Introduction
Hepatocellular carcinoma (HCC) is one of the most
prevalent malignant diseases worldwide (1). Many locoregional therapeutic
approaches, including surgical resection, radio-frequency ablation
(RFA), percutaneous ethanol injection (PEI), and transcatheter
hepatic arterial chemoembolization (TACE) have been applied in the
search for curative treatments for HCC. Although current advances
in therapeutic modalities have improved the prognosis of patients
with HCC, the survival rate is still unsatisfactory (2). One reason for the poor prognosis is
the high rate of recurrence after treatment (3–5).
Current therapeutic approaches do not prevent tumor recurrence
efficiently.
Patients with HCC demonstrate some dysfunctions in
their immune system, including abnormal innate and adaptive immune
responses (6). Therefore, one
strategy to reduce tumor recurrence is to enhance antitumor immune
responses that may induce sufficient inhibitory effects to prevent
tumor cell growth and survival. Dendritic cells (DCs) are
professional antigen presenting cells that play a central role in
the immune system by initiating an antigen-specific cytotoxic T
lymphocyte (CTL) response (7,8). DCs
acquire antigens through endocytosis and phagocytosis in peripheral
tissues in their immature state and become mature. Subsequently,
mature DCs migrate via blood and lymphatics to the secondary
lymphoid organs, where they prime T cells. Due to their unique
capacity to regulate T cell immunity, DCs are increasingly used as
adjuvants for vaccination strategies. Recently, several studies
have been performed using DC generated ex vivo from
peripheral blood, and no significant toxicities were observed in
the majority of patients. In addition, induction or enhancement of
cellular immune responses against tumor antigens was found after DC
vaccination (9,10).
Although immunotherapy strategies to eliminate HCC
have consistently demonstrated high efficacy in animal models, very
limited efficacy has been demonstrated in patients (11–20).
There are possible explanations that may explain this discrepancy,
but one major limitation for clinical trials is obtaining adequate
amounts of immunogenic tumor-associated antigens (TAAs). DC loaded
with autologous tumor or tumor lysates, which contain TAAs, are
most frequently used for clinical trials (11–13,16).
Another approach is to use apoptotic or necrotic tumor cells, which
are induced by the standard treatments for HCC, as tumor antigens.
Previous studies have shown that these cells effectively
cross-prime the T cell response and induce potent immunity
(14,15,18,20).However, ideal protocols to induce
antigen-specific immunity involve DC loaded with TAAs themselves if
such TAAs have been defined. Although many specific proteins have
been identified with differential expression profiles in HCC cells,
appropriate antigens for incorporation into DC vaccines for HCC
have not been defined. α-fetoprotein (AFP) is a potential candidate
antigen, and Butterfield et al reported that DC pulsed with
HLA-A0201-restricted peptides induced AFP-specific T cell
responses, though no clinical responses were observed (17).
To overcome these problems, we conducted a phase
I/II clinical study using DC vaccine prepared as follows: i)
TAA-pulsed mature DCs were used together with topical application
of toll-like receptor (TLR)-7 agonist; ii) recombinant proteins,
instead of epitope peptides, were used as a source of TAA to
overcome the restriction of HLA type; iii) 3 different HCC antigens
were used to cover the broad spectrum of HCC heterogeneity; iv) for
efficient delivery of antigens into the cytoplasm of DC,
cytoplasmic transduction protein (CTP)-mediated transduction system
(21) was used. The primary
objective of this study was to assess the safety, feasibility and
immune activity of multiple TAA-pulsed DC therapy. The efficacy of
this therapy was also evaluated.
Patients and methods
Patient selection
The clinical trial protocol was approved by the
Institutional Review Board of Ehime University Hospital (Approval
ID #0809003). Patients were informed of the investigative nature of
this study, and written consent in accordance with institutional
regulations was obtained prior to study entry. Eligibility criteria
included radiological diagnosis of primary HCC by computed
tomography (CT), classified in stage II and III according to the
tumor-node-metastasis (TNM) classification; age over 20 years/both
male and female; Eastern Cooperative Oncology Group scale 0–1; and
indicatiors of acceptable hematological (hemoglobin ≥8.5 g/dl,
white blood cells ≥2,000/mm3, platelet
≥50,000/mm3), hepatic (Child Pugh score ≤7, alanine
aminotransferase, aspartate aminotransferase ≤5x upper normal
limit) and renal (creatinine ≤1.5 mg/dl) function. Important
exclusion criteria consisted of organ transplantation; a medical
history of autoimmune disease, immunodeficiency, or autoimmune
disease that might be aggravated by immunotherapy; not exceeding 2
weeks after antibiotic treatment needed due to a serious infectious
disease; seropositivity for human immunodeficiency virus antigen;
use of immunosuppressive drug such as cyclosporin A and
azathioprine; any cardiopulmonary disability judged by the
investigator; a medical history of psychological disease or
epilepsy; and evidence of another active malignant neoplasm.
