Introduction
Osteoarthritis (OA) is one of the most common joint
diseases worldwide and its incidence in the world population
>60-years old is ~20%. The typical features of OA including
cartilage degeneration, osteophyte formation, subchondral bone
sclerosis and synovial inflammation (1,2).
Although a number of studies have shown that the sex, age and
obesity are the main risks factors of OA, the pathophysiology of OA
has not been fully demonstrated (3,4).
Increasing number of studies reveal that inflammation and
inflammatory cytokines seem to play a critical role in the
initiation and progression of OA. Among these inflammatory
cytokines, IL-1β is considered as the most important (5). IL-1β contributes to the cartilage
degradation via increasing the expression of matrix
metalloproteinases (MMPs), NO and prostaglandin E2 (6,7).
Moreover, some small molecule also play a marked role, such as high
mobility group box 1 (8), free
fatty acids (9) and advanced
glycation end products (AGEs) (10). For example, AGEs could induce the
activation of NLR family pyrin domain containing 3 (NLRP3)
inflammasome to promote IL-1β production and inflammatory response
in human chondrocytes (11).
These results indicate that the pathophysiology of OA is
complicated and the mechanism underlying the occurrence and
development of OA remains to be elucidated.
Cold-inducible RNA-binding protein (CIRP) is a
172-amino acid protein and belongs to a family of cold-shock
proteins. The mature CIRP peptide contains one RNA recognition
motif domain in the N-terminal and one glycine-rich C-terminal
domain. CIRP was first identified as an RNA binding protein and is
lowly expressed in various tissues (12,13). It is a stress-inducible protein
which responds to a number of stresses, including hypoxia and UV
radiation. The stress-induced CIRP regulates target genes
expression through binding and stabilizing them (14,15). It has been reported that CIRP is
a new damage-associated molecular pattern molecule (DAMP) and can
function as an inflammatory mediator (16). Qiang et al (17) found that hypoxia stress could
induce CIRP secretion, the extracellular CIRP working as a DAMP to
promote inflammatory response in shock and sepsis. Sakurai et
al (18) reported that CIRP
increases the expression of TNF-α and IL-32, linking inflammation
and tumorigenesis in colitis-associated cancer. In cerebral
ischemia, CIRP activates the NF-κB signaling pathway, promotes the
TNF-α expression and mediates neuroinflammation (19). NLRP3 inflammasomes are key
regulators of inflammatory response in the pathogenesis of OA
(20-22). The activation of the NLRP3
inflammasome leads to the cleavage of caspase-1, which processes
pro-IL-1β into its mature form, IL-1β. This mature cytokine is then
secreted and further amplifies the inflammatory response (23). It has been reported CIRP can
trigger an inflammatory response through the Toll-like receptor 4
(TLR4)/NF-κB signaling pathway (17,24). The assembly of TLR4/NF-κB and
NLRP3/ASC/procaspase-1 complexes contributes to the activation of
canonical NLRP3 inflammasome signaling (25). Furthermore, CIRP has been
evaluated in the synovial fluid of OA and rheumatoid arthritis,
indicating a positive relationship between CIRP and OA (26,27). However, the roles of CIRP in OA
progression and the underlying mechanism remain to be
elucidated.
MicroRNAs (miRNAs) are a cluster of short non-coding
RNAs with an average length of 22 nucleotides. They suppress target
genes expression by binding to their 3'-UTRs. Studies have
indicated that miRNAs play critical roles in maintaining cartilage
homeostasis as well as clinical treatment of OA (28,29). For example, miR-92a-3p enhances
chondrogenesis and suppresses cartilage degradation by targeting
WNT5A (30). As cell-secreted
nanovesicles, exosomes contain multiple types of cargo, including
non-coding RNA molecules, DNA, lipids and functional proteins.
Recent evidence has suggested that exosomes play a novel function
as attractive carriers for gene therapy or drug delivery (31). For example, exosome-mediated
miR-144-3p promotes ferroptosis to inhibit osteosarcoma
proliferation, migration and invasion by regulating ZEB1 (32). Moreover, umbilical cord
blood-derived M1 macrophage exosomes loaded with cisplatin reversed
cisplatin resistance of ovarian cancer in vivo (33). These studies indicate that
exosomes are novel carriers capable of delivering RNA molecules or
drugs to target cells, representing a potential strategy for OA
treatment.
The present study aimed to investigate the function
and molecular mechanisms of CIRP in OA progression. It also sought
to explore how CIRP regulates the inflammatory response and
extracellular matrix (ECM) degradation in human chondrocytes
through the TLR4/NF-κB/NLRP3 inflammasome. Additionally, it
identified CIRP as a target of miRNA-145-5p (miR-145-5p), a
microRNA previously shown to have protective effects in
chondrocytes and nucleus pulposus cells (34-36). This led the present study to
further examine whether miR-145-5p could prevent OA development by
targeting CIRP.
Materials and methods
Chemicals and reagents
Human recombinant CIRP peptides were purchased from
Abcam. NF-κB inhibitor BAY 11-7082 and cell counting kit-8 (CCK-8)
was purchased from Beyotime Institute of Biotechnology. TLR4
inhibitor TAK-242 was purchased from Shanghai Xianding
Biotechnology Co., Ltd. Primary antibodies directed against
inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2),
MMP1, MMP3, MMP13, Lamin B1, CD63, CD9, 130 kDa cis-Golgi matrix
protein 1 (GM130) and GAPDH were purchased from Proteintech Group,
Inc., ADAM metallopeptidase with thrombospondin type 1 motif 5
(ADAMTS5) was purchased from ABclonal. Human Reactive Inflammasome
Antibody Sampler Kit II, CIRP, IκBα and p65 were purchased from
Cell Signaling Technology, Inc. Collagen II was purchased from
Beyotime Institute of Biotechnology. Anti-rabbit IgG, HRP-linked
antibody and Anti-mouse IgG, HRP-linked antibody were purchased
from Beyotime Institute of Biotechnology.
Preparation and culture of human primary
chondrocytes
The collection of cartilage tissues from patients
was approved by the Ethics Committee of Shenzhen Second People's
Hospital (2023-157-01YJ). Samples used in this study were collected
after all patients received detailed explanation of the research
and submitted written informed consents. Human articular cartilage
tissues were obtained from volunteer donors who had undergo joint
replacement or joint surgery (December 2023-September 2024). Normal
chondrocytes were obtained from the articular cartilage of patients
who underwent joint replacement surgery due to femoral neck
fractures. Notably, these patients did not have a diagnosis of OA.
The control group comprised 11 patients (5 males and 6 females),
all aged from 60-74 years old. By contrast, OA chondrocytes were
sourced from the articular cartilage of individuals diagnosed with
OA, comprising 11 patients (3 males and 8 females), also aged from
60 to 81 years old. Washing the cartilage samples 3 times with
phosphate buffer saline (PBS) containing penicillin and
streptomycin and then cutting them into small fragments (1-3
mm3). The minced samples were placed in 1 mg/ml
collagenase II (MilliporeSigma) for digestion at 37°C for 16 h in a
shaker, digesting the samples multiple times until collagen was
completely dissolved. After digestion, the suspension was filtered
with a 100-mm nylon cell strainer to remove matrix debris and the
cells collected via centrifugation at 108 × g for 10 min at 24°C.
After centrifugation, cells were resuspended and subcultures with
chondrocyte growth medium (DMEM/F12; 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.) and 100 IU/ml penicillin and 100 μg/ml
streptomycin (Gibco; Thermo Fisher Scientific, Inc.). The cells
were cultured in an incubator with 5% CO2 atmosphere at
37°C. The medium was changed when the cell was attached and the
cells were passaged at 80% confluence. Only the passage 1(P1) and
passage 2 (P2) chondrocytes were used for the experiments.
Toluidine blue staining was used to assess chondrocyte morphology.
Briefly, the samples were stained with toluidine blue solution for
5 min at room temperature, followed by the addition of an equal
volume of distilled water and mixed thoroughly. After allowing the
mixture to stand for 10 min at room temperature, images were
captured using an Olympus microscope (Olympus Corporation).
Cell viability
Human primary chondrocytes cells were seeded in
96-well cell culture plates at a density of 1×104/well
and cultured overnight. The concentration range of BAY 11-7082 and
TAK-242 used in this assay was based previous studies (37,38). The concentration range of CIRP
used in this assay was from 0-8 μg/ml. After incubation with
CIRP for 48 h at 37°C, the cell viability was measured using CCK-8
following the standard protocol. Briefly, the culture medium and
CCK-8 solution were mixed at a ratio of 10:1 and 100 μl
mixture was added into each well. After 1 h incubation at 37°C in a
cell culture incubator, the absorbance at 450 nm was detected using
a microplate reader.
RNA isolation, reveres transcription and
reverse transcription-quantitative (RT-q) PCR
Total RNA was extracted from human chondrocytes
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. In
brief, the cells were first lysed in TRIzol® to release
the RNA, and then chloroform was added to facilitate phase
separation. After mixing and centrifugation at 12,000 × g for 15
min at 4°C, the upper aqueous phase containing the RNA was
transferred to a new tube. Isopropanol was subsequently added to
precipitate the RNA. The RNA pellet was washed with 75% ethanol,
dried and finally resuspended in RNase-free water. Then cDNA was
synthesized using PrimeScript RT Reagent Kit with gDNA Eraser
(Takara Biotechnology Co., Ltd.) according to the manufacturer's
instructions. RT-qPCR was conducted with SYBR Green Supermix
(Beyotime Institute of Biotechnology) using aViiA 7 Real-Time PCR
System (Applied Biosystems; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. The PCR cycling
conditions consisted of an initial denaturation at 95°C for 10 min,
followed by 35 cycles of denaturation at 95°C for 15 sec, annealing
at 60°C for 1 min, and extension at 72°C for 30 sec. Transcript
levels were normalized to GAPDH (for mRNA) or the small U6 RNA (for
miRNA). The specific primers used for these analyses are listed in
Table I. Gene expression was
calculated using the 2−ΔΔCq method (39) and each experiment was performed
in triplicate.
 | Table ISequences of primers for PCR. |
Table I
Sequences of primers for PCR.
| Gene | Forward primer
(5'-3') | Reverse primer
(5'-3') |
|---|
| iNOS |
CCTTACGAGGCGAAGAAGGACAG' C |
AGTTTGAGAGAGGAGGCTCCG |
| COX-2 |
GAGAGATGTATCCTCCCACAGTCA |
GACCAGGCACCAGACCAAAG |
| IL-6 |
ACTCACCTCTTCAGAACGAATTG CC |
ATCTTTGGAAGGTTCAGGTTG |
| TNF-a |
GTCAGATCATCTTCTCGAACC |
CAGATAGATGGGCTCATACC |
| IL-1b |
ATGATGGCTTATTACAGTGGCAA |
GTCGGAGATTCGTAGCTGGA |
| MMP1 C |
TGTTCAGGGACAGAATGTGC |
TTGGACTCACACCATGTGTT |
| MMP3 |
TGCGTGGCAGTTTGCTCAGCC |
GAATGTGAGTGGAGTCACCTC |
| MMP13 |
GGCTCCGAGAAATGCAGTCTTTCTT |
ATCAAATGGGTAGAAGTCGCCATGC |
| Adamts5 |
GAACATCGACCAACTCTACTCCG C |
AATGCCCACCGAACCATCT |
| Aggrecan |
GTGCCTATCAGGACAAGGTCT |
GATGCCTTTCACCACGACTTC |
| Collagen II |
CTGTCCTTCGGTGTCAGGG |
CGGCTTCCACACATCCTTAT |
| CIRP C |
AAGTACGGACAGATCTCTG |
CGGATCTGCCGTCCATCTA |
| miR-145-5p |
GTCCAGTTTTCCCAGGAATC |
AGAACAGTATTTCCAGGAAT |
| U6 |
CTCGCTTCGGCAGCACATATACT |
ACGCTTCACGAATTTGCGTGTC |
| GAPDH |
GGAGCGAGATCCCTCCAAAAT |
GGCTGTTGTCATACTTCTCATGG |
Western blotting
The whole cell proteins were extracted from
chondrocytes using lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM
NaCl and 0.1% sodium dodecyl sulfate) with protease inhibitors
supplement. Separate extraction of nuclear and cytoplasmic proteins
was conducted using a Nuclear and Cytoplasmic Protein Extraction
Kit (Beyotime Institute of Biotechnology). The determination of
protein concentration was conducted using the BCA protein
concentration detection kit (Thermo Fisher Scientific, Inc.).
Denatured protein solution (40 μg) was subjected to 12%
SDS-PAGE followed by immunoblot analysis. The sample was then
transferred to a PVDF membrane (Merck KGaA) that had been incubated
in methanol for 1 min at room temperature. After removing the
methanol, the membrane was equilibrated in transfer buffer until it
was ready for use. Following a transfer at 100 V for 1 h at 4°C,
the membrane was blocked with 5% skimmed milk for 1 h at room
temperature before being incubated with primary antibodies against
GADPH (1:50,000; cat. no. 60004-1-Ig; ProteinTech Group, Inc.),
MMP1 (1:1,000, 10371-2-AP, ProteinTech Group, Inc.), MMP3 (1:5,000;
cat. no. 66338-1-Ig; ProteinTech Group, Inc.), MMP13 (1:1,000; cat.
no. 18165-1-AP; ProteinTech Group, Inc.), a disintegrin and
metalloproteinase with thrombospondin motif-5 (Adamts5; 1:500; cat.
no. A2836; ABclonal Biotech Co., Ltd.), iNOS (1:500; cat. no.
