Contents
Introduction
What is epimutation?
Germline epimutation and disease
Epimutation and cancer
Epimutation and Lynch syndrome
Conclusion
Introduction
Epimutation includes epigenetic repression of active
genes for which expression is not suppressed, or epigenetic
activation of genes for which expression is normally repressed.
Disease susceptibility is generally determined not only by DNA
sequence mutation, but also by changes in the activities of genes
and chromosomal regions. Epigenetic repression has attracted
attention as a mechanism underlying these changes in activities.
Epigenetic modification causes repression of gene activity and is
required for cell division and histogenesis. Phenotypic variation
in genetically identical cells is caused by differences in
epigenetic profiles. In addition, epimutation may be the first step
of tumorigenesis in specific cancers and this provides a direct
link to predisposition for cancer. In this article, we review the
relationship between epimutation and cancer.
What is epimutation?
Epimutation was first used as a term for epigenetic
changes, including methylation, that have no effect on DNA sequence
(1). It is currently defined as
abnormal transcriptional repression in active genes and/or abnormal
activation of usually repressed genes caused by errors in
epigenetic gene repression (1–3).
Epimutation affects one or both alleles of a gene. By preventing
transcription of the affected allele, epimutation effectively
reduces the level of the gene product. Tumor cells are a typical
consequence of epimutation in mammals. Epimutation in cancer
generally occurs in somatic cells and manifests as tumor
progression. Different kinds of epimutation occur in many cancers,
but epimutation is particularly common in tumor suppressor genes.
Epimutation may also arise in the germline, and constitutional
epimutation has been shown, but it is thought that epigenetic
mutation generally occurs stochastically in genes. Epimutation is
also found in non-cancerous tissues in the stage prior to
tumorigenesis. Methylation in normal tissues is referred to as a
field effect and has been shown to have a relationship with
clonally-related malignant cells in tissues.
Germline epimutation is defined as an event that
occurs in the genes of germ cells and is maintained in
fertilization and embryonic development, resulting in persistence
in all adult somatic cells. In this manner, epigenetic
characteristics are transmitted through generations, and therefore,
individuals with the same germline epimutation have a similar
oncogenic risk. However, epimutation is not always inherited and
epimutation has been shown to have a hereditary form that does not
follow Mendelian inheritance (4–7).
Epimutation has also been shown to disappear in spermatogenesis
(8). Previous studies have
indicated epimutation is inherited from the mother alone, which
suggests that epimutation is unlikely to disappear during oogenesis
(7,8). Some somatic aberrations of genomic
imprinting are also considered to be germline epimutations
(3). Constitutional epimutation is
defined as that occurring in the stage of early embryonic
development and in all human tissues in the stage prior to
triploblastic differentiation. Epimutation is mosaic in somatic
cells and is not found in all cells, with no study confirming
transmission from the previous generation. Epimutation is also the
first step of tumorigenesis and can be a direct cause of
carcinogenesis.
Germline epimutation and disease
Epimutation is involved in genomic imprinting
(Table I), in which a gene
inherited from one parent is selectively expressed and genetic
disease develops if this gene has a deficiency, even if the other
allele is normal. Phenotypes specific to genomic imprinting are
regulated by imprinting control centers (ICs). ICs are short
sequences in an imprinted gene. Only one allele of an IC is
methylated and transcribed, permitting regulation of imprinting.
Various genetic changes including microdeletion have been found in
ICs and these are considered to be caused by epimutation in
Angelman syndrome (AS), Prader-Willi syndrome (PWS) and
Beckwith-Wiedemann syndrome (BWS). BWS is a congenital disorder
with a high risk of embryonal tumors such as Wilms’ tumor,
hepatoblastoma and rhabdomyosarcoma. Chromosome 11p15.5 has been
identified as the disease locus. The 11p15.5 locus contains two
imprinted domains, the cyclin-dependent kinase inhibitor
1C/KCNQ1 opposite antisense transcript 1
(CDKN1C/KCNQ1OT1) and insulin-like growth factor 2
(IGF2)/H19 domains, through which expression of
imprinted genes surrounding the imprinting regulation regions is
controlled. In 30–50% of cases of BWS, expression of CDKN1C
decreases due to DNA hypomethylation, while expression of
IGF2 increases due to DNA hypermethylation of the
IGF2/H19 domain (9).