Preparation of recombinant
hepatocellular carcinoma antigens
cDNAs encoding AFP, MAGE-1 or glypican-3 (GPC3) were
cloned into the pCTP vector (21).
These 3 antigens were expressed in the form of 6x-His-attached
fusion proteins in E. coli BL21 (DE3) and purified using
nickel-nitrilotriacetic acid (Ni-NTA) column chromatography
(Qiagen, Hilden, Germany). The recombinant antigen production and
purification were performed at Good Manufacturing Practice
(GMP)-compliant facility following the Korean Food and Drug
Administration (KFDA) guideline. Each antigen was certified through
the process of quality control: purity >95% in SDS-PAGE analysis
and endotoxin <1.0 EU/μg in Limulus amebocyte lysate
test.
Autologous DC generation
DCs were generated from blood monocytes, as reported
previously (22), with
modifications. White blood cells obtained from the HCC patients
through leukapheresis. DCs were prepared in a GMP-compliant
facility at Ehime University Hospital (Ehime, Japan). Peripheral
blood mononuclear cells (PBMCs) were separated from WBC by
Ficoll-Paque™ PLUS (Amersham Biosciences, Uppsala, Sweden) density
gradient centrifugation. PBMCs were stored in a liquid nitrogen
tank until necessary for DC generation. PBMCs thawed, washed with
Hanks’ Balanced Salt Solutions, resuspended in RPMI-1640 medium
(Lonza, Basel, Switzerland) supplemented with autologous
heat-inactivated plasma, and then incubated in CellSTACK Culture
Chambers (Corning, Corning, NY, USA). After 0.5–1 h incubation at
37°C in a 5% CO2 incubator, non-adherent cells were
removed by gentle washes.
The adherent monocytes were cultured in X-VIVO15
(Cambrex, East Rutherford, NJ, USA) supplemented with 100 ng/ml of
granulocyte macrophage-colony stimulating factor (GMP grade: LG
Life Science, Seoul, Korea) and 300 ng/ml of interleukin (IL)-4 (JW
CreaGene Inc., Seongnam, Korea) for 5 days. On day 5, nonattached
immature DCs were harvested and pulsed with CTP-fused human AFP,
MAGE-1 and GPC-3 recombinant proteins at a final concentration of 5
μg/ml each. Antigen-pulsed dendritic cells were matured in
the presence of cytokine cocktail, IL-6 (Peprotech, Rocky Hill, NJ,
USA), IL-1β (Peprotech), tumor necrosis factor (TNF)-α (Peprotech),
prostaglandin E2 (PGE2) (Sigma Chemical Co., St. Louis,
MO, USA), interferon (IFN)-γ (LG Life Science), OK432 (Chugai
Pharmaceutical Co., Tokyo, Japan), and poly I:C (Sigma) for 1 or 2
days depending on surface phenotypes and cell population. On day
6–7, the DCs were harvested, washed, and resuspended in 1.2 ml of
cryopreserving solution containing 5% dimethyl sulfoxide (Bioniche
Pharma USA, Lake Forest, IL, USA). Finally fully equipped DCs were
packed into a sterile glass vial (4×107 cells/vial),
sealed with a snap-cap, and stored at an ultralow freezer for
>12 h.
Quality control of dendritic cell
vaccine
Safety test
For safety, endotoxin, germ-free and mycoplasma-free
tests were performed according to the KFDA-approved JW CreaGene
standard and test guidelines. Endotoxin was evaluated using
gel-clot techniques. The endotoxin of the product should be less
than 10 EU/ml per 1.2-ml vial. Mycoplasma test was performed by
both direct culture and PCR methods using e-Myco™ Mycoplasma PCR
detection kit (Intron Biotechnology, Seongnam, Korea), which
contains primer sets specifically designed to detect major
contaminants of Mycoplasma in cell cultures such as M.
arginini, M. faucium, M. fermentans, M.
hyorhinis, M. orale, and A. laidlawii as well as
other broad spectrum of mycoplasma.