18985-1-AP; ProteinTech Group, Inc.), cyclooxygenase-2 (COX-2;
1:500; cat. no. 12375-1-AP; ProteinTech Group, Inc.), lamin B1
(1:5,000; cat. no. 12987-1-AP; ProteinTech Group, Inc.), CD63
(1:500; cat. no. 32151-1-AP, ProteinTech Group, Inc.), CD9 (1:500;
cat. no. 84801-13-RR, ProteinTech Group, Inc.), GM130 (1:500; cat.
no. 11308-1-AP, ProteinTech Group, Inc.), Human Reactive
Inflammasome Antibody Sampler Kit II (1:1,000; cat. no. 25620T;
Cell Signaling Technology Inc.), CIRP (1:1,000; cat. no. 68522;
Cell Signaling Technology Inc.), p65 (1:1,000; cat. no. 8242; Cell
Signaling Technology Inc.), IκBα (1:1,000, cat. no. 4814; Cell
Signaling Technology Inc.), and collagen II (1:500; cat. no.
AF6528, Beyotime Institute of Biotechnology) overnight at 4°C.
Subsequently, the membranes were incubated with a HRP-conjugated
goat anti-rabbit (1:1,000; cat. no. A0208; Beyotime Institute of
Biotechnology Inc.) or goat anti-mouse IgG (1:1,000; cat. no.
A0216; Beyotime Institute of Biotechnology Inc.) secondary antibody
for 2 h at room temperature. ECL reagent (Thermo Fisher Scientific,
Inc.) was used for development according to the manufacturer's
protocol. The semi-quantitatively analysis of immunoreactive bands
was performed using ImageJ software 1.53e (National Institutes of
Health).
Immunofluorescence
For immunofluorescence, cells were treated with CIRP
for 24 h. The cells were fixed with 4% paraformaldehyde for 15 min
at room temperature. Fixed samples were permeabilized with 0.3%
Triton X-100 for 10 min at room temperature and then blocked with
5% BSA for 30 min. Chondrocytes were incubated with p65 (1:50)
primary antibodies overnight at 4°C. Afterwards, they were washed
with PBS 3 times and incubated with Cy3-conjugated secondary
antibodies for 1 h at room temperature in the dark. Finally, the
cells were incubated with DAPI for nucleuses stain at room
temperature for 10 min. A fluorescence microscope was used to
acquire the images at a magnification of 400×. Collagen II (1:50)
and MMP13 (1:50) primary antibody were used to detect its
expression in human chondrocytes using the same immunofluorescence
protocol, and a fluorescence microscope was used to acquire the
images at a magnification of 100×. The cell density seeded onto the
slide was 1×105 cells per well.
Purification of exosomes
Exosomes were harvested through a sequential
centrifugation method. Cells were cultured in serum-free medium for
48 h. The medium was collected and centrifuged sequentially at 300
× g for 10 min, at 2,000 × g for 15 min and at 10,000 × g for 30
min at 4°C. The supernatant was filtered using a 0.22 mm filter
(Beyotime Institute of Biotechnology) to remove remaining cells and
debris and then centrifuged at 120,000 × g for 70 min at 4°C to
harvest exosomes. PBS was used to re-suspend the purified exosomes.
Exosomes were kept either at −80°C for long term preservation or at
−20°C for short term preservation.
Characterization of exosomes
Exosomes were entrusted to Wuhan Servicebio
Technology Co., Ltd. for transmission electron microscopy. In
brief, fix the exosomes with 1 ml of 2% paraformaldehyde for 5 min
at room temperature. Then, 10 μl of exosomes was applied to
carbon-coated copper grids for 1 min, then washed with
double-distilled water and dried using filter paper. Afterwards,
~15 μl of 2% uranyl acetate staining solution was added for
1 min at room temperature. The grids were examined using a Hitachi
HT7700 transmission electron microscope (Hitachi High-Technologies
Corporation). The particle size and particle concentration were
determined by a Nano sight Tracking Analysis (NTA) system NanoSight
NS300 (Malvern Panalytical Instruments Corp.). Briefly, exosomes
were re-suspended in PBS at a concentration of ~3 μg protein
content/ml. Then the solution was further diluted by 100 to 500
hundred folds for analysis. A 1 min video was recorded by the NTA
software (NTA 3.4-Sample Assistant Build 3.4.003-SA, Malvern
Panalytical Ltd.) for further analysis. Western blotting was used
to detect the exosome marker protein expression.
Construction of a Venn diagram of CIRP
targeting micro-RNAs
To predict the miRNAs that could potentially
regulate CIRP expression, the miRWalk (http://mirwalk.umm.uni-heidelberg.de/), TargetScan
(http://www.targetscan.org/), StarBase
(http://starbase.sysu.edu.cn/), miRDIP
(http://ophid.utoronto.ca/mirDIP/) and a
chondrogenesis-related microRNA databases (GSE135588) were used to
find CIRP targeting miRNAs. A Venn diagram (http://bioinformatics.psb.ugent.be/cgi-bin/liste/Venn/calculate_venn.htpl)
was created based on comparisons of these miRNAs.
Dual-luciferase reporter assay
To study the interaction between miR-145-5p and the
3' untranslated region (UTR) of the CIRP gene, plasmid vectors
containing the wild-type CIRP 3' UTR that included the predicted
binding sites for miR-145-5p were constructed. These plasmids were
transfected into 293 cells using GP-transfect-Mate (GenePharma,
Inc.). To evaluate transfection efficiency, a Renilla
luciferase vector was co-transfected in each experiment. After
transfecting the cells, incubate them in a 37°C incubator for 24 h,
the luciferase activity was measured in relative light units, with
the firefly luciferase activity normalized against the
Renilla luciferase activity using Dual-Luciferase Reporter
Assay System (Promega Corporation). In brief, 250 μl of 1X
cell lysis buffer was added to each well, and 50 μl of the
lysate transferred to a microplate, setting up three replicates.
Then, 10 μl of firefly luciferase reaction solution was
added, gently mixed, and the firefly luciferase activity measured
within 30 min. Finally, 30 μl of Renilla luciferase
reaction solution was added, mixed immediately and the
Renilla luciferase activity measured.
Loading exosomes with miRNA mimic
Electroporation method was used to load miR-145-5p
mimics into exosomes. Exosomes at a final concentration of 100
μg/μl protein (measured by BCA) were mixed with
electroporation buffer (in a 1:1 ratio) and 100 pmol of synthetic
miR-145-5p mimics (sense 5'-GUCCAGUUUUCCCAGGAAUCCCU-3', antisense
5'-GGAUUCCUGGGAAAACUGGACUU-3') or negative control (mimics NC,
sense 5'-UUCUCCGAACGUGUCACGUTT-3', antisense
5'-ACGUGACACGUUCGGAGAATT-3') and electroporated at 0.400 kV using
an electroporation instrument. After the loading of miR-145-5p
mimics, the exosome samples were diluted with PBS and centrifuged
at 10,000 × g for 30 min and 100,000 × g for 70 min at 4°C to
remove unloaded miR-145-5p mimics.
Establishment of a mice model of OA
All procedures were approved by the Animal Research
Committee of Southwest Medical University (approval no.
20220307-004). A total of 20 six-week-old male C57BL/6J mice were
randomized into four groups: normal group, OA group, miR-NC-Exos OA
group and miR-145-Exos OA group, with each group comprising five
mice. The OA model was built on the instability of the medial
meniscus (DMM) caused by surgical therapy (40). The normal and OA group were given
articular cavity injections of PBS. MiR-NC-Exos OA group were given
articular cavity injections of 100 μg exosomes from cells
overexpressed miR-NC mimics and miR-145-Exos OA group were given
articular cavity injections of 100 μg exosomes from cells
overexpressed miR-145 mimics. The injections were carried out twice
a week for four consecutive weeks. All animals were sacrificed via
cervical dislocation while under anesthesia induced by an
intraperitoneal injection of pentobarbital sodium (50 mg/kg).
Mortality was confirmed through the observation of respiratory
arrest. a total of 10 cartilage samples were collected for
safranin-O staining, hematoxylin and eosin staining and
immunohistochemical (IHC) staining.
Histopathological analysis
The calcification of the knee joint was removed with
a 10% EDTA solution after it had been fixed with 4%
paraformaldehyde (24 h; 4°C). The tissue was then dehydrated by
immersing it in 70% ethanol for 30 min, followed by 80% ethanol for
30 min, 95% ethanol for 30 min, and finally 100% ethanol for 1 h to
ensure complete dehydration. After dehydration, the tissue was
cleared and placed in melted paraffin wax at approximately 60°C for
3 h, which was then transferred into a mold filled with fresh
paraffin wax to cool and solidify. The paraffin blocks were sliced
into frontal serial slices with a thickness of 5 mm. Hematoxylin
and eosin (H&E) staining was performed at room temperature,
with hematoxylin applied for 10 min, and eosin applied for 3 min.
Safranin O-fast green (S-O) staining was also performed at room
temperature, with safranin O staining lasting 5 min, and fast green
staining for 15 sec. And a light microscope was used to identify
the degree of cartilage degeneration.
Immunohistochemical assay
MMP3 and collagen II expression levels were detected
in the cartilage by immunohistochemistry. Briefly, the slides were
dewaxed and rehydrated. The slide was sealed with 5% BSA for 30 min
after a 20 min incubation with 0.4% pepsin (Sangon Biotech Co.,
Ltd.) in 5 mM HCl to repair the antigen. Then, with primary
antibodies specific to MMP3 (1:500) and collagen II (1:50)
overnight (at 4°C), incubation of the slides was performed. After
50 min (room temperature) of incubation, the slides were subjected
to incubation with HRP-conjugated goat anti-rabbit (1:50; cat. no.
A0208; Beyotime Institute of Biotechnology, Inc.) and images were
captured with an Olympus BX51 microscope (Olympus Corporation).
RNA sequencing (RNA-seq)
RNA-seq was conducted and analyzed by Genedenov Co.,
Ltd. Briefly, total RNA extraction from the cells was performed
using TRIzol® Reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and genomic DNA removal was performed with DNase
I (Takara Biotechnology Co., Ltd.). After preparation of the
RNA-seq transcriptome library, the Illumina HiSeq xten/NovaSeq 6000
sequencer (Illumina, Inc.) was used to perform RNA-seq. Gene
Ontology (GO) functional enrichment and Kyoto Encyclopedia of Genes
and Genomes (KEGG) pathway analysis were conducted using omicshare
(https://www.omicshare.com). The RNA-seq
dataset has been deposited in the BioProject database under the
accession number PRJNA1302182 and the URL is https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1302182.
Statistical analysis
The data were expressed as the mean ± SD and all
experiments were repeated three times with independent cultures.
Prior to statistical analysis, the normality of the data
distribution was verified using the Shapiro-Wilk test. The
statistical significance of differences for the mean values between
two groups was determined with an unpaired two-tailed Student's
t-test and multiple groups compared using one-way analysis of
variance (ANOVA) with Bonferroni post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Overexpression and secretion of CIRP in
OA human chondrocytes
To assess the effect of CIRP on OA progression,
chondrocytes were first collected from patients with fractures or
OA undergoing joint replacement surgery to evaluate its expression.
As shown in Fig. 1A, the
extracted human chondrocytes were stained with toluidine blue,
confirming that the isolated cells were indeed human chondrocytes
and suitable for further study. In Fig. 1B, qPCR analysis revealed that the
expression of CIRP was markedly increased, while collagen Ⅱ levels
decreased in OA chondrocytes compared with normal chondrocytes.
Additionally, the expression of MMP1, MMP3, MMP13 and ADAMTS5 were
also overexpressed in OA chondrocytes. The present study then
analyzed the protein levels of these genes and the data presented
in Fig. 1C and D supported the
mRNA findings. These results indicated that CIRP is overexpression
in OA chondrocytes and may contribute to the progression of OA.
