
ferroptosis (38). This indicates
that the oxidation of PUFA by LOXs through a PHKG2-dependent iron
pool is required for ferroptosis, and that the covalent inhibition
of the catalytic selenocysteine in GPX4 prevents the elimination of
PUFA hydroperoxides. In a multicenter case-control study on 83
patients newly diagnosed with UC, the authors found that the risk
of developing UC was significantly decreased in patients who
consumed n-6/n-3 PUFAs, as compared to those who consumed larger
amounts of docosahexaenoic, eicosapentaenoic and docosapentaenoic
acid (39). However, an earlier
study reported that in addition to n-3 PUFAs, both eicosapentaenoic
acid and docosahexaenoic acid confer a statistically significant
protective effect against the risk of developing UC in a cohort
aged >45 years (40). It has
also been reported that the PUFA biosynthesis pathway determines
ferroptosis sensitivity in gastric cancer, as the expression of
elongation of very long-chain fatty acid protein (ELOVL)5 and fatty
acid desaturase 1 (FADS1) is upregulated in mesenchymal-type cancer
cells, and leads to ferroptosis sensitization (41). These findings implicate PUFAs as
key mediators of ferroptosis.
GSH/GPX4
The excessive production and accumulation of ROS
induce lipid peroxidation, a key mechanism of ferroptosis. GSH, the
reducing substrate of GPX4 activity, is indispensable for
preventing ferroptosis (42). The
combined effect of GSH/GPX4 protects cells from ferroptotic death,
with GPX4 serving as the cornerstone of the antiperoxidative
defense. As GSH inhibits the accumulation of ROS, its depletion
turns has been shown to be associated with marked lipid
peroxidation, dysregulated cellular functions and cell death
(43). GSH is synthesized from
glycine, L-cysteine and L-glutamate, and the system
XC− cystine/glutamate antiporter, which
comprises a light-chain subunit (SLC7A11, xCT) and a heavy-chain
subunit (SLC3A2, CD98hc), mainly regulates the exchange of
intracellular L-glutamate and extracellular L-cystine. However,
certain molecules, such as erastin and sulfasalazine can inhibit
system XC− and cause the depletion of
cellular cysteine, leading to the impairment of the intracellular
GSH homeostasis and ultimately, intracellular GSH deficiency and
ferroptosis (44,45). GPX4 modulates ferroptosis by
preventing lipid peroxide-induced toxicity and protecting the
membrane lipid bilayers through the conversion of toxic lipid
hydroperoxides into non-toxic lipid alcohols (46). On the other hand, RSL3 potently
drive ferroptosis by directly targeting and inhibiting GPX4
activity and promoting ROS production (14), as well as through NF-κB pathway
activation and GPX4 depletion (47).
Iron
Due to the central role played by iron in cell
viability and death, cellular iron homeostasis is subjected to
exquisite control, mainly through a post-transcriptional network
dictated by iron-regulatory protein (IRP)1 and IRP2, which modulate
intracellular iron import/export and storage/release (48). Cellular iron is involved in
ferroptosis through two major routes. Firstly, the metabolic
enzymes implicated in the peroxidation of phospholipids, LOXs and
POR, require iron for catalysis; iron is also crucial for a
plethora of metabolic enzymes involved in the generation of
cellular ROS. LOXs and POR can catalyze enzymatic lipid
peroxidation in a Fe2+-dependent manner and are
implicated in the generation of PLOOH, factors necessary for
ferroptosis (49). Secondly, the
iron-dependent Fenton chain reaction, a non-enzymatic chemical
reaction, is critical for ferroptosis; the inhibition of GPX4
causes PLOOHs to persist longer, triggering the Fenton reaction to
rapidly amplify PLOOHs, the hallmark of ferroptosis (25). Moreover, PLOOHs can interact with
both ferric and ferrous ions to produce the free radicals, PLOO·
and PLO· respectively, which subsequently react with PUFA-PLs to
further drive PLOOH production. Therefore, mechanisms that lead to
improved ion homeostasis by an enhanced cellular iron export result
in a more ferroptosis-resistant cell (50). A previous multicenter,
hospital-based case-control study on patients newly diagnosed with
UC indicated that the highest intake of iron led to an increased
odds ratio for UC in a multivariate analysis, implicating that a
high intake of iron has a certain effect on the development of UC
(51). It has also been reported
that the characteristic iron overload in hereditary
hemochromatosis, causes multiple metabolic disturbances, disrupts
colonic homeostasis and colon-microbiome interaction, and
exacerbates the development and progression of colonic inflammation
and colon cancer (52). This may
be related to the involvement of excess iron in ROS generation in
the intestinal mucosa, enhancing cellular damage and loss of
barrier integrity.
Organic peroxides
Organic peroxides are highly reactive and thermally
unstable compounds containing one or more oxygen-oxygen bonds
(ROOR) and can undergo self-accelerating decomposition. In this
process, the O-O linkage can easily be broken down, releasing free
radicals in the form of RO· (alkoxy anions). Organic peroxides are
mostly exploited in research models to generate oxidative damage in
cells. For instance, tert-butyl hydroperoxide is a typical lipid
peroxide analog generally known to stimulate cellular damage due to
lipid peroxidation-dependent ferroptosis in human and murine cell
lines (53,54). Artemisinin and its derivative
compounds, such as artesunate and dihydroartemisinin are
1,2,4-trioxane-based organic peroxides, and effectively sensitize
cancer cells to ferroptosis by regulating iron homeostasis
(55,56). FINO2, a ferroptosis-inducing
organic peroxide containing a 1,2-dioxolane skeleton, indirectly
inhibits GPX4 and oxidizes iron. It decreases GPX4 activity and
protein levels in vitro, but does not act as an active site,
allosteric, or covalent inhibitor of GPX4 or alter GPX homeostasis
(57). In other words, FINO2 has
a dual induction mechanism for ferroptosis, involving direct iron
oxide or indirect inhibition of GPX4 activity. The role of the key
mediators of ferroptosis, as described above is illustrated in
Fig. 2.
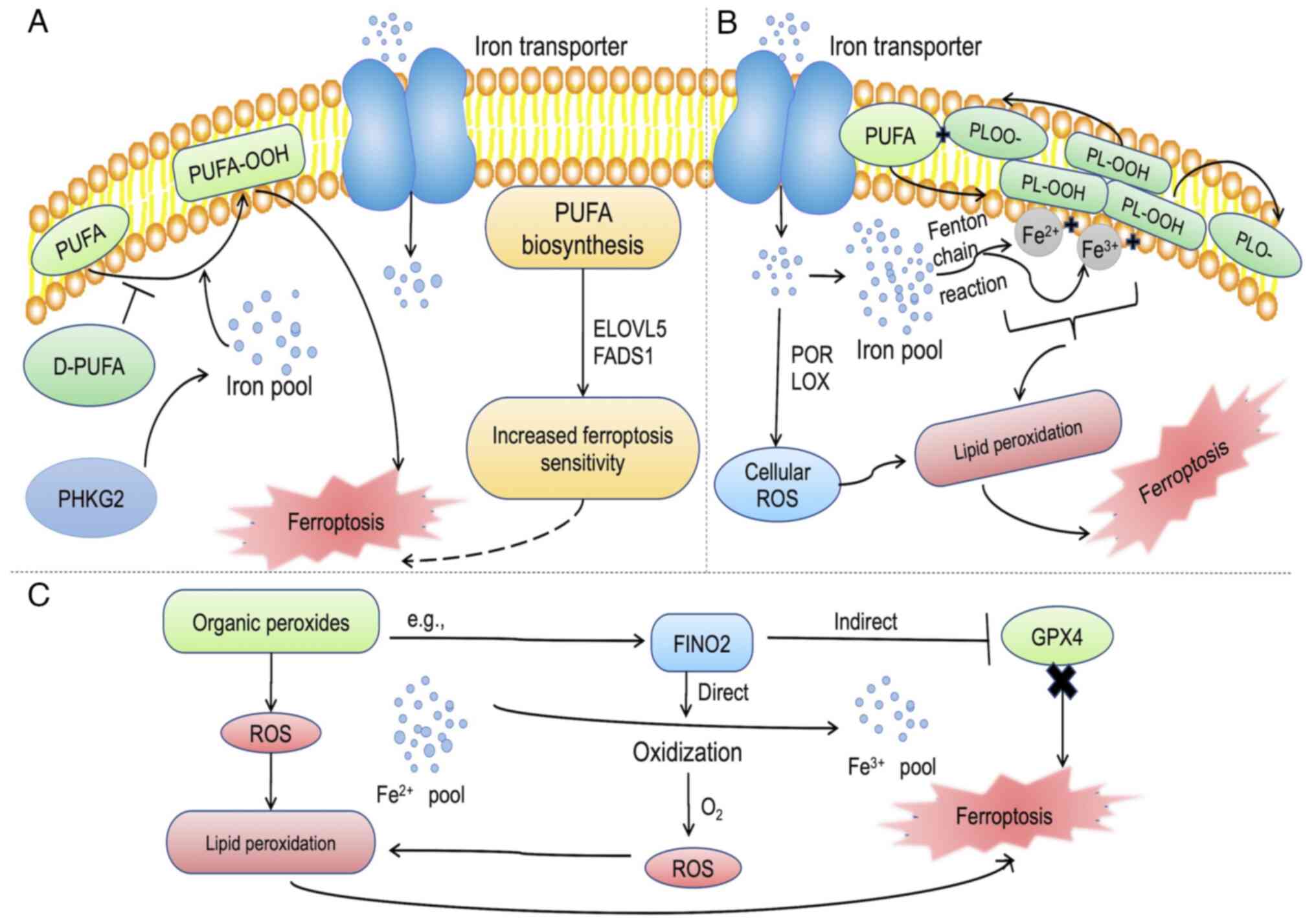 | Figure 2Role of key negative mediators of
ferroptosis. (A) PUFA serves as one of the drivers of ferroptosis.