Cell size and granularity
During the differentiation from monocytes to
dendritic cells, cell size and granularity increase. Based on these
principles, the cell size and granularity of each DC vaccine were
assessed by flow cytometric analysis. PBMCs were used for gating
control.
Phenotypic analysis
The phenotype of DC vaccine was determined by flow
cytometry using a FACSCalibur™ flow cytometer (Becton Dickinson,
Franklin Lakes, NJ, USA). The following monoclonal antibodies were
used: i) fluorescein isothiocyanate-conjugated mouse antihuman
IgG2a isotype control; ii) phycoerythrin-conjugated mouse antihuman
IgG1 isotype control; iii) anti-CD14, anti-CD19, anti-CD40,
anti-CD80, anti-D86, anti-HLA-ABC, and anti-HLA-DR (BD Pharmingen,
San Diego, CA, USA).
Viability
The viability of DC vaccine was assessed by
propidium iodide (PI) staining. PI (BD Pharmingen) was added to a
sample and kept in the dark at room temperature for 20 min. Cell
viability was examined by flow cytometry using a FACSCalibur™
(Becton Dickinson). Viability was represented as
100-[(PI+ of sample)−(PI+ of control)]
(%).
Lymphocyte proliferation assay
One vial from each DC vaccine lot was used to test
of T cell stimulation capacity according to the standard lymphocyte
proliferation assay. T cells were isolated from cryopreserved PBMC
using nylon wool column (Polysciences, Warrington, PA, USA).
Purified T cells (1×105) were cultured with serially
diluted DC vaccine (starting from 1×104 cells to
0.33×103 cells) at 37°C for 5 days. T cell proliferation
was assessed by
3-(4,5-di-methylthiazol-2-yl)-2,5-diphenyltetrazolium bromide,
yellow tetrazole: MTT) assay following manufacturer’s protocol
(CellTiter 96 Non-Radioactive proliferation assay kit; Promega,
Madison, WI, USA). R2 represent the standard curve of MTT assay for
the validation of a data set.
Cytokine production assay
Either culture supernatant of each antigen-pulsed DC
or co-cultured medium of T cells/DC at the ratio of 10:1 was
collected and stored at −80°C until this assay. The concentration
of IL-12p70, IL-10, IFN-γ, and IL-4 was measured with corresponding
human immunoassay kits (BD OptEIA™ kit, BD Pharmingen)
based on the manufacturer’s instruction. Each experiment was
performed 3 times and the result was described as the mean ±
standard deviation.
Treatment protocol (Fig. 1)
The screening evaluation was performed 3 weeks
before the start of immunotherapy and consisted of the following:
complete history, thorough physical examination, chest X-ray,
electrocardiogram, urine analysis, hematological and immunological
parameters, serum chemistry, tumor markers [AFP and protein induced
by vitamin K absence or antagonists-II (PIVKA-II)], ultrasonography
and abdominal CT scan. Eligible patients underwent TACE 2 weeks
before the start of the vaccination. PBMC collection by
leukapheresis was performed 1 week before the first planned
vaccination. Tumor antigen-pulsed DCs were injected subcutaneously
into the thigh near the inguinal lymph nodes. Topical TLR-7 agonist
(imiquimod; Aldara ™ Cream; Mochida Pharmaceutical Co.,
Tokyo, Japan) applied around the injection site from 2 consecutive
days before injection. During the first cycle, 4 vaccinations were
administered at biweekly intervals. Medical history and standard
blood tests and urine analysis were performed at each vaccination.
Vital signs were monitored during and after each injection.
Response evaluation was performed 4 weeks after fourth vaccination
(10 weeks after first vaccination), and TACE was repeated. Two
further vaccinations were administered at biweekly intervals, and
final response evaluation was performed at 18 weeks after first
vaccination. Tumor markers and serological tests for
autoantibodies, including anti-nuclear antibody, were evaluated
every 4 weeks.
