Contents
Introduction
Hepatocellular carcinoma, a common cancer
worldwide
Genomic deletion and tumor suppressor genes
Genomic overrepresentation and oncogenes
Genomic and related cancer gene alterations on
chromosome 8 in hepatocellular carcinoma
DLC1 a potent tumor suppressor gene in
hepatocellular carcinoma
MYC, a major oncogene in cancer
Critical role of MYC in the pathogenesis of human
and mouse hepatocellular carcinoma
Modulation of DLC1 and MYC in hepatocellular
carcinoma: prospects for combined pharmacologic interventions
Introduction
Genomic instability and several other hallmarks of
cancer underline the complexity of neoplastic disease (1). During the process of cancer
development, the expression output of a variety of genes can be
modulated by mutations, amplifications, deletions, and chromosome
translocations; these changes accumulate in time and materialize in
the appearance of an incipient abnormal cell with the capacity to
perpetually proliferate (2).
Advances in cytogenetics and molecular biology have led to the
establishment of the genetic basis of neoplasia and to the
recognition that chromosomal alterations affect genes critical in
the pathogenesis of human cancer. The detection of recurrent
chromosomal alterations has greatly facilitated the identification
of genes important in oncogenesis and provided the basis for
development of highly efficient cancer therapeutic agents. The best
known examples are the development of Imatinib and Herceptin
therapies for chronic myelocytic leukemia (CML) and breast cancer,
respectively. Imatinib was consequential to identification of a
bcr-abl fusion protein resulting from specific chromosome
translocation between chromosomes 9 and 22 in CML, whereas
development of Herceptin followed the detection of recurrent
HER/Neu (ErbB2) amplification on abnormal chromosomes in breast
cancer cells (3,4). Both examples underline the importance
of searching for gene alterations at the sites of recurrent
chromosome abnormalities.
Epigenetic changes are equally important to the
process of cancer development. In many types of cancer, tumor
suppressor genes (TSGs) are frequently downregulated or silenced by
promoter hypermethylation or histone deacetylation (5). In the past few years a new trend
emerged in cancer therapy with focus on functional alterations
mediated by epigenetic mechanisms. Therapeutic approaches that
reverse the adverse effect of epigenetic modifications have already
been developed, such as Vidaza and Decitabine, both potent DNA
methyltransferase inhibitors, or Vorinostat, a histone deacetylase
inhibitor. These therapeutics are applied either alone or in
combination (6,7).
Hepatocellular carcinoma, a common cancer
worldwide
Human hepatocellular carcinoma (HCC) is one of the
most common cancers worldwide, accounting for 90% of all liver
neoplasias. It is the third leading cause of cancer death, and its
incidence is increasing in several countries, including the United
States (8,9). Infection with hepatitis B (HBV) and C
viruses (HCV), consumption of aflatoxin-contaminated foods and/or
alcohol, and exposure to other chemical carcinogens have been
implicated in the etiology of HCC (8). In the multistep process of HCC
development, both the chemical carcinogens and the oncogenic
viruses can cause DNA damage, which is manifested at the chromosome
level as deletions, duplications, and translocations, frequently
affecting the structure and expression of cancer-related genes.
With the use of a combined approach based on integration of
molecular cytogenetics and molecular biology, our investigations
focused on identification and characterization of recurrent
chromosome alterations that have led to the discovery of new
cancer-related genes as well as to the detection of alterations in
known ones. Our group and many others searched for recurrent and
specific genomic and gene alterations in HCC and examined both the
karyotypes and the quantitative profiles of genomic imbalances and
gene expression of cell lines or primary tumors. A recent article
underlining the role of TSGs as metabolic regulators stated that
cancer therapy is increasingly shifting toward therapeutics based
on genetic alterations displayed by cancer cells (10).
Here we summarize the progress and our contribution
toward uncovering genomic and related gene alterations as targets
for therapeutic interventions, and focus on the role of two genes,
MYC oncogene and deleted in liver cancer 1 (DLC1) gene, in
hepatocarcinogenesis.
In our early studies, we identified novel nonrandom
chromosome rearrangements and a distinct pattern of multiple
recurrent DNA copy-number gains and losses, some of which mimicked
alterations frequently seen in other neoplasias and those
potentially specific for HCC (11,12).
Such analyses significantly expanded the map of genomic changes in
HCC and led to the identification of genes that may play an
important role in the pathogenesis of HCC. We examined the
karyotypes of a number of HCC cell lines and, by using approaches
such as spectral karyotyping (SKY), comparative genomic
hybridization (CGH), and array-based CGH (aCGH) analyses, we were
able to identify nonrandom chromosomal alterations and recurrent
breakpoints in balanced and unbalanced rearrangements, as well as
to precisely map recurrent genomic amplifications and deletions
(11,12). It became apparent that not all the
genomic sites were equally affected by gain or loss of DNA and that
the regions known as fragile sites (FSs) were most frequently
involved. In the past several years, molecular and cytogenetic
evidence for cancer-specific translocations, amplification of
oncogenes, deletion of TSGs, and viral integration at FSs firmly
implicated these regions of fragility and recombination in cancer
development (13). A half of all
microRNA genes, whose transcripts (mRNAs), may act as either
oncogenes or TSGs, and whose expression is deregulated by
amplification, deletion, mutation, and epigenetic modifications in
a variety of cancers, are located within or near FSs (14).
Genomic deletion and tumor suppressor
genes
Molecular cytogenetic analysis of HCC cell lines
revealed nonrandom deletions and unbalanced translocations of
chromosomes 1 and 3, with the breakpoints clustered at regions 1p36
and 3p14-21, close to the loci of p73 and FHIT TSGs, respectively
(11). This report was the first
to describe unbalanced translocations of chromosomes 1 and 3 with
the breakpoints nonrandomly involving these loci in HCC. Unlike in
balanced abnormalities, unbalanced alterations may result in loss
of genes. Cytogenetic abnormalities of chromosome 1 are common in
HCC, and several lines of evidence implicate alterations of
specific sites on its short arm in the pathogenesis of HCC
(15,16). Region 1p36 is a region of fragility
and recombination and is suspected to harbor multiple TSGs. Because
several oncogenes and at least two cell senescence genes are
located on chromosome 1, it has been postulated that structural
alterations and the consequential imbalance of chromosome 1 may be
important for gene dosage in certain types of cancer, including HCC
(12). Recently, using an approach
based on integrated genomic data of DNA copy number and gene
expression profiles, researchers identified several potential
driver genes on chromosome 1 in HCC (17).
In addition to 1p36, another site of recurrent
translocation involves the region 3p14-21 where the FHIT gene is
located. The FHIT gene is expressed in normal hepatocytes but is
either abnormally expressed or inactivated in HCC cells. We
detected recurrent chromosome 3p rearrangements, a decrease or
absence of FHIT mRNA expression, intragenic deletions, and an
absence of protein expression. These observations strongly
suggested that FHIT alterations might be pathologically relevant to
HCC. Chemical carcinogens or HBV integration at FRA3B may initiate
a background of genetic instability early in the process of
hepatocarcinogenesis (18). In HCC
cell lines, we also identified recurrent DNA loss on the short arm
of chromosome 3 at sites other than 3p21 (12).
Among other candidate TSGs, located at regions of
deletion in 3p in various cancers, is the TMEM7 gene, which encodes
a transmembrane protein (19).
This gene is expressed specifically in the liver, and its protein
shares substantial sequence homology with human and mouse 28-kDa
interferon-α (IFN-α) response modifier protein. We investigated the
role of TMEM7 in the development of human HCC and demonstrated
that, in the absence of genomic deletion and mutation, the
downregulation or silencing of the gene is due to aberrant DNA
hypermethylation and histone deacetylation. Ectopic expression of
TMEM7 inhibited HCC cell proliferation, colony formation, and cell
migration in vitro, and reduced tumor formation in nude
mice. Treatment of two highly invasive HCC cell lines with IFN-α
significantly increased TMEM7 expression and inhibited cell
migration. These observations implicate loss of TMEM7 expression in
hepatocarcinogenesis, and suggest that modification of TMEM7
expression by IFN-α may have potential therapeutic relevance in a
subset of HCC (20).
During the neoplastic process, certain tumor cells
acquire resistance to the antiproliferative signaling of TGFβ. We
examined a human HCC cell line sensitive to TGFβ1 (Hep3B-TS), and
its derivative, Hep3B-TR, rendered resistant to TGFβ1 by stepwise
exposure to the agent (21). SKY
and aCGH analysis showed that the TGFβ resistance resulted from a
loss of TGFβ receptor II (TGFβRII) gene, which occurred when the
only remaining apparently normal chromosome 3 in Hep3B-TR cells,
underwent microdeletion encompassing the site of the gene. A
comparative differential gene expression analysis of the
above-mentioned cell lines, using an oligonucleotide microarray,
identified six genes in Hep3BTR cells that are downstream targets
of the TNF gene, suggesting that loss of TGFβRII triggered the
activation of the tumor necrosis factor network known to be
regulated by the TGFβ1 pathway. Functionally, loss of TGFβRII in
cells resistant to TGFβ1 significantly enhanced cell migration and
anchorage-independent growth in vitro, and also increased
in vivo tumorigenicity compared with parental sensitive
cells (21).
Deletion and loss of heterozygosity (LOH) at
specific regions of the long arm of chromosome 16 are common in
several forms of cancer, including HCC (8). We have focused on region 16q24 that
harbors tumor suppressor gene WWOX, which spans FRA16D, the second
most common FS (22). The status
of WWOX genomic DNA, as well as of the transcribed RNA and
translated protein, was examined in HCC cell lines, and recurrent
alterations of the gene have been identified. Loss of DNA copy
number confined to band 16q23 was detected by CGH in several cell
lines (12). Although homozygous
deletions of WWOX were not detected, WWOX mRNA was either absent or
reduced in 60% of the cell lines examined. The detection of
aberrant RT-PCR products of WWOX transcripts, with deletion of
exons 6 to 8, correlated significantly with altered WWOX
expression. All of the cell lines showing downregulation of WWOX
mRNA, also had a reduced or undetectable level of WWOX protein.
Furthermore, in a majority of the HCC cell lines, the overall
amount of WWOX protein was markedly reduced or undetectable in
comparison with that of a normal liver. These results show that
WWOX is frequently altered in HCC, and therefore implicate it in
hepatocarcinogenesis (23).
Because carcinogenic agents preferentially target common FSs, it is
possible that breakage of WWOX locus at FRA16D and of FHIT gene at
FRA3B occurs concomitantly in certain HCCs.
Genomic overrepresentation and
oncogenes
In our CGH analysis of HCC cell lines, several
regions of recurrent DNA copy-number gains have been identified
(12). The detection of two
regions of DNA overrepresentation on 11q13 and 5q31, both
overlaping with the locations of common FSs, led us to take a
closer look at two genes, EMS1 and SMAD5, that reside at/or close
to those chromosomal spots. Region 11q13 harbors EMS1 oncogene and
FRA11H. We found that EMS1 is amplified in primary HCC and
overexpressed in HCC cell lines in the absence of gene
amplification. This oncogene encodes cortactin, a cortical
actin-associated protein that is a substrate for the tyrosine
kinase Src and contributes to reorganization of the actin
cytoskeleton. Alterations of EMS1 that lead to the overproduction
of cortactin may thus be important in the development of HCC. EMS1
amplification and overexpression are indicative of an unfavorable
prognosis in several cancers and may have similar prognostic
implications in liver cancer (24). The other earlier-mentioned minimal
region of DNA copy-number gain in HCC, identified at chromosome
5q31, overlaps with the location of FRA5C and with the locus of the
SMAD5 gene (25). Deletions at
this location, unbalanced translocations with breakpoints near the
SMAD5 locus, recurrent formation of isochromosome 5q resulting in
selective loss of 5p and gain of 5q, and intrachromosomal
amplification of SMAD5 at FRA5C have been detected; all point to
this locus as relevant in the development of HCC. High-level
amplification of SMAD5 at FRA5C is one of the few examples of gene
amplification at a common FS mediated by breakage-fusion-bridge
cycles (13). These observations
show that SMAD5 undergoes gain in copy number, high-level
amplification, and overexpression rather than loss of expression,
suggesting that it does not function as a TSG in HCC (25).
Genomic and related cancer gene alterations
on chromosome 8 in hepatocellular carcinoma
The genes thus far identified in our studies, which
promote or suppress tumor cell growth, most likely represent only a
fraction of possible future therapeutic targets. In spite of wide
heterogeneity of genomic alterations in HCC, certain chromosomes or
chromosomal sites are more commonly deleted or amplified, which
results in deregulation of critical genes that, ultimately, may
trigger transformation of normal hepatocytes into malignant
phenotype. The best example of the above, perhaps, are genomic
alterations of chromosome 8 in HCC. This chromosome shows a
distinctive pattern of both recurrent of DNA copy number loss on
8p, and gain on 8q (12). Due to
the high frequency of large genomic deletion and LOH in HCC and
several other cancers, chromosome 8p has been long suspected to
carry TSGs. Moreover, LOH on chromosome 8p is among the most common
alterations in HCC and is associated with liver dysplastic nodules
and primary and metastatic HCC (26–32).