Silver-Russell syndrome (SRS) is a disorder characterized by
intrauterine growth retardation and severe postnatal growth
retardation, and is caused by epimutation of the H19 gene in
the 11p15.5 region (10). Thus,
these diseases develop due to abnormality of the respective
ICs.
 | Table IEpimutation and disease. |
Table I
Epimutation and disease.
| Gene name | Epimutation
type | Disease | Reference |
|---|
| hMLH1 | Germline,
constitutional | Lynch syndrome | (4,24,43) |
| hMSH2 | Germline | Lynch syndrome | (28) |
| DAPK1 | Unknown | B-cell CLL | (29) |
| HBA2 | Unknown | α-thalassemia | (12) |
| BRCA2 | Constitutional | Sporadic breast
cancer | (40) |
|
KIP2/LIT1 | Unknown | Beckwith-Wiedemann
syndrome | (9) |
| IGF2 | Unknown | Beckwith-Wiedemann
syndrome | (9) |
| H19 | Unknown | Silver-Russell
syndrome | (10) |
Epimutation is also involved in onset of
α-thalassemia. Epimutations occur due to variations such as genomic
insertion and deletion, and changes in the length of tandem
repeats, which are referred to as copy number variation (CNV)
(11). In α-thalassemia, the
deletion locus of the LUC7-like (LUC7L) gene is
co-located with Hemoglobin α 2 (HBA2), an α globin
gene, resulting in methylation of the HBA2 promoter
(12).
Epimutation and cancer
Studies of familial cancer have shown that specific
gene groups inactivated by mutation cause a predisposition to
cancer. The tumor suppressor gene retinoblastoma (RB)
is a disease gene for hereditary cancer that was initially
identified through mutations found in cases of retinoblastoma
(13). Subsequently, Nishisho
et al found mutations in adenomatous polyposis coli
(APC) in familial adenomatous polyposis (14), and Hussussian et al
identified mutations in cyclin-dependent kinase inhibitor 2A
(CDKN2A) in familial melanoma (15). The mutations in different tumor
suppressor genes indicate various underlying mechanisms as causes
of cancer. The DNA mismatch repair genes breast cancer
susceptibility gene 1 (BRCA1), human MutL homologue
1 (hMLH1) and human MutS homologue 2
(hMSH2) have also been linked to a predisposition for
familial cancer. Inactivation of genes induced by mutation in
hereditary cancer is recessive inheritance and most carriers have
no abnormal phenotype. However, mutation, inactivation and loss of
heterozygosity are likely to occur in other normal alleles, and
consequently, cancer morbidity is frequently dominantly
inherited.
RB methylation was identified as the first
example of epimutation in a cancer-related gene (16,17).
Subsequently, many other oncogenes, including Von
Hippel-Lindau (VHL), hMLH1, APC and
BRCA1, have been shown to be methylated in sporadic cancers
(18–20). Among DNA mismatch genes,
hMLH1 and hMSH2 methylation causes a predisposition
for endometrial, small intestine and ovarian cancers, in addition
to colon cancer. hMLH1 and hMSH2 encode mismatch
repair proteins and inactivation of these genes causes
microsatellite instability (MSI) in tumor cells (21). hMLH1 is also methylated in
cases of sporadic colorectal cancer (19) with the same phenotype of mismatch
repair defect and clinicopathologic characteristics similar to
those of hereditary tumors. Such sporadic colorectal cancer also
has a close relationship with cancer with a CpG island methylator
phenotype (CIMP). CIMP-positive cancers frequently have methylation
in CpG islands in a specific promoter region (22). These cancers usually occur in the
ascending colon and are particularly common in elderly women.
