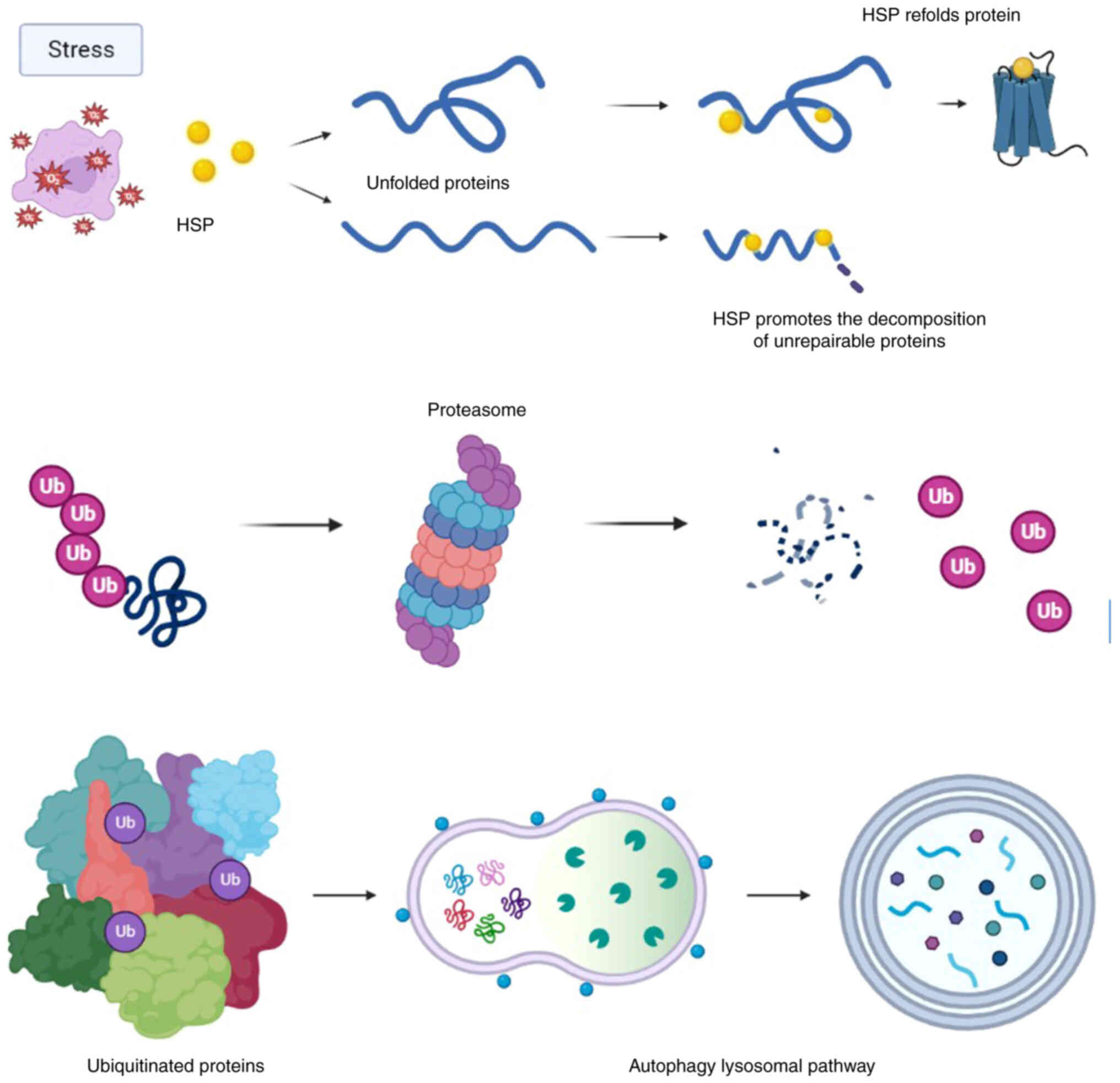
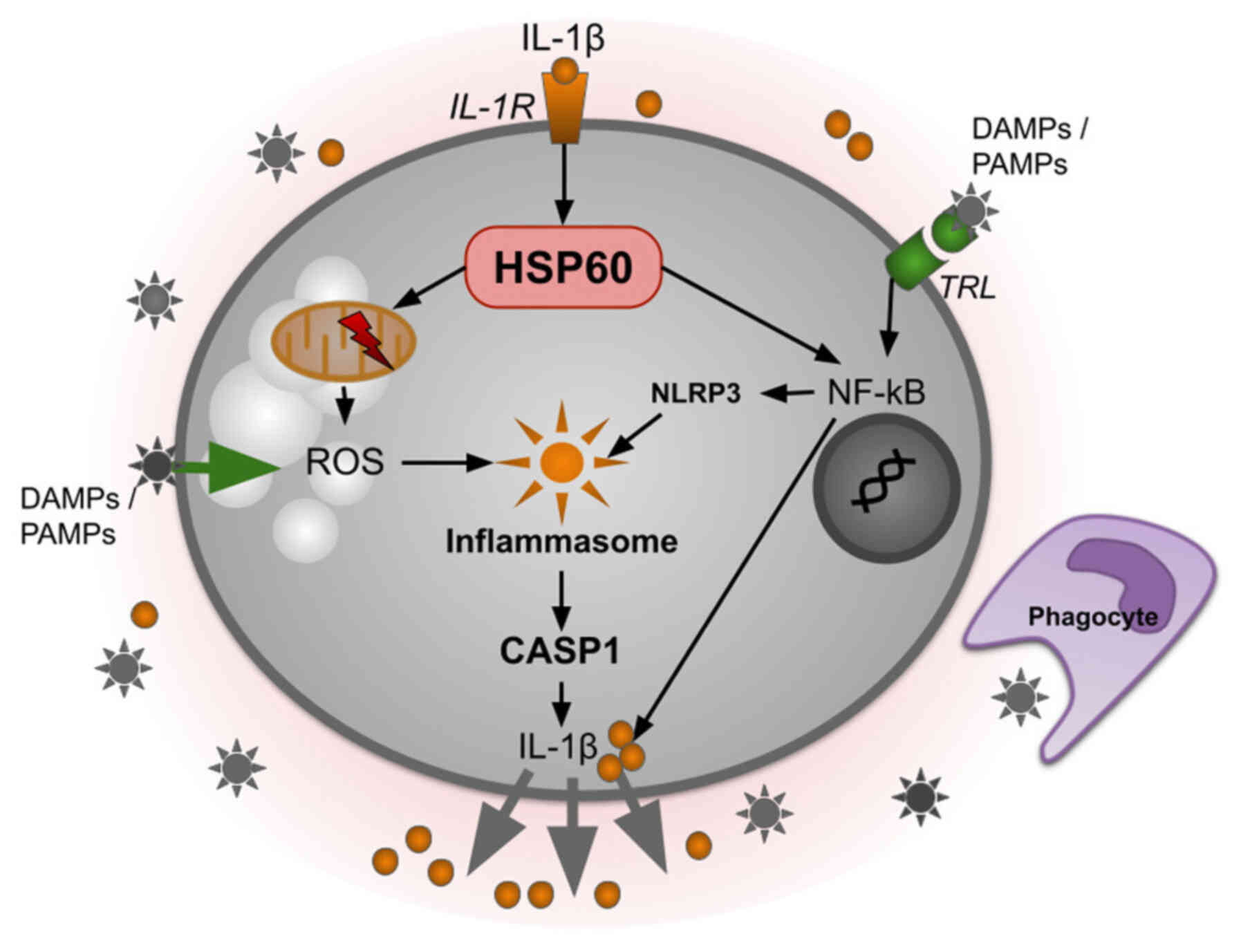
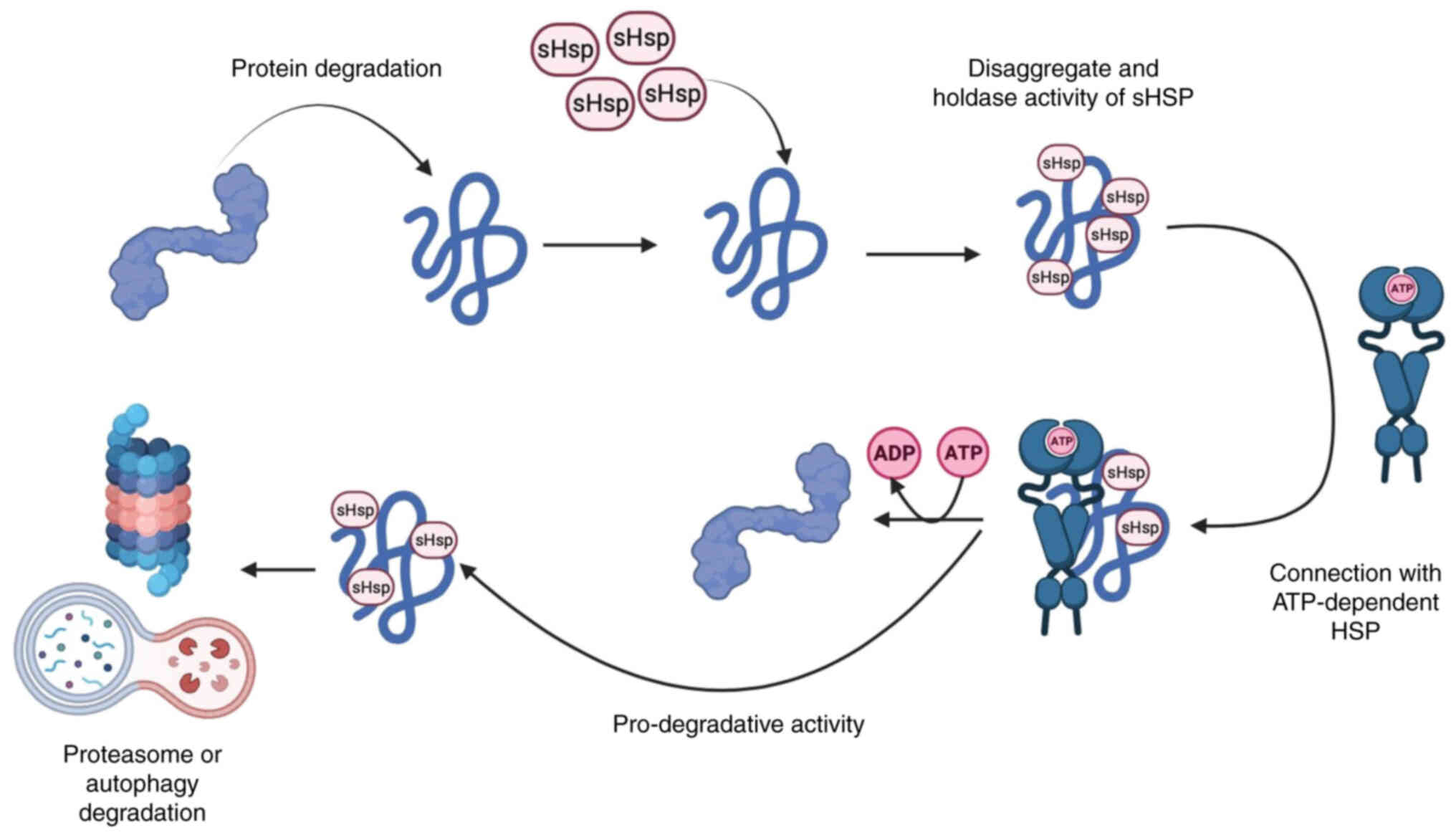
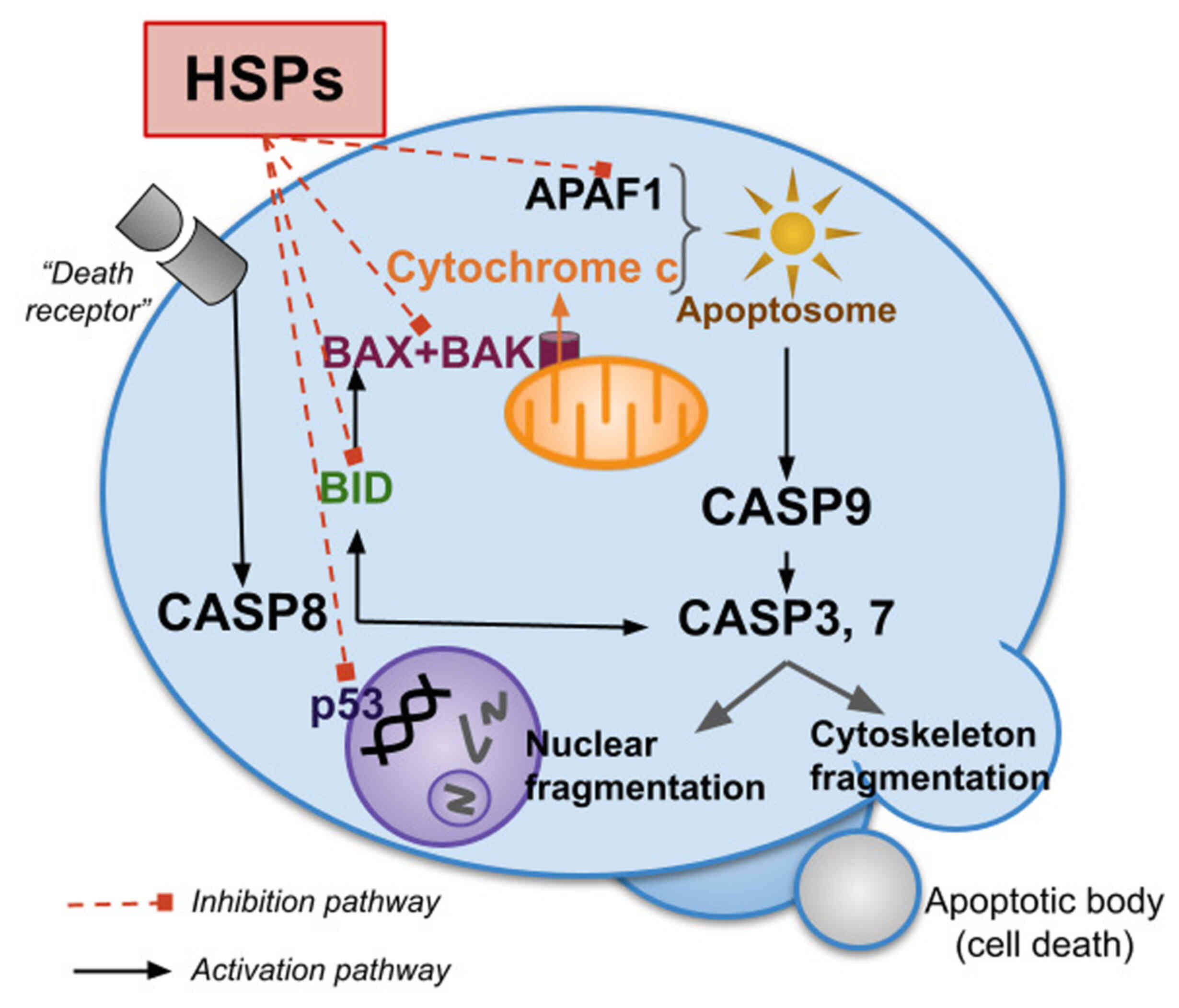
The reliability of cellular protein composition is
important for proper cellular function in various tissues,
especially in the myocardium, as cardiac myocytes (CMCs) are
terminally differentiated cells with very limited regenerative
potential (1). At the same time,
the metabolic demands of the heart require a tight control of
protein quality (2). Cellular
stress in the myocardium, which occurs during ischemia (3), hypertension (4) and metabolic disorders including
diabetes mellitus (DM) (5), can
disrupt protein homeostasis and cause abnormal folding of cellular
proteins. Intracellular accumulation of toxic, misfolded proteins
and their aggregates may contribute to the development of heart
failure (6). Cells have an innate
mechanism for detecting protein misfolding, which they can use to
repair or remove these proteins. This mechanism is called 'protein
quality control' and involves three main pathways (Fig. 1) (7,8).
First, misfolded proteins are exposed to heat shock proteins
(HSPs), which are characterized as molecular chaperones. If the
chaperone system fails to refold denatured proteins, they are
ubiquitinated by an activated second system and delivered to the
proteasome mechanism for subsequent degradation. When these systems
are disrupted or overloaded, such ubiquitinated proteins accumulate
in agresomes, perinuclear structures which are ultimately utilized
by autophagy, the third mechanism (9-12).
HSPs are a family of conserved proteins that can
contribute to proper protein folding, maintain protein stability
and act as molecular chaperones to regulate cellular metabolism,
tissue composition and other processes (13). HSPs are also called stress-induced
proteins because of their ability to stabilize and repair proteins
during their synthesis in the cell under the action of unfavorable
agents on the cell (14,15). Meanwhile, they are also involved
in the modulation of inflammatory response, oxidative stress and
metabolism (16-18).
The effect of HSPs on the regulated cell death of
CMCs is of great interest, as they contribute significantly to the
implementation of compensatory and adaptive mechanisms, such as in
the case of cardiac damage (19-21). HSPs mediate a number of activation
mechanisms of the apoptotic cascade, playing both pro- and
anti-apoptotic roles depending on their cellular localization
(22). In some cases, autophagy
can lead to cell death, while in other cases it acts as a mechanism