Chromosome 8p contains two FSs and is rich in candidate and
validated TSGs, some of which have been implicated in the
pathogenesis of HCC (33,34). Region 8p22 alone harbors six TSGs
(Table I). Among the candidate and
bona fide TSGs implicated in HCC are PDGFRI, DLC1, LFIRE/HFREP-1,
SFRP1, MSRA, and newly identified SCARA5 gene (35–41).
 | Table I
|
Table I
| A, Candidate or
bonafide tumor suppressor genes on chromosome 8p |
|
| Name | Location | Type of cancer |
| MSRA | p23 | Liver |
| DLC1 | p22 | Liver and other
cancers |
| N33 | p22 | Prostate |
| PDGFRL | p22 | Liver |
| MTUS1 | p22 | Liver and other
cancers |
| LFIRE-1 | p22 | Liver |
| LZTS1 | p22 | Various
cancers |
| TRAIL-R1 | p21 | Various
cancers |
| TRAIL-R2 | p21 | Various
cancers |
| DBC2 | p21 | Breast |
| RHOBTB2 | p21 | Breast |
| DOK | p21 | Lung |
| SORBS3 | p21 | Liver |
| SHRBS3 | p21 | Liver |
| SCARA5 | p12-11.1 | Liver |
| NRG1 | p11-12 | Breast,
pancreas |
| PROSC | p11 | Liver |
| FGFR | p11 | Myeloproliferative
syndrome |
| TACC | p11 | Breast |
| SFRP1 | p11 | Liver, breast |
| MOZ | p11 | Acute myeloid
leukemia |
| B, Protooncogenes
on chromosome 8q |
|
| Name | Location | Type of cancer |
| MOS | q11 | Lung and other
cancers |
| MDTH | q22.1 | Breast |
| MYC | q24 | Liver and other
cancers |
| HSF1 | q24.3 | Liver |
| LYN | q13-qter | Ewing’s
sarcoma |
Liver fibrinogen-related gene-1, LFIRE-1/HFREP-1,
specifically expressed in normal human liver tissue, is recurrently
downregulated or inactivated and has an antiproliferative effect on
HCC cells both in vitro and in vivo(37).
SFRP1, a secreted Wnt antagonist, and SCARA5, a
plasma membrane protein, which activate the focal adhesion kinase
(FAK) signaling pathway and inhibit the tyrosine phosphorylation
cascade of the FAK-Src-Cas pathway, are strong candidate tumor
suppressor genes due to their antiproliferative effect on HCC cells
(39,41). Since its isolation, SFRP1 gene was
predicted to be a TSG because of its location at 8p11-12, a site of
LOH in HCC (42). However, in many
types of cancer, including HCC, SFRP1 has been found to be silenced
primarily by hypermethylation, and not by genomic deletion
(39,43). In a few instances, restoration of
SFRP1 expression resulted in reversal of the malignant phenotype
(39,43). However, one study reports that
SFRP1 is upregulated in metastatic renal cell carcinoma and that it
contributes to the invasiveness of the tumor cells (44). The same researchers and others had
reported that SFRP1 is frequently downregulated in primary renal
cell carcinoma (45). The
probability that SFRP1 downregulation contributes to
hepatocarcinogenesis (39) is
rather high, given the role and importance of Wnt signaling in the
pathogenesis of HCC (46,47). A study demonstrating the
suppressive effect of SCARA5 on tumor cell proliferation in
vitro and on metastasis in vivo(41), indicates that this gene also may
play an important role in initiation and development of HCC.
Other characterized TSGs such as N33, TRAIL-R1,
TRAIL-R2, LZTS2, hBD-1, DBC2, DOK, and NRG1, are also located on
8p, but none of them was examined in HCC. Three new genes located
on 8p, SORBS3, SHRBS3 and PROSC, have been characterized quite
recently and their function will be discussed later in the text,
within the contest of TSG role in HCC. A number of oncogenes are
located on 8q, but only MYC is closely associated with the
development of HCC (Table I).
Among all cancer-related genes on chromosome 8, DLC1 and MYC play a
major role in hepatocarcinogenesis, and evidence for their
involvement in the development of HCC is presented next.
DLC1 a potent tumor suppressor gene in
hepatocellular carcinoma
The DLC1 gene was isolated from a chromosomal
fragment deleted in a human HCC, and was suspected to be a TSG
(36). Since its isolation, the
interest in this gene has grown considerably worldwide. Progress in
understanding of its tumor-suppressive function in multiple
cancers, particularly in HCC, will be discussed here. Two closely
related genes, DLC2 and DLC3, also were isolated and were found to
be frequently deregulated and to elicit oncosuppressive effects in
HCC (48,49). Until now, three DLC1 isoforms,
called variants 1, 2, and 3 (or α, β and γ), were isolated
(50). Most recently, a new
isoform 4 (DLC1-i4) was identified and shown to suppress tumor cell
growth, with its functional promoter regulated by p53 (51).
In recent years, DLC1 has emerged as a major tumor
suppressor gene and a strong candidate metastasis suppressor gene
in HCC and other cancers (50,52–55).
DLC1 is also a contributing factor in processes affecting normal
functions, or disorders unrelated to cancer. DLC1 is vital for
normal development because null mutations in the gene result in
embryonic lethality in mice and flies. It is also involved in
insulin signaling and the pathogenesis of coronary spastic angina
and is among four genes that negatively regulate periodontal
ligament cell proliferation (56–59).
The DLC1 gene is located on chromosome 8p22, a
region (Table I) of recurrent LOH
in HCC (36). Although region 8p22
does not correspond to an FS, its propensity for deletion is
similar to that of the most unstable and vulnerable FSs.
Downregulation or inactivation of TSGs has profound consequences in
the genesis and progression of cancer, and DLC1 is one of the most
frequently deregulated genes in the cancer genome. Downregulation
or inactivation of the DLC1 gene in many forms of cancer, is
commonly mediated at the genomic level by heterozygous and
homozygous deletion, and at the transcriptional level by aberrant
promoter methylation or histone deacetylation (50). Initially, we detected lack of DLC1
expression in over 40% of human primary HCC and in 90% of HCC cell
lines (36). These results were
independently confirmed, as 40% of HCC samples had no detectable
DLC1 expression (60). More
recently, using representational oligonucleotide microarray,
heterozygous deletions of DLC1 were found in 59 of 86 primary HCCs.
Furthermore, deletions of DLC1 occurred in other cancers more
frequently than of some other well-known TSGs (53). In contrast with the early
observation that mutations in the coding region of DLC1 are rare in
HCC, recent genome-wide sequencing analyses of several cancers have
identified missense mutation of DLC1 (61–64).
In an early study of HCC cell lines, we identified a number of
single nucleotide polymorphisms in the DLC1 genomic DNA sequence,
which may be associated with an increased risk for development of
HCC (61). Only recently, a
comprehensive genotyping analysis of a large number of cases in the
Chinese population provided convincing evidence for an association
between DLC1 polymorphism and susceptibility to HCC. Furthermore,
evidence shows that the DLC1 polymorphism and risk for HCC
development are consistent with its biological function and
strongly suggest that the DLC1 polymorphism may directly confer
susceptibility to HCC (65).
Promoter hypermethylation appears to be associated
with human cancer at least as frequently as is disruption of TSGs
by mutation or deletion. TSGs that are downregulated or silenced by
promoter hypermethylation are often located in genomic regions that
are frequently deleted in cancer (13,66).
Despite a high rate of deletion, DLC1 is predominantly silenced or
down-regulated by epigenetic mechanisms, frequently by promoter
methylation (50,67,68).
An expression profile of the three DLC1 isoforms - variants 1, 2,
and 3 - in normal and malignant hepatic tissue, showed that only
variant 1 was silenced by promoter methylation; variants 1 and 2
(but not variant 3) localize at focal adhesions and suppress stress
fiber formation and in vitro HCC cell proliferation
(69). DLC1 variant 1 is not the
only one silenced by promoter methylation. As recently demonstrated
with the newly identified isoform 4, DLC-i4 is expressed in normal
tissues and normal immortalized cell lines but is significantly
downregulated in a high number of nasopharyngeal, esophageal,
gastric, breast, colorectal, cervical, lung carcinoma cell lines as
well as in primary tumors. Methylation of DLC1-i4 was detected in
38% of primary HCCs (51).
In addition to deletion, mutation, and promoter
methylation, DLC1 can be downregulated or silenced by other
mechanisms. DLC1 did not escape the action of certain miRNAs that
target TSG expression (70). A new
study of significant interest and with implications for the role of
HCV in chronic liver disease and HCC, has demonstrated that
efficient HCV replication requires miR-141-mediated suppression of
DLC1. In primary human hepatocytes, HCV infection stimulated cell
proliferation that was reversed by transduction of DLC1 (71).
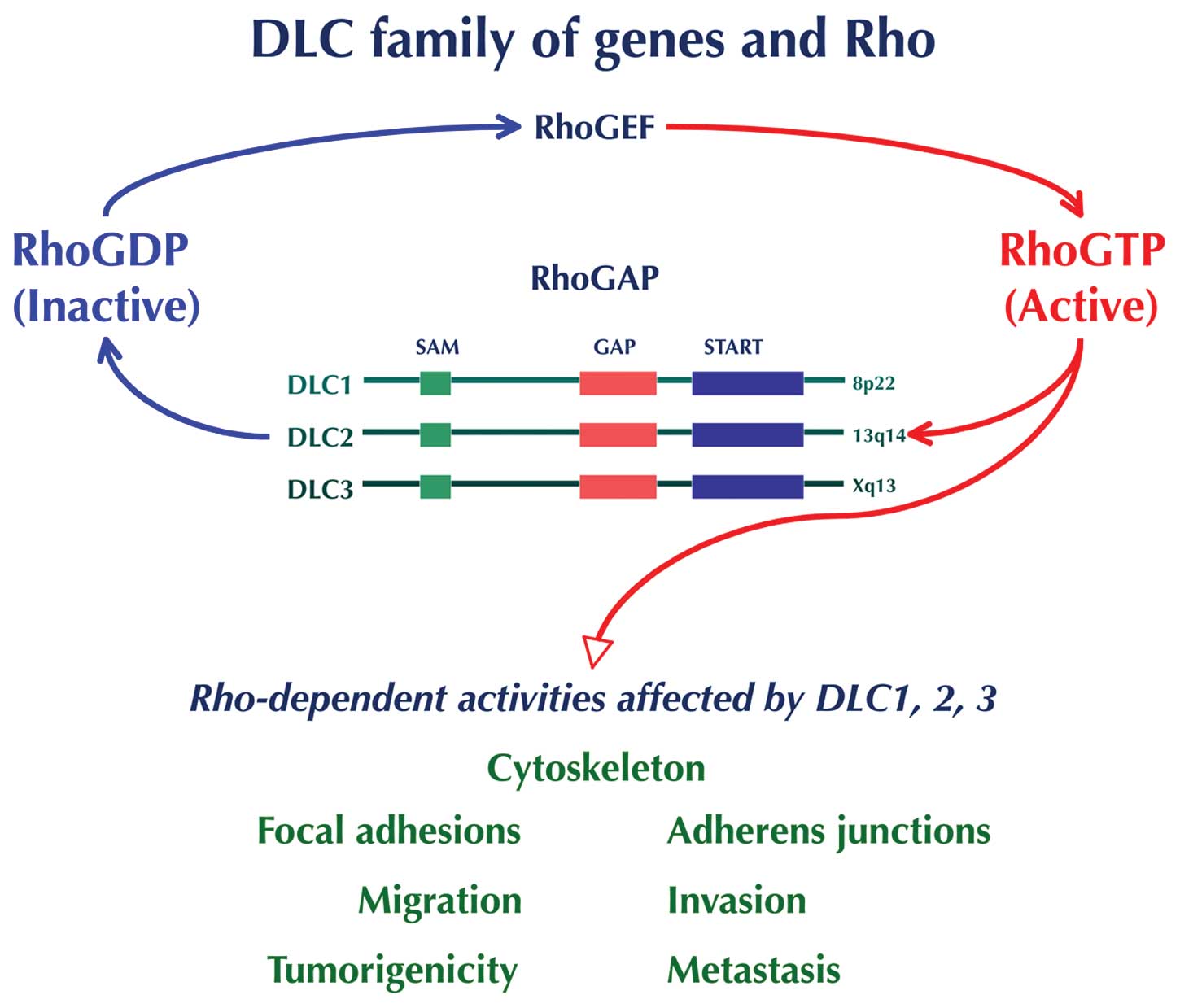
DLC1 encodes a RhoGAP protein that catalyzes the
conversion of active GTP-bound Rho GTPase (Rho) to the inactive
GDP-bound form (Fig. 1).