The increased accumulation of cellular irons triggers the oxidation
of PUFA to PUFA-OOH, which is promoted by PHKG2, leading to
ferroptosis. ELOVLS and FADS1 enhance cellular sensitivity to
ferroptosis, while D-PUFA inhibits the oxidation of PUFAs. (B)
Iron, both Fe2+ and Fe3+, induces lipid
peroxidation through the Fenton chain reaction by interacting with
PL-OOH to produce PLOO· and PLO·, respectively, and through the
enzymatic route, POR and LOX, to produce cellular ROS that drives
ferroptosis. (C) Organic peroxides contribute to ferroptosis by ROS
production. FINO2, a typical organic peroxide drives ferroptosis by
either inhibiting GPX4 or oxidizing iron to release ROS, causing
lipid peroxidation and subsequently, ferroptosis. PUFA,
polyunsaturated fatty acid; PUFA-OOH, polyunsaturated fatty acid
containing-phospholipid hydroperoxides; PHKG2, phosphorylase kinase
G2; PLOOH, phospholipid hydroperoxides; LOX, lipoxygenase; POR,
cytochrome P450 oxidoreductase; ROS, reactive oxygen species; GPX4,
glutathione peroxidase 4. |
4. Role of ferroptosis in the pathogenesis
of IBD
Studies have indicated that the fundamental
characteristics of ferroptosis, including increased lipid
peroxidation, the depletion of GSH, the inactivation of GPX4 and
elevated iron deposition, are manifested in the inflamed intestinal
tissue of patients with IBD and in animal models of IBD (19,58,59). Ferroptosis is involved in IBD,
particularly intestinal epithelial cell death (5). As a characteristic of several
intestinal diseases, such as IBD, an aberrant elevation in the rate
of intestinal epithelial cell death underlies instances of
extensive epithelial erosion. The mode of programmed cell death
influences intestinal tissue restitution responses with
implications for chronic inflammation, and ultimately the long-term
risks of intestinal fibrosis and colorectal cancer (60). Ferroptosis participates in driving
the chronic aberrant inflammation in IBD by a lethal accumulation
of ROS, iron overload and uncontrolled lipid peroxidation, leading
to intestinal epithelial cell death and epithelial erosion
(15,58). Moreover, the dysregulation of
critical ferroptosis-related genes has been shown to alter the
progression, severity, or even morbidity of mice with experimental
colitis (61,62). Compared with the control samples,
colitis specimens exhibit significantly higher malondialdehyde
(MDA) (58) and iron
(particularly, ferrous iron) levels (63), and increased cellular iron levels
during ferroptosis induce transcriptional upregulation of ferritin
(34), which may contribute to
the exacerbation of colitis.
A previous study found that ferroptosis is
significantly induced in the intestinal epithelial cells of both
patients with UC and mice with colitis, and is mediated by a number
of signaling, including the endoplasmic reticulum (ER) stress
signaling. In intestinal epithelial cells, the deletion of NF-κBp65
upregulates ferroptosis and exacerbates colitis, whereas
phosphorylated-NF-κBp65 significantly inhibits ER stress signaling
by directly binding eukaryotic translation initiation factor 2α
(5). These observations indicate
that ferroptosis can participate in UC through ER stress-mediated
intestinal epithelial cell death, and the phosphorylation of
NF-κBp65 inhibits ER stress-mediated ferroptosis in intestinal
epithelial cells to alleviate UC. Genetic analysis has also
revealed primary aberrance in several ER homeostasis-associated
genes, including anterior gradient protein 2 homolog, X-box binding
protein 1 and sphingolipid biosynthesis regulator 3 in patients
with UC (64). The inflamed
tissue of patients with IBD exhibits a reduced expression of
aryl-hydrocarbon receptor repressor (Ahrr) and the loss of
intestinal intraepithelial lymphocytes (IELs) in Ahrr−/−
mice increases the susceptibility to dextran sulfate sodium
(DSS)-induced colitis and Clostridium difficile
infection. Mechanistically, Ahrr deficiency results in ferroptosis
in Ahrr−/− IELs through the AHR-induced expression of
CYP1A1, a monooxygenase that generates ROS, increasing oxidative
stress, redox imbalance and lipid peroxidation (65). Thus, a tight regulation that
prevents oxidative stress and ferroptosis, preserves intestinal
immune responses and inflammation. Moreover, it has been reported
that the deficiency in GSH antioxidant function may participate in
the pathogenesis of UC-related liver injury, since the reduction of
hepatic GSH synthesis and reduced GSH function occurs earlier than
liver injury in UC. Moreover, a significant positive correlation
has been between the severity of colonic lesions and the degree of
liver injury (66).
Several ferroptosis-associated genes, including
ACSL4, acyl-CoA synthetase family member 2, GPX4,
LPCAT3, NCOA4, solute carrier family 38, member 1
(SLC38A1) and glucose-6-phosphate dehydrogenase, all of
which participate in the regulation of lipid or iron metabolism
(10,67), have been shown to be markedly
dysregulated (downregulated or upregulated) in UC specimens
(5). In the analysis of
ferroptosis-related genes in patients with UC, 26 differentially
expressed genes have been identified that are significantly
enriched in energy pathways and metabolism, with the top 10 hub
genes from the protein-protein interaction network as interleukin 6
(IL6), prostaglandin-endoperoxide synthase 2 (PTGS2),
hypoxia inducible factor 1 alpha (HIF1A), cluster of
differentiation 44 (CD44), mucin 1 (MUC1), caveolin 1
(CAV1), nitric oxide synthase 2 (NOS2), C-X-C motif
chemokine ligand 2 (CXCL2), stearoyl-CoA desaturase
(SCD), and acyl-CoA synthetase long chain family member 4
(ACSL4) (62). Moreover,
GSE94648, CD44 and MUC1 were highly expressed and
consistent with the expression trend in GSE75214 (62). Results from animal model have also
revealed the significantly increased expression of CD44 in
the colon. It was thus concluded that CD44 and MUC1
may be ferroptosis-related markers in UC (62), and thus, serve as new directions
for UC diagnosis and treatment. In a similar study, the analysis of
97 UC and 75 CD samples alongside 22 normal controls revealed six
differently expressed long non-coding RNAs (lncRNAs) associated
with ferroptosis and immunity in IBD. A constructed lncRNA-mRNA
regulatory network further identified potential miRNAs and
transcription factors based on trans and competing endogenous RNA
mechanisms, concluding that ferroptosis and immune-related lncRNA
are involved in the regulation of aberrant immune response in IBD
(59). The implication of
ferroptosis in IBD pathology, as outlined above, is illustrated in
Fig. 3.
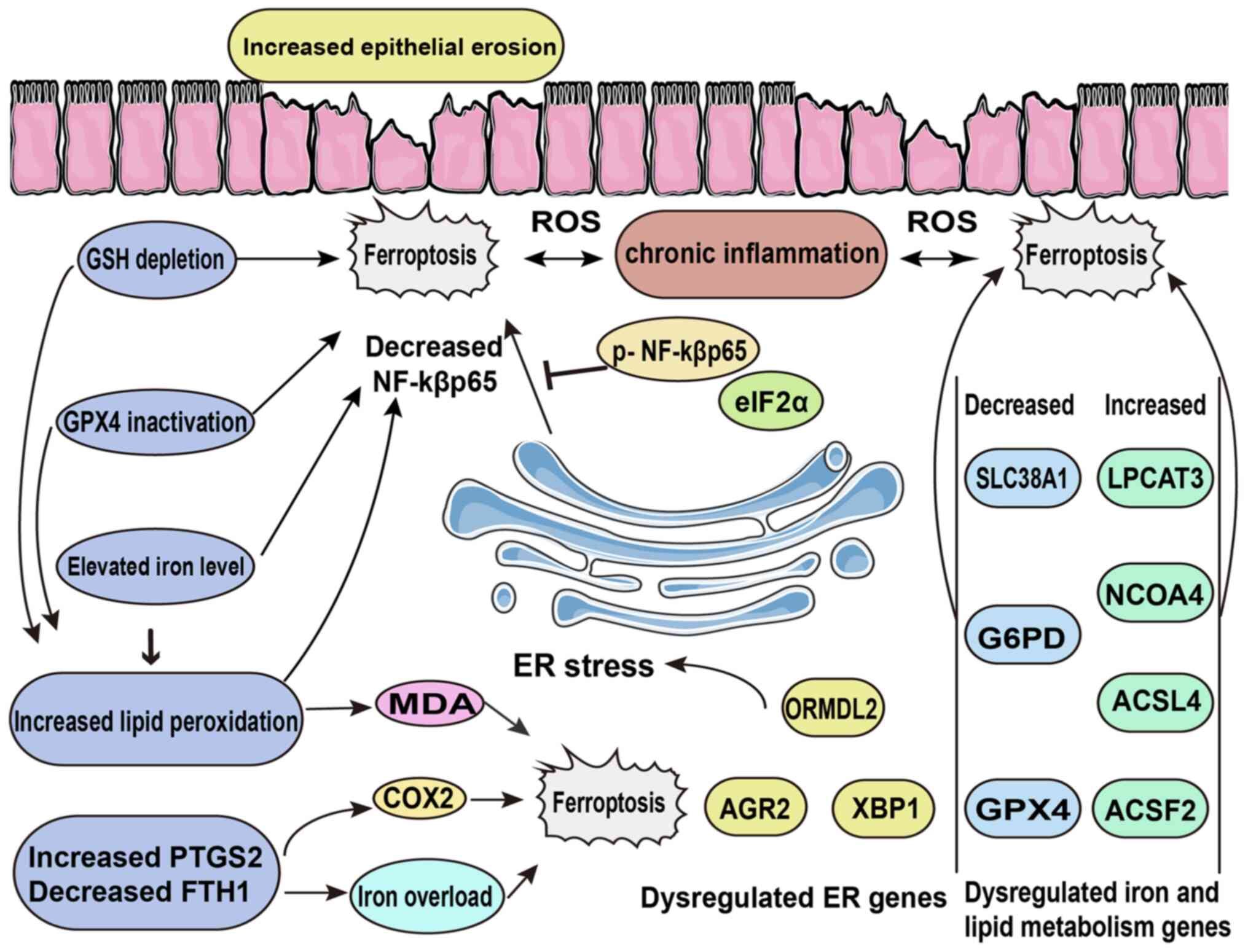 | Figure 3Role of ferroptosis in the pathology
of inflammatory bowel disease. In the gut, lipid peroxidation,
which is driven by depleted GSH, inactive GPX4 and elevated iron
levels, leads to increased ferroptosis that consequently promotes
epithelial cell erosion. Other factors, such as ER stress and
dysregulated genes contribute to chronic inflammation, ROS
generation and ferroptosis. GSH, glutathione; GPX4, glutathione
peroxidase 4; ROS, reactive oxygen species; ER, endoplasmic
reticulum, MDA, malondialdehyde; COX2, cyclooxygenase 2; eIF2α,
eukaryotic translation initiation factor 2α; ORMDL2, ORMDL
sphingolipid biosynthesis regulator 2; AGR2, anterior gradient
protein 2 homolog; XBP1, X-box binding protein 1; SLC38A1, solute
carrier family 38, member 1; G6PD, glucose-6-phosphate
dehydrogenase; GPX4, glutathione peroxidase 4; LPCAT3,
lysophosphatidylcholine acyltransferase 3; NCOA4, nuclear receptor
coactivator 4; ACSL4, acyl-CoA synthetase long-chain family member
4; ACSF2, acyl-CoA synthetase family member 2. |
5. Role of ferroptosis in the pathogenesis
of other intestinal diseases
Research on ferroptosis continues in the quest to
provide a more in-depth understanding of intestinal disease
pathogenesis in order to improve clinical treatment and diagnosis.