Gazzoli et al first found that hMLH1
may be methylated in peripheral blood, as well as in tumor cells,
in patients with colorectal cancer (23). In a study of 14 patients with Lynch
syndrome with MSI, hypermethylated hMLH1 was found in normal
blood DNA in one 25-year-old female patient (23). Allele methylation in tissues
derived from an embryologically discrete germ layer suggests the
presence of a constitutional or germline methylation pattern. Since
no mutation was found in specimens of her parents, hereditary
evidence for epimutation was not obtained; however, it is of
interest that methylation occurred in a young patient. It has also
been shown that patients with colorectal cancer with methylation in
one allele of hMLH1 have constitutional methylation
(24). Suter et al showed
hMLH1 methylation in a phenotype derived from a
triploblastic origin in 2 patients with colorectal cancer (24). Tissues of their parents were not
examined, but no methylation was found in tissues in 4 of their 5
children.
Much controversy exists regarding constitutional
epimutation; i.e., whether this is transmitted from a mother or
father, or occurs de novo in early embryonic development.
Miyakura et al showed that complete methylation in the
hMLH1 promoter region played an important role in
hMLH1 inactivation in patients with sporadic colorectal
cancer with high MSI (25). This
methylation occurred in both alleles and methylation in the upper
hMLH1 promoter region was also found in normal colonic
mucosa adjacent to cancer tissue in one-third of patients with
colorectal cancer associated with complete methylation (26). Miyakura et al subsequently
examined methylation in the hMLH1 promoter region of
peripheral blood lymphocytes (PBLs) in 30 patients with early-onset
sporadic colorectal cancer or multiple primary cancer. Four of
these patients (2 with early-onset sporadic colon cancer, 1 with
colon cancer and 1 with multiple cancer including endometrial
cancer) had complete methylation in the hMLH1 promoter
region in PBLs (27). Methylation
was found only in one allele. No methylation was detected in PBLs
of the sister of a patient with early-onset sporadic colorectal
cancer. MSI was found in all patients and methylation was also
detected in normal tissues of the large intestine, digestive
mucosa, endometrium and bone marrow of 3 patients. It is of
interest that loss of heterozygosity (LOH) in both alleles of
hMLH1, loss of G alleles in somatic cells of the
hMLH1 locus and methylation of both alleles of hMLH1
were detected in cases of colon cancer. This finding is consistent
with the mechanism of carcinogenesis based on germline epimutation
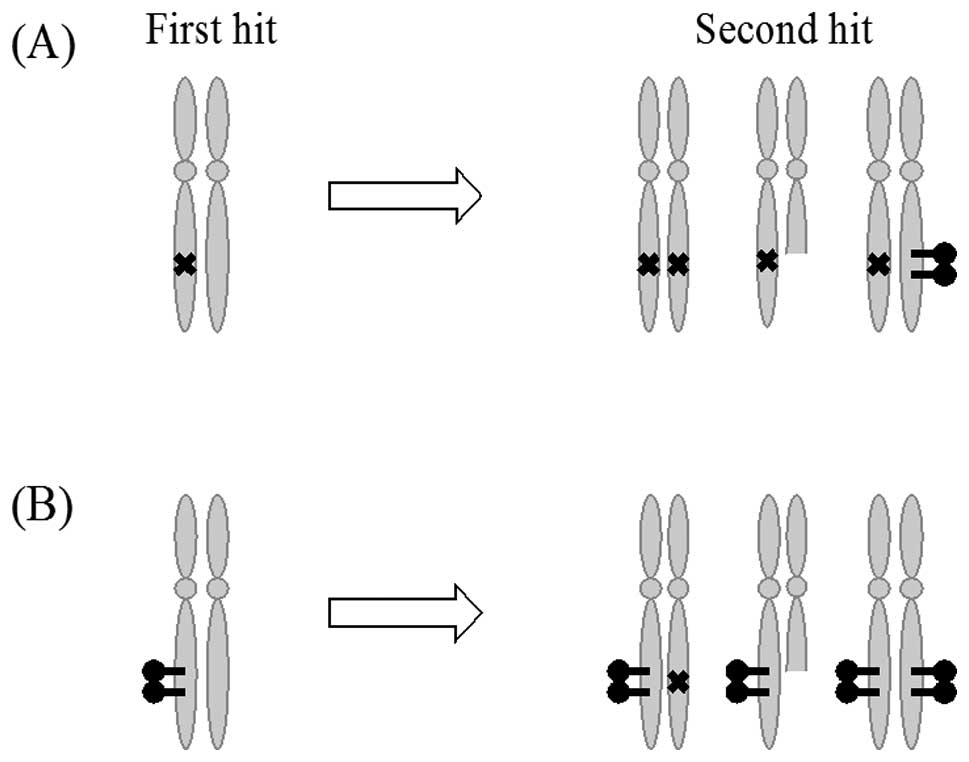
proposed by Suter et al based on Knudson’s ‘two hit’
hypothesis (Fig. 1) (24).