for their survival (23,24).
Despite the availability of numerous data,
concerning the role of HSPs and factors involved in programmed cell
death in response to pathological agents, there is currently no
clear idea of their role in the pathogenesis of heart failure
caused by various types of cardiovascular pathology.
HSPs are involved in inflammation, which mediates
various pathological processes in the myocardium. Schroder and
Tschopp (16) and other
scientists paid special attention to the role of HSPs in the
formation of intracellular platforms; inflammasomes, which are
activated by 'danger' signals and induce inflammatory caspases,
mainly caspase-1, which contributes to the production of
pro-inflammatory cytokine IL-1β (25,26). A number of other cytokines from
the family of pro-inflammatory mediators are also related to these
processes (27). TNF is
associated with the induction of necrosis, cell damage and tearing
of membrane molecules, membrane modularity (28,29). Some members of the HSP family
support inflammasome activation, while others inhibit it. In
particular, HSP60 (Fig. 2) is
required for the phosphorylation and nuclear localization of NF-kB
after stimulation by IL-1β in microglia (30). Knockdown of the HSP60 gene leads
to inhibition of phosphorylation of the p65 NF-kB subunit and
consequently to suppression of NF-kB nuclear translocation.
Presumably, HSP60 promotes p65 phosphorylation (31). This activation of the NF-kB
pathway leads to the overexpression of both pro-IL-1β and
nucleotide-binding oligomerization domain-containing protein 3
(NLRP3), corresponding to the 'priming' step of NLRP3 inflammasome
activation (32). HSP60 also
induces mitochondrial damage, as evidenced by a decrease in their
membrane potential after HSP60 overexpression and treatment with
IL-1β. This is accompanied by an increase in reactive oxygen
species (ROS) production, which contributes to oxidative stress,
which in turn activates the NLRP3 inflammasome (33).
Platelets serve as regulators of hemostasis. Upon
interaction with extracellular matrix proteins, such as collagen,
platelets are activated, leading to shape change, secretion,
filopodia formation and ultimately aggregation (34-36). Platelet aggregation is largely
mediated by activation of the platelet integrin αIIbβ3, which
undergoes conformational changes in response to stimulation of
soluble fibrinogen binding (37,38). The role of HSPs in the activation
of thrombus formation is of great interest, particularly in
relation to the problem of preventing coronary artery
thrombosis.