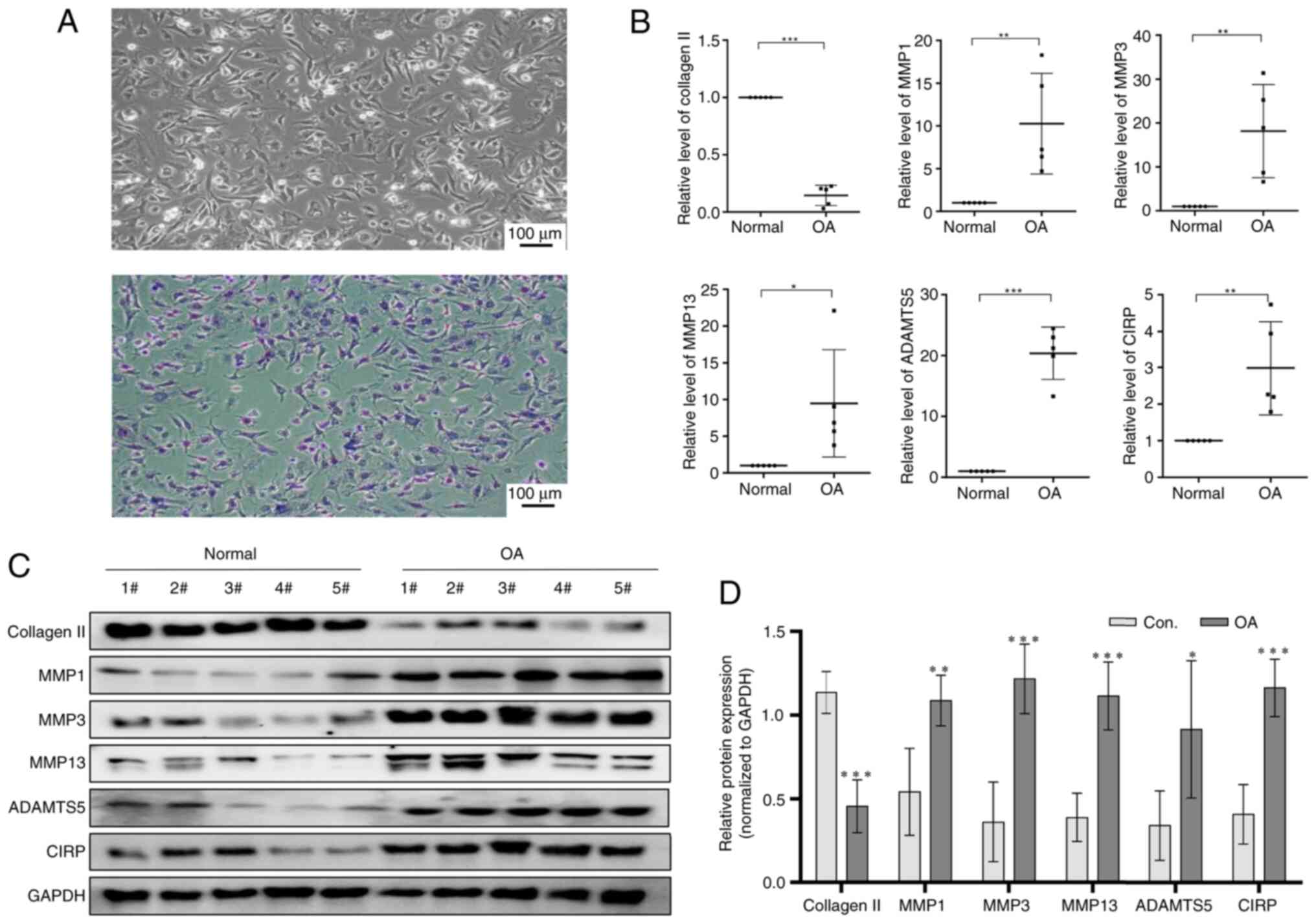 | Figure 1CIRP expression in chondrocytes from
healthy individuals and OA patients. (A) Trypan blue staining of
chondrocytes. (B) Representative quantitative PCR showing the
expression of CIRP, collagen Ⅱ, MMP1, MMP3, MMP13 and ADAMTS5 in
chondrocytes from healthy individuals and OA patients. (C and D)
Western blotting showing the expression of CIRP, collagen Ⅱ, MMP1,
MMP3, MMP13 and ADAMTS5 in chondrocytes from healthy individuals
and OA patients. (n=5), *P<0.05,
**P<0.01, ***P<0.001. CIRP,
cold-inducible RNA-binding protein; OA, osteoarthritis; MMPs,
matrix metalloproteinases; ADAMTS5, ADAM metallopeptidase with
thrombospondin type 1 motif 5. |
Secretion of CIRP in OA human
chondrocytes via exosomes
Recent studies revealed that CIRP can be released
into extracellular space by various cell types, including
cardiomyocytes (41-43). The exosomes released by OA
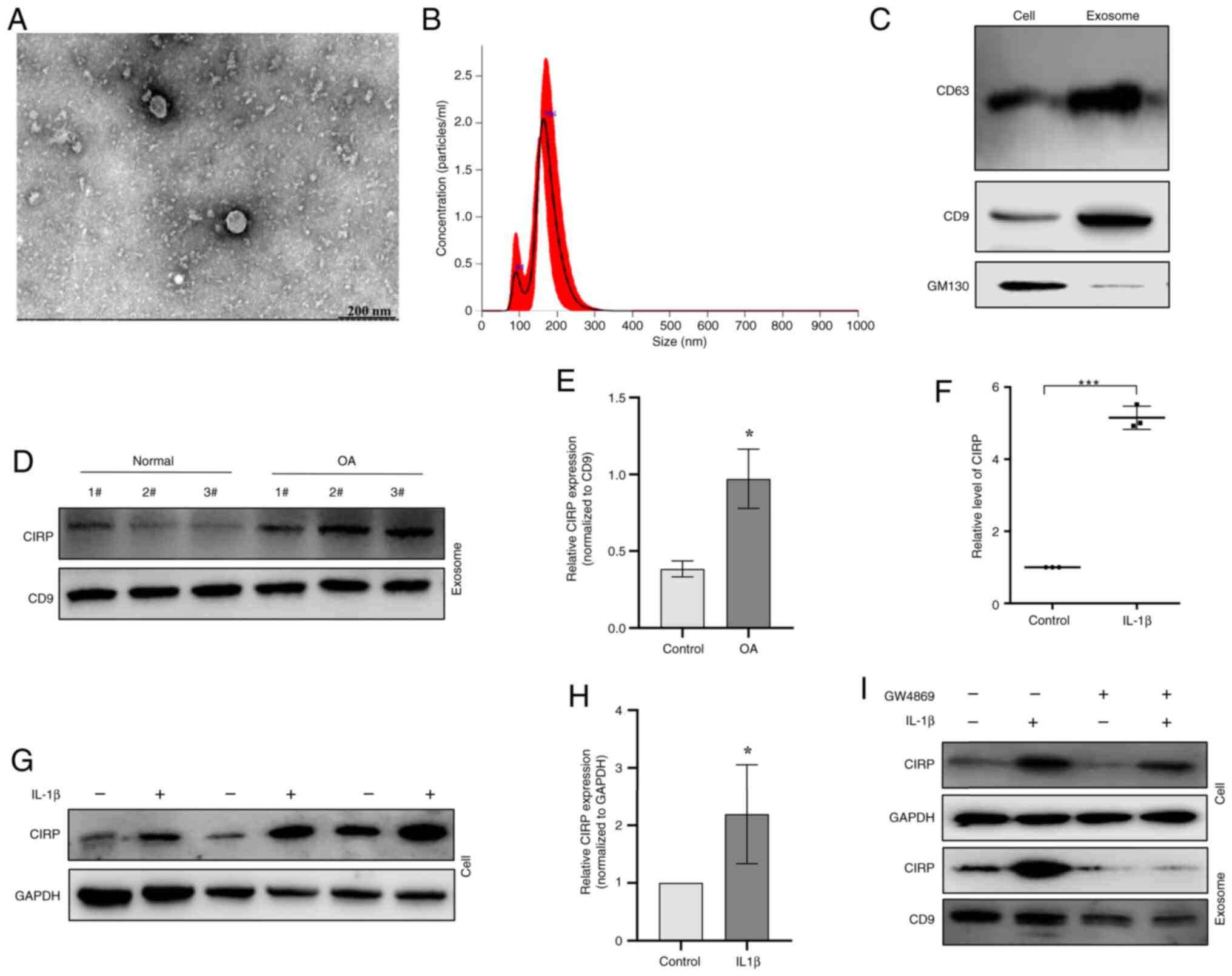
chondrocytes were next collected and analyzed using TEM (Fig. 2A). The observations indicated
that the exosomes derived from chondrocytes were round or
oval-shaped with uneven sizes and ranging in diameter from 150-200
nm (Fig. 2B). Western blotting
revealed positive expression of CD63 and CD9, along with negative
expression of GM130 in the exosomes (Fig. 2C). It was then examined whether
CIRP was present in chondrocyte-derived exosomes. The western
blotting results showed that chondrocyte exosomes indeed contained
CIRP and its expression was markedly increased in OA chondrocytes
compared with normal chondrocytes (Fig. 2D and E). Moreover, treatment of
chondrocytes with 10 ng/ml IL-1β also resulted in an increase in
CIRP expression (Fig. 2F-H).
Furthermore, the exosome release inhibitor GW4869 suppressed the
secretion of CIRP in human chondrocytes (Fig. 2I). These results indicate that
CIRP is overexpressed and released in an exosomal manner in OA
chondrocytes, which may contribute to the progression of OA.
CIRP enhances inflammatory cytokine
expression and ECM degradation in chondrocytes
From the aforementioned results, it was observed
that the expression of CIRP was upregulated in human OA
chondrocytes. This upregulation was associated with increased
expression of inflammatory cytokines and MMPs, suggesting that CIRP
may play a role in progression of OA. OA progression is strongly
associated with inflammatory responses and ECM degradation in
chondrocytes. To study the role of CIRP in OA, chondrocytes were
treated with CIRP concentrations ranging from 0-8 μg/ml. It
was found that 4 μg/ml CIRP did not affect cell viability,
while 8 μg/ml CIRP did (data not shown). Therefore, the
present study chose to use 4 μg/ml CIRP to treat
chondrocytes and analyze its effect on inflammatory cytokines
expression. The results demonstrated that CIRP treatment increased
the expression of IL-6, IL-1β and TNFα (Fig. 3A). Additionally, the expression
of iNOS and COX-2, two crucial inflammatory response factors in OA,
was also markedly upregulated following CIRP treatment (Fig. 3A and B). The present study then
examined the effect of CIRP on the expression of extracellular
matrix proteins. As shown in Fig.
3C, treatment with 4 μg/ml CIRP markedly decreased
collagen Ⅱ expression and increased the expression of MMP1, MMP3,
MMP13 and ADAMTS5 at mRNA level. The western blotting results in
Fig. 3D also showed that CIRP
induced the upregulation of MMP1, MMP3, MMP13 and ADAMTS5, along
with the downregulation of collagen II, with IL-1β treatment
serving as a positive control. Furthermore, Immunofluorescence
results indicated that collagen Ⅱ expression was markedly reduced,
while MMP13 expression was induced by CIRP treatment (Fig. 3E).
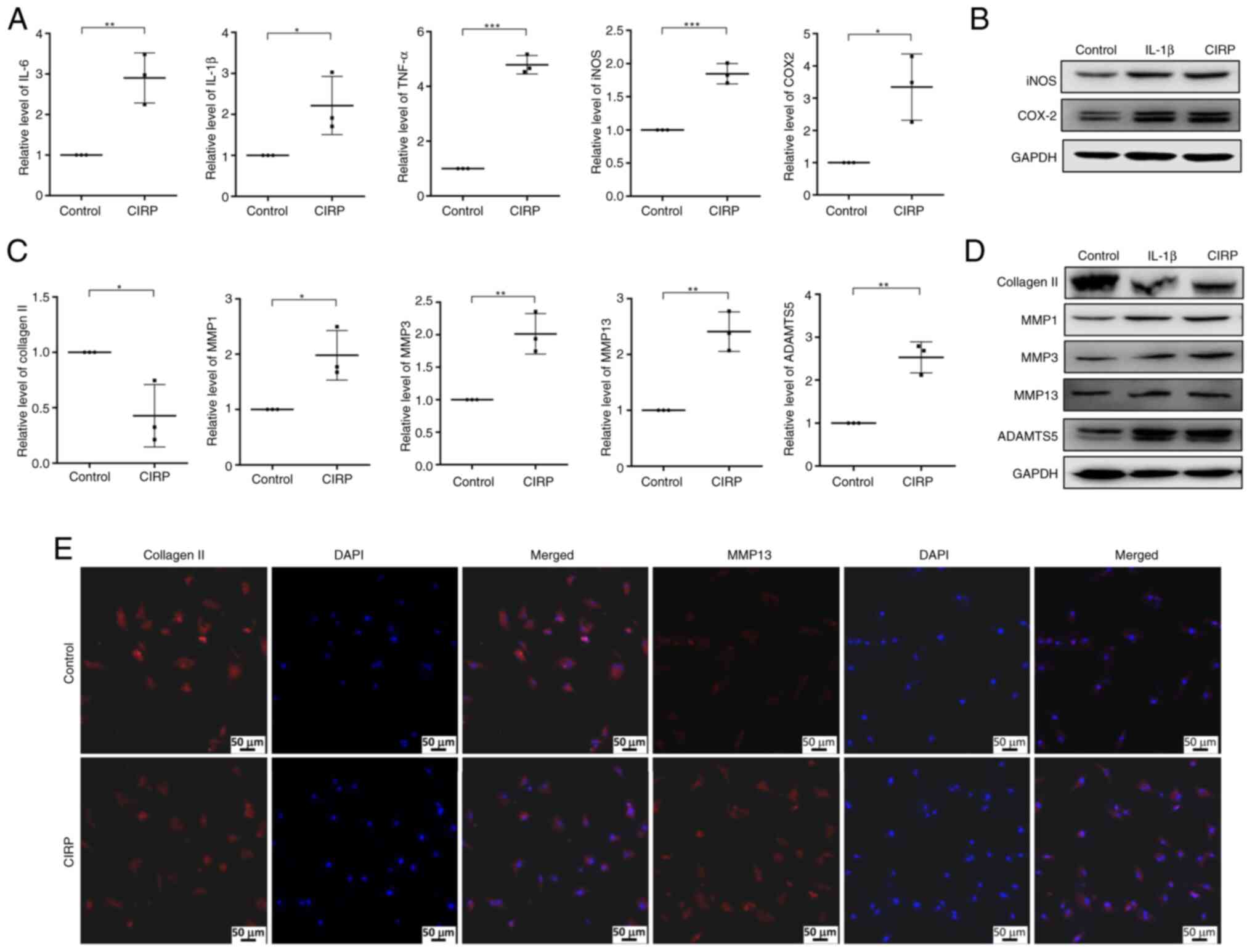 | Figure 3CIRP promotes inflammatory response
and ECM degradation in human chondrocytes. (A) Quantitative PCR
analysis showing the effect of CIRP on the expression of
inflammatory cytokines, including IL-6, IL-1β, TNF-α, iNOS and
COX-2. (B) Western blotting showing the protein expression of iNOS
and COX-2. (C and D) Quantitative PCR and western blotting showing