Epimutation is not always inherited and has also
been shown to have a hereditary form that does not follow Mendelian
inheritance (4–7). Epimutation disappears in
spermatogenesis, and may by inherited from the mother alone,
suggesting that disappearance during oogenesis is unlikely
(7,8). In a cohort study of 160 patients with
Lynch syndrome without germline mismatch repair gene mutations,
constitutional methylation of hMLH1 was found in only one
patient. No methylation of hMLH1 was detected in this
patient’s parents or siblings. This finding suggests that
clinicopathologic characteristics are a better indicator than
family history if constitutional epimutation of a tumor suppressor
gene is identified in a cancer patient (4).
Epimutation is known to have a relationship with
chronic lymphocytic leukemia (CLL). In CLL, apoptosis in leukemia
cells is inhibited by enhanced production of B-cell lymphoma 2
(BCL2) and methylation of the promoter region of
death-associated protein kinase 1 (DAPK1) (29). DAPK1 was identified as a
familial tumor suppressor gene and methylation is found in the
DAPK1 promoter region in CLL (29). Expression of homeobox B7 (HOXB7)
proteins connected with the upper promoter region increases due to
methylation, and 75% of DAPK1 genes are downregulated.
Inactivation of DAPK1 due to methylation is a cause of both
familial and sporadic CLL, and hypomethylation of DAPK1 has
been found in peripheral blood mononuclear cells (PBMCs) of healthy
individuals (30); however, the
relationship with hypomethylation in CLL is unclear.
Many studies have examined the relationship between
breast cancer and BRCA1 mutation. Armes and Venter (31) and Lakhani et al(32) showed that breast cancer in patients
with germline BRCA1 mutations had histological
characteristics including a high mitotic index and lymphatic
infiltration. This form is currently referred to as the basal-like
type. Foulkes et al showed that 80–90% of cancers that
developed in carriers of germline mutations in BRCA1 were of
the basal-like type (32).
Subsequently, methylation in the promoter region of BRCA1
has been found in sporadic breast cancer (34). The relationship between
BRCA1 mutation and methylation has been examined based on
the hypothesis that a sporadic tumor with BRCA1 methylation
should be similar to a tumor with BRCA1 mutation, if
BRCA1 methylation causes tumorigenesis. Catteau and Morris
showed that sporadic tumors with BRCA1 methylation had
pathological characteristics similar to those of hereditary breast
cancer (35) and Hedenfalk et
al found that the phenotypes of the cancers were similar to
each other (34). Many studies
have shown that tumors with BRCA1 methylation have
high-grade, estrogen receptor-negative and progesterone
receptor-negative characteristics and a high incidence in young
women (36). These characteristics
are referred to as BRCA1-like. Esteller et al
detected BRCA1 methylation in 67% of medullary carcinomas
and 55% of mucinous adenocarcinomas, and showed that this phenotype
was frequently found in families with BRCA1 mutation
(34). Snell et al found
methylation in the promoter region of BRCA1 in normal
tissues of breast cancer patients with a specific BRCA1-like
tissue type (37). No germline
mutation of BRCA1 and BRCA2 was detected in these
patients. This finding suggests constitutional epimutation of
BRCA1 in breast cancer patients. Methylation of BRCA1
is considered to be the first hit and the histologically
characteristic type occurs due to deletion of both BRCA1
genes.