DLC1-mediated inhibitory effect of cell growth and tumorigenicity
is primarily due to its RhoGAP’s ability to inactivate the Rho
proteins - RhoA, RhoB, and RhoC, and to some degree Cdc42, but not
Rac - although RhoGAP-independent mechanisms also contribute to its
antioncogenic activity (72–80).
Rho GTPase activity is frequently deregulated in human cancers, and
experimental evidence implicated Rho GTPase signaling in promoting
growth and invasiveness through morphologic and functional cellular
alterations such as cell shape, actin cytoskeleton remodeling and
focal adhesion organization, as well as cell migration and invasion
(50,81,82).
Because the activity of Rho GTPases is elevated in many human
cancers, the ability of RhoGAPs to downregulate Rho proteins may
attenuate or inhibit tumorigenic and/or metastatic process
(83–85). The importance of altered Rho GTPase
signaling in the genesis and progression of HCC has been previously
covered in detail (85). Special
attention was given to DLC1, whose loss of function was considered
a driving event in the promotion and progression of HCC. DLC1 also
was recognized as the best example of a RhoGAP alteration in HCC
(85). Most recently, in an
integrative genomic and transcriptomic profiling of HCC from 76
patients with HBV infection designed to identify survival-related
driver genes, DLC1 and five other genes on 8p were found deleted in
patients with poor outcome. Interestingly, three of these genes,
SORBS3, SHRBS3 and PROSC act as TSGs and suppress HCC in
vitro and in vivo(86).
Indeed, highly recurrent alterations of DLC1 have
been found in solid tumors and hematologic malignancies, and a
meta-analysis of microarray experiments lists DLC1 as the fifth of
the top-50 genes implicated in multiple cancers (87).
DLC1-transduced HCC cells display a diffuse
cytoplasmic localization of the protein (72,88),
whereas in other cells DLC1 protein co-localizes with the tips of
actin stress fibers and in focal adhesions, which is critical for
cell migration (72,75,88–90).
Evidence that DLC1 functions as a TSG in HCC comes from experiments
in which DLC1 cDNA was ectopically expressed in cells with disabled
endogenous gene expression. In several independent studies,
restoration of DLC1 expression invariably resulted in inhibition of
cell proliferation, colony formation, and cell migration in
vitro, and it reduced the development of tumors after
xenografting of HCC cells in athymic nude mice (60,72,74,88).
Two well-characterized cell lines, derived from patients with
aggressive HCC, and negative for endogenous DLC1 activity, were
used to test the antiproliferative effect of the DLC1 ectopic
expression. Restoration of DLC1 expression resulted in inhibition
of in vitro cell proliferation and in vivo
tumorigenicity, as well as in induction of apoptosis associated
with cleavage of caspase 3 and a reduced level of the antiapoptotic
Bcl-2 (88). Subsequent studies
with prostate cancer showed that tumor cells acquire sensitivity to
DLC1-induced apoptosis after treatment with HA14-1, a Bcl-2
inhibitor (76). It has been
demonstrated that subcellular locations of DLC1 protein determines
different manifestations of its antioncogenic action; in cytoplasm
DLC1 functions as an inhibitor of tumor cell proliferation and
migration, whereas in the nucleus it acts as an inducer of
apoptosis (91).
In the past few years, evidence supporting the
metastasis suppressor function of DLC1 has been generated with
breast cancer, and HCC cells (77,92,93).
The role of DLC1 in metastatic process will be covered in a
separate article.
A yeast two-hybrid screening was invaluable to
further dissect complex molecular machinery underlying DLC1
functions. While the multidomain structure of DLC1 merely indicated
its capacity to interact with other molecules, the yeast two-hybrid
analysis identified several potential binding partners of DLC1.
Those binding partners were later confirmed in human cells,
allowing the examination of consequences of their interactions with
DLC1. Such was the case with the tensin family of focal adhesion
proteins, which serve as a link between the actin cytoskeleton and
the cytoplasmic tails of integrins, whose relationship with DLC1
has been thoroughly investigated in different types of tumor cells
(94–100). In human lung cancer cells, it was
demonstrated that DLC1 binds to SH2 and PTB domains of the tensin
proteins and, most importantly, that cooperation between DLC1
RhoGAP action and its tensin-binding activity is required for
suppression of cancer cell migration, although the two functions
are not interdependent (75).
Another group reported that tensin 2 and DLC1 form a
complex, which binds to caveolin-1 (Cav-1), a structural component
of caveolae. Such binding brings DLC1 in close proximity to locally
enriched RhoGTPases and facilitates the inactivation of Rho
proteins through the RhoGAP activity of DLC1 and with repercussions
at the level of cytoskeleton. Interaction of this kind would
constitute a potential new mechanism of DLC1 action in hepatocytes,
through cytoskeleton reorganization (94). Cav-1 functions as a TSG, and
recently Cav-1 interaction with the START domain of DLC1 was
identified and shown to contribute to the tumor suppressor activity
of DLC1 (101). We also detected
DLC1 interactions with p120RasGAP, α-catenin, and S100A10 proteins
in human cells, and assessed their influence on DLC1’s tumor
suppressor function (102–104).
The interaction of DLC1 with p120RasGAP protein, which is
implicated in cross-talk between Ras and Rho, not only provided
relevant data for the negative modulation of DLC1 tumor suppressor
activity but may, also, lead to potential clinical interventions
(102). The interaction was
mapped to the RhoGAP catalytic domain of DLC1 and the SH3 domain of
p120RasGAP, and resulted in a dramatic reduction of DLC1’s RhoGAP
activity in vitro. Moreover, overexpression of p120RasGAP in
colon carcinoma cells that harbored mutant Ras and thus were
resistant to the negative regulation of Ras by p120RasGAP,
increased the level of endogenous active Rho in a DLC1-dependent
manner and antagonized the growth-suppressive effects of DLC1.
These data show that p120RasGAP can promote the growth of cells
containing mutant Ras, and are consistent with evidence suggesting
that it might represent a valid therapeutic target for tumors
carrying a mutant Ras, which account for approximately 30% of all
tumors, and for which an efficient cancer treatment to block Ras
function did not translate in clinical interventions (55). Thus, p120RasGAP-targeting therapy
might be effective also in a subset of HCC with mutated Ras
(102).
In contrast with the negative effect of interaction
with p120RasGAP on DLC1 tumor suppressor function, preliminary
evidence indicates that DLC1 interaction with α-catenin enhanced
DLC1 anti-oncogenic activity (103).
We reported DLC1 interaction with S100A10, an
inflammatory protein and a key cell surface receptor for
plasminogen that regulates pericellular proteolysis and tumor cell
invasion. DLC1 and annexin 2 share the same binding site at the
C-terminus of S110A10. DLC1 binding to S100A10 caused an
dose-dependent decrease in steady-state S100A10 expression by
displacing it from annexin 2 and making it accessible to
ubiquitin-dependent degradation. This process attenuated
plasminogen activation and was accompanied by inhibition of in
vitro cell migration, invasion, colony formation, and
anchorage-independent growth of aggressive lung cancer cells. DLC1
binding to S100A10 did not affect DLC1’s RhoGAP activity. Thus,
this study unraveled a novel GAP-independent mechanism that
contributes to the tumor suppressor activity of DLC1 (104).
Although transcriptional regulation of DLC1 through
genetic and epigenetic alterations was discussed in detail, only
recently have posttranslational modifications been reported,
mediated by phosphorylation of a serine-rich domain, an
unstructured region of DLC1 gene, and shown to negatively regulate
the anti-oncogenic function of DLC1 (93,105). Unstructured domains are preferred
sites for posttranslational modifications, including
phosphorylation, that target multiple sites in the DLC family
proteins (50,106). In an earlier study, Ser322 of rat
DLC1 was found to be phosphorylated in rat adipocytes after insulin
treatment, and posttranslationally, rat DLC1 has been shown to be
phosphorylated by Akt kinase (57). This finding set the stage for
investigations of the role of Akt phosphorylation of DLC1 on
suppression of tumorigenicity and metastasis in HCC (93).
The 14-3-3 protein is known to function as a
phosphorylation-dependent adaptor protein that facilitates
interactions with other proteins (107). It is interesting that activated
protein kinase D (PKD) phosphorylates DLC1 and in turn facilitates
interaction with 14-3-3 protein isoforms. Binding of 14-3-3 protein
inhibited DLC1 RhoGAP activity and prevented nucleocytoplasmatic
shuttling and, thus, may impede the DLC1 apoptotic function
(91,108). As a follow-up to this study, the
same group provided evidence showing that PKD phosphorylation of
Ser 807 within the GAP domain negatively regulates DLC1 function
(105).
In summary, the role of DLC1 is rather complex, and,
as recognized by others, intricacies of the DLC1 tumor suppressor
function and its modulation by multiple and elaborate interactions
should stimulate interest and new research in this TSG (109). DLC1 meets the criteria for a
gatekeeper gene because of its inhibitory effect on tumor cell
growth and cell death (110).
Given the unusually high number of TSGs on the short arm of
chromosome 8, the inhibitory effect of DLC1 on tumor cell growth
may compensate for loss of function of other TSGs. Evidence
presented here confirms DLC1 as an important gene in the
development and progression of HCC.
MYC, a major oncogene in cancer
MYC protooncogene, the human cellular homologue of
the avian myelocytic leukemia virus, a retroviral oncogene, is a
transcription factor that regulates cell proliferation, controls
the expression of many genes, and is implicated in the pathogenesis
of a plethora of human solid tumors, leukemias and lymphomas, as
well as in animal tumors. MYC is a powerful cancer-promoting gene
but, paradoxically, also shows oncosuppressive activity by inducing
apoptosis and cell senescence. Since its isolation 27 years ago,
MYC has commanded interest worldwide, and nearly 20,000 studies on
virtually every form of cancer have been published. A recent
comprehensive article reviewed the progress in understanding MYC
function; the mechanisms contributing to the induction of cell
transformation, apoptosis, gene deregulation, and control of
expression; and the identification of MYC target genes (111). Therefore, we will briefly outline
the general aspects of MYC’s role in cancer development and present
our encounter with this gene in various cancers, particularly in
HCC.
Abnormal MYC expression is a common denominator in
cancer, and its activation is mediated by insertional mutagenesis,
chromosomal translocations and gene amplification, but not by
mutations in the coding sequence (111). Insertional mutagenesis, the
process of insertion of new sequence(s) into a normal gene,
frequently occurs during integration of retroviral proviruses into
the host genome, resulting in chimeric sequences of both viral and
cellular origin. Retroviruses are obligate mutagens because their
life cycle require integration into the host’s chromosomal DNA (up
to 8% of human genome is of retroviral origin), an event that, in
addition to structural alterations at the site of impact, also may
activate adjacent cellular oncogenes (111–114). MYC activation by retroviral
insertion was the first demonstration of such phenomena (115). Unlike retroviruses, the
integration of certain DNA viruses into chromosomal DNA, being not
required for viral persistence, was long considered rather an
exception, than a rule. Abundant evidence for integration of
several oncogenic DNA viruses into the cellular genome, obtained
from a variety of human cancers, has challenged this view. As
already mentioned, their integration, apparently, is not a random
event, because they preferentially target FSs, which frequently
encompass the location of growth-regulatory genes (116). Integration of oncogenic DNA
viruses, such as of human papillomavirus (HPV) in most invasive
genital cancers, Epstein-Barr virus (EBV) in infected
lymphoblastoid cells or Burkitt’s lymphoma (BL), and
adeno-associated virus (AAV) targeting MYC locus, have been well
documented (13). The most recent
evidence of specific viral integration at FS was the localization
of simian virus 40 (SV40) in SV40-immortalized cells, at region
1q21.1, which corresponds with the location of a common FS (FRA1F).
SV40’s insertion in the cell’s genome caused deregulation of
adjecent genes involved in senescence and apoptosis and, thus,
played a critical role in cellular immortalization (117).
The specificity of viral integration is fundamental
to determining the biological significance of this phenomenon in
various pathologic conditions, including cancer. The introduction
of viral genetic material and its interaction with the cellular
genome may contribute to the induction of cell transformation, the
maintenance of the transformed phenotype, or tumor progression
(113,114,118). Undoubtedly, the most conclusive
evidence for the effect of viral integration at FSs in cancer
development was provided in a study of young patients with X-linked
severe combined immunodeficiency, when three of ten children
developed monoclonal acute lymphoblastic leukemia- like
lymphoproliferation after gene therapy that used murine leukemia
virus-derived vector as a gene delivery vehicle. It has been
unequivocally demonstrated that in two of those patients (and
probably in all three) the activating vector integrated within
FRA11E region, near the transcription start site of the LMO2
oncogene (119), thus causing
high levels of transcription and, consequently, of LMO2
protein.