Clinical response and toxicity
assessment
Clinical responses to vaccination were evaluated
according to the Response Evaluation Criteria in Solid Tumors
(RECIST) criteria (23). Complete
response was defined as disappearance of all target lesions.
Partial response was defined as 30% decrease in the sum of the
longest diameter of target lesions. Progressive disease was 20%
increase in the sum of the longest diameter of target lesions.
Stable disease was defined as small changes that do not meet above
criteria. Toxities were classified according to the National Cancer
Institute Common Toxicity Criteria.
Analysis of IFN-γ-producing cells
using enzyme-linked immunospot (ELISPOT) assay
The ELISPOT assay was adopted to detect and
enumerate individual cells that secrete IFN-γ in vitro upon
HCC-specific or -associated tumor antigens. Human IFN-γ ELISPOT
pair antibodies were purchased from BD Pharmingen, and ELISPOT
assay was performed according to the manufacturer’s instruction. In
brief, PBMC (2×105 cells) treated with each antigen (3–5
μg/ml) or antigen mixtures were loaded on a flat-bottomed
96-well ELISPOT plate (Millipore, Danvers, MA, USA) precoated with
capture antibody. The plate was incubated for 20 h at 37°C
CO2 incubator. After washing, detection antibody was
added to each well and incubated for 2 h at room temperature.
Avidinhorseradish peroxidase conjugate was added to each well, and
the plate was developed with 3-amino-9-ethyl-carbazole substrate
reagent set. Visible spots were enumerated using an automated
ELISPOT reader (CTL, USA) and default program.
Results
Patients
Treatment was performed at Ehime University, in 2009
(Ehime, Japan). Baseline characteristic of the 5 patients enrolled
are shown in Table I. The basis of
the diagnosis of HCC was histological and/or radiolgical for all
patients. All patients were male with age range 46–64 years. Two
and 3 patients were infected with hepatitis B virus and hepatitis C
virus, respectively. All patients were previously treated with
TACE.
 | Table I.Patient characteristics and
treatments. |