Metabolic syndrome is a risk factor for the
development of heart failure and DM. The features of the metabolic
syndrome are systemic inflammation and oxidative stress, which can
enhance the expression and release of HSP (44). HSPs play a role in cell signaling
and regulation of cell metabolism in conditions of insulin
resistance (45). In obesity, an
uncontrolled inflammatory reaction and a disorder of the body's
defense system play an important role in inhibiting the signaling
cascade of insulin receptors and, as a consequence, a disorder of
systemic metabolic homeostasis (41). Regulation of HSP expression at the
gene level is an important aspect of HSP72 activity in metabolic
syndrome. Overexpression of HSP72 leads to an increase in the
number of mitochondria in cells, the oxidative capacity and the
sensitivity of cells to insulin (44,45). Similarly, lack of HSP72 expression
results in mitochondrial dysfunction and insulin resistance
(46,47). In addition to increasing HSP72
levels and consequently the ability to improve mitochondrial
quality control, exercise also contributes to an increase in the
expression of peroxisome proliferator-activated receptor
γ-coactivator 1-α (PGC1a) (48).
PGC1a is the major transcriptional coactivator for mitochondrial
formation (49). The upstream
regulatory elements of the PPARGC1A gene were found to contain the
heat shock element (HSE) binding sequence (50). This HSE sequence provides a
docking site for the primary HSP transcription factor, heat shock
factor 1 (HSF1) (51). Numerous
HSF1 gene activation and knockdown experiments have convincingly
demonstrated that HSF1 is the master regulator of mitochondrial
biogenesis, enzymatic function and whole-body metabolism (48). These data illustrate a
coordination of HSF1 downstream targets (HSP1 and PGC1a) in the
regulation of mitochondrial biogenesis, quality control and
enzymatic function in metabolic demand and/or chronic disease
(52).
Elevated blood levels of HSPs have been found in
patients with coronary artery disease (CAD), although the source of
these proteins is still controversial (53). In general, the expression of HSPs
appears to be increased in response to ischemia and their
dysregulated production contributes to the development of
cardiovascular disease (54-57).
Extracellular HSP70 has been found to be an
independent predictive marker of mortality in patients with
progressive heart failure or sudden cardiac death (58-60). Its role in the development of
hypertension-induced cardiac hypertrophy and myocardial fibrosis
has also been demonstrated (61-63). It is also notable that HSP70 is a
ligand for damage-associated molecular pattern receptors, which can
induce inflammation in the myocardium (64,65).
The properties of extracellular HSP90 have been
demonstrated in a study by Ranek et al (6) in the context of progressive LV
hypertrophy. In pathological cardiac hypertrophy, LV mass increases
in parallel with extracellular matrix deposition, followed by
fibrosis and heart failure (66-68). The main mediator of this process
is TGF-β, which is secreted by CMCs and acts on collagen-secreting
fibroblasts (69-71). Extracellular HSP90 appears to play
some role in stabilizing the signal of TGF-β by influencing the
cascade of processes mediating TGF-β induction (72-74). Inhibition of extracellular HSP90
reduces collagen production and stimulation of the canonical TGF-ß
pathway (54,74). Since fibrosis is one of the major
factors influencing the pathogenesis of a number of chronic cardiac
diseases, including CAD and heart failure, targeting extracellular
HSP90 can be considered an important point of application for
therapeutic intervention with functions different from those
characteristics of the intracellular pool of this particular
HSP.
Small HSPs are ATP-independent proteins and
represent the first line of defense in preventing intracellular
protein aggregation under cellular stress when denaturation is
highly activated (75,76). At the same time, high molecular
weight HSPs, such as HSP60, HSP70 and HSP90, bind unfolded or
misfolded proteins and promote their refolding using the energy of
ATP hydrolysis (77-79). Therefore, under cellular stress
and energy deprivation, ATP-independent small HSPs are able to
rapidly and metabolically prevent protein aggregation, maintain
denaturing proteins in a folded state and inhibit the denaturation
process until ATP-dependent HSPs complete the refolding process
(80-82). If proper folding does not occur,
small HSPs contribute to the clearance of denatured proteins by
directing them to the pathway of degradation mechanisms (Fig. 3) (83-85).
Currently, the role of HSPs in inflammation and
their antioxidant capacity are of considerable interest, especially
in the aspect of modulating the progression of atherosclerosis by
altering its inflammatory mechanisms (86-88).
HSPs are also activated in response to the
appearance of oxidized low-density lipoproteins and mediate an
anti-inflammatory mechanism through the release of pro-inflammatory
IL-10 as well as through the activation of NFκB (89-91). The intracellular function of the
HSP27 chaperone is regulated by phosphorylation and
dephosphorylation in large aggregates that modulate the assembly of
an ATP-independent network. As a chaperone, HSP27 is involved in
DNA stabilization, supports antioxidant responses and also acts as
an anti-apoptotic factor (92-94). Extracellular release of HSP27 from
tissues where blood vessels are affected by atherosclerosis may
result from cellular injury or occur in association with secretory
lysosomes or exosomes. In its extracellular location, HSP27 binds
to a variety of cell membrane receptors on endothelial and
immunocompetent cells, including CD91, CD40, CD36, CD14, scavenger
receptor A (SR-A) and Toll-like receptors (TLRs) (95,96). The recombinant receptor HSP27
induces TLR-mediated NFκB activation with secretion of both pro-
and anti-inflammatory cytokines (55). According to the available data,
HSP27 provides protection against the progression of
atherosclerosis (22,97).
Induction of mitochondrial (mt) ROS during
hyperglycemia is a key event responsible for endothelial activation
and damage (99). HSP22 has been
shown to protect vascular endothelium from hyperglycemia-induced
damage by reducing mtROS production (100-103). Yu et al (101) performed a series of studies
using a high fat diet and a streptozotocin model to induce DM. They
also exposed human umbilical vein endothelial cells to a high
concentration of glucose after overexpressing or silencing HSP22 to
investigate the role of the latter. It was found that HSP22
significantly reduced endothelial cell activation and vascular
lesions by inhibiting endothelial adhesion, suppressing
mtROS-mediated endothelial activation and injury, abolishing a
hyperglycemia-induced increase in mtROS and also reducing cytokine
secretion. In addition, HSP22 attenuated mtROS and mitochondrial
dysfunction in hyperglycemia-stimulated endothelial cells (101).
In the case of cardiovascular pathology, cellular
stress may also be due to cardiac overload associated with
increased resistance to cardiac output caused by hypertension or
aortic stenosis. An increase in the autophagic flux of CMCs occurs
in association with their hypertrophy (116,117). Myocardial ischemia is associated
with a transient increase in autophagic flux, which, however,
decreases over time and falls below normal baseline levels
(19,118-120). In a model of acute focal LV
ischemia, it was shown that the content of Beclin-1 in the CMC
cytoplasm was significantly increased on day 1 and later, on days 3
and 5, it gradually decreased but remained above the control level
(20). In ischemia, the
intensification of CMC autophagy is aimed at replenishing metabolic
substrates and removing damaged organelles (121-123). Nutrient depletion stimulates
AMP-activated protein kinase, which in turn inhibits mTOR, thereby
removing the main inhibitory factor in this process (124-126). In this context, autophagy is
considered to be an adaptive response to ischemic injury with
cardioprotective effects. Thus, targeted activation of autophagy
may be a potential therapeutic approach to heat stress-induced
cardiovascular dysfunction (23).
A study showed that HSPB6 regulates the
ubiquitination and proteasomal degradation of BECN1. This effect
appears to be mediated by a direct interaction of HSPB6 with BECN1
(127). These ideas concerning
the role of HSPB6 in autophagy were generated in the context of the
description of a novel human mutation in the gene encoding HSPB6
(HSPB6S10F) identified in patients with dilated cardiomyopathy
(DCM) (128,129). Negative effects of HSPB6S10F
were associated with autophagy dysregulation. It is known that
constitutive autophagy in the heart under basal conditions is a
homeostatic mechanism aimed at maintaining myocyte size, structure
and function of the heart (130). It has also been found that the
activity of autophagy was significantly reduced in CMCs with the
mutation of the HSPB6S10F gene, as evidenced by the decreased
number of autophagosomes and inhibition of autophagic flux. Under
these conditions, the rate of CMC apoptosis increases and
hypertrophic remodeling develops, ultimately contributing to the
progression of heart failure (127). Similar negative effects were
associated with a decrease in the interaction of mutant HSPB6S10F
with Beclin1 (BECN1), leading to BECN1 ubiquitation and its
degradation by the proteasome. As a result, autophagy flux is
significantly suppressed and CMC apoptosis is enhanced. Conversely,
overexpression of wild-type HSPB6 (HSPB6 WT) contributed to
increased BECN1 levels and also competitively repressed BECN1
binding to Bcl2, thereby stimulating autophagy (127). These data reveal a novel
regulatory mechanism by which HSPB6 promotes cell survival through
its interaction with BECN1.
Depending on the specific cellular conditions,
autophagy can both protect cells from death and act as a means of
cellular self-destruction (131,132). The protective mechanism of
autophagy is implemented as follows: Organelles and/or parts of the
cytoplasm are engulfed by double-membrane autophagic vacuoles,
resulting in the physiological utilization of old or damaged
organelles (133,134). This provides cells with
metabolic substrates to meet energy demands during cellular stress
(135,136). However, the accumulation of
these vacuoles and further activation of the autophagic pathway
represents an alternative mechanism of cell death by atrophy and
functional collapse (type II cell death) (137-139). Autophagy can also activate
apoptotic (type I) or necrotic (type III) cell death programs by
activating common regulators such as Bcl-2 family proteins
(140,141).