the effect of CIRP on the expression of MMP1, MMP3, MMP13, ADAMTS5
and collagen Ⅱ. (E) Immunofluorescence analysis of collagen Ⅱ and
MMP13 expression following CIRP treatment, with DAPI staining for
nuclei (scale bar, 50 μm). n=3, *P<0.05,
**P<0.01, ***P<0.001. CIRP,
cold-inducible RNA-binding protein; ECM, extracellular matrix;
iNOS, nitric oxide synthase; COX-2, cyclooxygenase-2; MMPs, matrix
metalloproteinases; ADAMTS5, ADAM metallopeptidase with
thrombospondin type 1 motif 5. |
CIRP-induced inflammatory response and
ECM degradation dependent on TLR4/NF-κB/NLRP3 signaling
Next, chondrocytes were treated with 4 μg/ml
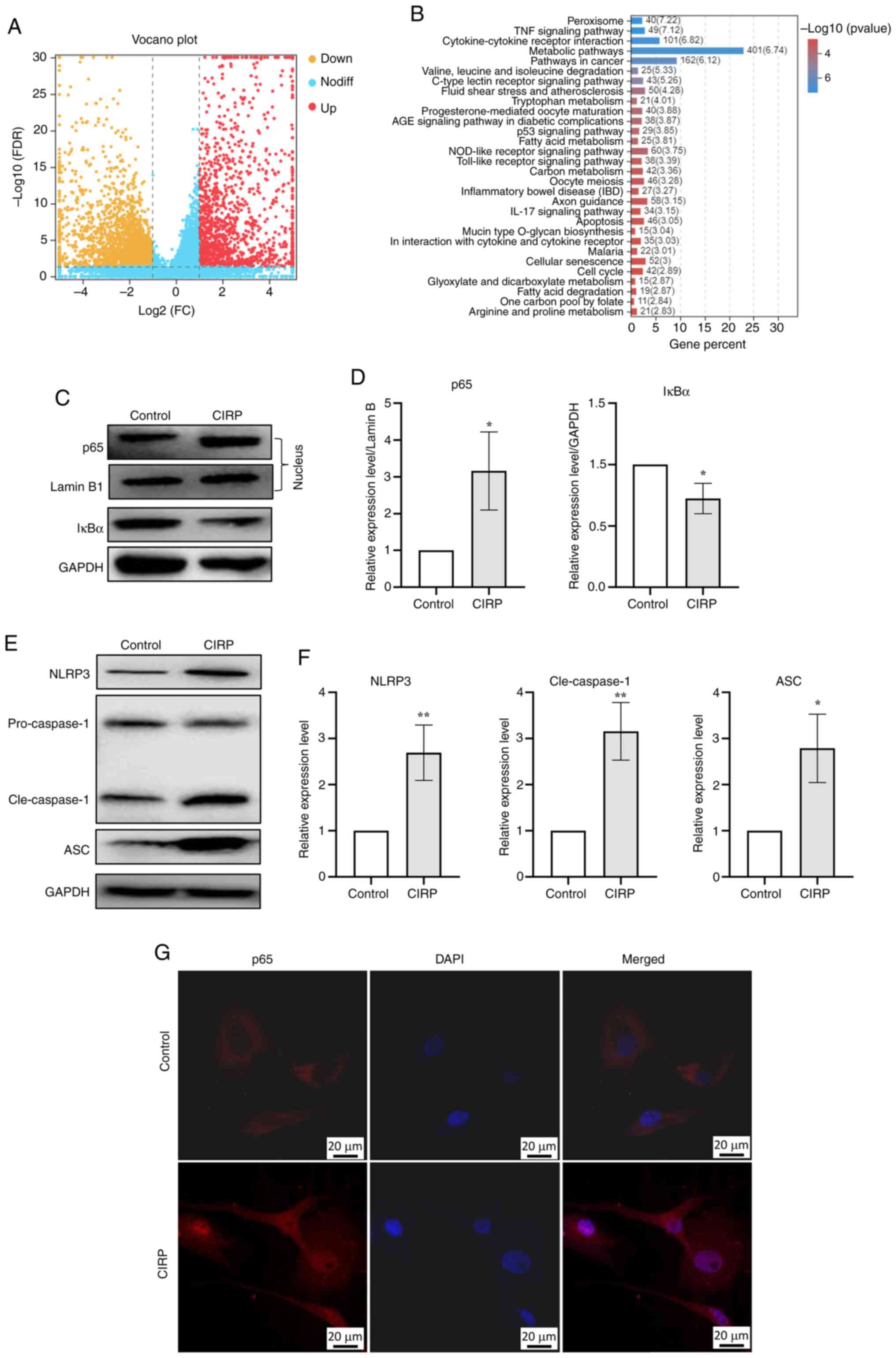
CIRP and cells were collected for RNA-seq. Fig. 4A shows the genes affected by CIRP
treatment. KEGG signaling pathway enrichment analysis indicated
that the differentially expressed genes were highly enriched in
functions related to inflammatory processes, including TNF-α
signaling, cytokine-cytokine receptor interaction, NOD-like
receptor signaling and Toll-like receptor signaling pathways
(Fig. 4B). The activation of
NF-κB signaling pathway plays crucial roles in the inflammatory
response and cartilage degradation associated with OA. Studies have
reported that CIRP can activate the NF-κB signaling pathway via
TLR4/MD2. The present study found that 4 μg/ml CIRP
increased the expression of p65 in the nucleus and promoted the
degradation of IκBα (Fig. 4C and
D). It was observed that CIRP markedly induced the nuclear
translocation of p65 in chondrocytes (Fig. 4G). These results demonstrate that
CIRP induces NF-κB activation in human chondrocytes. The present
study also analyzed the activation of NF-κB downstream NLRP3
inflammasome signaling pathway and the results showed that CIRP
induced the upregulation of NLRP3, ASC and cleaved-caspase1
(Fig. 4E and F).
It has been reported that extracellular CIRP
stimulates inflammatory responses by activating its receptors,
including TLR4, IL-6R and TREM-1 (41,44,45). Based on the RNA-seq analysis, the
present study sought to determine whether inhibition of TLR4/NF-κB
signaling pathway would eliminate CIRP's function. The inhibitors
BAY 11-7082 and TAK-242 were used to block the TLR4/NF-κB signaling
pathway. Fig. 5A shows their
toxicity on human chondrocytes. Next, chondrocytes were pretreated
with 5 μM BAY 11-7082 or TAK-242 and then treated with 4
μg/ml CIRP to analyze the effects of NF-κB signaling pathway
inhibition on CIRP. The results in Fig. 5B and C indicate that treatment
with 5 μM BAY 11-7082 and TAK-242 inhibited the activation
of the NF-κB signaling pathway. Furthermore, these two inhibitors
suppressed CIRP-induced activation of the NLRP3 signaling pathway
(Fig. 5D and E). Additionally,
these two inhibitors suppressed CIRP-induced downregulation of
collagen Ⅱ and upregulation of MMP1, MMP3, MMP13 and ADAMTS5
(Fig. 5F and G). These results
indicate that CIRP-induced inflammatory responses and ECM
degradation in human chondrocytes are dependent on the
TLR4/NF-κB/NLRP3 signaling pathway.
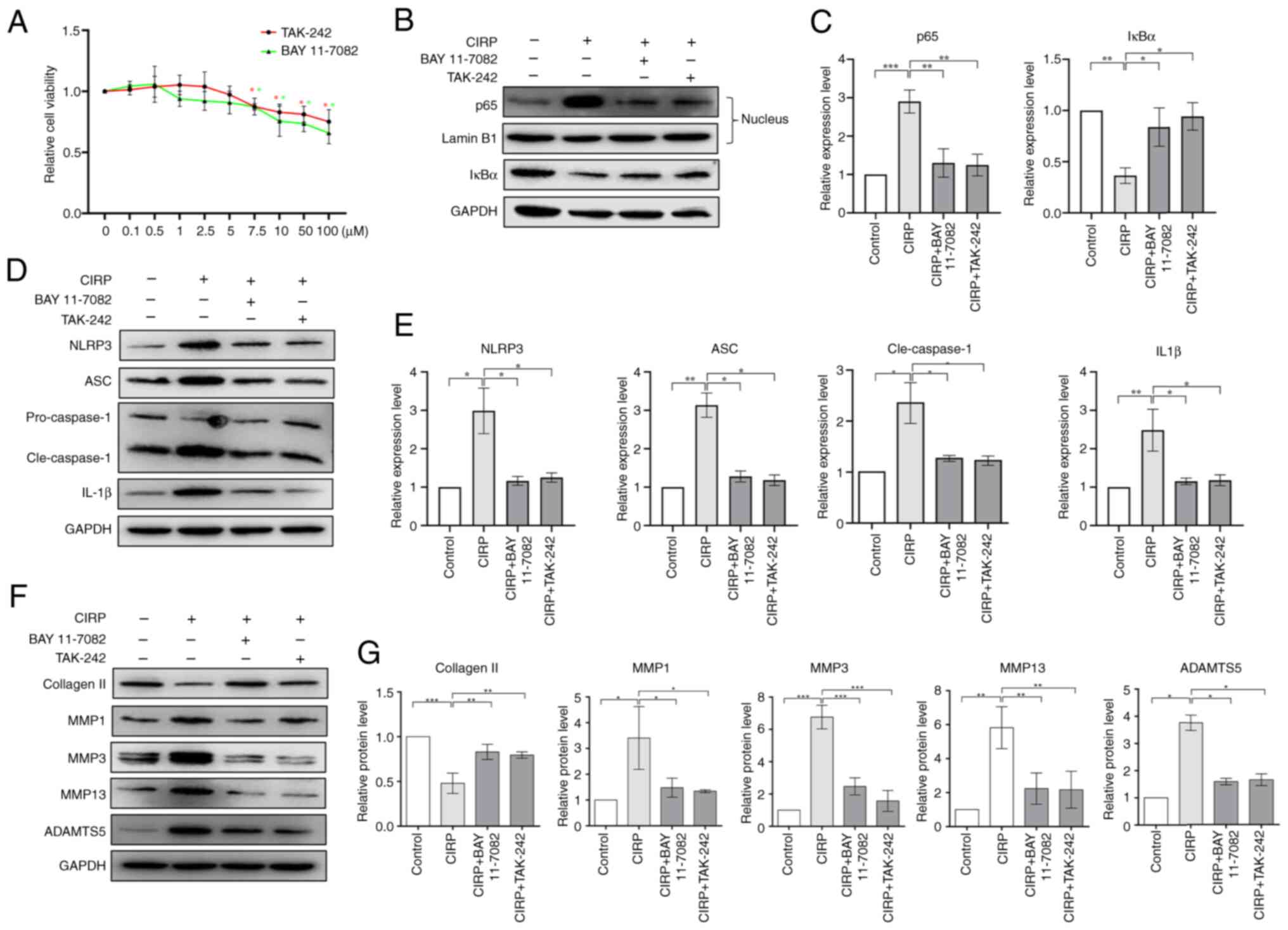 | Figure 5Dependence of CIRP-induced
chondrocyte damage on TLR4/NF-κB signaling. (A) Cytotoxicity of BAY
11-7082 and TAK-242 on chondrocytes at various concentration for 48
h, assessed using a CCK-8 assay. (B and C) Western blotting showing
the protein expression of IκBα in chondrocytes and p65 in the
nucleus of chondrocytes following treatment with the two
inhibitors. (D and E) Western blotting showing the protein
expression NLRP3, cleaved-caspase-1, ASC and IL-1β following
treatment with the two inhibitors. (F and G) Western blotting
showing the expression of extracellular matrix proteins in
chondrocytes following treatment with the two inhibitors. n=3,
*P<0.05, **P<0.01,
***P<0.001. CIRP, cold-inducible RNA-binding protein;
TLR4, Toll-like receptor 4; NLRP3, NLR family pyrin domain
containing 3; Cle, cleaved. |
CIRP as a putative target of
miR-145-5p
Subsequently, miRWalk, TargetScan, StarBase, miRDIP
and a chondrogenesis-related microRNA databases were applied to
predict the miRNAs that could potentially regulate CIRP expression.