Table I.
Patient characteristics and
treatments.
A
|
|---|
| Patient no. | Sex | Age (years) | Etiology | TNM stage | No. of tumors | Largest tumor | Child-Pugh |
|---|
| 1 | M | 65 | HCV | III | 2 | 22.1 | A |
| 2 | M | 58 | HBV | III | 2 | 15.9 | A |
| 3 | M | 59 | HCV | II | 1 | 6.6 | A |
| 4 | M | 64 | HBV | II | 1 | 12.4 | A |
| 5 | M | 46 | HCV | II | 9 | 30.3 | B |
B
|
|---|
| | AFP (<ng/m)
| PIVKA-II (mAU/ml)
| Outcome |
|---|
| Patient no. | Previous
treatment | Pre | Post | Pre | Post | |
|---|
| 1 | TACE | 23.7 | 56.7 | 842 | 3,109 | PD |
| 2 | TACE | 12.4 | 16.8 | 42 | 1,189 | PD |
| 3 | TACE | 27.4 | 23.2 | 53 | 67 | SD |
| 4 | TACE | 30.5 | 181.4 | 59 | 192 | PD |
| 5 | TACE | 854.1 | 660.0 | 10,707 | 30,615 | PD |
DC vaccine
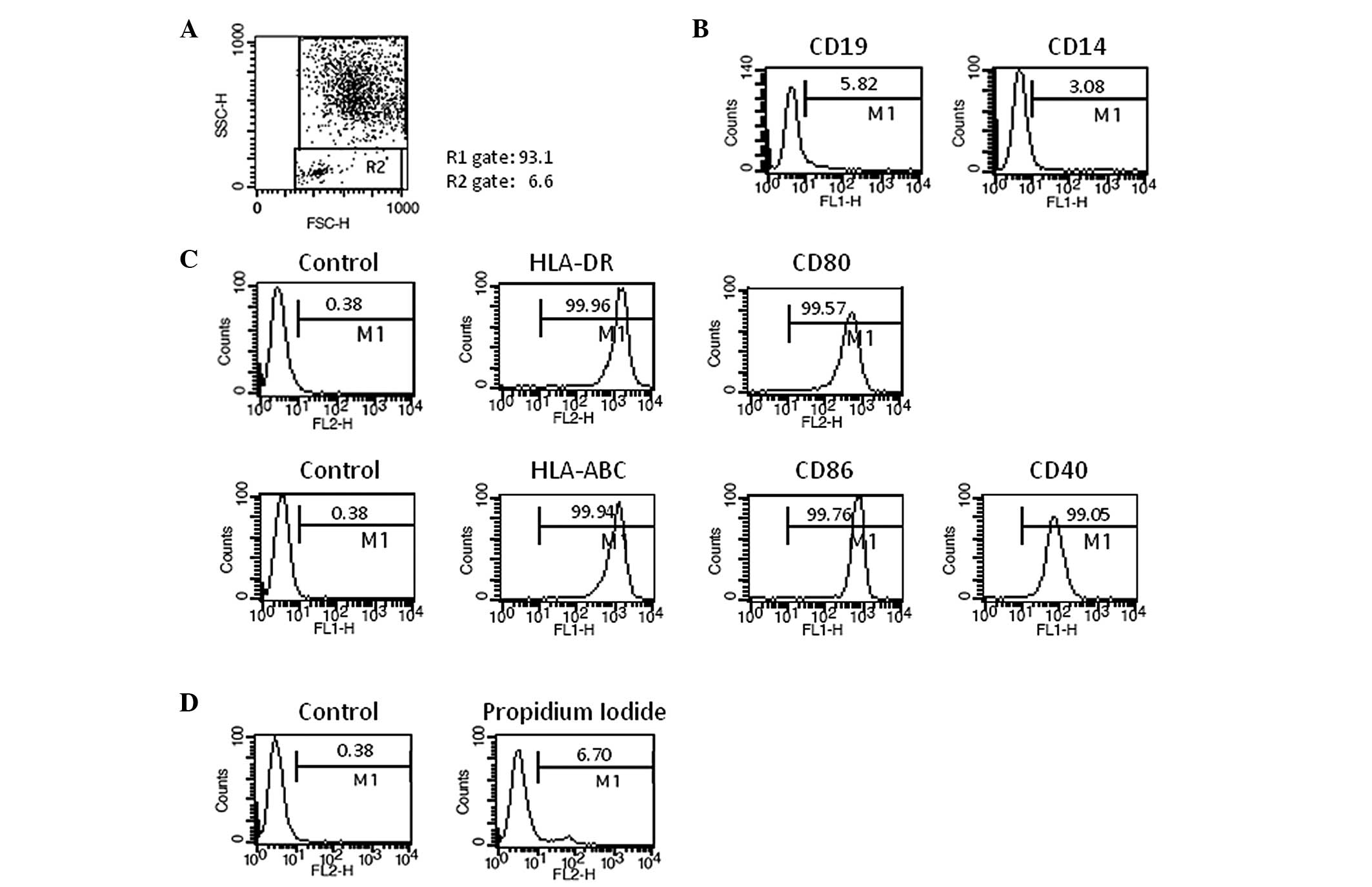
DC vaccine was generated successfully from the 5
patients with HCC. One vial from each lot of frozen DC vaccines was
thawed and used for quality control. DC vaccine demonstrated
typical features of mature DC morphology under a microscope. The
cell population in DC gate in the FACS analysis was over 90% on the
basis of the cell size and granularity, with a median value of
94.4% (Fig. 2A). The analysis of
lineage markers revealed that the contamination of B cells (CD19)
and monocytes (CD14) was less than 10% (Fig. 2B). Over 95% of DCs demonstrated MHC
class I (HLA-ABC) high, MHC class II (HLA-DR) high, and
costimulatory molecules (CD86, CD80, and CD40) high (Fig. 2C). These characteristics were
commonly maintained in all 5 different DC vaccines, which were
generated under the same culture conditions. Viability is one of
the most important issues in DC vaccine. The viabilities of DC
vaccines ranged from 86.2% to 93.5% and median value was 92.3%
(Fig. 2D), indicating that the
frozen DCs can be used as a therapeutic vaccine. The frozen DC
vaccine was stable for longer than 6 months (data not shown). The
purity, cell viability and surface phenotypes of 5 different DC
vaccines are summarized in Table
II.
| Table II.Quality control results of 5
different DC vaccines. |
Table II.