This fact indicates that the activation of these
proteins plays a maladaptive role in the activity of CMCs, which
can serve as an important marker for assessing the progression of
vascular damage in this type of pathology. A decrease in the
expression of the HSP60/HSP10 complex relative to Bax was also
found in all the pathological models studied, while this difference
was less pronounced in the combined hypertension with DM group
(43,98,143).
Thus, a decrease in the production of HSP60 can be
considered as one of the pathophysiological mechanisms of LV
myocardial damage caused by hypertension and/or DM. In the
mentioned study, the expression of HSP27 in LV myocardium was also
evaluated. In the group with longer duration of hypertension (SHR
rats aged 57 weeks), the ratio of Bax and Bcl-2 was the most
pronounced, while the level of HSP27 was also the highest in all
groups, which may indicate the activation of protein defense
mechanisms directed to Bax binding (43,98,143).
It can be concluded that the role of HSP in
autophagy regulation and modulation of the apoptotic cascade
requires further investigation.
Apoptosis, or programmed cell death, is
characterized by the activation of the caspase cascade: 'Initiator'
caspases induce a chain reaction of specific 'effector' caspases
(144-146). These, in turn, are cleaved and
thus activate each other. There are two main pathways of initiator
caspase activation (147,148).
The extrinsic pathway is associated with 'death receptors' on the
cell surface, while the intrinsic pathway is induced by
pro-apoptotic factors such as cytochrome c released from
mitochondria. Cytochrome c, which binds to apoptotic protease
activating factor 1 (Apaf-1) and pro-caspase-9, forms an apoptosome
that stimulates the activation of caspase-9, which then activates
the 'effector' caspase-3 and initiates the apoptotic protease
cascade (149,150).
HSPs have a wide range of functions in apoptotic
processes. Most of them are aimed at their suppression (151,152). Notably, the same cellular stress
signals that induce apoptosis also stimulate HSP synthesis and
release. However, when HSP production is increased, apoptosis is
inhibited due to the suppression of pro-apoptotic factors such as
р53, Bax, Bid, Akt, Apaf-1 and other members of the Bcl-2 family
(Fig. 4). HSPs promote cell
survival by protecting cells from changes in cellular redox
homeostasis and stabilizing the cytoskeleton (152). In addition, HSPs can directly
inhibit several steps of the apoptotic pathway (153). HSPs also inhibit the release of
pro-apoptotic molecules from mitochondria, thereby reducing caspase
activation (151,154-156).
During the execution of the apoptotic cascade and
under ATP deprivation, HSP60 protects mitochondrial proteins,
facilitates their folding and prevents their degradation (157,158). In particular, when the Hsp60
gene is deleted in CMCs of young mice, HSP60-dependent
mitochondrial proteins are degraded by LONP1 and mitochondrial
dysfunction develops, which is one of the mechanisms of DCM and
heart failure (159). Cytosolic
HSP60 co-localizes with Bax and plays an anti-apoptotic role in
cardiac myocytes. Loss of cytosolic HSP60 induces mitochondrial Bax
translocation, cytochrome c release and caspase-3 activation,
leading to apoptotic cell death (30). In addition, hypoxia triggers
apoptosis by inducing dissociation of the HSP60-Bax complex through
translocation of cytosolic HSP60 to the membrane and Bax to the
mitochondria (160).
It is also worth considering the possibility of
increasing the level of Bcl-2 in cells and reducing the activation
of the apoptotic cascade by HSPB1. Tian et al (161) showed that HSP27 can attenuate
oxidative stress-induced apoptosis in endothelial cells by
increasing the level of Bcl-2 and decreasing the content of cleaved
caspase-3 and Bax. In addition, under ER stress, HSP27 promotes
ERK-mediated phosphorylation and Bim degradation by inhibiting the
mechanisms of the intrinsic pathway of apoptosis initiation
(162). HSP27 can also directly
bind to cytochrome c in the cytosol, inhibiting apoptosome
formation and interfering with downstream caspase activation. HSP27
can also suppress apoptosis by inhibiting mitochondrial Smac
release and subsequent activation of the descending caspase cascade
(161). The anti-apoptotic
properties of HSP27 are attributed to its direct interaction with
caspase-3. HSP27 binds to the pro-domain of caspase-3 and inhibits
its proteolytic activation (161,163).
The role of HSP70 in the initiation of the apoptotic
cascade has also been demonstrated in a model of myocardial
ischemia/reperfusion injury (164-166). Overexpression of HSP70 in the
myocardium and endothelium has a cardio-protective effect and
increases myocardial injury tolerance. These mechanisms are
associated with inhibition of apoptosis and oxidative stress and
improvement of endothelial function (60,167). Myocardial HSP70 activates
mitochondrial superoxide dismutase Mn-SOD and inhibits nuclear
translocation of phosphorylated eukaryotic elongation factor 2 and
apoptosis inducing factor, resulting in improvement of
mitochondrial function and suppression of apoptosis (168,169). In addition, an increase in
mitochondrial aldehyde dehydrogenase 2 activity triggers the
accumulation of 4-hydroxynonenal during ischemic myocardial injury,
which initiates pro-apoptotic signaling by reducing HSP70 and
activating the JNK/p53 pathway. This mechanism ultimately
contributes to the development of heart failure, whereas the
aforementioned process can be reversed by overexpression of HSP70
(170). It has also been shown
that intracellular HSP70 has a cardioprotective effect, whereas
extracellular HSP70 appears to be pro-apoptotic (171).
Thus, cytosolic HSPs protect the myocardium from
ischemia/reperfusion injury by inhibiting the activation of the
apoptotic cascade in CMCs. In particular, the intracellular or
extracellular location of HSPs is very important for their function
in myocardial infarction or ischemia-reperfusion. Intracellular
HSPs have cardio-protective properties, whereas extracellular HSPs
exhibit cardio-toxic effects during ischemia-reperfusion.
HSPs are an important component of the protein
quality control system in the cell, both under normal and
pathological conditions. They ensure the correct assembly of a
number of intracellular proteins and regulate a significant part of
the synthesis processes. Insufficient or excessive production of
HSPs leads to disruption of cell homeostasis and may also
contribute to the activation of cellular stress. This can result in
endoplasmic reticulum damage, mitochondrial dysregulation and
modulation of regulated (programmed) cell death, including
apoptosis and autophagic flux.
Understanding the mechanisms of action of HSPs in
the cardiovascular system can serve as a basis for the development
of new methods of pharmacotherapy of cardiac pathology.
Data sharing is not applicable to this article, as
no data sets were generated or analyzed during the current
study.
AS and MB conceived the study and wrote the original
draft. SS, SC, AR, KA, DP, VG and EA participated in writing and
editing the manuscript. SS and EA edited the manuscript. All the
authors read and approved the final manuscript. Data authentication
is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
The authors thank the International Polyamines
Foundation ETS-ONLUS for their helpful suggestions and assistance
in reading the revised manuscripts.
The present study was supported by project no. 032532-0-000 of
RUDN University, Moscow, Russia.
|
1
|
Safari S, Malekvandfard F, Babashah S,
Alizadehasl A, Sadeghizadeh M and Motavaf M: Mesenchymal stem
cell-derived exosomes: A novel potential therapeutic avenue for
cardiac regeneration. Cell Mol Biol (Noisy-le-grand). 62:66–73.
2016.PubMed/NCBI
|
|
2
|
Tarone G and Brancaccio M: Keep your heart
in shape: Molecular chaperone networks for treating heart disease.
Cardiovasc Res. 102:346–361. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rabinovich-Nikitin I, Rasouli M, Reitz CJ,
Posen I, Margulets V, Dhingra R, Khatua TN, Thliveris JA, Martino
TA and Kirshenbaum LA: Mitochondrial autophagy and cell survival is
regulated by the circadian Clock gene in cardiac myocytes during
ischemic stress. Autophagy. 17:3794–3812. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cicalese SM, da Silva JF, Priviero F, Webb
RC, Eguchi S and Tostes RC: Vascular stress signaling in
hypertension. Circ Res. 128:969–992. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ma T, Huang X, Zheng H, Huang G, Li W, Liu
X, Liang J, Cao Y, Hu Y and Huang Y: SFRP2 improves mitochondrial
dynamics and mitochondrial biogenesis, oxidative stress, and
apoptosis in diabetic cardiomyopathy. Oxid Med Cell Longev.
2021:92650162021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ranek MJ, Stachowski MJ, Kirk JA and
Willis MS: The role of heat shock proteins and co-chaperones in
heart failure. Philos Trans R Soc Lond B Biol Sci.
373:201605302018. View Article : Google Scholar
|
|
7
|
Maejima Y: The critical roles of protein
quality control systems in the pathogenesis of heart failure. J
Cardiol. 75:219–227. 2020. View Article : Google Scholar
|
|
8
|
Schwabl S and Teis D: Protein quality
control at the Golgi. Curr Opin Cell Biol. 75:1020742022.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang X and Robbins J: Heart failure and
protein quality control. Circ Res. 99:1315–1328. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brownstein AJ, Ganesan S, Summers CM,
Pearce S, Hale BJ, Ross JW, Gabler N, Seibert JT, Rhoads RP,
Baumgard LH and Selsby JT: Heat stress causes dysfunctional
autophagy in oxidative skeletal muscle. Physiol Rep. 5:e133172017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hagymasi AT, Dempsey JP and Srivastava PK:
Heat-shock proteins. Curr Protoc. 2:e5922022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tedesco B, Vendredy L, Timmerman V and
Poletti A: The chaperone-assisted selective autophagy complex
dynamics and dysfunctions. Autophagy. 19:1619–1641. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yun CW, Kim HJ, Lim JH and Lee SH: Heat
Shock Proteins: Agents of cancer development and therapeutic
targets in anti-cancer therapy. Cells. 9:602019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Haslbeck M and Vierling E: A first line of
stress defense: Small heat shock proteins and their function in
protein homeostasis. J Mol Biol. 427:1537–1548. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jacob P, Hirt H and Bendahmane A: The
heat-shock protein/chaperone network and multiple stress
resistance. Plant. Biotechnol J. 15:405–414. 2017. View Article : Google Scholar :
|
|
16
|
Schroder K and Tschopp J: The
inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gomez-Pastor R, Burchfiel ET, Neef DW,
Jaeger AM, Cabiscol E, McKinstry SU, Doss A, Aballay A, Lo DC,
Akimov SS, et al: Abnormal degradation of the neuronal
stress-protective transcription factor HSF1 in Huntington's
disease. Nat Commun. 8:144052017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dowell J, Elser BA, Schroeder RE and
Stevens HE: Cellular stress mechanisms of prenatal maternal stress:
Heat shock factors and oxidative stress. Neurosci Lett.