In addition to IBD, studies have implicated ferroptosis in other
intestinal diseases, such as intestinal ischemia/reperfusion (I/R)
injury (68), necrotizing
enterocolitis (69),
colitis-associated colorectal cancer (CAC) (70), and other gastrointestinal
diseases, including pancreatitis (16), liver diseases (71) and gastrointestinal cancers, such
as colorectal, liver and gastric cancers (72-74). The hallmark characteristics of
ferroptosis, namely ROS generation, lipid peroxidation, depleted
GSH expression and increased MDA levels have been demonstrated in
intestinal I/R injury (6,68). In intestinal tissues from mice the
I/R model, the levels of pro-ferroptotic factors, such as iron and
ACSL4 are increased, while those of anti-ferroptotic factors, such
as GPX4, ferritin heavy chain 1 (FTH1) and GSH are decreased, with
the inhibition of ferroptosis ameliorating intestinal injury and
barrier dysfunction (68).
Ferroptosis has been implicated in the
pathophysiology of tumors, including colorectal cancer (75). The role of ferroptosis regulators,
such as LPCAT3, SLC7A11, erastin, GPX4 and ACSL4, among others have
been demonstrated in colorectal tumors. For example, the regulation
of GPX4 by SRSF9 serves as a crucial mechanism that drives
colorectal cancer tumorigenesis, as well as Erastin-induced
ferroptosis resistance (76), and
TP53-induced glycolysis and apoptosis regulator (TIGAR) functions
as a potential modulator of ferroptosis resistance in colorectal
cancer through the ROS/AMPK/SCD1 signal pathway, with the knockdown
of TIGAR causing elevated lipid peroxidation and sensitive to
erastin-induced ferroptosis (77). The induction of ferroptosis
continues to be explored a potential intervention strategy to the
treatment of cancers (78,79).
Studies have also identified distinct ferroptosis-associated
genetic or transcriptome clusters from 1,251 colorectal cancer bulk
samples, which are linked with varying biological pathways and
clinical outcomes. Ferroptosis-associated patterns linked with the
tumor microenvironment diversity and immune response phenotype have
been identified (80).
6. Targeting ferroptosis for the treatment
of IBD
Lipophilic/radical-trapping
antioxidants
This type of therapeutic agent serves as a scavenger
for chain-carrying radicals, thus breaking the self-oxidizing chain
reaction. The most common chain-breaking radical-trapping or
lipophilic antioxidants are aromatic amines and phenols. For the
aromatic amines, ferrostatin-1 and liproxstatin-1 have been
demonstrated to function as classical ferroptosis inhibitors in
vivo and in vitro (81,82). For example, a previous study
demonstrated that the ferroptosis inhibitory activity of
ferrostatin-1 and liproxstatin-1 is likely driven by their
reactivity as radical-trapping antioxidants rather than their
potency as inhibitors of lipoxygenases (81). As previously demonstrated, in a
mouse model of DSS-induced IBD, the expression levels of ACSL4 and
COX2 were significantly upregulated, while those of GPX4 and FTH1
were downregulated. Treatment with the ferroptosis inhibitors,
ferrostatin-1 or liproxstatin-1, mitigated intestinal inflammation
by decreasing ACSL4 and COX2, increasing GPX4 and FTH1, and
improving mouse body weight and colon length. Moreover, the
treatment reduced the expression of Nrf2 and heme oxygenase-1,
presenting the suppression of ferroptosis as an effective approach
in ameliorating DSS-induced UC in mice (58). Liproxstatin-1 mitigates intestinal
barrier injury caused by lipid peroxidation-mediated cell death by
inhibiting lipid peroxidation (16).
Ferroptosis has also been demonstrated to
participate in the pathophysiology of CD in both human and animal
models, indicated by the dysregulation of iron, lipid peroxidation
and redox homeostasis. While ACSL4 has been shown to be
significantly upregulated and GPX4 to be downregulated in colonic
biopsies of patients with CD, other ferroptosis biomarkers, such as
MDA, prostaglandin-endoperoxide synthase 2 (PTGS2) and FTH1 are
upregulated and GPX4 is downregulated in the colon of the animal
model. The administration of ferrostatin-1 in an animal model has
been found to alleviate the pathological phenotypes of
trinitrobenzene sulfonic acid (TNBS)-induced CD-like colitis
(17), providing ferroptosis as a
potential therapeutic strategy in treating CD. The role of signal
transducer and activator of transcription (STAT)3-mediated
ferroptosis, as a ferroptosis-related hub gene, has been reported
in in vitro and in vivo models of colitis, where
ferroptosis has been found to increase its expression in
DSS-induced colitis, Salmonella Typhimurium colitis and in
H2O2-induced IEC-6 cells. Treatment with
ferrostatin-1 reactivates the phosphorylation level of STAT3
(19). The expression of SLC6A14
is significantly elevated and positively correlates with the level
of PTGS2 in tissue samples from patients with UC. Moreover, a
series of in vivo and in vitro experiments have
indicated that the knockdown of SLC6A14 markedly suppresses
ferroptosis. Further analysis has revealed that SLC6A14 inhibits
the expression of p21 (RAC1)-activated kinase 6 (PAK6) and the
knockdown of PAK6 abolishes the effects of SLC6A14 on RSL3-induced
ferroptosis in Caco-2 cells. Mechanistically, SLC6A14 promotes
ferroptosis in UC by enhancing the expression of CCAAT enhancer
binding protein β (C/EBPβ) and its binding activity to inhibit PAK6
(83), providing the
SLC6A14-C/EBPβ-PAK6 axis-mediated ferroptosis a potential
therapeutic target for colitis.
α-tocopherol, a natural phenolic substance, is the
most active form of vitamin E and serves as an effective suppressor
of ferroptosis (84).
α-tocopherol specifically breaks both peroxidative chain
propagation and inhibits lipoxygenases (85). It has been shown that α-tocopherol
and GPX4 cooperatively maintain the lipid redox balance and prevent
ferroptosis (84). A previous
study demonstrated that the deprivation of vitamin E caused the
death of GPX4-deficient mice ~4 weeks after the vitamin E-enriched
diet was redrawn (86). These
findings indicate that the ferroptosis regulator, GPX4, is crucial
for cell survival and proper function, and that α-tocopherol can
compensate for the loss of GPX4 by protecting cells against
deleterious lipid peroxidation. In a preclinical trial, all 14
patients with IBD (with mild and moderately active UC) responded
clinically to d-α-tocopherol therapy, with remission induced in
nine of them (64%); of note, no adverse events or hospitalization
due to worsened disease activity were reported (87). Although several other studies have
reported the ability of α-tocopherol to mitigate experimental IBD
by protecting intestinal barrier function, modulating the gut
microbiota, accelerating intestinal tissue healing and modulating
the immune system (88-90), its inhibition of ferroptosis in
IBD requires further research. The anti-ferroptotic potential of
other synthetic phenolic compounds, such as
1,8-tetrahydronaphthyridinols has been demonstrated, where the
inherent radical-trapping antioxidant reactivity of
1,8-tetrahydronaphthyridinols was similarly effective as
ferrostatin-1 and liproxstatin-1 at subverting ferroptosis
triggered by either genetic or pharmacological inhibition of the
hydroperoxide-detoxifying enzyme GPX4 in mouse fibroblasts and
hippocampal cells (81). A
schematic summary of the various treatments that target ferroptosis
in IBD is presented in Fig.
4.
Iron chelators
Ion is involved in ferroptosis through either the
induction of a non-enzymatic iron-mediated Fenton reaction or the
activation of iron-containing lipid oxygenases, such as
CYP450/cytochrome P450 oxygenases and arachidonate lipoxygenase
(ALOX) (38,91). Thus, potent ion chelators,
including deferasirox, ciclopirox, deferoxamine and deferiprone,
effectively inhibit ferroptosis-induced cell death. The
anti-ferroptotic ability of these ion chelators has been largely
explored across many conditions, including intestinal cell injury
(37), traumatic spinal cord
injury (92), osteoarthritis
(93), type 2 diabetic
osteoporosis (94),
atherosclerosis (95),
neuroprotection (96) and acute
lung injury (97). In assessing
the pathogenic role of ROS in UC, Millar et al (98) examined the influence of exogenous
iron (ferric citrate) and iron chelators (desferrioxamine and
1,10-phenanthroline) on the colonic biopsies of patients with UC
and normal control subjects. Through luminol-amplified
chemiluminescence, they found that desferrioxamine and
phenanthroline decreased chemiluminescence by 47 and 26%,
respectively in inactive UC, and by 44 and 42% in active UC.
Notably, ferric citrate did not affect the chemiluminescence
produced by human colonic mucosa, suggesting that there is
sufficient free iron in inflamed biopsies already to drive the
Fenton reaction maximally (98).
Intestinal epithelial cell injury due to ionizing radiation-induced
ferroptosis is effectively abrogated by the iron chelator
deferoxamine in vitro and in vivo. Moreover, the
signs of ferroptosis in intestinal epithelial cells, including the
upregulation of intracellular iron levels and lipid peroxidation,
the increase in PTGS2 mRNA levels, the downregulation of GPX4 mRNA
and GSH levels, and significant mitochondrial damage are
significantly relieved by deferoxamine treatment (37).
Deferoxamine and deferiprone significantly alleviate
the macroscopic and/or pathological features of inflammation in
TNBS-induced colitis by reducing colon weight/length ratio, ulcer
index and the total colitis index (99). A recent study reported that
ferroptosis was induced in mice with UC, as evidenced by ferrous
iron accumulation, elevated ROS production, the depletion of
superoxide dismutase (SOD) and GSH, and the reduced expression of
GPX-4 and ferritin heavy chain (FTH), accompanied by the increased
expression of transferrin (TF). However, deferasirox treatment
significantly reversed the changes caused by ferroptotic
characteristics in DSS-induced mice and reshaped the composition of
intestinal microbiota (100). In
another study, the combined administration of deferasirox and
vitamin D3 mitigated ion induced-injury by decreasing the levels of
pro-inflammatory cytokines (IL-1β/IL-6/TNF-α), oxidative stress
(MDA/H2O2) and apoptotic markers, but
increasing IL-10, GSH, SOD1, catalase and GPX4 levels in rats
(101). This indicates that
deferasirox enhances cellular anti-inflammatory, anti-oxidative
stress and iron regulatory pathways in the inhibition of
ferroptosis. However, data on ion chelators in IBD therapy are
limited.