Epimutation and Lynch syndrome
Lynch syndrome (HNPCC) is a typical familial tumor
with autosomal dominant inheritance. A study of morbidity in
colorectal cancer showed that Lynch syndrome develops in
approximately 3% of patients with colorectal cancer (38). Aberrant mismatch repair (MMR) genes
are involved in carcinogenesis of Lynch syndrome. A total of 6 MMR
genes have been cloned to date: hMSH2, hMLH1,
human MutS homologue 3 (hMSH3) and 6
(hMSH6), and postmeiotic segregation increased 1
(PMS1) and 2 (PMS2). Lynch syndrome kindreds
have been confirmed to have mutations in 3 of these genes:
hMSH2, hMLH1 and hMSH6(37). hMLH1 and hMSH2
mutations are particularly considered to be a cause of Lynch
syndrome. Such mutations also cause a predisposition for
endometrial, small intestine and ovarian cancer, in addition to
rectal cancer. hMLH1 and hMSH2 encode MMR proteins
and inactivation of these genes may cause MSI in tumors (21). Microsatellites are short-tandem
repeats (STRs) that are typically in the non-coding region, and
therefore, have no relationship with production of aberrant
proteins with mutations. However, some STRs exist in regions
encoding important genes such as BCL2-associated X protein
(BAX), which is related to apoptosis induction, and
insulin-like growth factor 2 receptor (IGF2R), which
is involved in inhibition of cell proliferation. Mutation of these
genes is associated with carcinogenesis.
Recently, a new type of Lynch syndrome has been
found with no pathologic mutation in MMR genes, but epimutation in
the promoter region of hMLH1 or hMSH2(40). This finding suggested that
epimutation in germline hMLH1 can be a cause of Lynch
syndrome. In families with hMSH2 methylation, germline
mutation of epithelial cell adhesion molecule (EPCAM)
is a cause of epimutation in the upper hMSH2 promoter
(41). EPCAM is highly
expressed in epithelial tissues and cancer (42). A 3′ end deletion in EPCAM
causes readthrough with hMSH2, which leads to
hypermethylation of CpG islands in the hMSH2 promoter
(43). Furthermore, methylation of
hMSH2 in Lynch syndrome kindreds has been shown to be
transmitted genetically (28). It
is of interest that no methylation of hMSH2 has currently
been shown. In contrast to many patients with constitutional
methylation of hMLH1, for which allele methylation was
already confirmed, methylation of an hMSH2 allele was
detected in approximately 50%. The level of methylation also
depends on the tissues tested. Methylation of hMSH2 follows
Mendelian inheritance, in contrast to epimutation of hMLH1,
and patients with Lynch syndrome due to epimutation had a different
level of methylation from that of epimutation carriers in their
family and/or different levels among different tissues.
Conclusion
Epimutation has characteristics of inheritance,
deletion in embryonic development, and a hereditary form that does
not follow Mendelian inheritance. Cancers involved in epimutation
include Lynch syndrome (HNPCC), chronic lymphocytic leukemia,
breast cancer, and ovarian cancer. Tumors with epimutation may have
specific histological characteristics, and methylation patterns in
normal tissues may indicate the cancer tissue type. Use of this
information may reduce the need for current invasive tests,
including biopsy of tumor tissues. The cancer risk of healthy
individuals may also be accessible through a minimally invasive
procedure based on differences in methylation patterns between
healthy individuals and cancer patients. Development of such
techniques requires elucidation of the many factors causing
methylation. It is currently unknown how the many variations of
methylation occur in normal somatic tissues at an individual level;
however, methylation patterns are maintained in individuals.