709:1343682019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xie M, Kong Y, Tan W, May H, Battiprolu
PK, Pedrozo Z, Wang ZV, Morales C, Luo X, Cho G, et al: Histone
deacetylase inhibition blunts ischemia/reperfusion injury by
inducing cardiomyocyte autophagy. Circulation. 129:1139–1151. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Blagonravov ML, Korshunova AY, Azova MM,
Bondar' SA and Frolov VA: Cardiomyocyte autophagia and
morphological alterations in the left ventricular myocardium during
acute focal ischemia. Bull Exp Biol Med. 160:398–400. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang HL, Jia KY, Sun D and Yang M:
Protective effect of HSP27 in atherosclerosis and coronary heart
disease by inhibiting reactive oxygen species. J Cell Biochem.
120:2859–2868. 2019. View Article : Google Scholar
|
|
22
|
Shan R, Liu N, Yan Y and Liu B: Apoptosis,
autophagy and atherosclerosis: Relationships and the role of Hsp27.
Pharmacol Res. 166:1051692021. View Article : Google Scholar
|
|
23
|
Kovaleva OV, Shitova MS and Zborovskaya
IB: Autophagy: Cell death or a way of survival? Clin
Oncohematology. 7:103–113. 2014.
|
|
24
|
Del Re DP, Amgalan D, Linkermann A, Liu Q
and Kitsis RN: Fundamental mechanisms of regulated cell death and
implications for heart disease. Physiol Rev. 99:1765–1817. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Martine P and Rébé C: Heat shock proteins
and inflammasomes. Int J Mol Sci. 20:45082019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Choudhury A, Bullock D, Lim A, Argemi J,
Orning P, Lien E, Bataller R and Mandrekar P: Inhibition of HSP90
and activation of HSF1 diminish macrophage NLRP3 inflammasome
activity in alcohol-associated liver injury. Alcohol Clin Exp Res.
44:1300–1311. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jurisic V: Multiomic analysis of cytokines
in immuno-oncology. Expert Rev Proteomics. 17:663–674. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jurisic V, Srdic-Rajic V, Konjevic G,
Bogdanovic G and Colic M: TNF-α induced apoptosis is accompanied
with rapid CD30 and slower CD45 shedding from K-562 cells. J Membr
Biol. 239:115–122. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jurisic V, Terzic T, Colic S and Jurisic
M: The concentration of TNF-alpha correlate with number of
inflammatory cells and degree of vascularization in radicular
cysts. Oral Dis. 14:600–605. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Swaroop S, Sengupta N, Suryawanshi AR,
Adlakha YK and Basu A: HSP60 plays a regulatory role in
IL-1β-induced microglial inflammation via TLR4-p38 MAPK axis. J
Neuroinflammation. 13:272016. View Article : Google Scholar
|
|
31
|
Li XL, Wang YL, Zheng J, Zhang Y and Zhang
XF: Inhibiting expression of HSP60 and TLR4 attenuates
paraquat-induced microglial inflammation. Chem Biol Interact.
299:179–185. 2019. View Article : Google Scholar
|
|
32
|
Kelley N, Jeltema D, Duan Y and He Y: The
NLRP3 Inflammasome: An overview of mechanisms of activation and
regulation. Int J Mol Sci. 20:33282019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Swaroop S, Mahadevan A, Shankar SK,
Adlakha YK and Basu A: HSP60 critically regulates endogenous IL-1β
production in activated microglia by stimulating NLRP3 inflammasome
pathway. J Neuroinflammation. 15:1772018. View Article : Google Scholar
|
|
34
|
Aslan JE and McCarty OJ: Rho GTPases in
platelet function. J Thromb Haemost. 11:35–46. 2013. View Article : Google Scholar
|
|
35
|
Elvers M: RhoGAPs and Rho GTPases in
platelets. Hamostaseologie. 36:168–177. 2016. View Article : Google Scholar
|
|
36
|
Ngo ATP, Parra-Izquierdo I, Aslan JE and
McCarty OJT: Rho GTPase regulation of reactive oxygen species
generation and signaling in platelet function and disease. Small
GTPases. 12:440–457. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang L, Wu Y, Zhou J, Ahmad SS, Mutus B,
Garbi N, Hämmerling G, Liu J and Essex DW: Platelet-derived ERp57
mediates platelet incorporation into a growing thrombus by
regulation of the αIIbβ3 integrin. Blood. 122:3642–3650. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang J, Li X, Shi X, Zhu M, Wang J, Huang
S, Huang X, Wang H, Li L, Deng H, et al: Platelet integrin αIIbβ3:
Signal transduction, regulation, and its therapeutic targeting. J
Hematol Oncol. 12:262019. View Article : Google Scholar
|
|
39
|
Rigg RA, Healy LD, Nowak MS, Mallet J,
Thierheimer ML, Pang J, McCarty OJ and Aslan JE: Heat shock protein
70 regulates platelet integrin activation, granule secretion and
aggregation. Am J Physiol Cell Physiol. 310:C568–C575. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
De Maio A: Extracellular Hsp70: Export and
function. Curr Protein Pept Sci. 15:225–231. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Krause M, Heck TG, Bittencourt A,
Scomazzon SP, Newsholme P, Curi R and Homem de Bittencourt PI Jr:
The chaperone balance hypothesis: The importance of the
extracellular to intracellular HSP70 ratio to inflammation-driven
type 2 diabetes, the effect of exercise, and the implications for
clinical management. Mediators Inflamm. 2015:2492052015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jackson JW, Rivera-Marquez GM, Beebe K,
Tran AD, Trepel JB, Gestwicki JE, Blagg BSJ, Ohkubo S and Neckers
LM: Pharmacologic dissection of the overlapping impact of heat
shock protein family members on platelet function. J Thromb
Haemost. 18:1197–1209. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Blagonravov ML, Sklifasovskaya AP,
Korshunova AY, Azova MM and Kurlaeva AO: Heat shock protein HSP60
in left ventricular cardiomyocytes of hypertensive rats with and
without insulin-dependent diabetes mellitus. Bull Exp Biol Med.
170:10–14. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Henstridge DC, Whitham M and Febbraio MA:
Chaperoning to the metabolic party: The emerging therapeutic role
of heat-shock proteins in obesity and type 2 diabetes. Mol Metab.
3:781–793. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Archer AE, Von Schulze AT and Geiger PC:
Exercise, heat shock proteins and insulin resistance. Philos Trans
R Soc Lond B Biol Sci. 373:201605292018. View Article : Google Scholar
|
|
46
|
Drew BG, Ribas V, Le JA, Henstridge DC,
Phun J, Zhou Z, Soleymani T, Daraei P, Sitz D, Vergnes L, et al:
HSP72 is a mitochondrial stress sensor critical for Parkin action,
oxidative metabolism, and insulin sensitivity in skeletal muscle.
Diabetes. 63:1488–1505. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kitano S, Kondo T, Matsuyama R, Ono K,
Goto R, Takaki Y, Hanatani S, Sakaguchi M, Igata M, Kawashima J, et
al: Impact of hepatic HSP72 on insulin signaling. Am J Physiol
Endocrinol Metab. 316:E305–E318. 2019. View Article : Google Scholar
|
|
48
|
Xu L, Ma X, Bagattin A and Mueller E: The
transcriptional coactivator PGC1α protects against hyperthermic
stress via cooperation with the heat shock factor HSF1. Cell Death
Dis. 7:e21022016. View Article : Google Scholar
|
|
49
|
Jornayvaz FR and Shulman GI: Regulation of
mitochondrial biogenesis. Essays Biochem. 47:69–84. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Charos AE, Reed BD, Raha D, Szekely AM,
Weissman SM and Snyder M: A highly integrated and complex PPARGC1A
transcription factor binding network in HepG2 cells. Genome Res.
22:1668–1679. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ma X, Xu L, Alberobello AT, Gavrilova O,
Bagattin A, Skarulis M, Liu J, Finkel T and Mueller E: Celastrol
protects against obesity and metabolic dysfunction through
activation of a HSF1-PGC1α transcriptional axis. Cell Metab.
22:695–708. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dang X, Du G, Hu W, Ma L, Wang P and Li Y:
Peroxisome proliferator-activated receptor gamma
coactivator-1α/HSF1 axis effectively alleviates
lipopolysaccharide-induced acute lung injury via suppressing
oxidative stress and inflammatory response. J Cell Biochem.
120:544–551. 2019. View Article : Google Scholar
|
|
53
|
Meyer BA and Doroudgar S: ER
Stress-induced secretion of proteins and their extracellular
functions in the heart. Cells. 9:20662020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
García R, Merino D, Gómez JM, Nistal JF,
Hurlé MA, Cortajarena AL and Villar AV: Extracellular heat shock
protein 90 binding to TGFβ receptor I participates in TGFβ-mediated
collagen production in myocardial fibroblasts. Cell Signal.