Enzyme inhibitors
ACSL4, an enzyme that converts fatty acid to fatty
acyl-CoA esters, is known to dictate ferroptosis sensitivity by
shaping cellular lipid composition (27) and contributes to intestinal tissue
injury (68). Moreover, the
mitochondrial respiratory chain promotes lipid peroxidation by ALOX
or POR (cytochrome P450 reductase); thus, ALOX serves as a critical
enzyme of lipid peroxidation that leads to ferroptosis (102). Again, the NOXs (nicotinamide
adenine dinucleotide phosphate-oxidase) on the cell membranes are a
resource of cellular ROS for ferroptosis. Therefore, the inhibition
of ACSL4, ALOX and NOXs provides promising therapeutic targets for
preventing ferroptosis. Reported enzyme inhibitors include ACSL4
inhibitors, such as thiazolidinediones and triacsin C, ALOX
inhibitors such as baicalein,
cinnamyl-3,4-dihydroxya-cyanocinnamate, PD146176, zileuton, ML351,
AA-861 and NCTT-956, and NOX inhibitors, such as GKT137831,
diphenylene iodonium and 2-acetylphenothiazine (38,43). For example, the administration of
baicalein ameliorates UC by improving the intestinal epithelial
barrier (103) and enhances the
normalization of the levels of stress response protein and
inflammatory cytokine in the intestine, plasma, and marrow of
C57BL/6 mice undergoing mitigation of total-body irradiation
(104). Zileuton-fed mice
develop fewer intestinal polyps and exhibit a significant reduction
in systemic and polyp-associated inflammation (105). Furthermore, the NOX inhibitor,
diphenyleneiodonium, and the selective NOX1/4 isoform inhibitor,
GKT137831, significantly inhibit erastin-induced ROS production,
lipid ROS and cell death (106).
Data on the exploration of ferroptosis enzyme inhibition in IBD are
limited and provide direction for future studies.
Protein degradation inhibitors
As numerous ferroptosis activators possess the
capability of inducing GPX4 degradation, which leads to lipid
peroxidation, therapeutic agents that block the degradation of GPX4
enhance antioxidant capacity and inhibit ferroptosis. Typical
examples of protein degradation inhibitors are dopamine,
2-amino-5-chloro-N,3-dimethylbenzamide (CDDO) and
5-(tetradecyloxy)-2-furoic acid. These inhibitors block FIN56- or
erastin-induced GPX4 degradation to prevent ferroptosis (107,108). For instance, dopamine reduces
erastin-induced ferrous iron accumulation, GSH depletion and MDA
production through the increase in the protein stability of GPX4, a
phospholipid hydroperoxidase that protects cells against membrane
lipid peroxidation (108).
Therefore, a decrease in intestinal dopamine levels is closely
related to the development of IBD. Dopamine/dopamine receptor D5
signaling in colonic macrophages controls the development of
colitis by modulating M1/M2 macrophage polarization (109) and the activation of the dopamine
D2L receptor participates in suppressing colitis-induced weight
loss, colon shrinkage, and IL-17 secretion from mesenteric lymph
node lymphocytes in response to CD3/CD28 stimulation in mice
(110). Therefore, dopamine
serves not only as an anti-ferroptotic agent, but also as a potent
immune response regulator to ameliorate colitis. In other research,
CDDO, a compound known to inhibit heat shock protein 90 (HSP90),
was found to potently inhibit both ferroptosis and necroptosis
(111). Erastin-activated
ferroptosis was found to increase the levels of lysosome-associated
membrane protein 2a, also known as CD107b, to promote
chaperone-mediated autophagy (CMA), which in turn, promotes the
degradation of GPX4. Notably, the inhibition of CMA stabilized GPX4
and reduced ferroptosis (111).
These findings suggest that the use of agents that inhibit GPX4
degradation may provide an effective means of mitigating
ferroptosis-related lethal effects in IBD.
Stem cell-derived exosomes
Stem cells and their derived exosomes have been
largely explored as promising therapeutic agents in the treatment
of human IBD (112-114) and experimental IBD (115-117). As regards the the targetted
treatment of IBD by inhibiting ferroptosis, a recent study
confirmed that relative to healthy individuals, patients with UC
exhibit higher levels of iron, ACSL4 and MDA, whereas they exhibit
decreased levels of GSH and GPX in colon tissues (118). In that study, in the animal
model of IBD, exosomes derived from endometrial regenerative cells
attenuated the clinical symptoms of colitis and tissue damage by
enhancing the expression of GSH and GPX4, but reducing the levels
of iron, MDA and ACSL4 in the colons of mice with colitis.
Moreover, in vitro analysis revealed that the exosomes
rescued erastin-induced cell death and upregulated the levels of
GSH and GPX4, while downregulating the levels of iron, MDA and
ACSL4 (118). This illustrates
the involvement of ferroptosis in the pathogenesis of IBD and the
potential of stem cell-derived exosomes to mitigate colitis through
the downregulation of intestinal ferroptosis.
In other studies, mesenchymal stem cell-derived
exosomes (MSC-Ex) were protected against ferroptosis via the
exosomal-mediated stabilization of SLC7A11 in acute liver injury.
It was demonstrated that the MSC-Ex triggered the increased
expression of SLC7A11 protein, CD44 and OTUB1, and the activation
of system XC− to prevent ferroptosis
(119). The stability of SLC7A11
and the consequent activation of system XC−
may partly be attributed to CD44 expression, which suppresses
ferroptosis in an OTUB1-dependent manner (120). Moreover, MSC-Ex have been found
to inhibit the production of ROS and ferrous iron, upregulate the
expression of ferroptosis suppressor FSP1, and enhance the repair
of neurological function in mice. Mechanistically, MSC-Ex
lncGm36569 prevents ferroptosis-induced neuronal cell dysfunction
through the miR-5627-5p/FSP1 axis (121), and MSC-Ex also attenuate renal
ischemia/reperfusion injury by interacting with SRSF1 to regulate
ASCL4-mediated ferroptosis (122), attenuate myocardial injury by
inhibiting ferroptosis in acute myocardial infarction mice
(123), and inhibit the
ferroptosis of hepatic stellate cells by regulating the xCT/GPX4
axis (124). These observations
indicate the promising therapeutic outcomes of the MSC-Ex-induced
inhibition of ferroptosis in cell protection. This may provide
future direction for research in IBD therapy exploration.
Oral N-acetylcysteine and
glutathione
Replenishing cysteine/glutathione deficiency is also
an effective method with which to abrogate oxidative stress and its
associated lipoperoxidation that characterizes ferroptosis. Thus,
the administration of N-acetylcysteine, a cysteine prodrug,
replenishes intracellular GSH levels and serves as a safe and
well-tolerated antidote for cysteine/GSH deficiency (125). In addition to
N-acetylcysteine, oral GSH and sublingual form of GSH also
serves as potent GSH supplementation in humans (126). As previously demonstrated, the
in vivo administration of N-acetylcysteine attenuates
acute TNBS-induced colitis in rats through increased mucosal GSH
levels, reducing the extent of intestinal mucosal injury and
TNBS-induced ROS overgeneration (127). However, further studies are
required to further explore treatments that directly replenish
cysteine/glutathione stores as a potential therapy for IBD and
other ferroptosis-induced intestinal injuries.
Other agents targeting ferroptosis in
IBD
Curculigoside has been shown to alleviate colitis
in mice by reversing ferroptosis-related changes, such as iron
overload, GSH depletion, excessive ROS and MDA production, and the
decreased expression of SOD and GPX4. In IEC-6 cells, curculigoside
enhances selenium sensitivity and promotes GPX4 transcription. The
selective knockdown of GPX4 significantly inhibits the protective
effects of curculigoside on cell death and prevents its
downregulation of GSH, MDA, and lactate dehydrogenase activity in
ferroptotic IEC-6 (18). The
protease furin, a proteolytic enzyme found to exert protective
effects in several autoimmune diseases, inhibits DSS-induced
ferroptosis-like injury in colon epithelial cells in vivo
and in vitro by upregulating GPX4. Mechanistically, furin
alleviated the cell injury by activating the Nrf2-GPX4 signaling
pathway in mice and NCM460 cells (128). In a recent study, zero-valence
selenium-enriched Prussian blue nanozymes (Se-HMPB nanozymes),
multifunctional nanozymes capable of scavenging ROS and inhibiting
ferroptosis or T-cell differentiation, were explored for IBD
therapy (129). The researchers
reported that Se-HMPB nanozymes effectively scavenged various ROS
in inflammatory tissues and enhanced GPX activity, inhibiting
ferroptosis and reversing the lipid peroxidation of intestinal
epithelial cells to preserve intestinal barrier integrity in UC.
Additionally, the construct inhibited T-cell differentiation in a
CD model, regulating the intestinal immune barrier (129), and thus presenting great
potential for IBD therapy.
Dipeptidyl peptidase-4 (DPP4) has been implicated
in the pathogenesis of IBD and as a potential biomarker (130). It has been shown that TP53
limits erastin-induced ferroptosis by blocking DPP4 activity in a
transcription-independent manner. The loss of TP53 prevents the
nuclear accumulation of DPP4, and thus facilitates
plasma-membrane-associated DPP4-dependent lipid peroxidation, which
finally results in ferroptosis (131). In experimental IBD, the DPP4
inhibitor, linagliptin, mitigates colitis by inhibiting colonic
DPP4 activity, suppressing colonic IL-6, TNF-α, and
myeloperoxidase, and the upregulation of IL-10. Linagliptin also
reduces intestinal mucosal oxidative stress by inhibiting lipid
peroxides and augmenting GSH, GPX and total antioxidant capacity
(132). In similar studies,
linagliptin has exhibited promising anti-inflammatory activity
against acetic acid-induced colitis through the stimulation of the
AMPK/SIRT1/PGC-1α pathway, as well as the inhibition of the
JAK2/STAT3 signaling pathway, which may partly be mediated through
AMPK activation (133).
7. Targeting ferroptosis in other intestinal
diseases
Intestinal I/R injury
Similar to other gastrointestinal diseases,
intestinal I/R injury is associated with multiple types of
regulated cell death, including autophagy, apoptosis and
necroptosis, and the discovery of ferroptosis has triggered the
exploration of potential mechanisms of ferroptotic cell death in
intestinal I/R injury. ROS generation and lipid peroxidation are
associated with intestinal I/R injury and are primary contributors
to the initiation and execution of ferroptosis (134). A previous study found that the
inhibition of ACSL4, the key enzyme that regulates lipid
composition, prior to intestinal reperfusion, protected against
ferroptosis and cell death (68).
The authors of that study concluded that ferroptosis was closely
associated with intestinal I/R injury, and ACSL4 participated in
the lethal process, while specificityprotein 1 served as a crucial
transcription factor for the increased expression of ACSL4
(68). Depleted GSH levels and a
reduced SOD activity, as well as elevated MDA levels, have been
observed in animal models of intestinal tissue I/R injury (135,136). These observations implicate the
participation of ferroptosis in I/R-induced intestinal injury;
however, studies on the underlying mechanisms are largely
lacking.
Necrotizing enterocolitis
Necrotizing enterocolitis is the most common
gastrointestinal disease among newborns and research has
established the involvement of several types of regulated cell
death in the gut barrier dysfunction that characterizes the disease
(137,138). In a previous study, through
bioinformatics analyses, ferroptosis was identified to play a role
in necrotizing enterocolitis, where ACSL4 gene expression
levels were significantly upregulated and positively correlated
with ferroptosis, and the influx of multiple immune cells,
including macrophages, neutrophils, activated dendritic cells and
regulatory T-cells. The inhibition of ferroptosis significantly
attenuated necrotizing enterocolitis in newborn mice (69). In other studies, the inhibition of
ACSL4 has been proven to be a viable therapeutic approach to
preventing ferroptosis-related inflammation, tissue injury, and
even tumors (139,140), as ACSL4 dictates ferroptosis
sensitivity by shaping cellular lipid composition (27). Moreover, ROS, a product of
ferroptosis, has been found to accumulate in the damaged gut
tissues of infants with necrotizing enterocolitis and to serve as a
regulatory point (ROS-NF-κB axis) in ameliorating inflammation in
necrotizing enterocolitis (141).