Identical twins have different patterns of DNA methylation and
these differences are increased in twins living in different
environments (44). CpG islands
are methylated age-dependently, and methylation may influence
methyl group metabolism such as that involving folic acid,
methionine, choline, vitamin B12 and betaine due to metabolite
ingestion, which may change methylation patterns. Environmental
factors, particularly in early development, may be associated with
predisposition to diseases, including cancer, and other epigenomic
changes (45). The possible
association of methylation with environmental factors and aging, in
addition to genetic factors, indicates the need for additional
studies of the relationship between epimutation and these
factors.
Research efforts are also currently directed at
improvement of epigenetic abnormalities based on epimutation
studies. Epigenetic therapy to enhance re-expression of tumor
suppressor genes using DNA methyltransferase (DNMT) inhibitors
(azacitidine and decitabine) has some effect on hematologic
malignancies (44). However,
epigenetic therapy with a combination of a DNMT inhibitor and a
histone deacetylase (HDAC) inhibitor cannot completely remodel
chromosomes, and consequently has no effect on re-expression of
stable genes. Furthermore, many studies show that re-expressed
genes are inhibited again after withdrawal of epigenetic therapy.
Thus, epigenetic therapy still has many limitations and further
studies are needed to improve this approach.
Acknowledgements
The authors gratefully acknowledge the
grant support from the Japan Society for the Promotion of Science
(JSPS) through a Grant-in-Aid for Scientific Research (KAKENHI); a
Grant-in-Aid for Scientific Research (C) (22591866), and a
Grant-in-Aid for Young Scientists (B) (21791573); the Ichiro
Kanehara Foundation; Kobayashi Foundation for Cancer Research; and
the Keio University Medical Science Fund through a Research Grant
for Life Sciences and Medicine.
References
|
1
|
Holliday R: The inheritance of epigenetic
defects. Science. 238:163–170. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Das OP and Messing J: Variegated phenotype
and developmental methylation changes of a maize allele originating
from epimutation. Genetics. 136:1121–1141. 1994.PubMed/NCBI
|
|
3
|
Schofield PN, Joyce JA, Lam WK, Grandjean
V, Ferguson-Smith A, Reik W and Maher ER: Genomic imprinting and
cancer; new paradigms in the genetics of neoplasia. Toxicol Lett.
120:151–160. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hitchins M, Williams R, Cheong K, et al:
MLH1 germline epimutations as a factor in hereditary nonpolyposis
colorectal cancer. Gastroenterology. 129:1392–1399. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hitchins MP, Wong JJ, Suthers G, Suter CM,
Martin DI, Hawkins NJ and Ward RL: Inheritance of a
cancer-associated MLH1 germ-line epimutation. N Engl J Med.
356:697–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Valle L, Carbonell P, Fernandez V, Dotor
AM, Sanz M, Benitez J and Urioste M: MLH1 germline epimutations in
selected patients with early-onset non-polyposis colorectal cancer.
Clin Genet. 71:232–237. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Morak M, Schackert HK, Rahner N, et al:
Further evidence for heritability of an epimutation in one of 12
cases with MLH1 promoter methylation in blood cells clinically
displaying HNPCC. Eur J Hum Genet. 16:804–811. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hitchins MP and Ward RL: Erasure of MLH1
methylation in spermatozoa-implications for epigenetic inheritance.
Nat Genet. 39:12892007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cooper WN, Luharia A, Evans GA, et al:
Molecular subtypes and phenotypic expression of Beckwith-Wiedemann
syndrome. Eur J Hum Genet. 13:1025–1032. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schönherr N, Meyer E, Roos A, Schmidt A,
Wollmann HA and Eggermann T: The centromeric 11p15 imprinting
centre is also involved in Silver-Russell syndrome. J Med Genet.