Chromosomal translocations, isochromosomes, double
minute chromosomes (DM), and abnormally banded or homogeneously
stained regions (HSRs) are also responsible for MYC deregulation in
a variety of cancers. Chromosomal translocations are common in
leukemias and lymphomas, and the best known examples are reciprocal
translocations in BL involving the MYC locus at FRA8C to any of the
three immunoglobulin genes on chromosome 2, 14, or 22, resulting in
MYC gene deregulation. Juxtaposition of MYC with immunoglobulin
loci was causally related to development of lymphomas (120). In addition to the reciprocal
translocation t(8;14), in two well-characterized BL cell lines we
identified two more complex translocations, t(8;14;18) and
t(7;8;14). These novel rearrangements, a manifestation of genomic
instability, resulted in transposition of MYC sequences in a new
genomic context and, together with duplication of chromosome 1q,
which is the second most common alteration in BL, may have
contributed to the acquisition of an invasive tumor phenotype
(121).
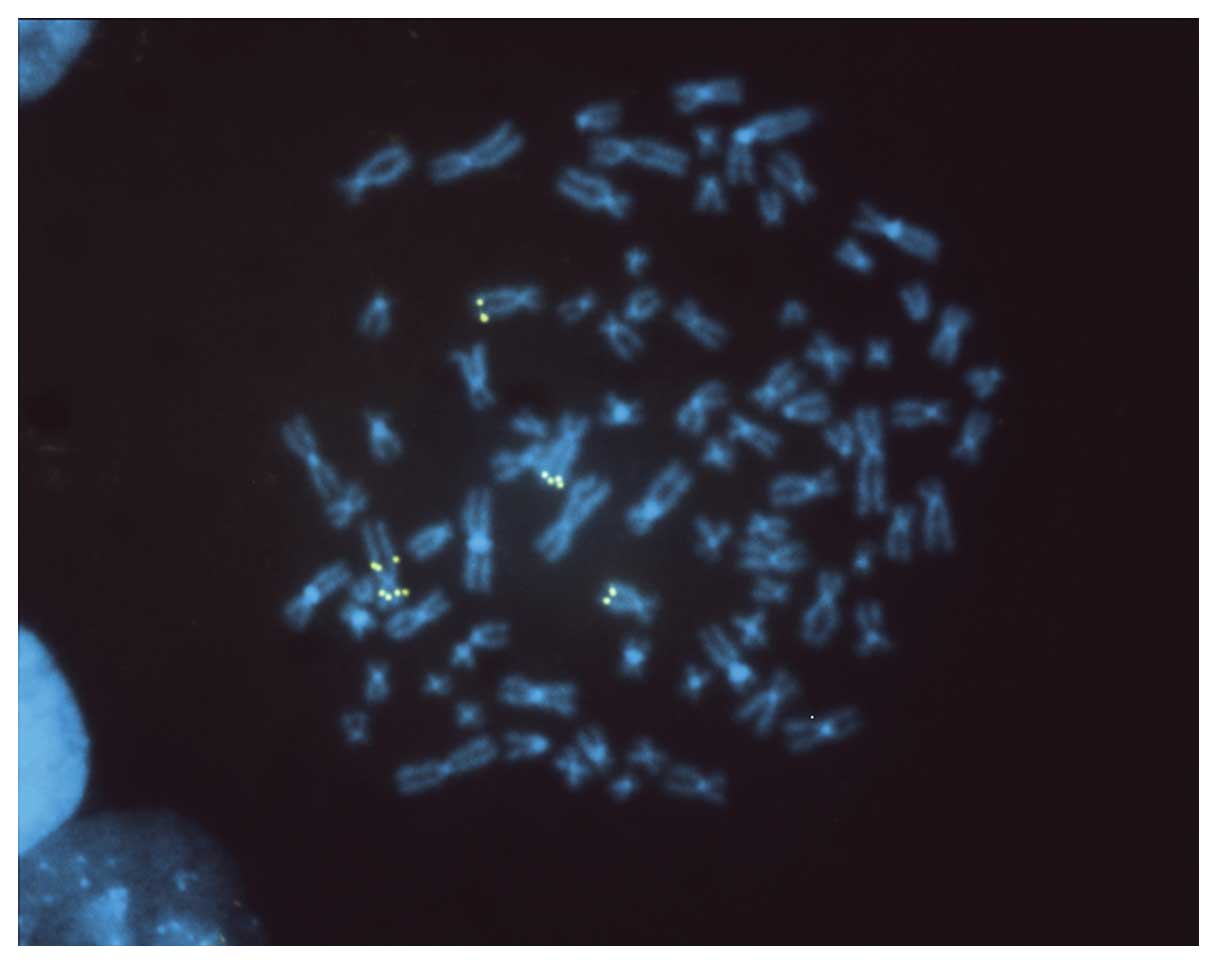
An example of an increased copy number of MYC on
normal and rearranged chromosome 8 in HCC is shown in Fig. 2.
Whereas abnormal isochromosomes cause a modest
increase in MYC copy number, DM and HSR may generate high-level
amplification, commonly found in human solid tumors (122,123). HSR may contain two or more
amplified genes, frequently oncogenes. In breast cancer cells, for
example, using DNA fibers hybridized with genomic probes, we
detected high-level amplification not only of MYC but of ERBB2, as
well (124). Nonrandom chromosome
alterations resulting in amplification of MYC gene also have been
identified in normal human mammary cells neoplastically transformed
by the controlled introduction of SV40LT, hTERT, and H-RAS genes,
thus implicating MYC in the early stages of neoplastic development
(125).
Critical role of MYC in the pathogenesis of
human and mouse hepatocellular carcinoma
Evidence from genetic analyses of human early liver
lesions, primary and metastatic liver tumors, tumor-derived cell
lines, and from different rodent models, unequivocally demonstrated
that MYC deregulation is a critical alteration in the initiation
and progression of HCC. A recent gene expression profiling of
cirrhotic and dysplastic nodules and early HCC, identified MYC as a
plausible driver gene and a central regulator of malignant
transformation in initial stages of hepatocarcinogenesis (126). In primary and advanced HCC and in
derived cell lines, MYC is commonly overexpressed because of
genomic amplification. Using a conventional CGH analysis in HCC
cell lines, we detected a DNA copy number increase throughout the
8q arm, and the minimal common region of amplification confined to
band 8q24.1, which corresponds with the site of FRA8C and with
location of MYC locus (12). In
this study and in several others, the CGH analyses did not
establish a correlation between genomic imbalances and the etiology
of HCC (127). It is interesting
that a CGH analysis and gene expression profiling established both
the copy-number gains and MYC overexpression in viral and
alcohol-related HCC, but not in cryptogenic, nonalcoholic,
steatohepatitis-induced HCC (128).
In an earlier minireview article, describing
chromosome-mediated alterations of MYC in various cancers, we
discussed the impact of viral integration in hepatocarcinogenesis,
and stressed the finding that the woodchuck hepatitis virus (WHV)
is known to integrate near MYC locus and cause enhanced MYC
expression (114). HBV has a
genomic structure similar to that of WHV, and both viruses exhibit
similarities to retroviruses in terms of integration and
requirements of RNA intermediate and reverse transcriptase for
replication. It is possible that HBV integrates at FRA8C and
activates the MYC gene (129).
However, due to HBV’s capacity to cause secondary genomic
rearrangements and virus relocation, the localization of HBV
integration sites in HCC may not represent the initial site of
integration (114).
Protooncogenes other than MYC often are amplified in HCC because of
HBV integration (130).
Mutagens-carcinogens also target FSs and induce genomic and gene
alterations (131). It is
possible that damage inflicted at FRA8C may explain the high
frequency of MYC overexpression in viral and alcohol-related HCC
(128), in addition to the
ability of HBV’s X protein to produce such an effect (132).
The development of mouse models for HCC provided an
ideal experimental approach for the detection of cellular and
genetic alterations and the discovery of candidate cancer genes
relevant to human hepatocarcinogenesis. Several new mouse models
for HCC have been developed in our laboratory, among which MYC
single-, and MYC-TGFα double-transgenic mice underscore the
critical role of the MYC gene in hepatocarcinogenesis (133).
Such models offer the opportunity to detect lesions
associated with the initial stages of neoplastic development, and
identify alterations related to the acquisition of malignancy or to
tumor progression. A conventional cytogenetic analysis revealed
that at 10 weeks of age, the level of structural chromosomal
aberrations was nearly tenfold higher in MYC-TGFα hepatocytes than
in wild-type hepatocytes, and that, as a sign of genomic
instability, early displastic lesions display breakage on several
chromosomes, whose locations correspond with regions of tumor
susceptibility (134). Also, it
has been postulated that increased generation of reactive oxygen
species might be responsible for the extensive chromosomal damage
and acceleration of hepatocarcinogenesis characteristic of MYC-TGFα
mice.
To determine whether vitamin E (VE), a potent
anti-oxidant, is able to protect against the chromosomal damage
triggered by oxidative stress caused by overexpression of c-myc and
TGF-α transgenes, the incidence of chromosomal and chromatid
aberrations was examined in primary hepatocyte cultures established
from 10-week-old MYC-TGFα mice maintained on either a control diet,
or a VE-supplemented diet. VE supplementation markedly decreased
the frequency of chromosomal damage at the early stage of
hepatocarcinogenesis before the development of morphologically
defined preneoplastic and neoplastic lesions (135). The cytogenetic constitution of
tumors developed in MYC-TGFα transgenic mice has been analyzed
using conventional G-banding analysis and FISH with several
chromosome painting and single-copy gene probes. Cells from all
such cytogenetically characterized tumors maintained ability to
form tumors after reinoculation in nude mice. The most frequently
observed stable alterations involved chromosomes 1, 4, 6, 7, and
12. A correspondence existed between the location of breakpoints in
stable alterations of these chromosomes, and the regions of
fragility identified in the early stage of hepatocarcinogenesis,
thus strongly suggesting the importance of these nonrandom changes
in both initiation and progression of neoplasia (136). The location of MYC transgene was
identified at chromosome 5G-ter; interestingly, in all tumors and
derivative HCC cell lines, a balanced translocation t(5;6) was
identified, with the breakpoint near the site of MYC transgene
integration. This is the first balanced translocation found in
either human or mouse liver tumors (137,138). Significantly, near the balanced
translocation t(5;6), we characterized a deletion that inactivated
a gene whose human homolog, GTF2IRD1, encodes a widely expressed,
multifunctional helix loop helix transcription factor that binds to
pRb and is one of the 16 genes present in approximately 1.6 Mb
interval commonly deleted in Williams-Beuren syndrome (WBS)
(139). Therefore, the c-myc
transgenic mice carrying the t(5;6) translocation represent, also,
the first ‘knockout’ of one of the genes present in the WBS
critical region. The Gtf2ird1-null mice exhibited growth
retardation and craniofacial and neurologic abnormalities similar
to clinical symptoms in WBS patients (140).
Our combined SKY and aCGH analysis of several cell
lines derived from HCC developed spontaneously in MYC transgenic
mice, identified recurrent chromosome rearrangements and genomic
imbalances. Among genomic imbalances, partial or complete gain of
chromosomes 15 and 19 and loss of chromosomes 4, 9, and 14 were the
most common alterations. These alterations are also recurrent in
HCC developed in other transgenic mouse models, in mouse
spontaneous HCC, in derivative cell lines, and in preneoplastic
liver lesions induced by chemical carcinogens. Overall, these
results demonstrate selective, nonrandom genomic changes associated
with development of HCC (141).
Among those changes, gains of chromosomes 15 and 19
are particularly important. Mouse chromosome 15 bears large regions
of homology with human chromosome 8q, and it is likely that
increased copy number of chromosome 15 might enhance the effect of
MYC in development of HCC, by providing more copies of the gene. DM
and HSR have been found in a variety of solid tumors and
hematologic malignancies but not in adult HCC or in HCC developed
in animals (137,138). In a cell line derived from HCC
developed in MYC transgenic mice, we detected a recurrent gain of
chromosome 19 and DM derived from chromosome 19. At the site of DNA
amplification are located the MYCS and MXI1 genes, both known to
interfere with MYC gene activity, and the mouse RelA gene that
promotes cell survival by inhibiting apoptosis and allowing MYC to
drive proliferation of transformed cells (142).
Other related studies demonstrated that coexpression
of MYC and TGF-α enhances the development of HCC through the
disruption of the pRb/EF2 pathway and that TGF-α might function as
a survival factor for neoplastic cells, thereby accelerating the
neoplastic process (143). In a
different mouse model, it has been demonstrated that MYC
inactivation induced regression of invasive HCC resulting in
differentiation into hepatocytes and biliary cells forming bile
duct structures. This suppressive effect was accompanied by a loss
of expression of the tumor markers and an increase in expression of
liver cell and liver stem cell markers (144).