A Venn diagram was created based on comparisons of these miRNAs
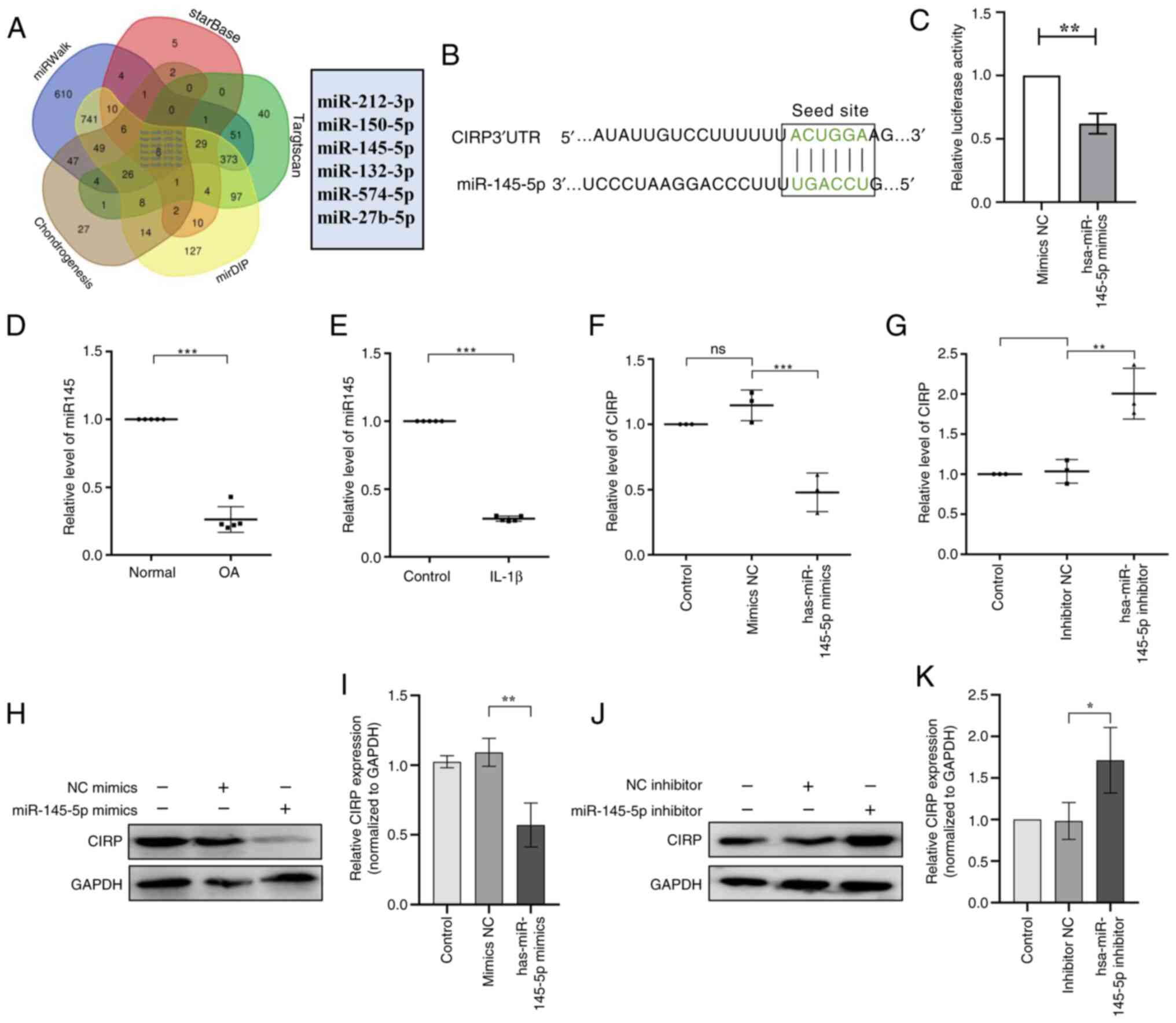
(Fig. 6A), which revealed six
candidate miRNAs of interest. Among them, has-miR-145-5p has been
reported to play an important role in chondrogenesis (34-36). Therefore, it was hypothesized
that miR-145-5p could influence OA progression by targeting CIRP.
To validate this hypothesis, online tools indicated the presence of
specific binding sites between CIRP mRNA and miR-145-5p (Fig. 6B), suggesting that CIRP might be
a downstream target gene of miR-145-5p. Moreover, a dual luciferase
reporter gene assay showed that, compared with the NC-mimic
control, luciferase activity decreased after the introduction of
miR-145-5p mimic and the CIRP-WT reporter plasmid (Fig. 6C). Next, the expression of
miR-145-5p in OA human chondrocytes and IL-1β treated human
chondrocytes was analyzed by qPCR. The results revealed that the
miR-145-5p expression was reduced in OA chondrocytes compared with
normal chondrocytes. Moreover, chondrocytes treated with IL-1β
exhibited lower miR-145-5p expression compared with untreated
chondrocytes (Fig. 6D and E).
Subsequently, the expression of CIRP in human chondrocytes treated
with miR-145-5p mimic or miR-145-5p inhibitor was assessed. The
results revealed that miR-145-5p mimic led to increased miR-145-5p
expression (Fig. S1A) and decreased CIRP expression (Fig. 6F, H and I), whereas the
miR-145-5p inhibitor decreased miR-145-5p (Fig. S1B) induced the opposite effects
(Fig. 6G, J and K). Taken
together, these results indicated that miR-145-5p is downregulated
in OA and specifically binds to and suppresses CIRP expression.
miR-145-5p maintains chondrocyte function
in vitro
Subsequently, to investigate the effect of
miR-145-5p on human chondrocytes, the cells were transfected with
miR-145-5p mimic, followed by overexpression of CIRP to induce the
inflammation and ECM degradation in chondrocytes. qPCR and western
blotting analysis were conducted to evaluate the mRNA and protein
expression of ECM-related proteins (collagen Ⅱ, ADAMTS5 and MMPs)
as well as pro-inflammatory factors (iNOS, COX-2, IL-6, TNF-α and
IL-1β). As shown in Fig. 7A, B and
E, the expression of pro-inflammatory factors was markedly
increased after CIRP overexpression but was reduced by miR-145-5p
mimic treatment. In Fig. 7C-E,
the expression of collagen Ⅱ was markedly decreased following CIRP
overexpression but was increased by miR-145-5p mimic treatment,
with the expression of other ECM-related proteins showing inverse
results. These findings indicated that miR-145-5p overexpression
markedly increased collagen Ⅱ expression, decreased the expression
of MMP1, MMP3, MMP13 and ADAMTS5 and reduced inflammatory response
in human chondrocytes. Moreover, the damage induced by CIRP
overexpression in human chondrocytes was alleviated by
overexpressed miR-145-5p overexpression. These results indicate
that miR-145-5p plays a key role in maintaining the function of
chondrocytes in vitro.
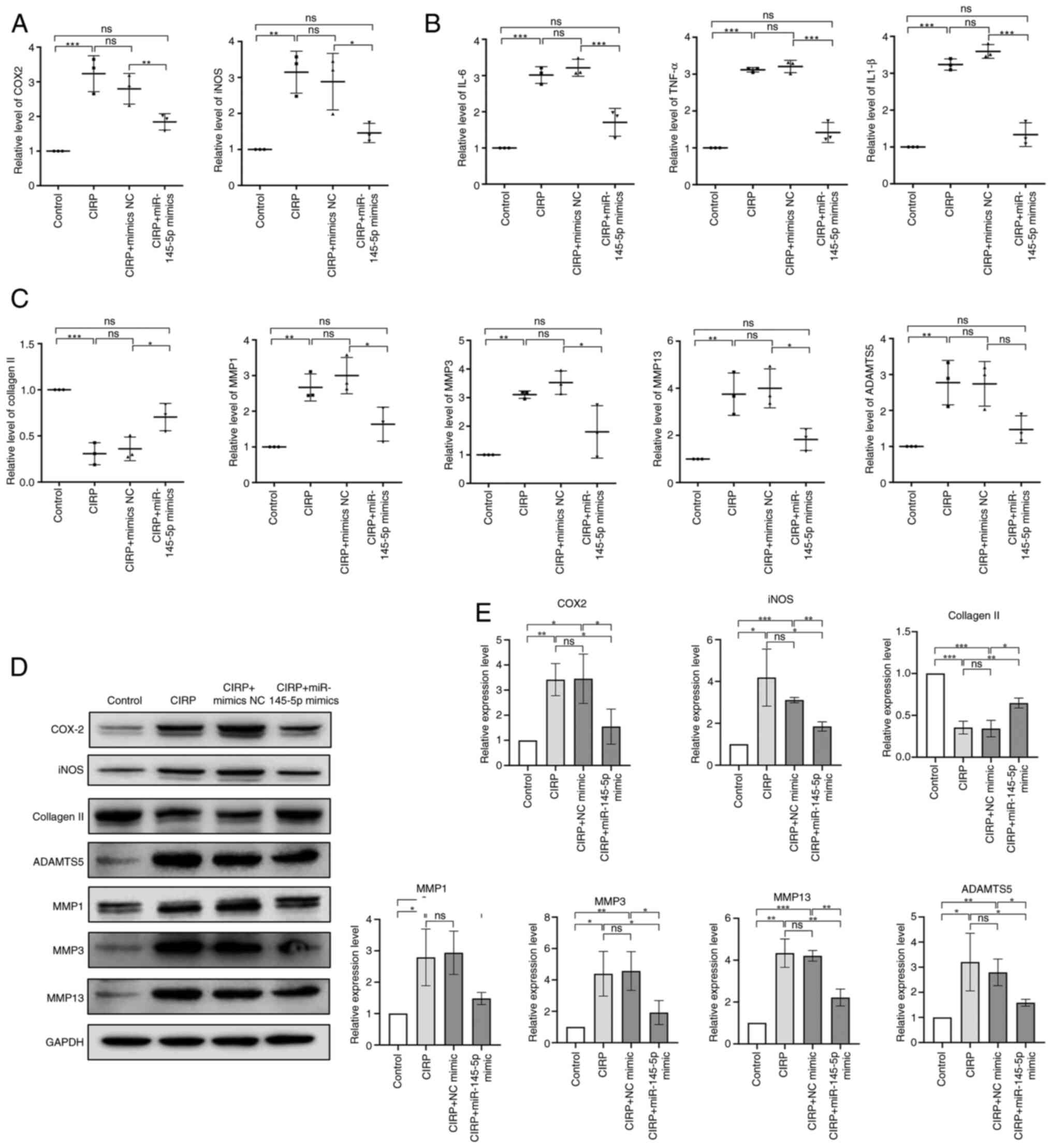 | Figure 7miR-145-5p elevation suppresses CIRP
induced damage of chondrocytes. (A and B) Chondrocytes were
transfected with miR-145-5p mimics or CIRP, IL-6, IL-1β, TNF-α,
iNOS and COX-2 expression were analyzed by qPCR. (C) Chondrocytes
were transfected with miR-145-5p mimics or CIRP, the mRNA
expression of extracellular matrix proteins of chondrocytes were
analyzed by qPCR. (D and E) Chondrocytes were transfected with
miR-145-5p mimics or CIRP, the protein expression of iNOS, COX-2
and extracellular matrix proteins of chondrocytes were analyzed by
western blotting. n=3, *P<0.05,
**P<0.01, ***P<0.001. miR, microRNA;
CIRP, cold-inducible RNA-binding protein; iNOS, inducible nitric
oxide synthase; COX-2, cyclooxygenase-2; qPCR, quantitative
PCR. |
miR-145-5p suppresses IL-1β-induced
CIRP/NF-κB/NLRP3 signaling pathway activation
The present study investigated the role of
miR-145-5p in IL-1β-induced activation of the CIRP/NF-kB/NLRP3
signaling pathway. It was found that treatment with 10 ng/ml IL-1β
increased the expression of p65 in the nucleus and promoted the
degradation of IκBα in human chondrocytes (Fig. 8A and B). Additionally, IL-1β
induced the nuclear translocation of p65 (Fig. 8C). However, the activation of the
NF-κB signaling pathway was suppressed by treatment with miR-145-5p
mimics (Fig. 8A-C). Moreover,
IL-1β-induced expression of CIRP and activated the downstream NLRP3
inflammasome signaling pathway, both of which were suppressed by
treatment with miR-145-5p mimics (Fig. 8D and E). These results indicated
that miR-145-5p exerts a protective function in chondrocytes by
inhibiting the CIRP/NF-κB/NLRP3 signaling pathway.