CAC
The inhibition of ferroptosis as a treatment
approach for CAC presents varying outcomes and warrants further
investigations. On the one hand, the inhibition of ferroptosis in
intestinal tissues by OTSSP167, a MELK inhibitor, alleviates
colitis and abrogates the occurrence and progression of CAC by
reducing M1 macrophage polarization, macrophage infiltration,
downregulating the secretion of pro-inflammatory factors, and
significantly repairing intestinal damage. Mechanistically, the
treatment inhibits the AKT/IKK/p65 and ERK/IKK/p65 signaling
cascades in vivo and in vitro to mitigate colitis and
its related colorectal cancer (142). On the other hand, the
administration of the ferroptosis inhibitor, ferrostatin-1, results
in an increased incidence of CAC. This effect is exacerbated in
mice fed a high-fat diet, as they exhibit an increased tumor number
and a higher degree of dysplasia following the repression of lipid
peroxidation and ferroptosis marker expression in mouse colon
tissue (70), providing a new
perspective for the prevention of CAC. Additional studies are
required in this field to provide clarity and explore the
underlying mechanisms. The therapeutic agents, key effects and
mechanisms of ferroptosis targets in IBD and other intestinal
diseases are summarized in Table
I.
 | Table ITherapeutic targets of ferroptosis in
IBD and other gastrointestinal diseases. |
Table I
Therapeutic targets of ferroptosis in
IBD and other gastrointestinal diseases.
| Therapeutic
agent | Disease/model | Mechanism | Key effects | (Refs.) |
|---|
| Ferrostatin-1,
liproxstatin-1, or deferiprone | DSS-induced UC in
mice | Ferroptosis
inhibition via the Nrf2/HO-1 signaling pathway | Decreased COX2 and
ACSL4 but increased GPX4 and FTH1 Reduced inflammation indexes and
malanoyl dialdehyde levels | (58) |
| Ferrostatin-1 | TNBS-induced
CD-like colitis in mice | Inhibition of
ferroptosis | Ameliorates the
pathological phenotypes of TNBS-induced colitis Reduced PTGS2, MDA
and FTH1, and increased GPX4 levels | (17) |
| Ferrostatin-1 | DSS-induced
colitis, Salmonella Typhimurium colitis, and
H2O2-induced IEC-6 cells | Ferroptosis is
closely associated with the development of colitis via the
STAT3 gene | Reactivates the
phosphorylation level of STAT3 | (19) |
| Desferrioxamine and
phenanthroline | Colonic mucosa
biopsies from patients with UC | Reduction of
chemiluminescence generated by reactive oxygen species | Decreased mucosal
reactive oxygen species production in both active and inactive
UC | (98) |
|
N-acetylcysteine | TNBS +
ethanol-induced colitis in rats | Increase of mucosal
GSH levels to mitigate acute colitis | Increased GSH
stores 2-fold, decreased extent of mucosal injury | (127) |
| Curculigoside | DSS-induced colitis
and IEC-6 cells | Ferroptosis
inhibition via the induction of GPX4 | Decreased ROS, MDA,
and ion but increased GSH, SOD and GPX4 Relieved colitis | (18) |
| Linagliptin | TNBS-induced
colitis in rats | Inhibits the
IL-6/JAK2/STAT3 pathway via downregulating p-JAK2/JAK2 and
p-STAT3/STAT3 proteins Inhibits the HMGB1/RAGE/NF-κB cascade via
lowering HMGB1, RAGE, and p-NF-κB p65/NF-κB p65 proteins | Suppresses colonic
IL-6, TNF-α, and myeloperoxidase and upregulates IL-10 Inhibits
mucosal oxidative stress by reducing lipid peroxides and augmenting
GSH, GPX, and total antioxidant capacity | (132) |
| Endometrial
regenerative cells-derived exosome | DSS-induced colitis
and erastin-treated NCM460 human intestinal epithelial cell
line | Inhibition of
ferroptosis | Increased GSH and
GPX4 but reduced iron, MDA, and ACSL4 in the colon | (118) |
| Furin | DSS-induced colitis
in mice and DSS-treated NCM460 cells | Inhibits
ferroptosis-like injury by activating the Nrf2-Gpx4 signaling
pathway | Upregulated GPX4
and prevented cell/tissue injury in vivo and in
vitro. Alleviates DSS-induced damage to mitochondrial
membranes | (128) |
| OTSSP167 | DSS-induced colitis
and CAC | Inhibits
ferroptosis via the AKT/IKK/p65 and ERK/IKK/p65 signaling cascades
in vivo and in vitro | Alleviation of
intestinal inflammation and the occurrence of CAC | (142) |
| Ferrostatin-1
(ferroptosis inhibitor) | AOM/DSS-induced
mouse CAC | CAC aggravates
through the evasion of ferroptosis in the ER stress-mediated
pathway | Ferrostatin-1
treatment increases the incidence of CAC. The induction of
ferroptosis partly abolished the pro-tumor effects of a high-fat
diet on CAC | (70) |
| Liproxstatin-1 | I/R-induced
intestinal injury | Inhibit ferroptosis
by reducing ACSL4 activation and function | Increases GPX4 and
reduces COX2, lipid peroxidation, serum LDH, TNF-α, and IL-6
levels, and alleviates intestinal I/R injury | (68) |
| Resveratrol,
dioscin | I/R-induced
intestinal injury | Inhibition of
oxidative stress | Reduces the levels
of MDA, MPO, and NO, but increase SOD activity, thus, ameliorating
intestinal I/R injury | (134, 135) |
| ACSL4
knockdown | Necrotizing
enterocolitis | ACSL4-mediated
ferroptosis | ACSL4 expression is
positively associated with ferroptosis, inflammation, and the
abundance of macrophage, activated dendritic cells, neutrophils,
and regulatory T-cells | (69) |
| Antioxidant peptide
from tuna backbone protein (APTBP) | Necrotizing
enterocolitis | ROS-NF-κB-mediated
inflammation | Reduces
intracellular ROS and inflammatory cytokines | (141) |
8. Conclusions and future perspectives
Ferroptosis is driven by iron-dependent
phospholipid peroxidation and is modulated by several cellular
metabolic activities, such as redox homeostasis, iron transport and
metabolism, mitochondrial activity, and the metabolism of lipids,
amino acids and sugars, as well as multiple signaling pathways
relevant to disease processes. The regulation of key mediators,
including GPX4, ACSL4, SLC7A11 and p53 is crucial in modulating
ferroptosis-associated intestinal diseases. Although the
pathophysiological role of ferroptosis continues to be explored,
its involvement in several human diseases, including IBD has been
documented. A number of studies, as those aforementioned, have
demonstrated the participation of ferroptosis in IBD and the
ability of several therapeutic agents to mitigate the lethal effect
associated with ferroptosis in IBD. Notably, the therapeutic
regulation of ferroptosis has exhibited promising outcomes as a
novel treatment avenue for IBD and other intestinal diseases.
However, the field is still relatively new and requires further
studies and the extensive exploration of related mechanisms. For
example, although the uncontrolled peroxidation of PUFA-PLs has
been identified as the most downstream step in the process of
ferroptosis, the exact mechanisms through which cells ultimately
die warrant further elucidation. Moreover, the discovery of precise
biomarkers for in vivo ferroptosis may enhance the current
understanding of the pathophysiological role of this cell death
modality. This will also provide more precise and effective targets
for the inhibition of ferroptosis. Again, the current experimental
practice of preventing cell death by a single ferroptosis inhibitor
may not be sufficient evidence that ferroptosis participates in a
given process, since the association between ferroptotic cell death
and iron/lipid peroxides remains largely unclear. Further evidence
is required in order to better understand these links, as well as
to provide transcriptional regulators and signaling pathways
associated with ferroptosis in IBD development and progression.
Availability of data and materials
Not applicable.
Authors' contributions
DKWO and JY were involved in the conception and
design of the study, as well as in the collection and/or assembly,
analysis and interpretation of data to be included in the review,
and in the writing of the manuscript. ZW was involved in the
provision of study materials, as well as in the interpretation and
analysis of data to be included in the review. FM and ZZ were
involved in the design of the study, as well as in the
interpretation and analysis of data to be included in the review,
and in the writing of the manuscript. All authors have read and
approved the final manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was sponsored by the National Natural Science
Fund of China (grant no. 82250410378), the 2022 Jiangsu Excellent
postdoctoral program (grant no. 2022ZB634) and the Project of
Suzhou Science and Technology (grant no. SKY2022027).
References
|
1
|
Hodson R: Inflammatory bowel disease.
Nature. 540:S972016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cai Z, Wang S and Li J: Treatment of
inflammatory bowel disease: A comprehensive review. Front Med.
8:7654742021. View Article : Google Scholar
|
|
3
|
Na SY and Moon W: Perspectives on current
and novel treatments for inflammatory bowel disease. Gut Liver.
13:604–616. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ocansey DKW, Xu X, Zhang L and Mao F:
Mesenchymal stem cell-derived exosome: The likely game-changer in
stem cell research. BIOCELL. 46:1169–1172. 2022. View Article : Google Scholar
|
|
5
|
Xu M, Tao J, Yang Y, Tan S, Liu H, Jiang
J, Zheng F and Wu B: Ferroptosis involves in intestinal epithelial
cell death in ulcerative colitis. Cell Death Dis. 11:862020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu S, He Y, Lin L, Chen P, Chen M and
Zhang S: The emerging role of ferroptosis in intestinal disease.
Cell Death Dis. 12:2892021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Otasevic V, Vucetic M, Grigorov I,
Martinovic V and Stancic A: Ferroptosis in different pathological
contexts seen through the eyes of mitochondria. Oxid Med Cell
Longev. 2021:55373302021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Battaglia AM, Chirillo R, Aversa I, Sacco
A, Costanzo F and Biamonte F: Ferroptosis and cancer: Mitochondria
meet the 'iron maiden' cell death. Cells. 9:15052020. View Article : Google Scholar
|
|
10
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell Death
Differ. 23:369–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu C, Liu Z and Xiao J: Ferroptosis: A
double-edged sword in gastrointestinal disease. Int J Mol Sci.