44:59–63. 2007.PubMed/NCBI
|
|
11
|
Conrad DF, Pinto D, Redon R, et al:
Origins and functional impact of copy number variation in the human
genome. Nature. 464:704–712. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tufarelli C, Stanley JA, Garrick D, Sharpe
JA, Ayyub H, Wood WG and Higgs DR: Transcription of antisense RNA
leading to gene silencing and methylation as a novel cause of human
genetic disease. Nat Genet. 34:157–165. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Friend SH, Bernards R, Rogelj S, Weinberg
RA, Rapaport JM, Albert DM and Dryja TP: A human DNA segment with
properties of the gene that predisposes to retinoblastoma and
osteosarcoma. Nature. 323:643–646. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nishisho I, Nakamura Y, Miyoshi Y, et al:
Mutations of chromosome 5q21 genes in FAP and colorectal cancer
patients. Science. 253:665–669. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hussussian CJ, Struewing JP, Goldstein AM,
et al: Germline p16 mutations in familial melanoma. Nat Genet.
8:15–21. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Greger V, Passarge E, Höpping W, Messmer E
and Horsthemke B: Epigenetic changes may contribute to the
formation and spontaneous regression of retinoblastoma. Hum Genet.
83:155–158. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sakai T, Toguchida J, Ohtani N, Yandell
DW, Rapaport JM and Dryja TP: Allele-specific hypermethylation of
the retinoblastoma tumor-suppressor gene. Am J Hum Genet.
48:880–888. 1991.PubMed/NCBI
|
|
18
|
Herman JG, Latif F, Weng Y, et al:
Silencing of the VHL tumor-suppressor gene by DNA methylation in
renal carcinoma. Proc Natl Acad Sci USA. 91:9700–9704. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kane MF, Loda M, Gaida GM, Lipman J,
Mishra R, Goldman H, Jessup JM and Kolodner R: Methylation of the
hMLH1 promoter correlates with lack of expression of hMLH1 in
sporadic colon tumors and mismatch repair-defective human tumor
cell lines. Cancer Res. 57:808–811. 1997.PubMed/NCBI
|
|
20
|
Dobrovic A and Simpfendorfer D:
Methylation of the BRCA1 gene in sporadic breast cancer. Cancer
Res. 57:3347–3350. 1997.PubMed/NCBI
|
|
21
|
de la Chapelle A: Genetic predisposition
to human disease: allele-specific expression and low-penetrance
regulatory loci. Oncogene. 28:3345–3348. 2009.PubMed/NCBI
|
|
22
|
Weisenberger DJ, Siegmund KD, Campan M, et
al: CpG island methylator phenotype underlies sporadic
microsatellite instability and is tightly associated with BRAF
mutation in colorectal cancer. Nat Genet. 38:787–793. 2006.
View Article : Google Scholar
|
|
23
|
Gazzoli I, Loda M, Garber J, Syngal S and
Kolodner RD: A hereditary nonpolyposis colorectal carcinoma case
associated with hypermethylation of the MLH1 gene in normal tissue
and loss of heterozygosity of the unmethylated allele in the
resulting microsatellite instability-high tumor. Cancer Res.
62:3925–3928. 2002.
|
|
24
|
Suter CM, Martin DI and Ward RL: Germline
epimutation of MLH1 in individuals with multiple cancers. Nat
Genet. 36:497–501. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miyakura Y, Sugano K, Konishi F, et al:
Extensive methylation of hMLH1 promoter region predominates in
proximal colon cancer with microsatellite instability.
Gastroenterology. 121:1300–1309. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Miyakura Y, Sugano K, Konishi F, et al:
Methylation profile of the MLH1 promoter region and their
relationship to colorectal carcinogenesis. Genes Chromosomes
Cancer. 36:17–25. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miyakura Y, Sugano K, Akasu T, et al:
Extensive but hemiallelic methylation of the hMLH1 promoter region
in early-onset sporadic colon cancers with microsatellite
instability. Clin Gastroenterol Hepatol. 2:147–156. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chan TL, Yuen ST, Kong CK, et al:
Heritable germline epimutation of MSH2 in a family with hereditary
nonpolyposis colorectal cancer. Nat Genet. 38:1178–1183. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Raval A, Tanner SM, Byrd JC, et al:
Downregulation of death-associated protein kinase 1 (DAPK1) in
chronic lymphocytic leukemia. Cell. 129:879–890. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reddy AN, Jiang WW, Kim M, Benoit N,
Taylor R, Clinger J, Sidransky D and Califano JA: Death-associated
protein kinase promoter hypermethylation in normal human
lymphocytes. Cancer Res. 63:7694–7698. 2003.PubMed/NCBI
|
|
31
|
Armes JE and Venter DJ: The pathology of
inherited breast cancer. Pathology. 34:309–314. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lakhani SR, Van De Vijver MJ, Jacquemier
J, Anderson TJ, Osin PP, MaGuffog L and Easton DF: The pathology of
familial breast cancer: predictive value of immunohistochemical
markers estrogen receptor, progesterone receptor, HER-2, and p53 in
patients with mutations in BRCA1 and BRCA2. J Clin Oncol.