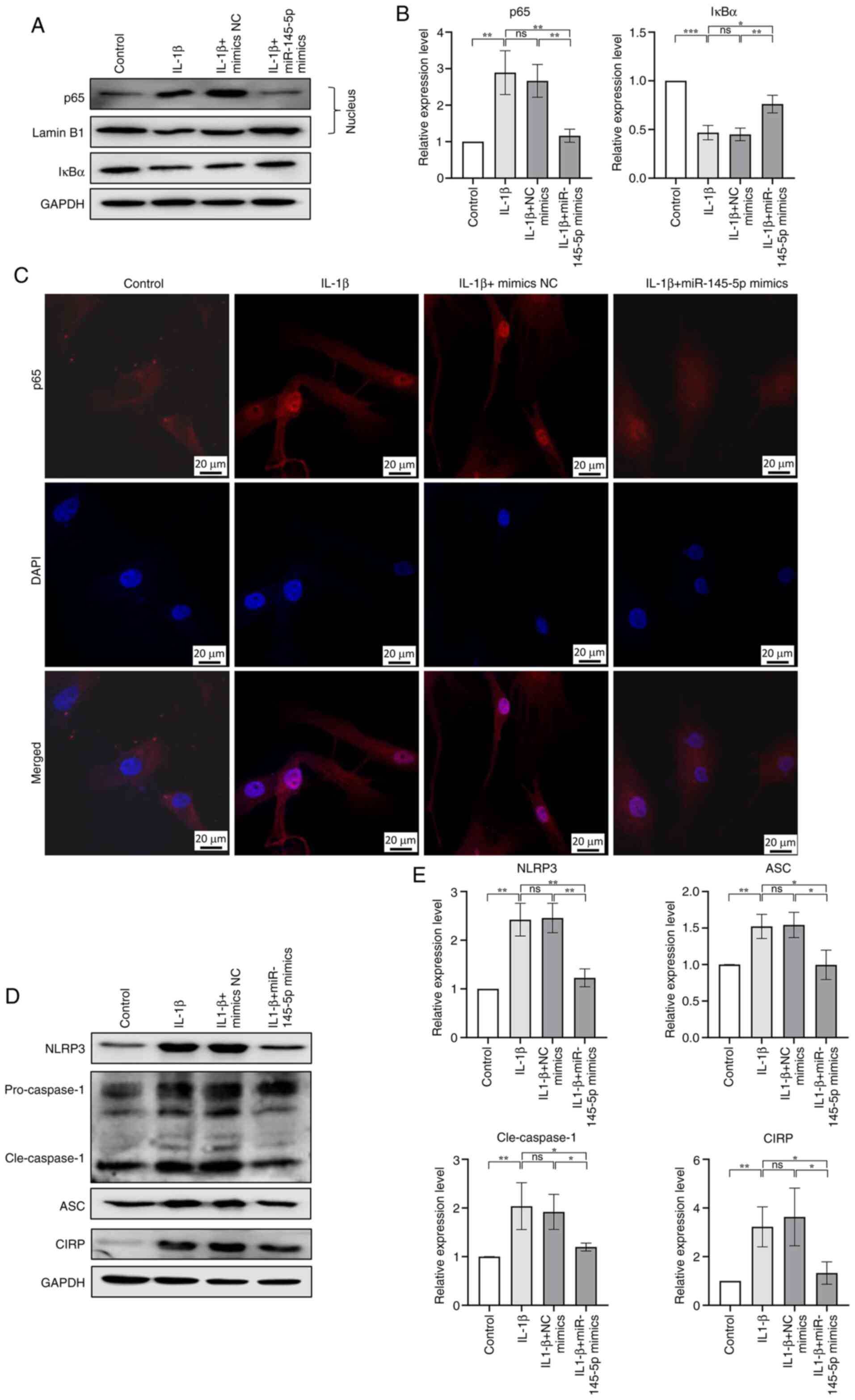 | Figure 8miR-145-5p elevation suppressed CIRP
induced NF-κB/NLRP3 activation. (A and B) Chondrocytes were
transfected with miR-145-5p mimics or IL-1β treatment, western
blotting results showed the protein expression of IκBα in the
cytoplasm and p65 in the nucleus of chondrocytes. (C) The nuclei
translocation of p65 was detected by the immunofluorescence
combined with DAPI staining for the nuclei. (D and E) western
blotting results showed the protein expression NLRP3,
cleaved-caspase-1, ASC and CIRP in chondrocytes transfected with
miR-145-5p mimics or IL-1β treatment. n=3, *P<0.05,
**P<0.01, ***P<0.001. miR, microRNA;
CIRP, cold-inducible RNA-binding protein; NLRP3, NLR family pyrin
domain containing 3; Cle, cleaved. |
miR-145-5p inhibits articular cartilage
degradation in vivo
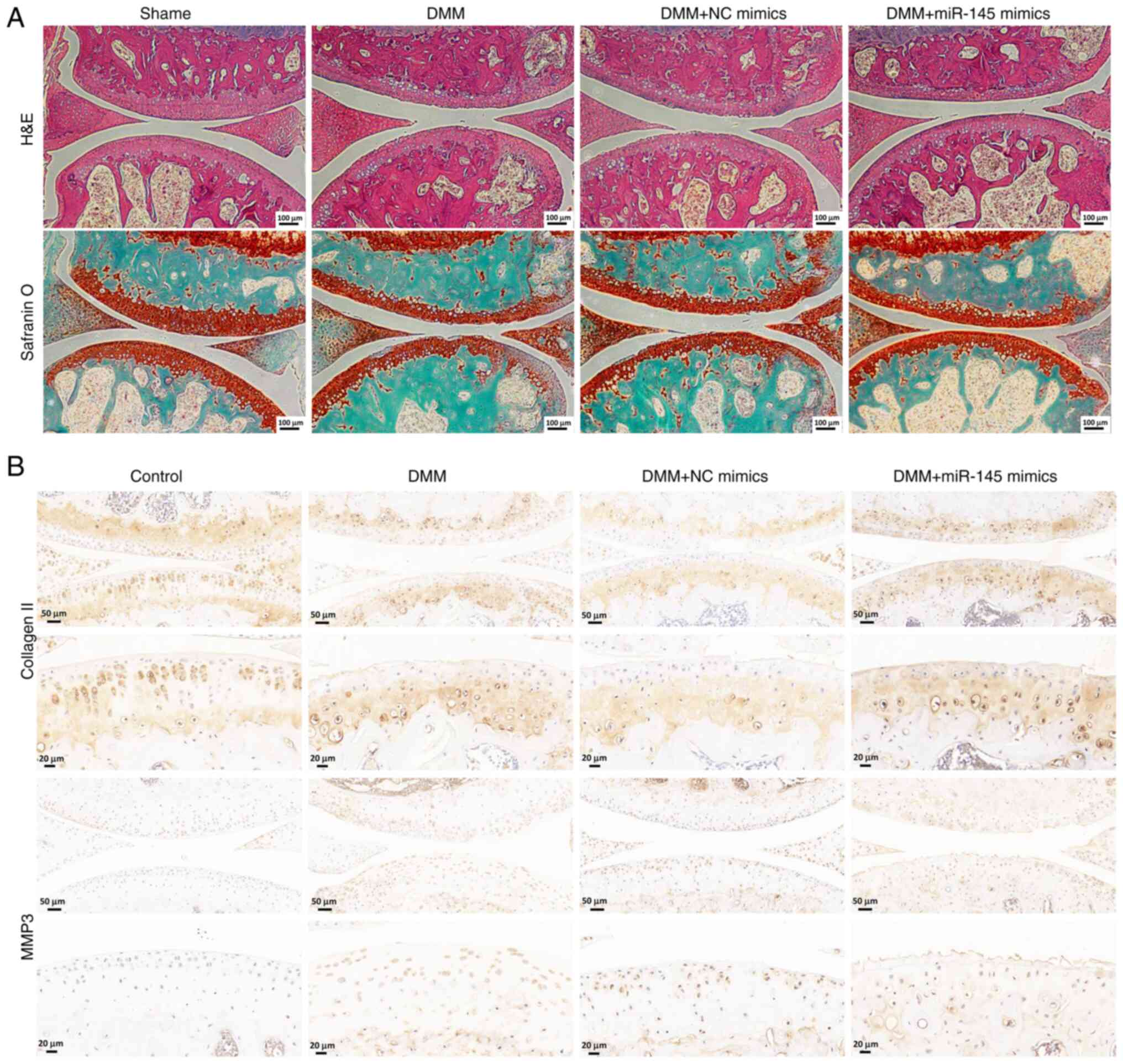
Finally, the effect of miR-145-5p on mice with OA
was evaluated in vivo. The mice were divided into four
groups: One sham group and three DMM groups (DMM group, DMM
injected with miR-NC exosomes group and DMM injected with
miR-145-5p mimic exosomes group). hematoxylin and eosin staining
was conducted to observe the pathological changes in the articular
cartilage of the knee joint and S/O staining was used to assess
articular cartilage degeneration. The S/O staining revealed that
the articular cartilage in the sham control group exhibited a
normal, red-dyed area and a smooth surface. By contrast, the
thickness of the articular cartilage was markedly reduced in both
the DMM group and the DMM injected with miR-NC exosomes group
compared with the sham control group. Notably, the DMM injected
with miR-145-5p mimic exosomes group showed a thicker red-dyed area
and a smoother surface of articular cartilage compared with the DMM
injected with miR-NC exosomes group (Fig. 9A), suggesting that miR-145-5p
plays a role in attenuating cartilage matrix degradation.
Additionally, immunohistochemical assays for MMP3 and collagen Ⅱ
were also performed in the OA model. In the DMM group and the DMM
injected with miR-NC exosomes group, the expression of MMP3 was
markedly increased compared with the sham group, while the DMM
injected with miR-145-5p mimic exosomes group exhibited decreased
expression of MMP3 (Fig. 9B).
Similarly, the expression of collagen Ⅱ was markedly reduced in the
DMM group and the DMM injected with miR-NC group compared with the
sham group, while the DMM injected with miR-145-5p mimic exosomes
group exhibited elevated expression levels (Fig. 9B). Therefore, these results
indicate that, as demonstrated by the in vivo experiments,
overexpressing of miR-145-5p can hinder OA-related damage.
Discussion
OA is a chronic, age-associated degenerative joint
disease featured by cartilage degeneration and inflammatory
response, which imposes a significant social and economic burden
(1,46). Currently, pain management and
end-stage joint replacement surgery are the most used strategies
for OA treatment. However, these approaches do not stop or reverse
cartilage degeneration and destruction (3,47). Therefore, research has shifted
its focus toward understanding the pathogenesis of OA and
identifying new strategies to prevent its early onset (4). An increasing number of studies have
highlighted that the diagnostic and therapeutic potential of miRNAs
in treatment of OA (28,48). Additionally, exosomes have
emerged as potential natural vehicles for miRNA delivery in disease
therapy. The current study aimed to investigate the effects of the
novel pro-inflammatory factor CIRP on OA progression through the
activation of the TLR4/NF-κB/NLRP3 inflammasome signaling pathway.
Furthermore, its key findings provided evidence that exosomes
overexpressing miR-145-5p can attenuate OA damage by repressing
CIRP.
CIRP is an RNA-binding protein known for its
tumor-promoting function and our research group has focused on it
for an extended period (15,49). CIRP has been identified as a
significant pro-inflammatory factor, playing important roles in
various inflammation diseases, including sepsis, neuroinflammation,
colitis-associated cancer and endothelial cell inflammation
(17,18,24,50). Two studies have been reported
that CIRP levels were evaluated in the synovial fluid of patients
with OA and rheumatoid arthritis, indicating a positive correlation
between CIRP and OA (26,27).
However, the specific roles of CIRP in OA progression and the
underlying mechanisms remain unclear. The present study aimed to
investigate the function of CIRP in OA progression. It first
assessed its expression and found that CIRP was markedly
upregulated in OA chondrocytes. Additionally, a previous study has
detected CIRP in the synovial fluid of OA patients and it has been
reported that CIRP can be released by macrophages during sepsis
(51). The present study then
examined the expression of CIRP in the exosomes derived from OA
chondrocytes and discovered that these chondrocytes released higher
levels of CIRP in an exosomal manner. Furthermore, treatment with
IL-1β increased the expression of CIRP in both whole chondrocyte
cells and their exosomes. These findings may help to explain the
elevated levels of CIRP observed in the synovial fluid of OA
patients.
Next, the present study investigated the influence
of CIRP on the expression of pro-inflammatory cytokines and MMP.
During OA progression, the balance of metabolic process is
disrupted. In response to inflammatory cytokines, such as iNOS,
chondrocytes secret many metalloproteinases that contribute to
extracellular matrix degradation (52,53). The results demonstrated that CIRP
markedly upregulated the expression of pro-inflammatory cytokines,
including iNOS and COX-2. Additionally, the expression of matrix
metalloproteinase, such as MMP1, MMP3, MMP13 and ADAMTS5, was
markedly elevated following CIRP treatment. Importantly, CIRP also
decreased the expression of collagen Ⅱ. These findings suggested
that CIRP is positively associated with inflammation and
extracellular matrix degradation in OA and the underlying molecular
mechanisms require further investigation.
Furthermore, the present study treated human
chondrocytes with CIRP and performed RNA-seq to explore the
downstream effects. The RNA-seq analysis revealed that CIRP
treatment induced differential gene expression enriched in pathways
related to TNF-α signaling, cytokine-cytokine receptor interaction,
IL-17 signaling, NOD-like receptor signaling and TLR signaling
pathway. These pathways are well-established contributors to OA
progression. For example, elevated circulating TNF-α levels are
associated with severe OA and cartilage loss and small molecules
that targeting TNF-α receptors have demonstrated protective effects
against OA (54,55). Previous studies have shown that
extracellular CIRP can stimulate inflammatory responses by
interacting with its receptors such as TLR4, IL-6R and triggering
receptor expressed on myeloid cells 1 (24,56,57). The RNA-seq data also suggested
that CIRP affects the TLR signaling pathway. Notably, TLR4 is
overexpressed in human OA chondrocytes (58,59) and is a well-studied upstream
activator of the NF-κB signaling pathway (60), implying that CIRP may activate
NF-κB through TLR4 signaling. To investigate this hypothesis, the
present study assessed the activation of NF-κB and found that CIRP
increased the nuclear translocation of p65, a hallmark of NF-κB
pathway activation. Additionally, CIRP upregulated the expression
of IL-1β, a key pro-inflammatory cytokine. The TLR4/NF-κB axis and
NLRP3/ASC/procaspase-1 inflammasome complex are critical for the
maturation and secretion of IL-1β (61,62). It was further confirmed that CIRP
activates the NLRP3 inflammasome. These findings suggested that
CIRP promotes inflammation and ECM degradation in OA by activating
NF-κB/NLRP3 inflammasome signaling pathway via TLR4. To validate
this mechanism, two inhibitors, BAY 11-7082 (an NF-κB inhibitor)
and TAK-242 (a TLR4 inhibitor), were used to inhibit the TLR4/NF-κB
signaling pathway. The two inhibitors effectively suppressed
CIRP-induced nuclear translocation of p65. Furthermore, they
inhibited the activation of downstream NLRP3 inflammasome and the
expression of IL-1β. Importantly, treatment with these inhibitors
also reversed CIRP-induced ECM degradation.
miRNAs, a type of conserved endogenous non-coding
RNAs, regulate target mRNA stability or translation by directly
binding to their targets. Growing evidence demonstrates that a
number of miRNAs exert protective functions in chondrocytes by
directly targeting multiple mRNAs involved in the development of
OA. miR-145 is a well-known non-coding RNA involved in the
pathogenesis of various inflammation-related diseases, such as
colitis and inflammation-associated colon cancer (63), rheumatoid arthritis (64) and myocardial ischemia/reperfusion
injury (65). The current study
found that the expression of miR-145 was decreased in the cartilage
of OA patients and in chondrocytes treated with IL-1β, highlighting
its possible role in the pathogenesis of OA. More importantly, CIRP
was identified as a target of miR-145-5p in chondrocytes. The
chondrocyte-protective function of miR-145-5p was assessed both
in vivo and in vitro. It was found that miR-145-5p
could suppress the inflammatory response, ECM degradation and
activation of NF-κB/NLRP3 signaling pathway induced by
overexpression of CIRP or IL-1β treatment in vitro. The
results were consistent with a previous study showing that miR-145
plays a protective role in chondrocytes during OA progression
(34). In vivo
experiments further supported the idea that exosomal delivery of
miR-145 could suppress OA progression. The present study found that
miR-145 suppresses the expression of CIRP in OA, suggesting that
miR-145 may play an important role in modulating the inflammatory
response and pathological progression of OA. While miR-145 is a
significant microRNA that suppresses CIRP expression, it is not the
only one identified in the literature. Previous study has reported
that miR-383-5p can target CIRP mRNA and inhibit its expression in
ovarian granulosa cells (66).