Modulation of DLC1 and MYC in hepatocellular
carcinoma: prospects for combined pharmacologic interventions
There is no question that downregulation or
silencing of DLC1 gene and/or amplification or upregulation of MYC
gene are major contributing factors in the pathogenesis of human
and murine HCC. A highly recurrent pattern of genomic imbalances in
HCC predicts that both DLC1 and MYC would be deregulated in a large
number of cases and that such synchronyzation may be consequential
in hepatocarcinogenesis. This was demonstrated in an in
vitro/in vivo reconstitution mouse model for HCC
(53). In this model, genetically
modified liver progenitor cells lacking p53, transduced with a
retrovirus expressing MYC and with another expressing a DLC1 shRNA,
were transplanted into the liver of syngeneic mice (53,145). DLC1 knockdown aided MYC in the
induction of HCC in mice, and tumors thus developed are similar to
aggressive human HCC. Ectopic overexpression of DLC1 in HCC cells
expressing oncogenic Ras and containing increased GTP-bound RhoA,
abolished the tumor formation. Conversely, suppression of RhoA
inhibited growth of DLC1-negative HCC cells, showing that loss of
DLC1 and RhoA activation has a similar effect in
hepatocarcinogenesis. The results of this study provide conclusive
evidence that loss of DLC1, combined with an oncogenic stimulus,
promotes HCC in vivo and that this oncogenic process is
associated with activation of RhoA, which is a key consequence of
loss of DLC1 tumor suppressor activity (53,54).
Given the fact that many types of cancer accumulate
multiple genetic alterations over time, the treatment of cancer by
targeting a single damaged gene was originally considered unlikely
to be effective. However, numerous experiments subsequently showed
that silencing of an oncogene or activation of a TSG can be
sufficient to abolish cell tumorigenicity (146). Despite the accumulation of a
multitude of genetic alterations, tumor cells may depend only on a
single oncogenic pathway to provide critical competitive advantage
for their survival and unlimited multiplication. This phenomenon,
known as ‘oncogene addiction,’ was described more than a decade ago
(147). Only in the past few
years, however, has this concept received renewed interest, and its
potential to translate into effective targeted cancer therapy and
the need for future investigations have been eloquently advocated
(148). Suppression of such a
critical oncogenic pathway may result in the death of tumor cells
without any harmful consequences in normal cells. Likewise,
reintroduction of the wild-type of TSGs, which are frequently
inactivated in cancer cells, referred to as ‘tumor suppressor
hypersensitivity,’ prevents tumor growth by inducing cell death or
cell cycle arrest, and in many instances, it yields a complete
response without toxicity to normal cells. These are fundamental
requirements for an efficient cancer therapy.
It is well documented that DLC1 and MYC are
deregulated in a large fraction of HCC. In a preclinical study
designed to provide ground for potential tailored therapeutics,
transplanting tumor tissue from HCC patients into nude mice has
generated a number of explant models. The analyses of gene copy
number, gene mutation, mRNA expression, and protein expression
profiles demonstrated that transplanted tumors preserve the
genotypic alterations of the original tumors. Consistent with the
concept of oncogene addiction and tumor suppressor
hypersensitivity, amplification of MYC and cyclin D1 oncogenes and
deletion of DLC1 and FHIT TSGs were detected in different
individual models, as well as in corresponding patients’ HCC
tissues (149).
Novel therapeutic intervention options targeting the
DLC1 pathway in HCC have been discussed and the clinical
therapeutic efficacy of various agents was rigorously evaluated in
several articles (53,54,85,150). Therapeutic interventions based on
DLC1 activity require a full understanding of signaling pathways
affected by loss of its function (54). The inhibition of RhoA pathway and
Rho kinase (ROCK), a downstream effector of Rho, tops the options
for therapeutic interventions (53,54,85,150). Currently, a major effort is
underway to develop small molecule Rho kinase inhibitors to treat
various disorders, including cancer. An effective therapeutic
agent, Y-27632 displays distinct antimetastatic activity in HCC
(151,152). Wf-536, a more potent derivative
of Y-27632, also inhibited the metastasis of melanoma in
vivo but has not yet been tested in HCC (153,154). Results with a new potent and
selective ROCK inhibitor that is superior to Y compounds will be
reported soon (Channing Der, personal communication).
A number of compounds that restore DLC1 expression,
extend the half-life of its protein, or mimic its function, in
different cancers, also might be effective in therapy for HCC. The
ability of flavone to restore DLC1 expression and, consequently, to
suppress metastatic breast cancer cell proliferation is not an
isolated example (155). Other
agents, such as all-trans retinoic acid and peroxisome
proliferator-activated receptor γ (PPARγ), significantly elevated
DLC1 expression in cancer cells (50). Morellofalvone, a biflavonoid, also
inhibited tumor growth and angiogenesis of prostate cancer
xenografts in mice by targeting RhoA and Rac1 GTPases (156). A newly developed synthetic
flavone derivative, 6f, induces apoptosis mediated through death
receptor and mitochondria-dependent pathway in human HCC (157). Thiazolidinedione, a synthetic
activator of PPARγ, inhibits the growth of PPARg-expressing human
colon cancer cells by inducing terminal differentiation and a
marked increase in p21 abundance (158). Because epigenetic modifications
are the main cause of DLC1 down-regulation and inactivation,
therapy based on the ability of DNA methyl-transferase and histone
deacetylase inhibitors (HDAC) to restore DLC1 expression in cancer
cells has been highlighted as an attractive therapeutic approach
(54). In HCC cells, a combined
treatment involving suberoylanilide hydroxamic acid, a powerful
HDAC inhibitor already used in clinical trials, and DLC1
transduction had a synergistic inhibitory effect on tumor cell
proliferation and anchorage-independent growth (159). Other unrelated agents, such as
ursodeoxycholic acid (UDCA), a hydrophilic bile acid, inhibited
in vitro HCC cell proliferation by increasing the half-life
of DLC1 protein and reducing RhoA activity (160). It remains to be seen whether UDCA
has a similar antiproliferative effect in xenografted HCC in
mice.
It is disappointing that after many years of
intense effort and despite major advances in understanding the
regulation and function of the MYC gene, the development of cancer
therapeutics that would target MYC itself remains elusive (111). Future pharmacologic interventions
targeting MYC should be guided by a new strategy that takes into
account the differential MYC roles of either tumor promoting or
tumor suppressing, depending on its level of expression, and also
should consider modulation of the two opposite functions of MYC
oncoprotein (161,162).
An informative review article outlined the modest
progress in development of MYC targeted therapeutics in HCC. For
example, small MYC inhibitors showed an antiproliferative effect on
HCC cells in vitro and, furthermore, sensitized those cells
to chemotherapeutic agents, but they were not equally effective
in vivo(163). Quarfloxin
CX-3453, which targets MYC expression through a four-stranded DNA
structure, reached clinical trials for neuroendocrine carcinoma
(164). Overall, there are few
treatment options for advanced HCC. The development of Sorafenib, a
multikinase inhibitor that marked a breakthrough in understanding
of the disease, has shown remarkable survival benefits in patients
with advanced HCC (165,166). Quarfloxin has the potential to be
effective in therapy for HCC because, like Sorafenib, it inhibits
vascular endothelial growth factor or may enhance the antitumor
effect of Sorafenib (163).
Numerous studies suggest that statins, the
cholesterol-lowering drugs, have an antiproliferative effect on
cancer cells. Significantly, an interesting recent study
demonstrated that inhibition of HMG-coenzyme A reductase by
Atorvastatin blocks MYC phosphorylation and activation, leading to
suppression of mouse and human HCC cell proliferation in
vivo and in vitro. Suppression of MYC phosphorylation
was, most likely, mediated through the inhibition of GTPases in the
Rac pathway in vitro(167). In a clinical trial, treatment
with Pravastatin improved survival in patients with HCC (168). Regarding the effects of statins,
there is a significant link with DLC1 gene, which in its
COOH-terminal regions harbors a START domain, thought to be
important in cholesterol binding (36,169,170). As discussed earlier, Cav1, a
cholesterol transporter, interacts with DLC1 START domain and
contributes to the tumor suppressor activity of DLC1 (101). It is possible that statins may
affect normal START function.
In summary, given the limitations of single-agent
therapy, a combination of molecular therapies that target different
pathways is expected to be more effective in treatment of HCC
(166). Cancer therapeutics
targeting gain and loss of function of oncogenes and TSGs,
respectively, could lead to the identification of novel
genotype-selective antitumor agents (171). In that respect, a combined
therapeutic approach, targeting both MYC and DLC1 signaling
networks, have realistic potential to improve the treatment of
liver cancer.
Acknowledgements
This study was supported by the
Intramural Research Program of the National Cancer Institute,
NIH.
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bishop JM: The molecular genetics of
cancer. Science. 235:305–311. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rowley JD: Ph1-positive leukaemia,
including chronic myelogenous leukaemia. Clin Haematol. 9:55–86.
1980.PubMed/NCBI
|
|
4
|
Kraus MH, Popescu NC, Amsbaugh SC and King
CR: Overexpression of the EGF receptor-related protooncogene erbB-2
in human mammary tumor cell lines by different molecular
mechanisms. EMBO J. 6:605–610. 1987.PubMed/NCBI
|
|
5
|
Jones PA and Baylin SB: The epigenomics of
cancer. Cell. 128:683–692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gore SD, Baylin S, Sugar E, Carraway H,
Miller CB, Carducci M, Grever M, Galm O, Dauses T, Karp JE, Rudek
MA, Zhao M, Smith BD, Manning J, Jiemjit A, Dover G, Mays A,
Zwiebel J, Murgo A, Weng LJ and Herman JG: Combined DNA
methyltransferase and histone deacetylase inhibition in the
treatment of myeloid neoplasms. Cancer Res. 66:6361–6369. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Richon VM, Garcia-Vargas J and Hardwick
JS: Development of vorinostat: current applications and future
perspectives for cancer therapy. Cancer Lett. 280:201–210. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thorgeirsson SS and Grisham JW: Molecular
pathogenesis of human hepatocellullar carcinoma. Nat Genet.
31:339–346. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Altekruse SF, McGlynn KA and Reichman ME:
Hepatocellular carcinima incidence, mortality and survival trends
in United States from 1975 to 2005. J Clin Oncol. 27:1485–1491.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jones RG and Thompson CB: Tumor suppressor
and cell metabolism: a recipe for cancer growth. Genes Dev.
23:537–548. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Keck CL, Zimonjic DB, Yuan BZ,
Thorgeirsson SS and Popescu NC: Nonrandom breakpoints of unbalanced
chromosome translocations in human hepatocellular carcinoma cell
lines. Cancer Genet Cytogenet. 111:37–44. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zimonjic DB, Keck CL, Thorgeirsson SS and
Popescu NC: Novel recurrent genetic imbalances in human
hepatocellular carcinoma cell lines identified by comparative
genomic hybridization. Hepatology. 29:1208–1214. 1999. View Article : Google Scholar
|
|
13
|
Popescu NC: Genetic alterations in cancer
as a result of breakage at fragile sites. Cancer Lett. 192:1–17.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Garzon R, Calin GA and Croce CM: MicroRNAs
in cancer. Annu Rev Med. 60:167–179. 2009. View Article : Google Scholar
|
|
15
|
Simon D, Knowles B and Weith A:
Abnormalities of chromosome 1 and loss of heterozygosity on 1p in
primary hepatomas. Oncogene. 6:765–770. 1991.PubMed/NCBI
|
|
16
|
Yeh SH, Chen PJ, Chen HL, Lai MY, Wang CC
and Chen DS: Frequent genetic alterations at the distal region of
chromosome 1p in human hepatocellular carcinomas. Cancer Res.
54:4188–4192. 1994.PubMed/NCBI
|
|
17
|
Woo HG, Park ES, Lee JS, Lee YH, Ishikawa
T, Kim YJ and Thorgeirsson SS: Identification of potential driver
genes in human liver carcinoma by genomewide screening. Cancer Res.
69:4059–4066. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yuan BZ, Keck-Waggoner C, Zimonjic DB,
Thorgeirsson SS and Popescu NC: Alterations of FHIT gene in human
hepatocellular carcinoma. Cancer Res. 60:1049–1053. 2000.PubMed/NCBI
|
|
19
|
Imreh S, Klein G and Zabarovsky ER: Search
for unknown tumor-antagonizing genes. Genes Chromosomes Cancer.
38:307–321. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou X, Popescu NC, Klein G and Imreh S:
The interferon-alpha responsive gene TMEM7 suppresses cell
proliferation and is downregulated in human hepatocellular
carcinoma. Cancer Genet Cytogenet. 177:6–15. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zimonjic DB, Zhou X, Lee JS,
Ullmannova-Benson V, Tripathi V, Thorgeirsson SS and Popescu NC:
Acquired genetic and functional alterations associated with
transforming growthfactor beta type I resistance in Hep3B human
hepatocellular carcinoma cell line. J Cell Mol Med. 13:3985–3992.