Quality control results of 5
different DC vaccines.
| Patient no. | No. 1 | No. 2 | No.3 | No. 4 | No. 5 |
|---|
| Sterility | | | | | |
| I | Pass | Pass | Pass | Pass | Pass |
| II | Pass | Pass | Pass | Pass | Pass |
| Mycoplasma | | | | | |
| I (PCR) | Pass | Pass | Pass | Pass | Pass |
| II (Direct
culture) | Pass | Pass | Pass | Pass | Pass |
| Endotoxin (<10
EU/ml) | Pass | Pass | Pass | Pass | Pass |
| Viability (%) | 86.2 | 91.2 | 92.7 | 93.5 | 92.3a |
| Identification | | | | | |
| Size &
granularity (%) | 93.7 | 94.8 | 94.4 | 97.3 | 90.0 |
| Cell surface
phenotypes (%) | | | | | |
| HLA-DR | 99.8 | 99.0 | 98.9 | 99.7 | 99.7 |
| HLA-ABC | 99.5 | 99.8 | 99.9 | 99.8 | 99.9 |
| CD86 | 99.6 | 98.9 | 99.4 | 99.9 | 99.8 |
| CD80 | 95.6 | 98.9 | 98.9 | 99.4 | 99.1 |
| CD40 | 87.9 | 97.2 | 98.9 | 95.0 | 97.9 |
| Purity test | | | | | |
| CD14 | 8.5 | 7.1 | 3.1 | 3.5 | 2.1 |
| CD19 | 0.9 | 0.6 | 1.6 | 0.8 | 1.3 |
| Total cell number
(×107) | 4.1 | 4.08 | 4.24 | 4.22 | 4.25 |
| T cell
proliferation | | | | | |
| DC
1×104 cells | Not tested | Not tested | 0.777 | 0.849 | 0.908 |
| DC
0.33×103 cells | | | 0.349 | 0.439 | 0.343 |
| Coefficient
factor (R2)* | | | 0.989 | 0.948 | 0.993 |
Cytokine production assay
To determine whether DC vaccine was functionally
active to induce Th1 immune responses, we examined IL-12 and IL-10
production from DC induced by each specific antigen such as AFP,
GPC-3, or MAGE-1. As a result, IL-12 was highly produced whereas
the amount of IL-10 production was almost a basal level (Table IIIA). Furthermore, predominant
IFN-γ level in T cell/DC co-cultured supernatant from those five
HCC patients was also confirmed, while the level of IL-4 production
was <15 pg/ml (Table IIIB).
| Table III.Cytokine production assay results of
5 different DC vaccines. |
Table III.
Cytokine production assay results of
5 different DC vaccines.
| A |
|---|
|
|---|
| Patient no. | Antigens | IL-12p70
(ng/ml) | IL-10 (ng/ml) |
|---|
| 1 | AFP | 35.3±3.5 | 0.13±0.03 |
| GPC-3 | 32.3±3.0 | 0.014±0.002 |
| MAGE-1 | 58.8±3.0 | 0.65±0.15 |
| 2 | AFP | 3.3±0.5 | 0.04±0.01 |
| GPC-3 | 5.5±0.6 | 0.05±0.01 |
| MAGE-1 | 31.1±4.9 | 0.34±0.15 |
| 3 | AFP | 9.0±0.8 | 0.013±0.006 |
| GPC-3 | 13.4±1.0 | 0.04±0.01 |
| MAGE-1 | 43.9±4.4 | 0.23±0.06 |
| 4 | AFP | 1.9±0.3 | 0.09±0.02 |
| GPC-3 | 2.0±0.4 | 0.09±0.04 |
| MAGE-1 | 11.0±1.4 | 0.37±0.07 |
| 5 | AFP | 3.1±0.5 | 0.53±0.06 |
| GPC-3 | 2.6±0.6 | 0.07±0.01 |
| MAGE-1 | 2.3±0.4 | 0.08±0.05 |
| B |
|---|
|
|---|
| Patient no. | IFN-γ (ng/ml) |
|---|
| 1 | 16.5±0.9 |
| 2 | 10.4±2.9 |
| 3 | 20.5±3.3 |
| 4 | 9.3±0.8 |
| 5 | 18.9±2.3 |
| Positive
control | 23.5±3.3 |
| Negative
control | 0.1±0.04 |
Toxicity assignment
Injection of DC vaccine was safe and well tolerated.