20:2310–2318. 2002. View Article : Google Scholar
|
|
33
|
Foulkes WD, Stefansson IM, Chappuis PO,
Bégin LR, Goffin JR, Wong N, Trudel M and Akslen LA: Germline BRCA1
mutations and a basal epithelial phenotype in breast cancer. J Natl
Cancer Inst. 95:1482–1485. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Esteller M, Silva JM, Dominguez G, et al:
Promoter hypermethylation and BRCA1 inactivation in sporadic breast
and ovarian tumors. J Natl Cancer Inst. 92:564–569. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Catteau A and Morris JR: BRCA1
methylation: a significant role in tumour development? Semin Cancer
Biol. 12:359–371. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hedenfalk I, Duggan D, Chen Y, et al:
Gene-expression profiles in hereditary breast cancer. N Engl J Med.
344:539–548. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Snell C, Krypuy M, Wong EM; kConFab
investigators; Loughrey MB and Dobrovic A: BRCA1 promoter
methylation in peripheral blood DNA of mutation negative familial
breast cancer patients with a BRCA1 tumour phenotype. Breast Cancer
Res. 10:R122008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hampel H, Frankel WL, Martin E, et al:
Feasibility of screening for Lynch syndrome among patients with
colorectal cancer. J Clin Oncol. 26:5783–5788. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Peltomäki P and Vasen H: Mutations
associated with HNPCC predisposition - Update of ICG-HNPCC/INSiGHT
mutation database. Dis Markers. 20:269–276. 2004.PubMed/NCBI
|
|
40
|
Hirata K, Kanemitsu S, Nakayama Y, Nagata
N, Itoh H, Ohnishi H, Ishikawa H and Furukawa Y; HNPCC registry and
genetic testing project of the Japanese Society for Cancer of the
Colon and Rectum (JSCCR): A novel germline mutation of MSH2 in a
hereditary nonpolyposis colorectal cancer patient with liposarcoma.
Am J Gastroenterol. 101:193–196. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Naruse H, Ikawa N, Yamaguchi K, et al:
Determination of splice-site mutations in Lynch syndrome
(hereditary nonpolyposis colorectal cancer) patients using
functional splicing assay. Fam Cancer. 8:509–517. 2009. View Article : Google Scholar
|
|
42
|
Winter MJ, Nagtegaal ID, van Krieken JH
and Litvinov SV: The epithelial cell adhesion molecule (Ep-CAM) as
a morpho-regulatory molecule is a tool in surgical pathology. Am J
Pathol. 163:2139–2148. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ligtenberg MJ, Kuiper RP, Chan TL, et al:
Heritable somatic methylation and inactivation of MSH2 in families
with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat
Genet. 41:112–117. 2009.PubMed/NCBI
|
|
44
|
Fraga MF, Ballestar E, Paz MF, et al:
Epigenetic differences arise during the lifetime of monozygotic
twins. Proc Natl Acad Sci USA. 102:10604–10609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Issa JP, Ottaviano YL, Celano P, Hamilton
SR, Davidson NE and Baylin SB: Methylation of the oestrogen
receptor CpG island links ageing and neoplasia in human colon. Nat
Genet. 7:536–540. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rose MG: Hematology: azacitidine improves
survival in myelodysplastic syndromes. Nat Rev Clin Oncol.
6:502–503. 2009. View Article : Google Scholar : PubMed/NCBI
|