28:1563–1579. 2016. View Article : Google Scholar
|
|
55
|
Shi C, Ulke-Lemée A, Deng J, Batulan Z and
O'Brien ER: Characterization of heat shock protein 27 in
extracellular vesicles: A potential anti-inflammatory therapy.
FASEB J. 33:1617–1630. 2019. View Article : Google Scholar
|
|
56
|
Liu P, Bao HY, Jin CC, Zhou JC, Hua F, Li
K, Lv XX, Cui B, Hu ZW and Zhang XW: Targeting extracellular heat
shock protein 70 ameliorates doxorubicin-induced heart failure
through resolution of toll-like receptor 2-mediated myocardial
inflammation. J Am Heart Assoc. 8:e0123382019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jan RL, Yang SC, Liu YC, Yang RC, Tsai SP,
Huang SE, Yeh JL and Hsu JH: Extracellular heat shock protein HSC70
protects against lipopolysaccharide-induced hypertrophic responses
in rat cardiomyocytes. Biomed Pharmacother. 128:1103702020.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang X, Xu Z, Zhou L, Chen Y, He M, Cheng
L, Hu FB, Tanguay RM and Wu T: Plasma levels of Hsp70 and
anti-Hsp70 antibody predict risk of acute coronary syndrome. Cell
Stress Chaperones. 15:675–686. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Jenei ZM, Gombos T, Förhécz Z, Pozsonyi Z,
Karádi I, Jánoskuti L and Prohászka Z: Elevated extracellular HSP70
(HSPA1A) level as an independent prognostic marker of mortality in
patients with heart failure. Cell Stress Chaperones. 18:809–813.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Song YJ, Zhong CB and Wang XB: Heat shock
protein 70: A promising therapeutic target for myocardial
ischemia-reperfusion injury. J Cell Physiol. 234:1190–1207. 2019.
View Article : Google Scholar
|
|
61
|
Yang J, Yu XF, Li YY, Xue FT and Zhang S:
Decreased HSP70 expression on serum exosomes contributes to cardiac
fibrosis during senescence. Eur Rev Med Pharmacol Sci.
23:3993–4001. 2019.PubMed/NCBI
|
|
62
|
Yoon S, Kim M, Min HK, Lee YU, Kwon DH,
Lee M, Lee S, Kook T, Joung H, Nam KI, et al: Inhibition of heat
shock protein 70 blocks the development of cardiac hypertrophy by
modulating the phosphorylation of histone deacetylase 2. Cardiovasc
Res. 115:1850–1860. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Rodriguez-Iturbe B, Johnson RJ,
Sanchez-Lozada LG and Pons H: HSP70 and primary arterial
hypertension. Biomolecules. 13:2722023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Mathur S, Walley KR, Wang Y, Indrambarya T
and Boyd JH: Extracellular heat shock protein 70 induces
cardiomyocyte inflammation and contractile dysfunction via TLR2.
Circ J. 75:2445–2452. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Birmpilis AI, Paschalis A, Mourkakis A,
Christodoulou P, Kostopoulos IV, Antimissari E, Terzoudi G,
Georgakilas AG, Armpilia C, Papageorgis P, et al: Immunogenic cell
death, DAMPs and prothymosin α as a putative anticancer immune
response biomarker. Cells. 11:14152022. View Article : Google Scholar
|
|
66
|
Bacmeister L, Schwarzl M, Warnke S,
Stoffers B, Blankenberg S, Westermann D and Lindner D: Inflammation
and fibrosis in murine models of heart failure. Basic Res Cardiol.
114:192019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Shah AK, Bhullar SK, Elimban V and Dhalla
NS: Oxidative stress as a mechanism for functional alterations in
cardiac hypertrophy and heart failure. Antioxidants (Basel).
10:9312021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kruszewska J, Cudnoch-Jedrzejewska A and
Czarzasta K: Remodeling and fibrosis of the cardiac muscle in the
course of obesity-pathogenesis and involvement of the extracellular
matrix. Int J Mol Sci. 23:41952022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Khalil H, Kanisicak O, Prasad V, Correll
RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, et al:
Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac
fibrosis. J Clin Invest. 127:3770–3783. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Tian J, Zhang M, Suo M, Liu D, Wang X, Liu
M, Pan J, Jin T and An F: Dapagliflozin alleviates cardiac fibrosis
through suppressing EndMT and fibroblast activation via
AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J Cell Mol
Med. 25:7642–7659. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ko T, Nomura S, Yamada S, Fujita K, Fujita
T, Satoh M, Oka C, Katoh M, Ito M, Katagiri M, et al: Cardiac
fibroblasts regulate the development of heart failure via
Htra3-TGF-β-IGFBP7 axis. Nat Commun. 13:32752022. View Article : Google Scholar
|
|
72
|
Cáceres RA, Chavez T, Maestro D, Palanca
AR, Bolado P, Madrazo F, Aires A, Cortajarena AL and Villar AV:
Reduction of cardiac TGFβ-mediated profibrotic events by inhibition
of Hsp90 with engineered protein. J Mol Cell Cardiol. 123:75–87.
2018. View Article : Google Scholar
|
|
73
|
Zhang X, Zhang Y, Miao Q, Shi Z, Hu L, Liu
S, Gao J, Zhao S, Chen H, Huang Z, et al: Inhibition of HSP90
S-nitrosylation alleviates cardiac fibrosis via TGFβ/SMAD3
signalling pathway. Br J Pharmacol. 178:4608–4625. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhong W, Chen W, Liu Y, Zhang J, Lu Y, Wan
X, Qiao Y, Huang H, Zeng Z, Li W, et al: Extracellular HSP90α
promotes cellular senescence by modulating TGF-β signaling in
pulmonary fibrosis. FASEB J. 36:e224752022. View Article : Google Scholar
|
|
75
|
Christians ES, Ishiwata T and Benjamin IJ:
Small heat shock proteins in redox metabolism: Implications for
cardiovascular diseases. Int J Biochem Cell Biol. 44:1632–1645.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Collier MP and Benesch JLP: Small
heat-shock proteins and their role in mechanical stress. Cell
Stress Chaperones. 25:601–613. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Nguyen VC, Deck CA and Pamenter ME: Naked
mole-rats reduce the expression of ATP-dependent but not
ATP-independent heat shock proteins in acute hypoxia. J Exp Biol.
222(Pt 22): jeb2112432019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Janowska MK, Baughman HER, Woods CN and
Klevit RE: Mechanisms of small heat shock proteins. Cold Spring
Harb Perspect Biol. 11:a0340252019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Alagar Boopathy LR, Jacob-Tomas S, Alecki
C and Vera M: Mechanisms tailoring the expression of heat shock
proteins to proteostasis challenges. J Biol Chem. 298:1017962022.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Carver JA, Ecroyd H, Truscott RJW, Thorn
DC and Holt C: Proteostasis and the regulation of intra- and
extracellular protein aggregation by ATP-independent molecular
chaperones: Lens α-crystallins and milk caseins. Acc Chem Res.
51:745–752. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Izumi M: Heat shock proteins support
refolding and shredding of misfolded proteins. Plant Physiol.
180:1777–1778. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Choudhary D, Mediani L, Carra S and
Cecconi C: Studying heat shock proteins through single-molecule
mechanical manipulation. Cell Stress Chaperones. 25:615–628. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Dokladny K, Myers OB and Moseley PL: Heat
shock response and autophagy-cooperation and control. Autophagy.
11:200–213. 2015. View Article : Google Scholar :
|
|
84
|
Shan Q, Ma F, Wei J, Li H, Ma H and Sun P:
Physiological functions of heat shock proteins. Curr Protein Pept
Sci. 21:751–760. 2020. View Article : Google Scholar
|
|
85
|
Hosaka Y, Araya J, Fujita Y and Kuwano K:
Role of chaperone-mediated autophagy in the pathophysiology
including pulmonary disorders. Inflamm Regen. 41:292021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wick G, Jakic B, Buszko M, Wick MC and
Grundtman C: The role of heat shock proteins in atherosclerosis.
Nat Rev Cardiol. 11:516–529. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Bakthisaran R, Tangirala R and Rao ChM:
Small heat shock proteins: Role in cellular functions and
pathology. Biochim Biophys Acta. 1854:291–319. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Hashikawa N, Ido M, Morita Y and
Hashikawa-Hobara N: Effects from the induction of heat shock
proteins in a murine model due to progression of aortic
atherosclerosis. Sci Rep. 11:70252021. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Cuerrier CM, Chen YX, Tremblay D, Rayner
K, McNulty M, Zhao X, Kennedy CR, de BelleRoche J, Pelling AE and
O'Brien ER: Chronic over-expression of heat shock protein 27
attenuates atherogenesis and enhances plaque remodeling: A combined
histological and mechanical assessment of aortic lesions. PLoS One.
8:e558672013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Liu A, Ming JY, Fiskesund R, Ninio E,
Karabina SA, Bergmark C, Frostegård AG and Frostegård J: Induction
of dendritic cell-mediated T-cell activation by modified but not
native low-density lipoprotein in humans and inhibition by annexin
a5: Involvement of heat shock proteins. Arterioscler Thromb Vasc
Biol. 35:197–205. 2015. View Article : Google Scholar
|
|
91
|
Gong R, Li XY, Chen HJ, Xu CC, Fang HY,
Xiang J and Wu YQ: Role of heat shock protein 22 in the protective
effect of geranylgeranylacetone in response to oxidized-LDL. Drug
Des Devel Ther. 13:2619–2632. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Nahomi RB, Palmer A, Green KM, Fort PE and
Nagaraj RH: Pro-inflammatory cytokines downregulate Hsp27 and cause
apoptosis of human retinal capillary endothelial cells. Biochim
Biophys Acta. 1842:164–174. 2014. View Article : Google Scholar :
|
|
93
|
Batulan Z, Pulakazhi Venu VK, Li Y,
Koumbadinga G, Alvarez-Olmedo DG, Shi C and O'Brien ER:
Extracellular release and signaling by heat shock protein 27: Role
in modifying vascular inflammation. Front Immunol. 7:2852016.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Zhou XY, Sun JY, Wang WQ, Li SX, Li HX,
Yang HJ, Yang MF, Yuan H, Zhang ZY, Sun BL and Han JX: TAT-HSP27
Peptide improves neurologic deficits via reducing apoptosis after
experimental subarachnoid hemorrhage. Front Cell Neurosci.