22:124032021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gao W, Zhang T and Wu H: Emerging
pathological engagement of ferroptosis in gut diseases. Oxid Med
Cell Longev. 2021:42462552021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dixon SJ and Stockwell BR: The hallmarks
of ferroptosis. Annu Rev Cancer Biol. 3:35–54. 2019. View Article : Google Scholar
|
|
14
|
Sui X, Zhang R, Liu S, Duan T, Zhai L,
Zhang M, Han X, Xiang Y, Huang X, Lin H and Xie T: RSL3 drives
ferroptosis through GPX4 inactivation and ROS production in
colorectal cancer. Front Pharmacol. 9:13712018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang J, Zhang J, Ma J, Ma J, Liu J, Wang
F and Tang X: Inhibiting ferroptosis: A novel approach for
ulcerative colitis therapeutics. Oxid Med Cell Longev.
2022:96786252022.PubMed/NCBI
|
|
16
|
Ma D, Jiang P, Jiang Y, Li H and Zhang D:
Effects of lipid peroxidation-mediated ferroptosis on severe acute
pancreatitis-induced intestinal barrier injury and bacterial
translocation. Oxid Med Cell Longev. 2021:66445762021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu J, Liu S, Cui Z, Wang X, Ning T, Wang
T, Zhang N, Xie S, Min L, Zhang S, et al: Ferrostatin-1 alleviated
TNBS induced colitis via the inhibition of ferroptosis. Biochem
Biophys Res Commun. 573:48–54. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang S, Liu W, Wang J and Bai X:
Curculigoside inhibits ferroptosis in ulcerative colitis through
the induction of GPX4. Life Sci. 259:1183562020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang F, Zhang S, Li X, Huang Y, He S and
Luo L: STAT3-mediated ferroptosis is involved in ulcerative
colitis. Free Radic Biol Med. 188:375–385. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cao JY and Dixon SJ: Mechanisms of
ferroptosis. Cell Mol Life Sci. 73:2195–2209. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tang D, Chen X, Kang R and Kroemer G:
Ferroptosis: Molecular mechanisms and health implications. Cell
Res. 31:107–125. 2021. View Article : Google Scholar :
|
|
22
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hu W, Liang K, Zhu H, Zhao C, Hu H and Yin
S: Ferroptosis and its role in chronic diseases. Cells.
11:20402022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Conrad M and Pratt DA: The chemical basis
of ferroptosis. Nat Chem Biol. 15:1137–1147. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dixon SJ, Winter GE, Musavi LS, Lee ED,
Snijder B, Rebsamen M, Superti-Furga G and Stockwell BR: Human
haploid cell genetics reveals roles for lipid metabolism genes in
nonapoptotic cell death. ACS Chem Biol. 10:1604–1609. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Doll S, Proneth B, Tyurina YY, Panzilius
E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A,
et al: ACSL4 dictates ferroptosis sensitivity by shaping cellular
lipid composition. Nat Chem Biol. 13:91–98. 2017. View Article : Google Scholar :
|
|
28
|
Bano I, Horky P, Abbas SQ, Majid M, Bilal
AHM, Ali F, Behl T, Hassan SSU and Bungau S: Ferroptosis: A new
road towards cancer management. Molecules. 27:21292022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ursini F, Maiorino M and Gregolin C: The
selenoenzyme phospholipid hydroperoxide glutathione peroxidase.
Biochim Biophys Acta. 839:62–70. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Skouta R, Dixon SJ, Wang J, Dunn DE, Orman
M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A and
Stockwell BR: Ferrostatins inhibit oxidative lipid damage and cell
death in diverse disease models. J Am Chem Soc. 136:4551–4556.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen X, Yu C, Kang R and Tang D: Iron
metabolism in ferroptosis. Front Cell Dev Biol. 8:5902262020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh
HJ III, Kang R and Tang D: Autophagy promotes ferroptosis by
degradation of ferritin. Autophagy. 12:1425–1428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gao M, Yi J, Zhu J, Minikes AM, Monian P,
Thompson CB and Jiang X: Role of mitochondria in ferroptosis. Mol
Cell. 73:354–363.e3. 2019. View Article : Google Scholar :
|
|
34
|
Gao M, Monian P, Pan Q, Zhang W, Xiang J
and Jiang X: Ferroptosis is an autophagic cell death process. Cell
Res. 26:1021–1032. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen X, Kang R, Kroemer G and Tang D:
Organelle-specific regulation of ferroptosis. Cell Death Differ.
28:2843–2856. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee H, Zandkarimi F, Zhang Y, Meena JK,
Kim J, Zhuang L, Tyagi S, Ma L, Westbrook TF, Steinberg GR, et al:
Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat
Cell Biol. 22:225–234. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou H, Zhou YL, Mao JA, Tang LF, Xu J,
Wang ZX, He Y and Li M: NCOA4-mediated ferritinophagy is involved
in ionizing radiation-induced ferroptosis of intestinal epithelial
cells. Redox Biol. 55:1024132022.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang WS, Kim KJ, Gaschler MM, Patel M,
Shchepinov MS and Stockwell BR: Peroxidation of polyunsaturated
fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci
USA. 113:E4966–E4975. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kobayashi Y, Ohfuji S, Kondo K, Fukushima
W, Sasaki S, Kamata N, Yamagami H, Fujiwara Y, Suzuki Y and Hirota
Y; Japanese Case-Control Study Group for Ulcerative Colitis:
Association of dietary fatty acid intake with the development of
ulcerative colitis: A multicenter case-control study in Japan.
Inflamm Bowel Dis. 27:617–628. 2021. View Article : Google Scholar
|
|
40
|
John S, Luben R, Shrestha SS, Welch A,
Khaw KT and Hart AR: Dietary n-3 polyunsaturated fatty acids and
the aetiology of ulcerative colitis: A UK prospective cohort study.
Eur J Gastroenterol Hepatol. 22:602–606. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lee JY, Nam M, Son HY, Hyun K, Jang SY,
Kim JW, Kim MW, Jung Y, Jang E, Yoon SJ, et al: Polyunsaturated
fatty acid biosynthesis pathway determines ferroptosis sensitivity
in gastric cancer. Proc Natl Acad Sci USA. 117:32433–32442. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ursini F and Maiorino M: Lipid
peroxidation and ferroptosis: The role of GSH and GPx4. Free. Radic
Biol Med. 152:175–185. 2020. View Article : Google Scholar
|
|
43
|
Chen X, Li J, Kang R, Klionsky DJ and Tang
D: Ferroptosis: Machinery and regulation. Autophagy. 17:2054–2081.
2021. View Article : Google Scholar :
|
|
44
|
Conrad M and Sato H: The oxidative
stress-inducible cystine/glutamate antiporter, system x (c) (-):
Cystine supplier and beyond. Amino Acids. 42:231–246. 2012.
View Article : Google Scholar
|
|
45
|
Ahmed I, Manno FAM, Manno SHC, Liu Y,
Zhang Y and Lau C: Detection of lithium in breast milk and in situ
elemental analysis of the mammary gland. Biomed Opt Express.
9:4184–4195. 2018. View Article : Google Scholar :
|
|
46
|
Ingold I, Berndt C, Schmitt S, Doll S,
Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T,
et al: Selenium utilization by GPX4 is required to prevent
hydroperoxide-induced ferroptosis. Cell. 172:409–422.e21. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li S, He Y, Chen K, Sun J, Zhang L, He Y,
Yu H and Li Q: RSL3 Drives ferroptosis through NF-κB pathway
activation and GPX4 depletion in glioblastoma. Oxid Med Cell
Longev. 2021:29150192021. View Article : Google Scholar
|
|
48
|
Zhang DL, Ghosh MC and Rouault TA: The
physiological functions of iron regulatory proteins in iron
homeostasis-an update. Front Pharmacol. 5:1242014. View Article : Google Scholar
|
|
49
|
Yang L, Cao LM, Zhang XJ and Chu B:
Targeting ferroptosis as a vulnerability in pulmonary diseases.
Cell Death Dis. 13:6492022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Brown CW, Amante JJ, Chhoy P, Elaimy AL,
Liu H, Zhu LJ, Baer CE, Dixon SJ and Mercurio AM: Prominin2 drives
ferroptosis resistance by stimulating iron export. Dev Cell.
51:575–586.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kobayashi Y, Ohfuji S, Kondo K, Fukushima
W, Sasaki S, Kamata N, Yamagami H, Fujiwara Y, Suzuki Y and Hirota
Y; Japanese Case-Control Study Group for Ulcerative Colitis:
Association between dietary iron and zinc intake and development of
ulcerative colitis: A case-control study in Japan. J Gastroenterol
Hepatol. 34:1703–1710. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sivaprakasam S, Ristic B, Mudaliar N,
Hamood AN, Colmer-Hamood J, Wachtel MS, Nevels AG, Kottapalli KR
and Ganapathy V: Hereditary hemochromatosis promotes colitis and
colon cancer and causes bacterial dysbiosis in mice. Biochem J.
477:3867–3883. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wenz C, Faust D, Linz B, Turmann C,
Nikolova T, Bertin J, Gough P, Wipf P, Schröder AS, Krautwald S and
Dietrich C: t-BuOOH induces ferroptosis in human and murine cell
lines. Arch Toxicol. 92:759–775. 2018. View Article : Google Scholar
|
|
54
|
Wenz C, Faust D, Linz B, Turmann C,
Nikolova T and Dietrich C: Cell-cell contacts protect against
t-BuOOH-induced cellular damage and ferroptosis in vitro. Arch
Toxicol. 93:1265–1279. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen X, Kang R, Kroemer G and Tang D:
Broadening horizons: The role of ferroptosis in cancer. Nat Rev
Clin Oncol. 18:280–296. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen GQ, Benthani FA, Wu J, Liang D, Bian
ZX and Jiang X: Artemisinin compounds sensitize cancer cells to
ferroptosis by regulating iron homeostasis. Cell Death Differ.
27:242–254. 2020. View Article : Google Scholar :
|
|
57
|
Gaschler MM, Andia AA, Liu H, Csuka JM,
Hurlocker B, Vaiana CA, Heindel DW, Zuckerman DS, Bos PH, Reznik E,
et al: FINO2 initiates ferroptosis through GPX4
inactivation and iron oxidation. Nat Chem Biol. 14:507–515. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chen Y, Zhang P, Chen W and Chen G:
Ferroptosis mediated DSS-induced ulcerative colitis associated with
Nrf2/HO-1 signaling pathway. Immunol Lett. 225:9–15. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hu Y, Sun S and Li H: Identification and
functional exploration of Ferroptosis and Immune Related Long
Non-Coding RNA in Inflammatory bowel disease. Res Sq. 2022.
|
|
60
|
Patankar JV and Becker C: Cell death in
the gut epithelium and implications for chronic inflammation. Nat
Rev Gastroenterol Hepatol. 17:543–556. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Luo L, Zhang S, Guo N, Li H and He S:
ACSF2-mediated ferroptosis is involved in ulcerative colitis. Life
Sci. 313:1212722023. View Article : Google Scholar
|
|
62
|
Cui DJ, Chen C, Yuan WQ, Yang YH and Han
L: Integrative analysis of ferroptosis-related genes in ulcerative
colitis. J Int Med Res. 49:30006052110429752021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ma H, Shu Q, Li D, Wang T, Li L, Song X,
Lou K and Xu H: Accumulation of intracellular ferrous iron in
inflammatory-activated macrophages. Biol Trace Elem Res.