Additionally, Lin et al (67) recently demonstrated that CIRP is
a direct target of miR-377-3p in nasopharyngeal carcinoma. Aside
from these two miRNAs, no other microRNAs have been documented to
inhibit CIRP expression in the literature. Analysis using databases
such as miRWalk, TargetScan, StarBase, miRDIP and
chondrogenesis-related microRNA resources revealed six candidate
microRNAs that may also regulate CIRP expression. Further
investigation demonstrated specific binding sites between CIRP mRNA
and miR-145-5p, with decreased expression of miR-145-5p observed in
OA chondrocytes. Consequently, the present study focused on miR-145
for more detailed study. The dual luciferase reporter gene assay
confirmed that miR-145-5p suppresses CIRP expression in OA
chondrocytes. Further research is needed to explore the potential
existence of other microRNAs that may regulate CIRP expression in
the context of OA.
In conclusion, the present study confirmed that CIRP
can be secreted in the form of exosomes, functions as a
pro-inflammatory factor and activates the TLR4/NF-κB/NLRP3
signaling pathway, thereby promoting the inflammatory response, ECM
degradation and the progression of OA (Fig. 10). It also established that CIRP
is a target of miR-145-5p, which inhibits CIRP expression in OA.
In vitro, miR-145-5p was shown to suppress the inflammatory
response, ECM degradation and the activation of the NF-κB/NLRP3
signaling pathway induced by either CIRP overexpression or IL-1β
treatment. Furthermore, in vivo delivery of miR-145-5p via
exosomes was effective in inhibiting OA progression, highlighting
the potential of exosome-based miR-145-5p delivery as a therapeutic
strategy. Overall, the findings suggested a novel strategy for OA
treatment by targeting pro-inflammatory factors such as CIRP, with
miR-145-5p acting as a key regulator of CIRP expression. However,
the clinical relevance of CIRP in alleviating OA is still in its
early stages. While there is promising potential for targeting CIRP
in OA treatment, this area remains an emerging field of research.
The therapeutic implications of CIRP inhibition warrant further
exploration through clinical trials to determine whether CIRP
suppression can indeed provide significant relief or disease
modification in OA patients. It is important to note the
limitations of the present study. It would be an oversimplification
to assert that suppressing CIRP expression will directly lead to
the suppression of OA. Although CIRP has been implicated in the
inflammatory response and cartilage degradation associated with OA,
the relationship between CIRP and OA is probably complex and
multifactorial. Therefore, while targeting CIRP may present
therapeutic potential, it is unlikely to be a sufficient strategy
for fully mitigating OA. A comprehensive understanding of
additional factors and pathways is essential when considering CIRP
as a therapeutic target for OA. Future investigations should focus
on identifying the underlying mechanisms at play in this
relationship to improve the informing of therapeutic
strategies.
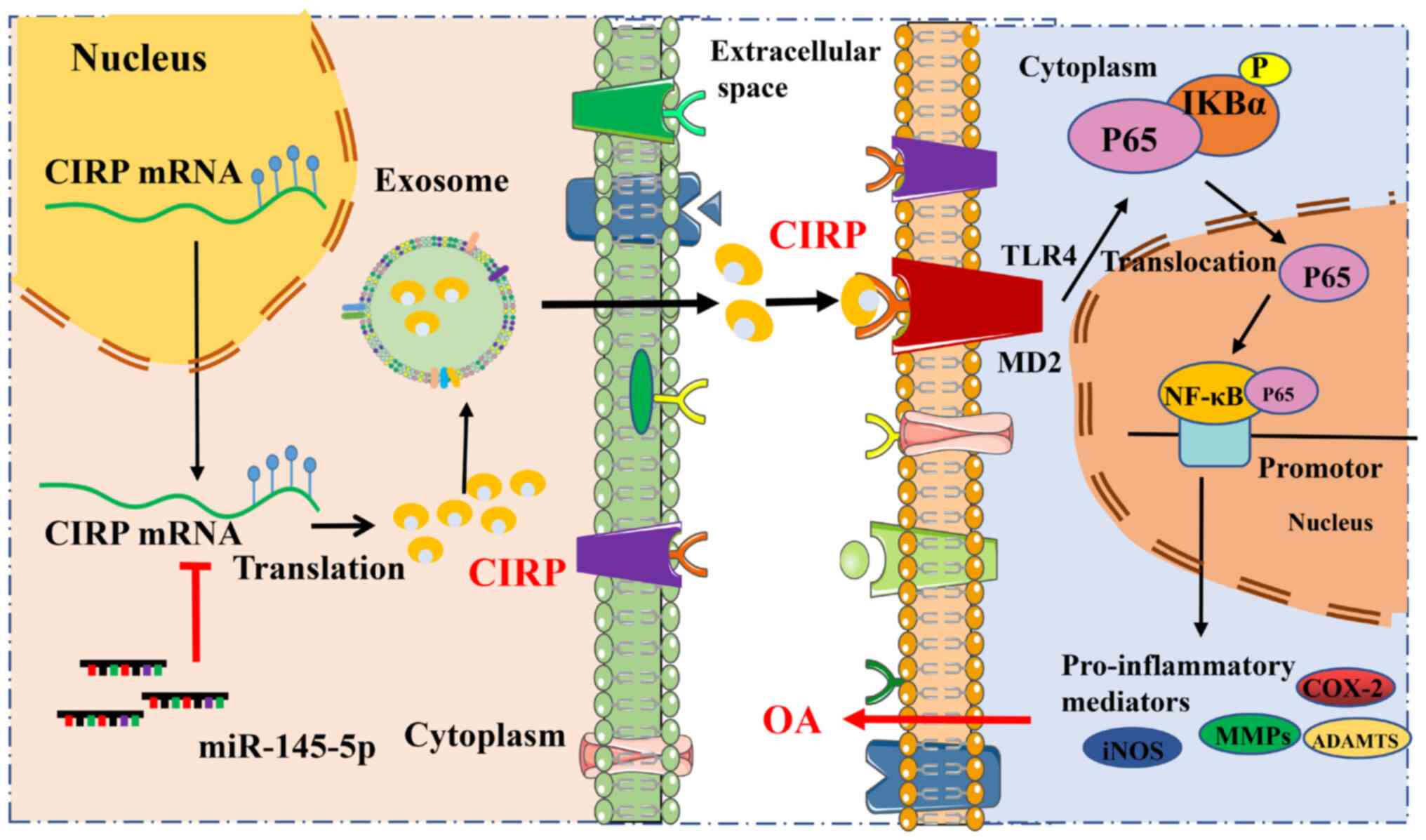 | Figure 10Schematic illustration of targeting
of CIRP attenuates osteoarthritis progression and the underlying
mechanism. CIRP can be secreted in the form of exosomes and acts as
a pro-inflammatory factor that activates the TLR4/NF-κB/NLRP3
signaling pathway, promoting the inflammatory response, ECM
degradation and the progression of OA. Additionally, CIRP is
identified as a target of miR-145, which inhibits its expression in
OA. CIRP, cold-inducible RNA-binding protein; TLR4, Toll-like
receptor 4; NLRP3, NLR family pyrin domain containing 3; OA,
osteoarthritis; ECM, extracellular matrix; miR, microRNA; iNOS,
inducible nitric oxide synthase; COX-2, cyclooxygenase-2; MMPs,
matrix metalloproteinases. |
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
WCS was responsible for data curation, formal
analysis, funding acquisition and original draft. YL was
responsible for data curation, formal analysis, the original draft
manuscript and methodology. JGF was responsible for data curation,
formal analysis and validation. JHL was responsible for formal
analysis and validation. QFH was responsible for formal analysis
and investigation. DXH and YXC were responsible for formal analysis
and investigation. HYS and WY were responsible for investigation
and validation. WS was responsible for conceptualization, funding
acquisition, formal analysis and validation. QY was responsible for
conceptualization, funding acquisition, formal analysis and
original draft. WCS and QY confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The collection of cartilage tissues from patients
was approved by the Ethics Committee of Shenzhen Second People's
Hospital (approval no. 2023-157-01YJ). Samples used in this study
were collected after all patients received detailed explanation of
the research and submitted written informed consents. All in
vivo experiments procedures were approved by the Animal
Research Committee of Southwest Medical University (approval no.
20220307-004).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
OA
|
osteoarthritis
|
|
MMPs
|
matrix metalloproteinases
|
|
AGEs
|
advanced glycation end products
|
|
CIRP
|
cold-inducible RNA-binding
protein
|
|
DAMP
|
damage-associated molecular pattern
molecule
|
|
TLR4
|
Toll-like receptor 4
|
|
NLRP3
|
NLR family pyrin domain containing
3
|
|
ECM
|
extracellular matrix
|
|
COX-2
|
cyclooxygenase-2
|
|
iNOS
|
inducible nitric oxide synthase
|
Acknowledgements
Not applicable.
Funding
The present study was supported by the joint foundation of
Luzhou Government and Southwest Medical University (grant no.
2024LZXNYDJ104); the Shenzhen Medical Research Fund (grant no.
A2403030); Natural Science Foundation of China (grant no.
82172831); Shenzhen Science and Technology Projects (grant no.
JSGG20220831110400001, KCXFZ20230731093059012) and Shenzhen
High-level Hospital Construction Fund (grant no. 1801024).
References
|
1
|
Martel-Pelletier J, Barr AJ, Cicuttini FM,
Conaghan PG, Cooper C, Goldring MB, Goldring SR, Jones G, Teichtahl
AJ and Pelletier JP: Osteoarthritis. Nat Rev Dis Primers.
2:160722016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Loeser RF, Goldring SR, Scanzello CR and
Goldring MB: Osteoarthritis: A disease of the joint as an organ.
Arthritis Rheum. 64:1697–1707. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bijlsma JW, Berenbaum F and Lafeber FP:
Osteoarthritis: An update with relevance for clinical practice.
Lancet. 377:2115–2126. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Glyn-Jones S, Palmer AJ, Agricola R, Price
AJ, Vincent TL, Weinans H and Carr AJ: Osteoarthritis. Lancet.
386:376–387. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang B, Kang X, Xing Y, Dou C, Kang F, Li
J, Quan Y and Dong S: Effect of microRNA-145 on IL-1beta-induced
cartilage degradation in human chondrocytes. FEBS Lett.
588:2344–2352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tu C, Huang X, Xiao Y, Song M, Ma Y, Yan
J, You H and Wu H: Schisandrin A inhibits the IL-1β-Induced
inflammation and cartilage degradation via suppression of MAPK and
NF-κB signal pathways in rat chondrocytes. Front Pharmacol.
10:412019. View Article : Google Scholar
|
|
7
|
Chabane N, Zayed N, Afif H, Mfuna-Endam L,
Benderdour M, Boileau C, Martel-Pelletier J, Pelletier JP, Duval N
and Fahmi H: Histone deacetylase inhibitors suppress
interleukin-1beta-induced nitric oxide and prostaglandin E2
production in human chondrocytes. Osteoarthritis Cartilage.
16:1267–1274. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou S, Liu G, Si Z, Yu L and Hou L:
Glycyrrhizin, an HMGB1 inhibitor, suppresses interleukin-1β-induced
inflammatory responses in chondrocytes from patients with
osteoarthritis. Cartilage. 13(2_Suppl): 947S–955S. 2021. View Article : Google Scholar
|
|
9
|
Frommer KW, Schaffler A, Rehart S, Lehr A,
Muller-Ladner U and Neumann E: Free fatty acids: Potential
proinflammatory mediators in rheumatic diseases. Ann Rheum Dis.
74:303–310. 2015. View Article : Google Scholar
|
|
10
|
Xu HC, Wu B, Ma YM, Xu H, Shen ZH and Chen
S: Hederacoside-C protects against AGEs-induced ECM degradation in
mice chondrocytes. Int Immunopharmacol. 84:1065792020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gao F and Zhang S: Loratadine alleviates
advanced glycation end Product-induced activation of NLRP3
inflammasome in human chondrocytes. Drug Des Devel Ther.
14:2899–2908. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liao Y, Tong L, Tang L and Wu S: The role
of cold-inducible RNA binding protein in cell stress response. Int
J Cancer. 141:2164–2173. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhong P and Huang H: Recent progress in
the research of cold-inducible RNA-binding protein. Future Sci OA.
3:FSO2462017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wellmann S, Buhrer C, Moderegger E, Zelmer
A, Kirschner R, Koehne P, Fujita J and Seeger K: Oxygen-regulated
expression of the RNA-binding proteins RBM3 and CIRP by a
HIF-1-independent mechanism. J Cell Sci. 117:1785–1794. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sun W, Liao Y, Yi Q, Wu S, Tang L and Tong
L: The mechanism of CIRP in regulation of STAT3 phosphorylation and
Bag-1/S expression Upon UVB radiation. Photochem Photobiol.
94:1234–1239. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aziz M, Brenner M and Wang P:
Extracellular CIRP (eCIRP) and inflammation. J Leukoc Biol.
106:133–146. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qiang X, Yang WL, Wu R, Zhou M, Jacob A,
Dong W, Kuncewitch M, Ji Y, Yang H, Wang H, et al: Cold-inducible
RNA-binding protein (CIRP) triggers inflammatory responses in
hemorrhagic shock and sepsis. Nat Med. 19:1489–1495. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sakurai T, Kashida H, Watanabe T, Hagiwara
S, Mizushima T, Iijima H, Nishida N, Higashitsuji H, Fujita J and
Kudo M: Stress response protein cirp links inflammation and
tumorigenesis in colitis-associated cancer. Cancer Res.