Toxicity was mild and no grade III/IV serious adverse events
occurred in a total of 30 instances of cell injection (Table IV). No hematological, hepatic or
renal toxicities or de novo autoantibody formation were
observed in any patient.
| Table IV.Toxicity profiles by patients. |
Table IV.
Toxicity profiles by patients.
| Toxities | Grade 1 | Grade 2 | Grade 3 | Grade 4 |
|---|
| Injection site
reaction | 5/5 | - | - | - |
| Fever | 4/5 | 1/5 | - | - |
Clinical response assessment
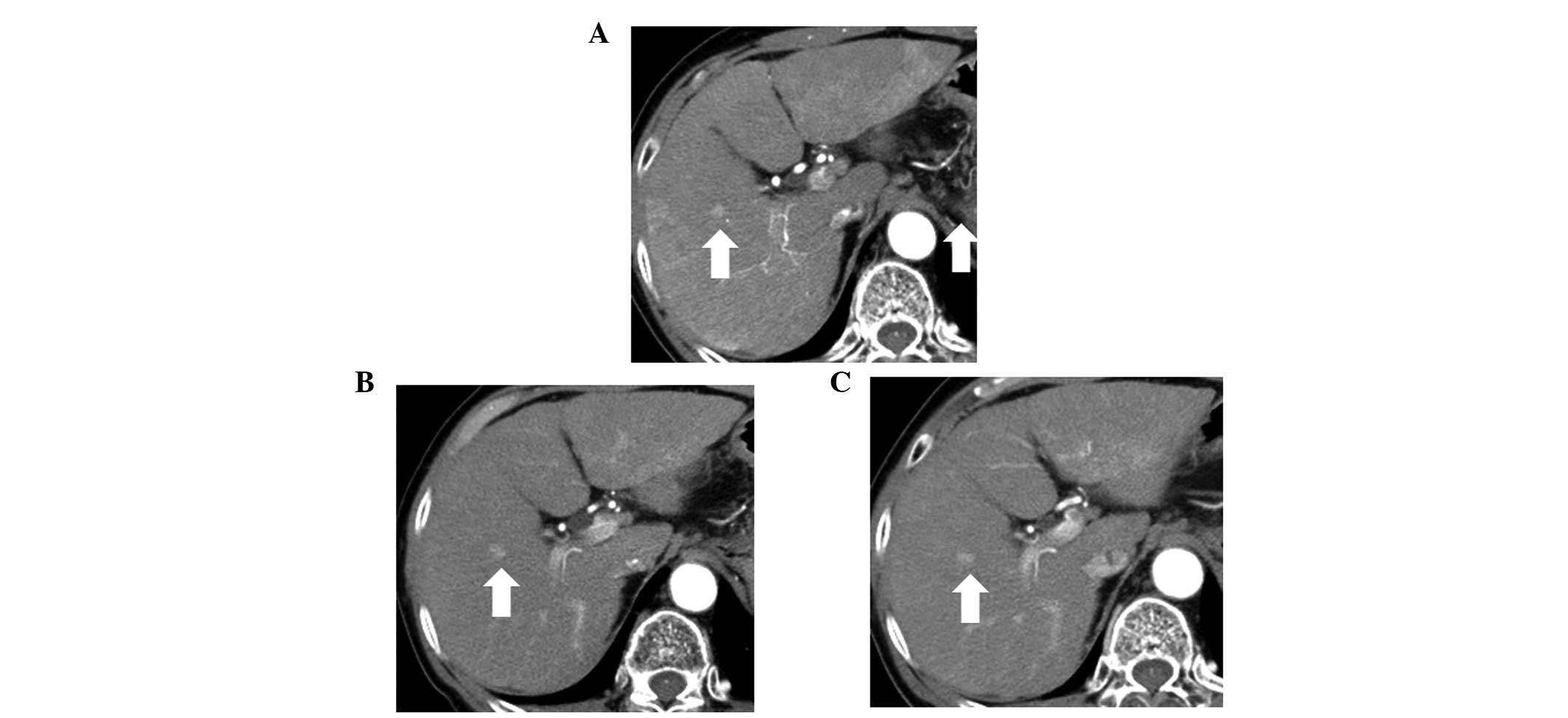
One patient (patient no. 3) achieved disease
stabilization during the follow-up period (Fig. 3), however, no tumor response was
observed in the other 4 patients (Table I). Serum AFP levels decreased in 2
patients; however, serum PIVKA-II levels increased in all
patients.