201:2303–2310. 2023. View Article : Google Scholar
|
|
64
|
Maloy KJ and Powrie F: Intestinal
homeostasis and its breakdown in inflammatory bowel disease.
Nature. 474:298–306. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Panda SK, Peng V, Sudan R, Ulezko Antonova
A, Di Luccia B, Ohara TE, Fachi JL, Grajales-Reyes GE, Jaeger N,
Trsan T, et al: Repression of the aryl-hydrocarbon receptor
prevents oxidative stress and ferroptosis of intestinal
intraepithelial lymphocytes. Immunity. 56:797–812.e4. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang L, Han R, Zang K, Yuan P and Qin H:
Deficiency in glutathione synthesis and reduction contributes to
the pathogenesis of colitis-related liver injury. Zhong Nan Da Xue
Xue Bao Yi Xue Ban. 47:271–279. 2022.In English, Chinese.
PubMed/NCBI
|
|
67
|
Lin JH, Walter P and Yen TSB: Endoplasmic
reticulum stress in disease pathogenesis. Annu Rev Pathol.
3:399–425. 2008. View Article : Google Scholar
|
|
68
|
Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian
D, Liu D, Zhang F, Ning S, Yao J and Tian X: Ischemia-induced ACSL4
activation contributes to ferroptosis-mediated tissue injury in
intestinal ischemia/reperfusion. Cell Death Differ. 26:2284–2299.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Dang D, Zhang C, Meng Z, Lv X, Li Z, Wei J
and Wu H: Integrative analysis links ferroptosis to necrotizing
enterocolitis and reveals the role of ACSL4 in immune disorders.
iScience. 25:1054062022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhang X, Li W, Ma Y, Zhao X, He L, Sun P
and Wang H: High-fat diet aggravates colitis-associated
carcinogenesis by evading ferroptosis in the ER stress-mediated
pathway. Free Radic Biol Med. 177:156–166. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Chen J, Li X, Ge C, Min J and Wang F: The
multifaceted role of ferroptosis in liver disease. Cell Death
Differ. 29:467–480. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhang X, Du L, Qiao Y, Zhang X, Zheng W,
Wu Q, Chen Y, Zhu G, Liu Y, Bian Z, et al: Ferroptosis is governed
by differential regulation of transcription in liver cancer. Redox
Biol. 24:1012112019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wang Y, Zhang Z, Sun W, Zhang J, Xu Q,
Zhou X and Mao L: Ferroptosis in colorectal cancer: Potential
mechanisms and effective therapeutic targets. Biomed Pharmacother.
153:1135242022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ouyang S, Li H, Lou L, Huang Q, Zhang Z,
Mo J, Li M, Lu J, Zhu K, Chu Y, et al: Inhibition of
STAT3-ferroptosis negative regulatory axis suppresses tumor growth
and alleviates chemoresistance in gastric cancer. Redox Biol.
52:1023172022. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W and
Wang J: Molecular mechanisms of ferroptosis and its role in cancer
therapy. J Cell Mol Med. 23:4900–4912. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Wang R, Su Q, Yin H, Wu D, Lv C and Yan Z:
Inhibition of SRSF9 enhances the sensitivity of colorectal cancer
to erastin-induced ferroptosis by reducing glutathione peroxidase 4
expression. Int J Biochem Cell Biol. 134:1059482021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhang L, Liu W, Liu F, Wang Q, Song M, Yu
Q, Tang K, Teng T, Wu D, Wang X, et al: Corrigendum to 'IMCA
induces ferroptosis mediated by SLC7A11 through the AMPK/mTOR
pathway in colorectal cancer'. Oxid Med Cell Longev.
2020:69014722020. View Article : Google Scholar
|
|
78
|
Xia Y, Liu S, Li C, Ai Z, Shen W, Ren W
and Yang X: Discovery of a novel ferroptosis
inducer-talaroconvolutin A-killing colorectal cancer cells in vitro
and in vivo. Cell Death Dis. 11:9882020. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Gao W, Huang Z, Duan J, Nice EC, Lin J and
Huang C: Elesclomol induces copper-dependent ferroptosis in
colorectal cancer cells via degradation of ATP7A. Mol Oncol.
15:3527–3544. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Luo W, Dai W, Li Q, Mo S, Han L, Xiao X,
Gu R, Xiang W, Ye L, Wang R, et al: Ferroptosis-associated
molecular classification characterized by distinct tumor
microenvironment profiles in colorectal cancer. Int J Biol Sci.
18:1773–1794. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zilka O, Shah R, Li B, Friedmann Angeli
JP, Griesser M, Conrad M and Pratt DA: On the mechanism of
cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of
lipid peroxidation in ferroptotic cell death. ACS Cent Sci.
3:232–243. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kajarabille N and Latunde-Dada GO:
Programmed cell-death by ferroptosis: Antioxidants as mitigators.
Int J Mol Sci. 20:49682019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Chen Y, Yan W, Chen Y, Zhu J, Wang J, Jin
H, Wu H, Zhang G, Zhan S, Xi Q, et al: SLC6A14 facilitates
epithelial cell ferroptosis via the C/EBPβ-PAK6 axis in ulcerative
colitis. Cell Mol Life Sci. 79:5632022. View Article : Google Scholar
|
|
84
|
Hu Q, Zhang Y, Lou H, Ou Z, Liu J, Duan W,
Wang H, Ge Y, Min J, Wang F and Ju Z: GPX4 and vitamin E
cooperatively protect hematopoietic stem and progenitor cells from
lipid peroxidation and ferroptosis. Cell Death Dis. 12:7062021.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Maiorino M, Conrad M and Ursini F: GPx4,
lipid peroxidation, and cell death: Discoveries, rediscoveries, and
open issues. Antioxid Redox Signal. 29:61–74. 2018. View Article : Google Scholar
|
|
86
|
Carlson BA, Tobe R, Yefremova E, Tsuji PA,
Hoffmann VJ, Schweizer U, Gladyshev VN, Hatfield DL and Conrad M:
Glutathione peroxidase 4 and vitamin E cooperatively prevent
hepatocellular degeneration. Redox Biol. 9:22–31. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Mirbagheri SA, Nezami BG, Assa S and
Hajimahmoodi M: Rectal administration of d-alpha tocopherol for
active ulcerative colitis: A preliminary report. World J
Gastroenterol. 14:5990–5995. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Rachmawati H, Pradana AT, Safitri D and
Adnyana IK: Multiple functions of D-α-tocopherol polyethylene
glycol 1000 succinate (TPGS) as curcumin nanoparticle stabilizer:
In vivo kinetic profile and anti-ulcerative colitis analysis in
animal model. Pharmaceutics. 9:242017. View Article : Google Scholar
|
|
89
|
Liu KY, Nakatsu CH, Jones-Hall Y, Kozik A
and Jiang Q: Vitamin E alpha- and gamma-tocopherol mitigate
colitis, protect intestinal barrier function and modulate the gut
microbiota in mice. Free Radic Biol Med. 163:180–189. 2021.
View Article : Google Scholar
|
|
90
|
Wang Y, Shen W, Shi X, Fu F, Fan Y, Shen
W, Cao Y, Zhang Q and Qi R: Alpha-tocopheryl succinate-conjugated
G5 PAMAM dendrimer enables effective inhibition of ulcerative
colitis. Adv Healthc Mater. 6:2017. View Article : Google Scholar
|
|
91
|
Zou Y, Li H, Graham ET, Deik AA, Eaton JK,
Wang W, Sandoval-Gomez G, Clish CB, Doench JG and Schreiber SL:
Cytochrome P450 oxidoreductase contributes to phospholipid
peroxidation in ferroptosis. Nat Chem Biol. 16:302–309. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX,
Sun C, Li B, Fan BY, Wang X, Li WX, et al: Deferoxamine promotes
recovery of traumatic spinal cord injury by inhibiting ferroptosis.
Neural Regen Res. 14:532–541. 2019. View Article : Google Scholar :
|
|
93
|
Guo Z, Lin J, Sun K, Guo J, Yao X, Wang G,
Hou L, Xu J, Guo J and Guo F: Deferoxamine alleviates
osteoarthritis by inhibiting chondrocyte ferroptosis and activating
the Nrf2 pathway. Front Pharmacol. 13:7913762022. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Ma H, Wang X, Zhang W, Li H, Zhao W, Sun J
and Yang M: Melatonin suppresses ferroptosis induced by high
glucose via activation of the Nrf2/HO-1 signaling pathway in type 2
diabetic osteoporosis. Oxid Med Cell Longev. 2020:90676102020.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Bai T, Li M, Liu Y, Qiao Z and Wang Z:
Inhibition of ferroptosis alleviates atherosclerosis through
attenuating lipid peroxidation and endothelial dysfunction in mouse
aortic endothelial cell. Free Radic Biol Med. 160:92–102. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Rayatpour A, Foolad F, Heibatollahi M,
Khajeh K and Javan M: Ferroptosis inhibition by deferiprone,
attenuates myelin damage and promotes neuroprotection in
demyelinated optic nerve. Sci Rep. 12:196302022. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Liu X, Zhang J and Xie W: The role of
ferroptosis in acute lung injury. Mol Cell Biochem. 477:1453–1461.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Millar AD, Rampton DS and Blake DR:
Effects of iron and iron chelation in vitro on mucosal oxidant
activity in ulcerative colitis. Aliment Pharmacol Ther.
14:1163–1168. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Minaiyan M, Mostaghel E and Mahzouni P:
Preventive therapy of experimental colitis with selected iron
chelators and anti-oxidants. Int J Prev Med. 3(Suppl 1): S162–S169.
2012.PubMed/NCBI
|
|
100
|
Wu Y, Ran L, Yang Y, Gao X, Peng M, Liu S,
Sun L, Wan J, Wang Y, Yang K, et al: Deferasirox alleviates
DSS-induced ulcerative colitis in mice by inhibiting ferroptosis
and improving intestinal microbiota. Life Sci. 314:1213122023.
View Article : Google Scholar
|
|
101
|
Ghaith MM, El-Boshy M, Almasmoum H,
Abdelghany AH, Azzeh FS, Almaimani RA, Idris S, Ahmad J, Mahbub AA,
BaSalamah MA, et al: Deferasirox and vitamin D3
co-therapy mitigates iron-induced renal injury by enhanced
modulation of cellular anti-inflammatory, anti-oxidative stress,
and iron regulatory pathways in rat. J Trace Elem Med Biol.
74:1270852022. View Article : Google Scholar
|
|
102
|
Chen X, Comish PB, Tang D and Kang R:
Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol.
9:6371622021. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Li YY, Wang XJ, Su YL, Wang Q, Huang SW,
Pan ZF, Chen YP, Liang JJ, Zhang ML, Xie XQ, et al: Baicalein
ameliorates ulcerative colitis by improving intestinal epithelial
barrier via AhR/IL-22 pathway in ILC3s. Acta Pharmacol Sin.