74:6119–6128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou M, Yang WL, Ji Y, Qiang X and Wang P:
Cold-inducible RNA-binding protein mediates neuroinflammation in
cerebral ischemia. Biochim Biophys Acta. 1840:2253–2261. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vande Walle L, Van Opdenbosch N, Jacques
P, Fossoul A, Verheugen E, Vogel P, Beyaert R, Elewaut D,
Kanneganti TD, van Loo G and Lamkanfi M: Negative regulation of the
NLRP3 inflammasome by A20 protects against arthritis. Nature.
512:69–73. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
McAllister MJ, Chemaly M, Eakin AJ, Gibson
DS and McGilligan VE: NLRP3 as a potentially novel biomarker for
the management of osteoarthritis. Osteoarthritis Cartilage.
26:612–619. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen Z, Zhong H, Wei J, Lin S, Zong Z,
Gong F, Huang X, Sun J, Li P, Lin H, et al: Inhibition of Nrf2/HO-1
signaling leads to increased activation of the NLRP3 inflammasome
in osteoarthritis. Arthritis Res Ther. 21:3002019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu Q, Zhang D, Hu D, Zhou X and Zhou Y:
The role of mitochondria in NLRP3 inflammasome activation. Mol
Immunol. 103:115–124. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou K, Cui S, Duan W, Zhang J, Huang J,
Wang L, Gong Z and Zhou Y: Cold-inducible RNA-binding protein
contributes to intracerebral hemorrhage-induced brain injury via
TLR4 signaling. Brain Behav. 10:e016182020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Elliott EI and Sutterwala FS: Initiation
and perpetuation of NLRP3 inflammasome activation and assembly.
Immunol Rev. 265:35–52. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu L, Li QH, Deng F, Yu ZW, Luo XZ and Sun
JL: Synovial fluid concentrations of cold-inducible RNA-binding
protein are associated with severity in knee osteoarthritis. Clin
Chim Acta. 464:44–49. 2017. View Article : Google Scholar
|
|
27
|
Yoo IS, Lee SY, Park CK, Lee JC, Kim Y,
Yoo SJ, Shim SC, Choi YS, Lee Y and Kang SW: Serum and synovial
fluid concentrations of cold-inducible RNA-binding protein in
patients with rheumatoid arthritis. Int J Rheum Dis. 21:148–154.
2018. View Article : Google Scholar
|
|
28
|
Felekkis K, Pieri M and Papaneophytou C:
Exploring the feasibility of circulating miRNAs as diagnostic and
prognostic biomarkers in osteoarthritis: Challenges and
opportunities. Int J Mol Sci. 24:131442023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin Z, Jiang T, Zheng W, Zhang J, Li A, Lu
C and Liu W: N6-methyladenosine (m6A) methyltransferase
WTAP-mediated miR-92b-5p accelerates osteoarthritis progression.
Cell Commun Signal. 21:1992023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mao G, Zhang Z, Hu S, Zhang Z, Chang Z,
Huang Z, Liao W and Kang Y: Exosomes derived from
miR-92a-3p-overexpressing human mesenchymal stem cells enhance
chondrogenesis and suppress cartilage degradation via targeting
WNT5A. Stem Cell Res Ther. 9:2472018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen H, Yao H, Chi J, Li C, Liu Y, Yang J,
Yu J, Wang J, Ruan Y, Pi J and Xu JF: Engineered exosomes as drug
and RNA co-delivery system: New hope for enhanced therapeutics?
Front Bioeng Biotechnol. 11:12543562023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jiang M, Jike Y, Liu K, Gan F, Zhang K,
Xie M, Zhang J, Chen C, Zou X, Jiang X, et al: Exosome-mediated
miR-144-3p promotes ferroptosis to inhibit osteosarcoma
proliferation, migration, and invasion through regulating ZEB1. Mol
Cancer. 22:1132023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang X, Wang J, Liu N, Wu W, Li H, Lu W
and Guo X: Umbilical cord Blood-derived M1 macrophage exosomes
loaded with cisplatin target ovarian cancer in vivo and reverse
cisplatin resistance. Mol Pharm. 20:5440–5453. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hu G, Zhao X, Wang C, Geng Y, Zhao J, Xu
J, Zuo B, Zhao C, Wang C and Zhang X: MicroRNA-145 attenuates
TNF-α-driven cartilage matrix degradation in osteoarthritis via
direct suppression of MKK4. Cell Death Dis. 8:e31402017. View Article : Google Scholar
|
|
35
|
Zhou J, Sun J, Markova DZ, Li S, Kepler
CK, Hong J, Huang Y, Chen W, Xu K, Wei F and Ye W: MicroRNA-145
overexpression attenuates apoptosis and increases matrix synthesis
in nucleus pulposus cells. Life Sci. 221:274–283. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tian K, Deng B, Han X, Zheng H, Lin T,
Wang Z, Zhang Y and Wang G: Over-expression of microRNA-145
elevating autophagy activities via downregulating FRS2 expression.
Comb Chem High Throughput Screen. 27:127–135. 2024. View Article : Google Scholar
|
|
37
|
Wang X, Lu W, Xia X, Zhu Y, Ge C, Guo X,
Zhang N, Chen H and Xu S: Selenomethionine mitigate PM2.5-induced
cellular senescence in the lung via attenuating inflammatory
response mediated by cGAS/STING/NF-κB pathway. Ecotoxicol Environ
Saf. 247:1142662022. View Article : Google Scholar
|
|
38
|
Chen Z, Zhang M, Zhao Y, Xu W, Xiang F, Li
X, Zhang T, Wu R and Kang X: Hydrogen sulfide contributes to
uterine quiescence through inhibition of NLRP3 inflammasome
activation by suppressing the TLR4/NF-κB signalling pathway. J
Inflamm Res. 14:2753–2768. 2021. View Article : Google Scholar :
|
|
39
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
40
|
Wang S, Li W, Zhang P, Wang Z, Ma X, Liu
C, Vasilev K, Zhang L, Zhou X, Liu L, et al: Mechanical overloading
induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis
via Piezo1 channel facilitated calcium influx. J Adv Res. 41:63–75.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gong T, Wang QD, Loughran PA, Li YH, Scott
MJ, Billiar TR, Liu YT and Fan J: Mechanism of lactic
acidemia-promoted pulmonary endothelial cells death in sepsis: Role
for CIRP-ZBP1-PANoptosis pathway. Mil Med Res. 11:712024.PubMed/NCBI
|
|
42
|
Zhou M, Aziz M, Yen HT, Ma G, Murao A and
Wang P: Extracellular CIRP dysregulates macrophage bacterial
phagocytosis in sepsis. Cell Mol Immunol. 20:80–93. 2023.
View Article : Google Scholar
|
|
43
|
Zhang P, Bai L, Tong Y, Guo S, Lu W, Yuan
Y, Wang W, Jin Y, Gao P, Liu J, et al: CIRP attenuates acute kidney
injury after hypothermic cardiovascular surgery by inhibiting
PHD3/HIF-1α-mediated ROS-TGF-β1/p38 MAPK activation and
mitochondrial apoptotic pathways. Mol Med. 29:612023. View Article : Google Scholar
|
|
44
|
Ye L, Tang X, Liu F, Wei T, Xu T, Jiang Z,
Xu L, Xiang C, Yuan X, Shen L, et al: Targeting CIRP and
IL-6R-mediated microglial inflammation to improve outcomes in
intracerebral hemorrhage. J Adv Res. Sep 9–2025. View Article : Google Scholar : Epub ahead of
print.
|
|
45
|
Han J, Zhang Y, Ge P, Dakal TC, Wen H,
Tang S, Luo Y, Yang Q, Hua B, Zhang G, et al: Exosome-derived CIRP:
An amplifier of inflammatory diseases. Front Immunol.
14:10667212023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Palazzo C, Nguyen C, Lefevre-Colau MM,
Rannou F and Poiraudeau S: Risk factors and burden of
osteoarthritis. Ann Phys Rehabil Med. 59:134–138. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Carr AJ, Robertsson O, Graves S, Price AJ,
Arden NK, Judge A and Beard DJ: Knee replacement. Lancet.
379:1331–1340. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu J, Wu X, Lu J, Huang G, Dang L, Zhang
H, Zhong C, Zhang Z, Li D, Li F, et al: Exosomal transfer of
osteoclast-derived miRNAs to chondrocytes contributes to
osteoarthritis progression. Nat Aging. 1:368–384. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sun W, Bergmeier AP, Liao Y, Wu S and Tong
L: CIRP sensitizes cancer cell responses to ionizing radiation.
Radiat Res. 195:93–100. 2021.PubMed/NCBI
|
|
50
|
Zhang F, Yang WL, Brenner M and Wang P:
Attenuation of hemorrhage-associated lung injury by adjuvant
treatment with C23, an oligopeptide derived from Cold-inducible
RNA-binding protein. J Trauma Acute Care Surg. 83:690–697. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Murao A, Tan C, Jha A, Wang P and Aziz M:
Exosome-mediated eCIRP release from macrophages to induce
inflammation in sepsis. Front Pharmacol. 12:7916482021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ying X, Peng L, Chen H, Shen Y, Yu K and
Cheng S: Cordycepin prevented IL-β-induced expression of
inflammatory mediators in human osteoarthritis chondrocytes. Int
Orthop. 38:1519–1526. 2014. View Article : Google Scholar
|
|
53
|
Schmidt N, Pautz A, Art J, Rauschkolb P,
Jung M, Erkel G, Goldring MB and Kleinert H: Transcriptional and
post-transcriptional regulation of iNOS expression in human
chondrocytes. Biochem Pharmacol. 79:722–732. 2010. View Article : Google Scholar
|
|
54
|
Stannus O, Jones G, Cicuttini F,
Parameswaran V, Quinn S, Burgess J and Ding C: Circulating levels
of IL-6 and TNF-α are associated with knee radiographic
osteoarthritis and knee cartilage loss in older adults.
Osteoarthritis Cartilage. 18:1441–1447. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhao Y, Li Y, Qu R, Chen X, Wang W, Qiu C,
Liu B, Pan X, Liu L, Vasilev K, et al: Cortistatin binds to TNF-α
receptors and protects against osteoarthritis. EBioMedicine.
41:556–570. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhou M, Aziz M, Denning NL, Yen HT, Ma G
and Wang P: Extracellular CIRP induces macrophage endotoxin
tolerance through IL-6R-mediated STAT3 activation. JCI Insight.
5:e1337152020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Denning NL, Aziz M, Murao A, Gurien SD,
Ochani M, Prince JM and Wang P: Extracellular CIRP as an endogenous
TREM-1 ligand to fuel inflammation in sepsis. JCI Insight.
5:e1341722020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gomez R, Villalvilla A, Largo R, Gualillo
O and Herrero-Beaumont G: TLR4 signalling in osteoarthritis-finding
targets for candidate DMOADs. Nat Rev Rheumatol. 11:159–170. 2015.
View Article : Google Scholar
|
|
59
|
Barreto G, Senturk B, Colombo L, Brück O,
Neidenbach P, Salzmann G, Zenobi-Wong M and Rottmar M: Lumican is
upregulated in osteoarthritis and contributes to TLR4-induced
pro-inflammatory activation of cartilage degradation and macrophage
polarization. Osteoarthritis Cartilage. 28:92–101. 2020. View Article : Google Scholar
|
|
60
|
Liu L, Gu H, Liu H, Jiao Y, Li K, Zhao Y,
An L and Yang J: Protective effect of resveratrol against
IL-1β-induced inflammatory response on human osteoarthritic
chondrocytes partly via the TLR4/MyD88/NF-κB signaling pathway: An
'in vitro study'. Int J Mol Sci. 15:6925–6940. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhang A, Wang P, Ma X, Yin X, Li J, Wang
H, Jiang W, Jia Q and Ni L: Mechanisms that lead to the regulation
of NLRP3 inflammasome expression and activation in human dental
pulp fibroblasts. Mol Immunol. 66:253–262. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wang C, Gao Y, Zhang Z, Chen C, Chi Q, Xu
K and Yang L: Ursolic acid protects chondrocytes, exhibits
anti-inflammatory properties via regulation of the NF-κB/NLRP3
inflammasome pathway and ameliorates osteoarthritis. Biomed
Pharmacother. 130:1105682020. View Article : Google Scholar
|
|
63
|
Dougherty U, Mustafi R, Zhu H, Zhu X, Deb
D, Meredith SC, Ayaloglu-Butun F, Fletcher M, Sanchez A, Pekow J,
et al: Upregulation of polycistronic microRNA-143 and microRNA-145
in colonocytes suppresses colitis and Inflammation-associated colon
cancer. Epigenetics. 16:1317–1334. 2021. View Article : Google Scholar
|
|
64
|
Tang J, Yi S and Liu Y: Long non-coding
RNA PVT1 can regulate the proliferation and inflammatory responses
of rheumatoid arthritis fibroblast-like synoviocytes by targeting
microRNA-145-5p. Hum Cell. 33:1081–1090. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Liu Z, Tao B, Fan S, Pu Y, Xia H and Xu L:
MicroRNA-145 protects against myocardial ischemia reperfusion
injury via CaMKII-Mediated antiapoptotic and Anti-inflammatory
pathways. Oxid Med Cell Longev. 2019:89486572019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Li Y, Wu X, Miao S and Cao Q: MiR-383-5p
promotes apoptosis of ovarian granulosa cells by targeting CIRP
through the PI3K/AKT signaling pathway. Arch Gynecol Obstet.
306:501–512. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lin TY, Jia JS, Luo WR, Lin XL, Xiao SJ,
Yang J, Xia JW, Zhou C, Zhou ZH, Lin SJ, et al:
ThermomiR-377-3p-induced suppression of Cirbp expression is
required for effective elimination of cancer cells and cancer
stem-like cells by hyperthermia. J Exp Clin Cancer Res. 43:622024.
View Article : Google Scholar : PubMed/NCBI
|