2009. View Article : Google Scholar
|
|
22
|
Ludes-Meyers JH, Bednarek AK, Popescu NC,
Bedford M and Aldaz CM: WWOX, the common chromosomal fragile site,
FRA16D, cancer gene. Cytogenet Genome Res. 100:101–110. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Park SW, Ludes-Meyers J, Zimonjic DB,
Durkin ME, Popescu NC and Aldaz CM: Frequent downregulation and
loss of WWOX gene expression in human hepatocellular carcinoma. Br
J Cancer. 91:753–759. 2004.PubMed/NCBI
|
|
24
|
Yuan BZ, Zhou X, Zimonjic DB, Durkin ME
and Popescu NC: Amplification and overexpression of the EMS 1
oncogene, a possible prognostic marker, in human hepatocellular
carcinoma. J Mol Diagn. 5:48–53. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zimonjic DB, Durkin ME, Keck-Waggoner CL,
Park SW, Thorgeirsson SS and Popescu NC: SMAD5 gene expression, re
arrangements, copy number, and amplification at fragile site FRA5C
in human hepatocellular carcinoma. Neoplasia. 5:390–396. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Emi M, Fujiwara Y, Ohata H, Tsuda H,
Hirohashi S, Koike M, Miyaki M, Monden M and Nakamura Y: Allelic
loss at chromosome band 8p21.3-p22 is associated with progression
of hepatocellular carcinoma. Genes Chromosomes Cancer. 7:152–157.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pineau P, Nagai H, Prigent S, Wei Y,
Gyapay G, Weissenbach J, Tiollais P, Buendia MA and Dejean A:
Identification of three distinct regions of allelic deletions on
the short arm of chromosome 8 in hepatocellular carcinoma.
Oncogene. 18:3127–3134. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Qin LX, Tang ZY, Sham JS, Ma ZC, Ye SL,
Zhou XD, Wu ZQ, Trent JM and Guan XY: The association of chromosome
8p deletion and tumor metastasis in human hepatocellular carcinoma.
Cancer Res. 59:5662–5665. 1999.PubMed/NCBI
|
|
29
|
Chan KL, Lee JM, Guan XY, Fan ST and Ng
IO: High-density allelotyping of chromosome 8p in hepatocellular
carcinoma and clinicopathologic correlation. Cancer. 94:3179–3185.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kahng YS, Lee YS, Kim BK, Park WS, Lee JY
and Kang CS: Loss of heterozygosity of chromosome 8p and 11p in the
dysplastic nodule and hepatocellular carcinoma. J Gastroenterol
Hepatol. 18:430–436. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pang JZ, Qin LX, Ren N, Hei ZY, Ye QH, Jia
WD, Sun BS, Lin GL, Liu DY, Liu YK and Tang ZY: Loss of
heterozygosity at D8S298 is a predictor for long-term survival of
patients with tumor-node-metastasis stage I of hepatocellular
carcinoma. Clin Cancer Res. 13:7363–7369. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yam JW, Wong CM and Ng IO: Molecular and
functional genetics in hepatocellular carcinoma. Front Biosci
(Schol Ed). 2:117–134. 2010. View
Article : Google Scholar
|
|
33
|
Birnbaum D, Adélaïde J, Popovici C,
Charafe-Jauffret E, Mozziconacci MJ and Chaffanet M: Chromosome arm
8p and cancer: a fragile hypothesis. Lancet Oncol. 4:639–642. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Popescu NC: Fragile sites and cancer genes
on the short arm of chromosome 8. Lancet Oncol. 5:772004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fujiwara Y, Ohata H, Kuroki T, Koyama K,
Tsuchiya E, Monden M and Nakamura Y: Isolation of a candidate tumor
suppressor gene on chromosome 8p21.3-p22 that is homologous to an
extracellular domain of the PDGF receptor beta gene. Oncogene.
10:891–895. 1995.PubMed/NCBI
|
|
36
|
Yuan BZ, Miller MJ, Keck CL, Zimonjic DB,
Thorgeirsson SS and Popescu NC: Cloning, characterization, and
chromosomal localization of a gene frequently deleted in human
liver cancer (DLC-1) homologous to rat RhoGAP. Cancer Res.
58:2196–2199. 1998.PubMed/NCBI
|
|
37
|
Yan J, Yu Y, Wang N, Chang Y, Ying H, Liu
W, He J, Li S, Jiang W, Li Y, Liu H, Wang H and Xu Y:
LFIRE-1/HFREP-1, a liver-specific gene, is frequently downregulated
and has growth suppressor activity in hepatocellular carcinoma.
Oncogene. 23:1939–1949. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shih YL, Shyu RY, Hsieh CB, Lai HC, Liu
KY, Chu TY and Lin YW: Promoter methylation of the secreted
frizzled-related protein 1 gene SFRP1 is frequent in hepatocellular
carcinoma. Cancer. 107:579–590. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang J, Zhang YL, Teng XM, Lin Y, Zheng
DL, Yang PY and Han ZG: Down-regulation of SFRP1 as a putative
tumor suppressor gene can contribute to human hepatocellular
carcinoma. BMC Cancer. 7:1262007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lei KF, Wang YF, Zhu XQ, Lu PC, Sun BS,
Jia HL, Ren N, Ye QH, Sun HC, Wang L, Tang ZY and Qin LX:
Identification of MSRA gene on chromosome 8p as a candidate
metastasis suppressor for human hepatitis B virus-positive
hepatocellular carcinoma. BMC Cancer. 7:1722007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Huang J, Zheng DL, Qin FS, Cheng N, Chen
H, Wan BB, Wang YP, Xiao HS and Han ZG: Genetic and epigenetic
silencing of SCARA5 may contribute to human hepatocellular
carcinoma by activating FAK signaling. J Clin Invest. 120:223–241.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Finch PW, He X, Kelley MJ, Uren A,
Schaudies P, Popescu NC, Rudicoff S, Aaronson SA, Varmus HE and
Rubin JS: Purification and molecular cloning of a secreted,
Frizzled-related antagonist of Wnt action. Proc Natl Acad Sci USA.
94:6670–6675. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rubin JS, Barshishat-Kupper M,
Feroze-Merzoug F and Xi ZF: Secreted WNT antagonists as tumor
suppressors: pro and con. Front Biosci. 11:2093–2105. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Saini S, Liu J, Yamamura S, Majid S,
Kawakami K, Hirata H and Dahiya R: Functional significance of
secreted Frizzled-related protein 1 in metastatic renal cell
carcinomas. Cancer Res. 69:6815–6822. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kawamoto K, Hirata H, Kikuno N, Tanaka Y,
Nakagawa M and Dahiya R: DNA methylation and histone modifications
cause silencing of Wnt antagonist gene in human renal cell
carcinoma cell lines. Int J Cancer. 123:535–542. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Thompson MD and Monga SP: WNT/beta-catenin
signaling in liver health and disease. Hepatology. 45:1298–1305.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Takigawa Y and Brown AM: Wnt signaling in
liver cancer. Curr Drug Targets. 9:1013–1024. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ching YP, Wong CM, Chan SF, Leung TH, Ng
DC, Jin DY and Ng IO: Deleted in liver cancer (DLC) 2 encodes a
RhoGAP protein with growth suppressor function and is
underexpressed in hepatocellular carcinoma. J Biol Chem.
278:10824–10830. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Durkin EM, Ullmannova V, Guan M and
Popescu NC: Deleted in liver cancer 3(DLC-3), a novel
RhoGTPase-activating protein, is downregulated in cancer and
inhibits tumor cell growth. Oncogene. 26:4580–4589. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Durkin ME, Yuan BZ, Zhou X, Zimonjic DB,
Lowy DR, Thorgeirsson SS and Popescu NC: DLC-1: a Rho
GTPase-activating protein and tumor suppressor. J Cell Mol Med.
11:1185–1207. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Low JS, Tao Q, Ng KM, Goh HK, Shu XS, Woo
WL, Ambinder RF, Srivastava G, Shamay M, Chan AT, Popescu NC and
Hsieh WS: A novel isoform of the 8p22 tumor suppressor gene DLC1
suppresses tumor growth and is frequently silenced in multiple
common tumors. Oncogene. 30:1923–1935. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liao YC and Lo SH: Deleted in liver
cancer-1 (DLC-1): a tumor suppressor not just for liver. Int J
Biochem Cell Biol. 40:843–847. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xue W, Krasnitz A, Lucito R, Sordella R,
Vanaelst L, Cordon-Cardo C, Singer S, Kuehnel F, Wigler M, Powers
S, Zender L and Lowe SW: DLC1 is a chromosome 8p tumor suppressor
whose loss promotes hepatocellular carcinoma. Genes Dev.
22:1439–1444. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lahoz A and Hall A: DLC1: a significant
GAP in the cancer genome. Genes Dev. 22:1724–1730. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Vigil D, Cherfils J, Rossman KL and Der
CJ: Ras superfamily GEFs and GAPs: validated and tractable targets
for cancer therapy? Nat Rev Cancer. 12:842–857. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Durkin ME, Avner MR, Huh CG, Yuan BZ,
Thorgeirsson SS and Popescu NC: DLC-1, a Rho GTPase-activating
protein with tumor suppressor function, is essential for embryonic
development. FEBS Lett. 579:1191–1196. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hers I, Wherlock M, Homma Y, Yagisawa H
and Tavaré JM: Identification of p122RhoGAP (deleted in liver
cancer-1) Serine 322 as a substrate for protein kinase B and
ribosomal S6 kinase in insulin-stimulated cells. J Biol Chem.
281:4762–4770. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Murakami R, Osanai T, Tomita H, Sasaki S,
Maruyama A, Itoh K, Homma Y and Okumura K: p122 protein enhances
intra-cellular calcium increase to acetylcholine: its possible role
in the pathogenesis of coronary spastic angina. Arterioscler Thromb
Vasc Biol. 30:1968–1975. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wu J, Li Y, Fan X, Zhang C, Wang Y and
Zhao Z: Analysis of gene expression profile of periodontal ligament
cells subjected to cyclic compressive force. DNA Cell Biol.
30:865–873. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ng IO, Liang ZD, Cao L and Lee TK: DLC1 is
deleted in primary hepatocellular carcinoma and exerts inhibitory
effects on the proliferation of hepatoma cell lines with deleted
DLC1. Cancer Res. 60:6581–6584. 2000.PubMed/NCBI
|
|
61
|
Park SW, Durkin ME, Thorgeirsson SS and
Popescu NC: DNA variants of DLC-1, a candidate tumor suppressor
gene in human hepatocellular carcinoma. Int J Oncol. 23:133–137.
2003.PubMed/NCBI
|
|
62
|
Liao YC, Shih YP and Lo SH: Mutations in
the focal adhesion targeting region of deleted in liver cancer-1
attenuate their expression and function. Cancer Res. 68:7718–7722.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Jones S, Zhang X, Parsons DW, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A,
Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya
T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP,
Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR,
Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G,
Vogelstein B, Velculescu VE and Kinzler KW: Core signaling pathways
in human pancreatic cancers revealed by global genomic analyses.
Science. 321:1801–1806. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yachida S, Jones S, Bozic I, Antal T,
Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA,
Velculescu VE, Kinzler KW, Vogelstein B and Iacobuzio- Donahue CA:
Distant metastasis occurs late during the genetic evolution of
pancreatic cancer. Nature. 467:1114–1117. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Dong X, Zhou G, Zhai Y, Zhang H, Yang H,
Zhi L, Zhang X, Chu J and He F: Association of DLC1 gene
polymorphism with susceptibility to hepatocellular carcinoma in
Chinese hepatitis B virus carriers. Cancer Epidemiol. 33:265–270.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Teodoridis JM, Hardie C and Brown R: CpG
island methylator phenotype (CIMP) in cancer: causes and
implications. Cancer Lett. 268:177–186. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yuan BZ, Durkin ME and Popescu NC:
Promoter hypermethylation of DLC-1, a candidate tumor suppressor
gene, in several common human cancers. Cancer Genet Cytogenet.