43:1495–1507. 2022. View Article : Google Scholar
|
|
104
|
Thermozier S, Hou W, Zhang X, Shields D,
Fisher R, Bayir H, Kagan V, Yu J, Liu B, Bahar I, et al:
Anti-ferroptosis drug enhances total-body irradiation mitigation by
drugs that block apoptosis and necroptosis. Radiat Res.
193:435–450. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Gounaris E, Heiferman MJ, Heiferman JR,
Shrivastav M, Vitello D, Blatner NR, Knab LM, Phillips JD, Cheon
EC, Grippo PJ, et al: Zileuton, 5-lipoxygenase inhibitor, acts as a
chemopreventive agent in intestinal polyposis, by modulating polyp
and systemic inflammation. PLoS One. 10:e01214022015. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Dächert J, Ehrenfeld V, Habermann K,
Dolgikh N and Fulda S: Targeting ferroptosis in rhabdomyosarcoma
cells. Int J Cancer. 146:510–520. 2020. View Article : Google Scholar
|
|
107
|
Shimada K, Skouta R, Kaplan A, Yang WS,
Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ and
Stockwell BR: Global survey of cell death mechanisms reveals
metabolic regulation of ferroptosis. Nat Chem Biol. 12:497–503.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Wang D, Peng Y, Xie Y, Zhou B, Sun X, Kang
R and Tang D: Antiferroptotic activity of non-oxidative dopamine.
Biochem Biophys Res Commun. 480:602–607. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Liu L, Wu Y, Wang B, Jiang Y, Lin L, Li X
and Yang S: DA-DRD5 signaling controls colitis by regulating
colonic M1/M2 macrophage polarization. Cell Death Dis. 12:5002021.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Kawano M, Saika K, Takagi R, Matsui M and
Matsushita S: Tannic acid acts as an agonist of the dopamine D2L
receptor, regulates immune responses, and ameliorates
experimentally induced colitis in mice. Brain Behav Immun Health.
5:1000712020. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M,
Shan B, Pan H and Yuan J: Chaperone-mediated autophagy is involved
in the execution of ferroptosis. Proc Natl Acad Sci USA.
116:2996–3005. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Panés J, García-Olmo D, Van Assche G,
Colombel JF, Reinisch W, Baumgart DC, Dignass A, Nachury M,
Ferrante M, Kazemi-Shirazi L, et al: Expanded allogeneic
adipose-derived mesenchymal stem cells (Cx601) for complex perianal
fistulas in Crohn's disease: A phase 3 randomised, double-blind
controlled trial. Lancet. 388:1281–1290. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Ocansey DKW, Pei B, Yan Y, Qian H, Zhang
X, Xu W and Mao F: Improved therapeutics of modified mesenchymal
stem cells: An update. J Transl Med. 18:422020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Ocansey DKW, Qiu W, Wang J, Yan Y, Qian H,
Zhang X, Xu W and Mao F: The achievements and challenges of
mesenchymal stem cell-based therapy in inflammatory bowel disease
and its associated colorectal cancer. Stem Cells Int.
2020:78198242020. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Ocansey DKW, Zhang Z, Xu X, Liu L, Amoah
S, Chen X, Wang B, Zhang X and Mao F: Mesenchymal stem cell-derived
exosome mitigates colitis via the modulation of the gut
metagenomics-metabolomics-farnesoid X receptor axis. Biomater Sci.
10:4822–4836. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Wang G, Joel MDM, Yuan J, Wang J, Cai X,
Ocansey DKW, Yan Y, Qian H, Zhang X, Xu W and Mao F: Human
umbilical cord mesenchymal stem cells alleviate inflammatory bowel
disease by inhibiting ERK phosphorylation in neutrophils.
Inflammopharmacology. 28:603–616. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Ocansey DKW, Zhang L, Wang Y, Yan Y, Qian
H, Zhang X, Xu W and Mao F: Exosome-mediated effects and
applications in inflammatory bowel disease. Biol Rev Camb Philos
Soc. 95:1287–1307. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Zhu Y, Qin H, Sun C, Shao B, Li G, Qin Y,
Kong D, Ren S, Wang H, Wang Z, et al: Endometrial regenerative
cell-derived exosomes attenuate experimental colitis through
downregulation of intestine ferroptosis. Stem Cells Int.
2022:30141232022. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Lin F, Chen W, Zhou J, Zhu J, Yao Q, Feng
B, Feng X, Shi X, Pan Q, Yu J, et al: Mesenchymal stem cells
protect against ferroptosis via exosome-mediated stabilization of
SLC7A11 in acute liver injury. Cell Death Dis. 13:2712022.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Liu T, Jiang L, Tavana O and Gu W: The
deubiquitylase OTUB1 mediates ferroptosis via stabilization of
SLC7A11. Cancer Res. 79:1913–1924. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Shao C, Chen Y, Yang T, Zhao H and Li D:
Mesenchymal stem cell derived exosomes suppress neuronal cell
ferroptosis via lncGm36569/miR-5627-5p/FSP1 axis in acute spinal
cord injury. Stem Cell Rev Rep. 18:1127–1142. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Sun Z, Wu J, Bi Q and Wang W: Exosomal
lncRNA TUG1 derived from human urine-derived stem cells attenuates
renal ischemia/reperfusion injury by interacting with SRSF1 to
regulate ASCL4-mediated ferroptosis. Stem Cell Res Ther.
13:2972022. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Song Y, Wang B, Zhu X, Hu J, Sun J, Xuan J
and Ge Z: Human umbilical cord blood-derived MSCs exosome attenuate
myocardial injury by inhibiting ferroptosis in acute myocardial
infarction mice. Cell Biol Toxicol. 37:51–64. 2021. View Article : Google Scholar
|
|
124
|
Tan Y, Huang Y, Mei R, Mao F, Yang D, Liu
J, Xu W, Qian H and Yan Y: HucMSC-derived exosomes delivered BECN1
induces ferroptosis of hepatic stellate cells via regulating the
xCT/GPX4 axis. Cell Death Dis. 13:3192022. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Atkuri KR, Mantovani JJ and Herzenberg LA
and Herzenberg LA: N-acetylcysteine-a safe antidote for
cysteine/glutathione deficiency. Curr Opin Pharmacol. 7:355–359.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Schmitt B, Vicenzi M, Garrel C and Denis
FM: Effects of N-acetylcysteine, oral glutathione (GSH) and a novel
sublingual form of GSH on oxidative stress markers: A comparative
crossover study. Redox Biol. 6:198–205. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Ardite E, Sans M, Panés J, Romero FJ,
Piqué JM and Fernández-Checa JC: Replenishment of glutathione
levels improves mucosal function in experimental acute colitis. Lab
Invest. 80:735–744. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Dong S, Lu Y, Peng G, Li J, Li W, Li M,
Wang H, Liu L and Zhao Q: Furin inhibits epithelial cell injury and
alleviates experimental colitis by activating the Nrf2-Gpx4
signaling pathway. Dig Liver Dis. 53:1276–1285. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Zhu D, Wu H, Jiang K, Xu Y, Miao Z, Wang H
and Ma Y: Zero-valence selenium-enriched prussian blue nanozymes
reconstruct intestinal barrier against inflammatory bowel disease
via inhibiting ferroptosis and T cells differentiation. Adv Healthc
Mater. 12:e22031602023. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Perry C, Kapur N and Barrett TA: DPP-4 as
a novel biomarker for inflammatory bowel disease: Is it ready for
clinical use? Inflamm Bowel Dis. 26:1720–1721. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J,
Zhong M, Yuan H, Zhang L, Billiar TR, et al: The tumor suppressor
p53 limits ferroptosis by blocking DPP4 activity. Cell Rep.
20:1692–1704. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Arab HH, Eid AH, Mahmoud AM and Senousy
MA: Linagliptin mitigates experimental inflammatory bowel disease
in rats by targeting inflammatory and redox signaling. Life Sci.
273:1192952021. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
El-Ghannam MS, Saad MA, Nassar NN,
El-Yamany MF and El-Bahy AAZ: Linagliptin ameliorates acetic
acid-induced colitis via modulating AMPK/SIRT1/PGC-1α and
JAK2/STAT3 signaling pathway in rats. Toxicol Appl Pharmacol.
438:1159062022. View Article : Google Scholar
|
|
134
|
Hu Y, Mao Z, Xu L, Yin L, Tao X, Tang Z,
Qi Y, Sun P and Peng J: Protective effect of dioscin against
intestinal ischemia/reperfusion injury via adjusting
miR-351-5p-mediated oxidative stress. Pharmacol Res. 137:56–63.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Ozkan OV, Yuzbasioglu MF, Ciralik H,
Kurutas EB, Yonden Z, Aydin M, Bulbuloglu E, Semerci E, Goksu M,
Atli Y, et al: Resveratrol, a natural antioxidant, attenuates
intestinal ischemia/reperfusion injury in rats. Tohoku J Exp Med.
218:251–258. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Stefanutti G, Pierro A, Parkinson EJ,
Smith VV and Eaton S: Moderate hypothermia as a rescue therapy
against intestinal ischemia and reperfusion injury in the rat. Crit
Care Med. 36:1564–1572. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Meister AL, Doheny KK and Travagli RA:
Necrotizing enterocolitis: It's not all in the gut. Exp Biol Med
(Maywood). 245:85–95. 2020. View Article : Google Scholar
|
|
138
|
Yin Y, Wu X, Peng B, Zou H, Li S, Wang J
and Cao J: Curcumin improves necrotising microscopic colitis and
cell pyroptosis by activating SIRT1/NRF2 and inhibiting the TLR4
signalling pathway in newborn rats. Innate Immun. 26:609–617. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Xu Y, Li X, Cheng Y, Yang M and Wang R:
Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary
ischemia-reperfusion. FASEB J. 34:16262–16275. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Cui Y, Zhang Y, Zhao X, Shao L, Liu G, Sun
C, Xu R and Zhang Z: ACSL4 exacerbates ischemic stroke by promoting
ferroptosis-induced brain injury and neuroinflammation. Brain Behav
Immun. 93:312–321. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Zhang L, Fan J, He J, Chen W, Jin W, Zhu
Y, Sun H, Li Y, Shi Y, Jing Y, et al: Regulation of ROS-NF-κB axis
by tuna backbone derived peptide ameliorates inflammation in
necrotizing enterocolitis. J Cell Physiol. 234:14330–14338. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Tang B, Zhu J, Fang S, Wang Y, Vinothkumar
R, Li M, Weng Q, Zheng L, Yang Y, Qiu R, et al: Pharmacological
inhibition of MELK restricts ferroptosis and the inflammatory
response in colitis and colitis-propelled carcinogenesis. Free
Radic Biol Med. 172:312–329. 2021. View Article : Google Scholar : PubMed/NCBI
|