16:8786732022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Jin C, Cleveland JC, Ao L, Li J, Zeng Q,
Fullerton DA and Meng X: Human myocardium releases heat shock
protein 27 (HSP27) after global ischemia: The proinflammatory
effect of extracellular HSP27 through toll-like receptor (TLR)-2
and TLR4. Mol Med. 20:280–289. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Inia JA and O'Brien ER: Role of Heat Shock
Protein 27 in Modulating Atherosclerotic Inflammation. J Cardiovasc
Transl Res. 14:3–12. 2021. View Article : Google Scholar
|
|
97
|
Forouzanfar F, Butler AE, Banach M,
Barreto GE and Sahbekar A: Modulation of heat shock proteins by
statins. Pharmacol Res. 134:134–144. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Sklifasovskaya AP and Blagonravov ML:
Small heat shock proteins HSP10 and HSP27 in the left ventricular
myocardium in rats with arterial hypertension and insulin-dependent
diabetes mellitus. Bull Exp Biol Med. 170:699–705. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Sada K, Nishikawa T, Kukidome D, Yoshinaga
T, Kajihara N, Sonoda K, Senokuchi T, Motoshima H, Matsumura T and
Araki E: Hyperglycemia induces cellular hypoxia through production
of mitochondrial ROS followed by suppression of aquaporin-1. PLoS
One. 11:e01586192016. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Yu L, Chen S, Liang Q, Huang C, Zhang W,
Hu L, Yu Y, Liu L, Cheng X and Bao H: Rosiglitazone reduces
diabetes angiopathy by inhibiting mitochondrial dysfunction
dependent on regulating HSP22 expression. iScience. 26:1061942023.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Yu L, Liang Q, Zhang W, Liao M, Wen M,
Zhan B, Bao H and Cheng X: HSP22 suppresses diabetes-induced
endothelial injury by inhibiting mitochondrial reactive oxygen
species formation. Redox Biol. 21:1010952019. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Li X, Fang P, Yang WY, Chan K, Lavallee M,
Xu K, Gao T, Wang H and Yang X: Mitochondrial ROS, uncoupled from
ATP synthesis, determine endothelial activation for both
physiological recruitment of patrolling cells and pathological
recruitment of inflammatory cells. Can J Physiol Pharmacol.
95:247–252. 2017. View Article : Google Scholar
|
|
103
|
Fang H, Hu N, Zhao Q, Wang B, Zhou H, Fu
Q, Shen L, Chen X, Shen F and Lyu J: mtDNA haplogroup N9a increases
the risk of type 2 diabetes by altering mitochondrial function and
intracellular mitochondrial signals. Diabetes. 67:1441–1453. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Rodríguez ME, Cogno IS, Milla Sanabria LS,
Morán YS and Rivarola VA: Heat shock proteins in the context of
photodynamic therapy: Autophagy, apoptosis and immunogenic cell
death. Photochem Photobiol Sci. 15:1090–1102. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Penke B, Bogár F, Crul T, Sántha M, Tóth
ME and Vígh L: Heat shock proteins and autophagy pathways in
neuroprotection: From molecular bases to pharmacological
interventions. Int J Mol Sci. 19:3252018. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Kanugovi Vijayavittal A, Kumar P, Sugunan
S, Joseph C, Devaki B, Paithankar K and Amere Subbarao S: Heat
shock transcription factor HSF2 modulates the autophagy response
through the BTG2-SOD2 axis. Biochem Biophys Res Commun. 600:44–50.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Cuervo AM and Wong E: Chaperone-mediated
autophagy: Roles in disease and aging. Cell Res. 24:92–104. 2014.
View Article : Google Scholar :
|
|
108
|
Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M,
Shan B, Pan H and Yuan J: Chaperone-mediated autophagy is involved
in the execution of ferroptosis. Proc Natl Acad Sci USA.
116:2996–3005. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Hale BJ, Hager CL, Seibert JT, Selsby JT,
Baumgard LH, Keating AF and Ross JW: Heat stress induces autophagy
in pig ovaries during follicular development. Biol Reprod.
97:426–437. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Ganesan S, Pearce SC, Gabler NK, Baumgard
LH, Rhoads RP and Selsby JT: Short-term heat stress results in
increased apoptotic signaling and autophagy in oxidative skeletal
muscle in Sus scrofa. J Therm Biol. 72:73–80. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Roths M, Freestone AD, Rudolph TE, Michael
A, Baumgard LH and Selsby JT: Environment-induced heat stress
causes structural and biochemical changes in the heart. J Therm
Biol. 113:1034922023. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Li DL, Wang ZV, Ding G, Tan W, Luo X,
Criollo A, Xie M, Jiang N, May H, Kyrychenko V, et al: Doxorubicin
blocks cardiomyocyte autophagic flux by inhibiting lysosome
acidification. Circulation. 26(133): 1668–1687. 2016. View Article : Google Scholar
|
|
113
|
Packer M: Role of impaired nutrient and
oxygen deprivation signaling and deficient autophagic flux in
diabetic CKD development: Implications for understanding the
effects of sodium-glucose cotransporter 2-inhibitors. J Am Soc
Nephrol. 31:907–919. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Gu S, Tan J, Li Q, Liu S, Ma J, Zheng Y,
Liu J, Bi W, Sha P, Li X, et al: Downregulation of LAPTM4B
contributes to the impairment of the autophagic flux via unopposed
activation of mTORC1 signaling during myocardial
ischemia/reperfusion injury. Circ Res. 127:e148–e165. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Sciarretta S, Maejima Y, Zablocki D and
Sadoshima J: The role of autophagy in the heart. Annu Rev Physiol.
80:1–26. 2018. View Article : Google Scholar
|
|
116
|
Lavandero S, Troncoso R, Rothermel BA,
Martinet W, Sadoshima J and Hill JA: Cardiovascular autophagy:
Concepts, controversies, and perspectives. Autophagy. 9:1455–1466.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Ott C, Jung T, Brix S, John C, Betz IR,
Foryst-Ludwig A, Deubel S, Kuebler WM, Grune T, Kintscher U and
Grune J: Hypertrophy-reduced autophagy causes cardiac dysfunction
by directly impacting cardiomyocyte contractility. Cells.
10:8052021. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Zhang Y, Liu D, Hu H, Zhang P, Xie R and
Cui W: HIF-1α/BNIP3 signaling pathway-induced-autophagy plays
protective role during myocardial ischemia-reperfusion injury.
Biomed. Pharmacother. 120:1094642019. View Article : Google Scholar
|
|
119
|
Liu W, Chen C, Gu X, Zhang L, Mao X, Chen
Z and Tao L: AM1241 alleviates myocardial ischemia-reperfusion
injury in rats by enhancing Pink1/Parkin-mediated autophagy. Life
Sci. 272:1192282021. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Sui Z, Wang MM, Xing Y, Qi J and Wang W:
Targeting MCOLN1/TRPML1 channels to protect against
ischemia-reperfusion injury by restoring the inhibited autophagic
flux in cardiomyocytes. Autophagy. 18:3053–3055. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Liu L, Jin X, Hu CF, Li R, Zhou Z and Shen
CX: Exosomes derived from mesenchymal stem cells rescue myocardial
ischaemia/reperfusion injury by inducing cardiomyocyte autophagy
via AMPK and Akt pathways. Cell Physiol Biochem. 43:52–68. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Xiang M, Lu Y, Xin L, Gao J, Shang C,
Jiang Z, Lin H, Fang X, Qu Y, Wang Y, et al: Role of oxidative
stress in reperfusion following myocardial ischemia and its
treatments. Oxid Med Cell Longev. 2021:66140092021. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Xing Y, Sui Z, Liu Y, Wang MM, Wei X, Lu
Q, Wang X, Liu N, Lu C, Chen R, et al: Blunting TRPML1 channels
protects myocardial ischemia/reperfusion injury by restoring
impaired cardiomyocyte autophagy. Basic Res Cardiol. 117:202022.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Kim YC and Guan KL: mTOR: A pharmacologic
target for autophagy regulation. J Clin Invest. 125:25–32. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Wang Y and Zhang H: Regulation of
autophagy by mTOR signaling pathway. Adv Exp Med Biol. 1206:67–83.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Al-Bari MAA and Xu P: Molecular regulation
of autophagy machinery by mTOR-dependent and -independent pathways.
Ann N Y Acad Sci. 1467:3–20. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Liu GS, Zhu H, Cai WF, Wang X, Jiang M,
Essandoh K, Vafiadaki E, Haghighi K, Lam CK, Gardner G, et al:
Regulation of BECN1-mediated autophagy by HSPB6: Insights from a
human HSPB6S10F mutant. Autophagy. 14:80–97. 2018.
View Article : Google Scholar :
|
|
128
|
Nicolaou P, Knöll R, Haghighi K, Fan GC,
Dorn GW II, Hasenfub G and Kranias EG: Human mutation in the
anti-apoptotic heat shock protein 20 abrogates its cardioprotective
effects. J Biol Chem. 283:33465–33471. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Shatov VM and Gusev NB: Physico-chemical
properties of two point mutants of small heat shock protein HspB6
(Hsp20) with abrogated cardioprotection. Biochimie. 174:126–135.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Lavandero S, Chiong M, Rothermel BA and
Hill JA: Autophagy in cardiovascular biology. J Clin Invest.