140:113–117. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wong CM, Lee JM, Ching YP, Jin DY and Ng
IO: Genetic and epigenetic alterations of DLC-1 gene in
hepatocellular carcinoma. Cancer Res. 63:7646–7651. 2003.PubMed/NCBI
|
|
69
|
Ko FC, Yeung YS, Wong CM, Chan LK, Poon
RT, Ng IO and Yam JW: Deleted in liver cancer 1 isoforms are
distinctly expressed in human tissues, functionally different and
under differential transcriptional regulation in hepatocellular
carcinoma. Liver Int. 30:139–148. 2010. View Article : Google Scholar
|
|
70
|
Croce MC: Causes and consequences of
microRNA dysregulation in cancer. Nature Rev Genet. 10:704–714.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Banaudha K, Kaliszewski M, Korolnek T,
Florea L, Yeung ML, Kuan KT and Kumar A: MicroRNA silencing of
tumor suppressor DLC-1 promotes efficient hepatitis C virus
replication in primary human hepatocytes. Hepatology. 53:53–61.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wong CM, Yam JW, Ching YP, Yau TO, Leung
TH, Jin DY and Ng IO: Rho GTPase-activating protein deleted in
liver cancer suppresses cell proliferation and invasion in
hepatocellular carcinoma. Cancer Res. 65:8861–8868. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Healy KD, Hodgson L, Kim TY, Shutes AT,
Maddileti S, Juliano RL, Hahn KM, Harden TK, Bang YJ and Der CJ:
DLC1 suppresses non-small lung cancer growth and invasion by
RhoGAP-dependent and independent mechanisms. Mol Carcinog.
47:326–337. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Kim TY, Lee JW, Kim HP, Jong HS, Kim TY,
Jung M and Bang YJ: DLC-1, a GTPase-activating protein for Rho, is
associated with cell proliferation, morphology and migration in
human hepatocellular carcinoma. Biochem Biophys Res Commun.
355:72–77. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Qian X, Li G, Asmussen HK, Asnaghi L, Vass
WC, Braverman R, Yamada KM, Popescu NC, Papageorge AG and Lowy DR:
Oncogenic inhibition by a deleted in liver cancer gene requires
cooperation between tensin binding and Rho-specific
GTPase-activating protein activities. Proc Natl Acad Sci USA.
104:9012–9017. 2007. View Article : Google Scholar
|
|
76
|
Guan M, Tripathi V, Zhou X and Popescu NC:
Adenovirus-mediated restoration of the expression of the tumor
suppressor gene DLC1 inhibits the proliferation and tumorigenicity
of aggressive, androgen-independent human prostate cancer cell
lines: Prospects for gene therapy. Cancer Gene Ther. 15:371–381.
2008. View Article : Google Scholar
|
|
77
|
Zhou X, Zimonjic DB, Park SW, Yang XY,
Durkin ME and Popescu NC: DLC1 suppresses distant dissemination of
human hepatocellular carcinoma cells in nude mice through reduction
of RhoA GTPase activity, actin cytoskeletal disruption and
down-regulation of genes involved in metastasis. Int J Oncol.
32:1258–1291. 2008.
|
|
78
|
Holeiter G, Heering J, Erlmann P, Schmid
S, Jähne R and Olayioye MA: Deleted in liver cancer 1 controls
migration through a Dia1-dependent signaling pathway. Cancer Res.
68:8743–8751. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Erlmann P, Schmid S, Horenkamp FA, Geyer
M, Pomorski TG and Olayioye M: DLC1 activation requires lipid
interation through a polybasic region preceding the RhoGap domain.
Mol Biol Cell. 20:4400–4411. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhong D, Zhang J, Yang S, Soh UJ,
Buschdorf JP, Zhou YT, Yang D and Low BC: The SAM domain of the
RhoGAP DLC1 binds EF1A1 to regulate cell migration. J Cell Sci.
122:414–424. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Sahai E and Marshall CJ: RHO-GTPases and
cancer. Nat Rev Cancer. 2:133–142. 2002. View Article : Google Scholar
|
|
82
|
Gómez del Pulgar T, Benitah SA, Valerón
PF, Espina C and Lacal JC: Rho GTPase expression in tumourigenesis:
evidence for a significant link. Bioessays. 27:602–613.
2005.PubMed/NCBI
|
|
83
|
Jaffe AB and Hall A: Rho GTPases in
transformation and metastasis. Adv Cancer Res. 84:57–80. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Ridley AJ: Rho proteins and cancer. Breast
Cancer Res Treat. 84:13–19. 2004. View Article : Google Scholar
|
|
85
|
Grise F, Bidaud A and Moreau V: Rho
GTPases in hepatocellular carcinoma. Biochim Biophys Acta.
1795:137–151. 2009.PubMed/NCBI
|
|
86
|
Roessler S, Long EL, Budhu A, Chen Y, Zhao
X, Ji J, Walker R, Jia HL, Ye QH, Qin LX, Tang ZY, He P, Hunter KW,
Thorgeirsson SS, Meltzer PS and Wang XW: Integrative genomic
identification of genes on 8p associated with hepatocellular
carcinoma progression and patient survival. Gastroenterology. Dec
24–2011.(Epub ahead of print).
|
|
87
|
Pihur V and Datta S and Datta S: Finding
common genes in multiple cancer types through meta-analysis of
microarray experiments: a rank aggregation approach. Genomics.
92:400–403. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zhou X, Thorgeirsson SS and Popescu NC:
Restoration of DLC-1 gene expression induces apoptosis and inhibits
both cell growth and tumorigenicity in human hepatocellular
carcinoma cells. Oncogene. 23:1308–1313. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Kawai K, Yamaga M, Iwamae Y, Kiyota M,
Kamata H, Hirata H, Homma Y and Yagisawa H: A PLCdelta1-binding
protein, p122RhoGAP, is localized in focal adhesions. Biochem Soc
Trans. 32:1107–1109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kim TY, Vigil D, Der CJ and Juliano RL:
Role of DLC-1, a tumor suppressor protein with RhoGAP activity, in
regulation of the cytoskeleton and cell motility. Cancer Metastasis
Rev. 28:77–83. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Yuan BZ, Jefferson AM, Millecchia L,
Popescu NC and Reynolds SH: Morphological changes and nuclear
translocation of DLC1 tumor suppressor protein precede apoptosis in
human non-small cell lung carcinoma cells. Exp Cell Res.
313:3868–3880. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Goodison S, Yuan J, Sloan D, Kim R, Li C,
Popescu NC and Urquidi V: The RhoGAP protein DLC-1 functions as a
metastasis suppressor in breast cancer cells. Cancer Res.
65:6042–6053. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Ko FC, Chan LK, Tung EK, Lowe SW, Ng IO
and Yam JW: Akt phosphorylation of deleted in liver cancer 1
abrogates its suppression of liver cancer tumorigenesis and
metastasis. Gastroenterology. 139:1397–1407. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Yam JW, Ko FC, Chan CY, Jin DY and Ng IO:
Interaction of deleted in liver cancer 1 with tensin2 in caveolae
and implications in tumor suppression. Cancer Res. 66:8367–8372.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Liao YC, Si L, deVere White RW and Lo SH:
The phosphotyrosine-independent interaction of DLC-1 and the SH2
domain of cten regulates focal adhesion localization and growth
suppression activity of DLC-1. J Cell Biol. 176:43–49. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Hall EH, Daugherty AE, Choi CK, Horwitz AF
and Brautigan DL: Tensin1 requires protein phosphatase-1alpha in
addition to RhoGAP DLC-1 to control cell polarization, migration,
and invasion. J Biol Chem. 284:34713–34722. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Chan LK, Ko FC, Ng IO and Yam JW: Deleted
in liver cancer 1 (DLC1) utilizes a novel binding site for Tensin2
PTB domain interaction and is required for tumor-suppressive
function. PLoS One. 4:e55722009. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Hafizi S, Sernstad E, Swinny JD, Gomez MF
and Dahlbäck B: Individual domains of Tensin2 exhibit distinct
subcellular localisations and migratory effects. Int J Biochem Cell
Biol. 42:52–61. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Clark K, Howe JD, Pullar CE, Green JA,
Artym VV, Yamada KM and Critchley DR: Tensin 2 modulates cell
contractility in 3D collagen gels through the RhoGAP DLC1. J Cell
Biochem. 109:808–817. 2010.PubMed/NCBI
|
|
100
|
Kawai K, Kitamura SY, Maehira K, Seike J
and Yagisawa H: START-GAP1/DLC1 is localized in focal adhesions
through interaction with the PTB domain of tensin2. Adv Enzyme
Regul. 50:202–215. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Du X, Qian X, Papageorge A, Vass WC,
Braverman R and Lowy DR: Complex formation between DLC START domain
and Cav1 contributes to the tumor suppressor function of DLC1. Proc
Am Assoc Cancer Res. 52:5232011.
|
|
102
|
Yang XY, Guan M, Vigil D, Der CJ, Lowy DR
and Popescu NC: p120Ras-GAP binds the DLC1 Rho-GAP tumor suppressor
protein and inhibits its RhoA GTPase and growth-suppressing
activities. Oncogene. 28:1401–1409. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Tripathi V, Zimonjic DB and Popescu NC:
DLC1 and α-catenin protein interaction enhances DLC1 antioncogenic
activity by stabilizing adherens junctions and suppressing NFκB
signaling. Proc Am Assoc Cancer Res. 52:9622011.
|
|
104
|
Yang X, Popescu NC and Zimonjic DB: DLC1
interaction with S100A10 mediates inhibtion of in vitro cell
invasion and tumorigenicity of lung cancer cells through a
RhoGAP-indpendent mechanism. Cancer Res. 71:2916–2925.
2011.PubMed/NCBI
|
|
105
|
Scholz RP, Gustafsson JO, Hoffmann P,
Jaiswal M, Ahmadian MR, Eisler S, Erlmann P, Schmid S, Hausser A
and Olayioye MA: The tumor suppressor protein DLC1 is regulated by
PKD-mediated GAP domain phosphorylation. Exp Cell Res. 317:496–503.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Tompa P: Intrinsically unstructured
proteins. Trends Biochem Sci. 27:527–533. 2002. View Article : Google Scholar
|
|
107
|
Hermeking H: The 14-3-3 cancer connection.
Nat Rev Cancer. 3:931–943. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Scholz RP, Regner J, Theil A, Erlmann P,
Holeiter G, Jähne R, Schmid S, Hausser A and Olayioye MA: DLC1
interacts with 14-3-3 proteins to inhibit RhoGAP activity and block
nucleocytoplasmic shuttling. J Cell Sci. 122:92–102. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Wuestefeld T and Zender L: DLC1 and liver
cancer: the Akt connection. Gastroenterology. 139:1093–1096. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Oliveira AM, Ross JS and Fletcher JA:
Tumor suppressor genes in breast cancer: the gatekeepers and the
caretakers. Am J Clin Pathol. 124:S16–S28. 2005.PubMed/NCBI
|
|
111
|
Meyer N and Penn LZ: Reflecting on 25
years with MYC. Nat Rev Cancer. 8:976–990. 2008.PubMed/NCBI
|
|
112
|
Varmus H: Retroviruses. Science.
240:1427–1435. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Peters G: Oncogenes at viral integration
sites. Cell Growth Differ. 1:503–510. 1990.PubMed/NCBI
|
|
114
|
Popescu NC and Zimonjic DB:
Chromosome-mediated alterations of the MYC gene in human cancer. J
Cell Mol Med. 6:151–159. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Payne GS, Bishop JM and Varmus HE:
Multiple arrangements of viral DNA and an activated host oncogene
in bursal lymphomas. Nature. 295:209–214. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Popescu NC, Zimonjic DB and DiPaolo JA:
Viral integration, fragile sites and proto-oncogenes in human
neoplasia. Hum Genet. 84:383–386. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Liu J, Kaur G, Zhawar VK, Zimonjic DB,
Popescu NC, Kandpal R and Athwal RS: Role of SV40 integration site
at chromosomal interval 1q21.1 in immortalized CRL2504 cells.
Cancer Res. 69:7819–7825. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Weinberg RA: Integrated genomes of animal
viruses. Annu Rev Biochem. 49:197–226. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Bester AC, Schwartz M, Schmidt M, Garrigue
A, Hacein-Bey-Abina S, Cavazzana-Calvo M, Ben-Porat N, Von Kalle C,
Fischer A and Kerem B: Fragile sites are preferential targets for
integrations of MLV vectors in gene therapy. Gene Ther.
13:1057–1059. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Croce CM and Nowell PC: Molecular basis of
human B cell neoplasia. Blood. 65:1–7. 1985.
|
|
121
|
Zimonjic DB, Keck-Waggoner C and Popescu
NC: Novel genomic imbalances and chromosome translocations
involving c-myc gene in Burkitt’s lymphoma. Leukemia. 15:1582–1588.
2001.PubMed/NCBI
|
|
122
|
Alitalo KM and Schwab M: Oncogene
amplification in tumor cells. Adv Cancer Res. 47:235–281. 1986.
View Article : Google Scholar
|
|
123
|
Alitalo K, Schwab M, Lin CC, Varmus HE and
Bishop JM: Homogeneously staining chromosomal regions contain
amplified copies of an abundantly expressed cellular oncogene
(c-myc) in malignant neuroendocrine cells from a human colon
carcinoma. Proc Natl Acad Sci USA. 80:1707–1711. 1983. View Article : Google Scholar
|
|
124
|
Zimonjic DB, Keck-Waggoner CL, Yuan BZ,
Kraus MH and Popescu NC: Profile of genetic alterations and
tumorigenicity of human breast cancer cells. Int J Oncol.