125:55–64. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Parzych KR and Klionsky DJ: An overview of
autophagy: Morphology, mechanism, and regulation. Antioxid Redox
Signal. 20:460–473. 2014. View Article : Google Scholar :
|
|
132
|
Cao W, Li J, Yang K and Cao D: An overview
of autophagy: Mechanism, regulation and research progress. Bull
Cancer. 108:304–322. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Zhou Y, Manghwar H, Hu W and Liu F:
Degradation mechanism of autophagy-related proteins and research
progress. Int J Mol Sci. 23:73012022. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Li W, He P, Huang Y, Li YF, Lu J, Li M,
Kurihara H, Luo Z, Meng T, Onishi M, et al: Selective autophagy of
intracellular organelles: Recent research advances. Theranostics.
11:222–256. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Li Y, Li S and Wu H:
Ubiquitination-proteasome system (UPS) and autophagy two main
protein degradation machineries in response to cell stress. Cells.
11:8512022. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Popov SV, Mukhomedzyanov AV, Voronkov NS,
Derkachev IA, Boshchenko AA, Fu F, Sufianova GZ, Khlestkina MS and
Maslov LN: Regulation of autophagy of the heart in ischemia and
reperfusion. Apoptosis. 28:55–80. 2023. View Article : Google Scholar
|
|
137
|
Dong Y, Chen H, Gao J, Liu Y, Li J and
Wang J: Molecular machinery and interplay of apoptosis and
autophagy in coronary heart disease. J Mol Cell Cardiol. 136:27–41.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Denton D and Kumar S: Autophagy-dependent
cell death. Cell Death Differ. 26:605–616. 2019. View Article : Google Scholar :
|
|
139
|
Mahapatra KK, Mishra SR, Behera BP, Patil
S, Gewirtz DA and Bhutia SK: The lysosome as an imperative
regulator of autophagy and cell death. Cell Mol. Life Sci.
78:7435–7449. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Xu HD and Qin ZH: Beclin 1, Bcl-2 and
Autophagy. Adv Exp Med Biol. 1206:109–126. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Liu J, Liu W and Yang H: Balancing
apoptosis and autophagy for Parkinson's disease therapy: Targeting
BCL-2. ACS Chem. Neurosci. 10:792–802. 2019.
|
|
142
|
Blagonravov ML, Sklifasovskaya AP, Demurov
EA and Karimov AA: Beclin-1-dependent autophagy of left ventricular
cardiomyocytes in SHR and Wistar-Kyoto rats with type 1 diabetes
mellitus. Bull Exp Biol Med. 171:23–27. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Sklifasovskaya AP, Blagonravov ML,
Ryabinina AY, Azova MM and Goryachev VA: Expression of Bax and
Bcl-2 Proteins in Left-Ventricular Cardiomyocytes in Wistar-Kyoto
and SHR Rats with Insulin-Dependent Diabetes Mellitus. Bull Exp
Biol Med. 171:576–581. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Van Opdenbosch N and Lamkanfi M: Caspases
in cell death, inflammation, and disease. Immunity. 50:1352–1364.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Araya LE, Soni IV, Hardy JA and Julien O:
Deorphanizing caspase-3 and caspase-9 substrates in and out of
apoptosis with deep substrate profiling. ACS Chem Biol.
16:2280–2296. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Green DR: Caspase activation and
inhibition. Cold Spring Harb Perspect Biol. 14:a0410202022.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Kashyap D, Garg VK and Goel N: Intrinsic
and extrinsic pathways of apoptosis: Role in cancer development and
prognosis. Adv Protein Chem Struct Biol. 125:73–120. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Lossi L: The concept of intrinsic versus
extrinsic apoptosis. Biochem J. 479:357–384. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Tang D, Kang R, Berghe TV, Vandenabeele P
and Kroemer G: The molecular machinery of regulated cell death.
Cell Res. 29:347–364. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Obeng E: Apoptosis (programmed cell death)
and its signals-A review. Braz J Biol. 81:1133–1143. 2021.
View Article : Google Scholar
|
|
151
|
Kennedy D, Jäger R, Mosser DD and Samali
A: Regulation of apoptosis by heat shock proteins. IUBMB Life.
66:327–338. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Leung AM, Redlak MJ and Miller TA: Role of
heat shock proteins in oxygen radical-induced gastric apoptosis. J
Surg Res. 193:135–144. 2015. View Article : Google Scholar
|
|
153
|
Yu Y, Hu LL, Liu L, Yu LL, Li JP, Rao JA,
Zhu LJ, Bao HH and Cheng XS: Hsp22 ameliorates
lipopolysaccharide-induced myocardial injury by inhibiting
inflammation, oxidative stress, and apoptosis. Bioengineered.
12:12544–12554. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Ruan L, Zhou C, Jin E, Kucharavy A, Zhang
Y, Wen Z, Florens L and Li R: Cytosolic proteostasis through
importing of misfolded proteins into mitochondria. Nature.
543:443–446. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Koike N, Hatano Y and Ushimaru T: Heat
shock transcriptional factor mediates mitochondrial unfolded
protein response. Curr Genet. 64:907–917. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Verma A, Sumi S and Seervi M: Heat shock
proteins-driven stress granule dynamics: Yet another avenue for
cell survival. Apoptosis. 26:371–384. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Liyanagamage DSNK and Martinus RD: Role of
mitochondrial stress protein HSP60 in diabetes-induced
neuroinflammation. Mediators Inflamm. 2020:80735162020. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Kumar R, Chaudhary AK, Woytash J, Inigo
JR, Gokhale AA, Bshara W, Attwood K, Wang J, Spernyak JA, Rath E,
et al: A mitochondrial unfolded protein response inhibitor
suppresses prostate cancer growth in mice via HSP60. J Clin Invest.
132:e1499062022. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Duan Y, Tang H, Mitchell-Silbaugh K, Fang
X, Han Z and Ouyang K: Heat shock protein 60 in cardiovascular
physiology and diseases. Front Mol Biosci. 7:732020. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Song E, Tang S, Xu J, Yin B, Bao E and
Hartung J: Lenti-siRNA Hsp60 promote bax in mitochondria and
induces apoptosis during heat stress. Biochem Biophys Res Commun.
481:125–131. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Tian X, Zhao L, Song X, Yan Y, Liu N, Li
T, Yan B and Liu B: HSP27 inhibits homocysteine-induced endothelial
apoptosis by modulation of ROS production and mitochondrial
caspase-dependent apoptotic pathway. Biomed Res Int.
2016:48478742016. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Kennedy D, Mnich K, Oommen D, Chakravarthy
R, Almeida-Souza L, Krols M, Saveljeva S, Doyle K, Gupta S,
Timmerman V, et al: HSPB1 facilitates ERK-mediated phosphorylation
and degradation of BIM to attenuate endoplasmic reticulum
stress-induced apoptosis. Cell Death Dis. 8:e30262017. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Önay Uçar E and Şengelen A: Resveratrol
and siRNA in combination reduces Hsp27 expression and induces
caspase-3 activity in human glioblastoma cells. Cell Stress
Chaperones. 24:763–775. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Guo S, Gao C, Xiao W, Zhang J, Qu Y, Li J
and Ye F: Matrine protects cardiomyocytes from ischemia/reperfusion
injury by regulating HSP70 expression via activation of the
JAK2/STAT3 pathway. Shock. 50:664–670. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Xin BR, Li P, Liu XL and Zhang XF:
Visfatin relieves myocardial ischemia-reperfusion injury through
activation of PI3K/Akt/HSP70 signaling axis. Eur Rev Med Pharmacol
Sci. 24:10779–10789. 2020.PubMed/NCBI
|
|
166
|
Huang C, Deng H, Zhao W and Xian L:
Knockdown of miR-384-3p protects against myocardial
ischemia-reperfusion injury in rats through targeting HSP70. Heart
Surg Forum. 24:E143–E150. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Song N, Ma J, Meng XW, Liu H, Wang H, Song
SY, Chen QC, Liu HY, Zhang J, Peng K and Ji FH: Heat shock protein
70 protects the heart from ischemia/reperfusion injury through
inhibition of p38 MAPK Signaling. Oxid Med Cell Longev.
2020:39086412020. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Choudhury S, Bae S, Ke Q, Lee JY, Kim J
and Kang PM: Mitochondria to nucleus translocation of AIF in mice
lacking Hsp70 during ischemia/reperfusion. Basic Res Cardiol.
106:397–407. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Zhang C, Liu X, Miao J, Wang S, Wu L, Yan
D, Li J, Guo W, Wu X and Shen A: Heat shock protein 70 protects
cardiomyocytes through suppressing SUMOylation and nucleus
translocation of phosphorylated eukaryotic elongation factor 2
during myocardial ischemia and reperfusion. Apoptosis. 22:608–625.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Sun A, Zou Y, Wang P, Xu D, Gong H, Wang
S, Qin Y, Zhang P, Chen Y, Harada M, et al: Mitochondrial aldehyde
dehydrogenase 2 plays protective roles in heart failure after
myocardial infarction via suppression of the cytosolic JNK/p53
pathway in mice. J Am Heart Assoc. 3:e0007792014. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Jenei ZM, Széplaki G, Merkely B, Karádi I,
Zima E and Prohászka Z: Persistently elevated extracellular HSP70
(HSPA1A) level as an independent prognostic marker in
post-cardiac-arrest patients. Cell Stress Chaperones. 18:447–454.
2013. View Article : Google Scholar : PubMed/NCBI
|