16:221–230. 2000.PubMed/NCBI
|
|
125
|
Elenbaas B, Spirio L, Koerner F, Fleming
MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC and Weinberg RA:
Human breast cancer cells generated by oncogenic transformation of
primary mammary epithelial cells. Genes Dev. 15:50–65. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Kaposi-Novak P, Libbrecht L, Woo HG, Lee
YH, Sears NC, Coulouarn C, Conner EA, Factor VM, Roskams T and
Thorgeirsson SS: Central role of c-Myc during malignant conversion
in human hepatocarcinogenesis. Cancer Res. 69:2775–2782. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Farazi PA and DePinho RA: Hepatocellular
carcinoma pathogenesis: from genes to environment. Nat Rev Cancer.
6:674–687. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Schlaeger C, Longerich T, Schiller C,
Bewerunge P, Mehrabi A, Toedt G, Kleeff J, Ehemann V, Eils R,
Lichter P, Schirmacher P and Radlwimmer B: Etiology-dependent
molecular mechanisms in human hepatocarcinogenesis. Hepatology.
47:511–520. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Wei Y, Ponzetto A, Tiollais P and Buendia
MA: Multiple rearrangements and activated expression of c-myc
induced by woodchuck hepatitis virus integration in a primary liver
tumour. Res Virol. 143:89–96. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Tokino T and Matsubara K: Chromosomal
sites for hepatitis B virus integration in human hepatocellu lar
carcinoma. J Virol. 65:6761–6764. 1991.PubMed/NCBI
|
|
131
|
Yunis JJ, Soreng AL and Bowe AE: Fragile
sites are targets of diverse mutagens and carcinogens. Oncogene.
1:59–69. 1987.PubMed/NCBI
|
|
132
|
Yang L, He J, Chen L and Wang G: Hepatitis
B virus X protein upregulates expression of SMYD3 and C-MYC in
HepG2 cells. Med Oncol. 26:445–451. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Santoni-Rugiu E, Nagy P, Jensen MR, Factor
VM and Thorgeirsson SS: Evolution of neoplastic development in the
liver of transgenic mice co-expressing c-myc and transforming
growth factor. Am J Pathol. 149:407–428. 1996.PubMed/NCBI
|
|
134
|
Sargent LM, Sanderson ND and Thorgeirsson
SS: Ploidy and karyotypic alterations associated with early events
in the development of hepatocarcinogenesis in transgenic mice
harboring c-myc and transforming growth factor alpha transgenes.
Cancer Res. 56:2137–2142. 1996.
|
|
135
|
Factor VM, Laskowska D, Jensen MR, Woitach
JT, Popescu NC and Thorgeirsson SS: Vitamin E reduces chromosomal
damage and inhibits hepatic tumor formation in a transgenic mouse
model. Proc Natl Acad Sci USA. 97:2196–2201. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Sargent LM, Zhou X, Keck CL, Sanderson ND,
Zimonjic DB, Popescu NC and Thorgeirsson SS: Nonrandom cytogenetic
alterations in hepatocellular carcinoma from transgenic mice
overexpressing c-Myc and transforming growth factor-alpha in the
liver. Am J Path. 154:1047–1055. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Grisham JW: Interspecies comparison of
liver carcinogenesis: implications for cancer risk assessment.
Carcinogenesis. 18:59–81. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Grisham JW: Molecular genetic alterations
in primary hepatocellular meoplasm: hepatocellular adenoma,
hepatocellular carcinoma, and hepatoblastoma. The Molecular Basis
of Human Cancer. Coleman WB and Tsongalis GT: Humana Press; Totowa,
New Jersey, NJ: pp. 259–346. 2001
|
|
139
|
Durkin ME, Keck-Waggoner CL, Popescu NC
and Thorgeirsson SS: Integration of a c-myc transgene results in
disruption of the mouse Gtf2ird1 gene, the homologue of the human
GTF2IRD1 gene hemizygously deleted in Williams-Beuren syndrome.
Genomics. 73:20–27. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Tassabehji M, Hammond P, Karmiloff-Smith
A, Thompson P, Thorgeirsson SS, Durkin ME, Popescu NC, Hutton T,
Metcalfe K, Rucka A, Stewart H, Read AP, Maconochie M and Donnai D:
GTF2IRD1 in craniofacial development of humans and mice. Science.
310:1184–1187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Zimonjic DB, Ullmannova-Benson V, Factor
VM, Thorgeirsson SS and Popescu NC: Recurrent and nonrandom DNA
copy number and chromosome alterations in Myc transgenic mouse
model for hepatocellular carcinogenesis: implications for human
disease. Cancer Genet Cytogenet. 191:17–26. 2009. View Article : Google Scholar
|
|
142
|
Zimonjic DB, Zhang H, Shan Z, Factor VM,
Trent J, Thorgeirsson SS and Popescu NC: DNA amplification
associated with double minutes originating from chromosome 19 in
mouse hepatocellular carcinoma. Cytogenet Cell Genet. 93:114–116.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Murakami H, Sanderson ND, Nagy P, Marino
PA, Merlino G and Thorgeirsson SS: Transgenic mouse model for
synergistic effects of nuclear oncogenes and growth factors in
tumorigenesis: interaction of c-myc and transforming growth factor
alpha in hepatic oncogenesis. Cancer Res. 53:1719–1723. 1993.
|
|
144
|
Shachaf CM, Kopelman AM, Arvanitis C,
Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B,
Cardiff RD, Yang Q, Bishop JM, Contag CH and Felsher DW: MYC
inactivation uncovers pluripotent differentiation and tumour
dormancy in hepatocellular cancer. Nature. 431:1112–1117. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Zender L, Spector MS, Xue W, Flemming P,
Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, Mu D,
Lucito R, Powers S and Lowe SW: Identification and validation of
oncogenes in liver cancer using an integrative oncogenomic
approach. Cell. 125:1253–1267. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Roth JA and Grammer SF: Tumor suppressor
gene therapy. Tumor Supressor Genes: Regulation, Function and
Medicinal Applications. El-Deiry WS: 2. Humana Press; Totowa, NJ:
pp. 577–597. 2003, View Article : Google Scholar
|
|
147
|
Weinstein IB: Cancer. Addiction to
oncogenes - the Achilles heel of cancer. Science. 297:63–64. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Sharma SV and Settleman J: Oncogene
addiction: setting the stage for molecularly targeted cancer
therapy. Genes Dev. 21:3214–3231. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Zhang L, Zhang J, Xie L, Xie X, Guo Q, Lv
J, Gao Z, Qian Z, Yin X, Zheng L, Zhu G, Ji Q and Ren Z: Molecular
characterization of hepatocellular carcinoma (HCC) patient derived
explant models. Proc Am Assoc Cancer Res. 52:5772011.
|
|
150
|
Wong CC, Wong CM, Au SL and Ng IO:
RhoGTPases and Rho-effectors in hepatocellular carcinoma
metastasis: ROCK N’ Rho move it. Liver Int. 30:642–656.
2010.PubMed/NCBI
|
|
151
|
Takamura M, Sakamoto M, Genda T, Ichida T,
Asakura H and Hirohashi S: Inhibition of intrahepatic metastasis of
human hepatocellular carcinoma by Rho-associated protein kinase
inhibitor Y-27632. Hepatology. 33:577–581. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Ogawa T, Tashiro H, Miyata Y, Ushitora Y,
Fudaba Y, Kobayashi T, Arihiro K, Okajima M and Asahara T:
Rho-associated kinase inhibitor reduces tumor recurrence after
liver transplantation in a rat hepatoma model. Am J Transplant.
7:347–355. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Nakajima M, Hayashi K, Egi Y, Katayama K,
Amano Y, Uehata M, Ohtsuki M, Fujii A, Oshita K, Kataoka H, Chiba
K, Goto N and Kondo T: Effect of Wf-536, a novel ROCK inhibitor,
against metastasis of B16 melanoma. Cancer Chemother Pharmacol.
52:319–324. 2003. View Article : Google Scholar
|
|
154
|
McHenry PR and Vargo-Gogola T: Pleiotropic
functions of Rho GTPase signaling: a Trojan horse or Achilles’ heel
for breast cancer treatment? Curr Drug Targets. 11:1043–1058.
2010.
|
|
155
|
Ullmannova V and Popescu NC: Inhibition of
cell proliferation, induction of apoptosis, reactivation of DLC1,
and modulation of other gene expression by dietary flavone in
breast cancer cell lines. Cancer Detect Prev. 31:110–118. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Pang X, Yi T, Yi Z, Cho S G, Qu W, Pinkaew
D, Fujise K and Liu M: Morelloflavone, a biflavonoid, inhibits
tumor angiogenesis by targeting rho GTPases and extracellular
signal-regulated kinase signaling pathways. Cancer Res. 69:518–525.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Liu H, Dong A, Gao C, Tan C, Xie Z, Zu X,
Qu L and Jiang Y: New synthetic flavone derivatives induce
apoptosis of hepatocarcinoma cells. Bioorg Med Chem. 18:6322–6328.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Yoshizumi T, Ohta T, Ninomiya I, Terada I,
Fushida S, Fujimura T, Nishimura G, Shimizu K, Yi S and Miwa K:
Thiazolidinedione, a peroxisome proliferator-activated
receptor-gamma ligand, inhibits growth and metastasis of HT-29
human colon cancer cells through differentiation-promoting effects.
Int J Oncol. 25:631–639. 2004.
|
|
159
|
Zhou X, Yang XY and Popescu NC:
Synergistic antineoplastic effect of DLC1 tumor suppressor protein
and histone deacetylase inhibitor, suberoylanilide hydroxamic acid
(SAHA), on prostate and liver cancer cells: perspectives for
therapeutics. Int J Oncol. 36:999–1005. 2010.
|
|
160
|
Chung GE, Yoon JH, Lee JH, Kim HY, Myung
SJ, Yu SJ, Lee SH, Lee SM, Kim YJ and Lee HS: Ursodeoxycholic
acid-induced inhibition of DLC1 protein degradation leads to
suppression of hepatocellular carcinoma cell growth. Oncol Rep.
25:1739–1746. 2011.PubMed/NCBI
|
|
161
|
Murphy DJ, Junttila MR, Pouyet L, Karnezis
A, Shchors K, Bui DA, Brown-Swigart L, Johnson L and Evan GI:
Distinct thresholds govern Myc’s biological output in vivo. Cancer
Cell. 14:447–457. 2008.PubMed/NCBI
|
|
162
|
Larsson LG and Henricksson MA: The Yin and
Yang functions of the Myc oncoprotein in cancer development and as
targets for therapy. Exp Cell Res. 316:1429–1437. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Lin CP, Liu CR, Lee CN, Chan TS and Liu
HE: Targeting c-Myc as a novel approach for hepatocellular
carcinoma. World J Hepatol. 2:16–20. 2010.PubMed/NCBI
|
|
164
|
Brooks TA and Hurley LH: Targeting MYC
expression through G-quardruplexes. Genes Cancer. 1:641–649. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR,
Seitz JF, Borbath I, Haussinger D, Giannaris T, Shan M, Moscovici
M, Voliotis D and Bruix J: SHARP Investigators Study Group:
Sorafenib in advanced hepatocellular carcinoma. N Engl J Med.
359:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Llovet J M and Bruix J: Molecular targeted
therapies in hepatocellular carcinoma. Hepatology. 48:1312–1327.
2008.
|
|
167
|
Cao Z, Fan-Minogue H, Bellovin DI,
Yevtodiyenko A, Arzeno J, Yang Q, Gambhir SS and Felsher DW: MYC
phosphorylation, activation, and tumorigenic potential in
hepatocellular carcinoma are regulated by HMG-CoA reductase. Cance
Res. 71:2286–2297. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Kawata S, Yamasaki E, Nagase T, Inui Y,
Ito N, Matsuda Y, Inada M, Tamura S, Noda S, Imai Y and Matsuzawa
Y: Effect of pravastatin on survival in patients with advanced
hepatocellular carcinoma. A randomized controlled trial. Br J
Cancer. 84:886–891. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Homma Y and Emori Y: A dual functional
signal mediator showing RhoGAP and phospholipase C-delta
stimulating activities. EMBO J. 14:286–291. 1995.PubMed/NCBI
|
|
170
|
Ponting CP and Aravind L: START: a
lipid-binding domain in StAR, HD-ZIP and signalling proteins.
Trends Biochem Sci. 24:130–132. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Wang H, Han H, Mousses S and Von Hoff DD:
Targeting loss-of-function mutations in tumor-suppressor genes as a
strategy for development of cancer therapeutic agents. Semin Oncol.
33:513–520. 2006. View Article : Google Scholar : PubMed/NCBI
|