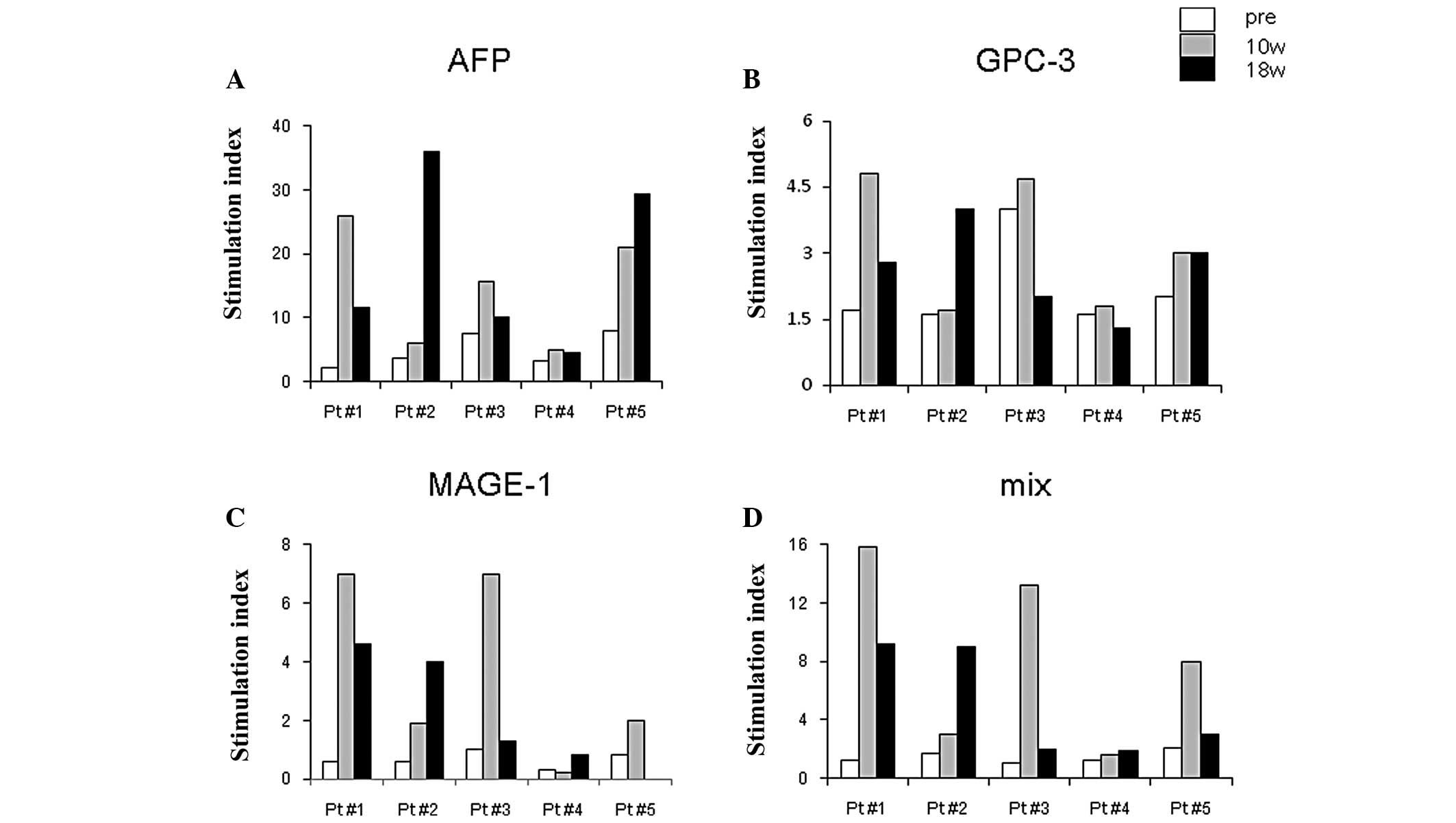
T cell responses after DC
vaccination
After DC vaccination, all 5 patients demonstrated
strong T cell responses against HCC antigens compared with the
samples obtained before vaccination. The stimulation index (SI)
shown in Fig. 4 illustrates the
high reactivity of AFP antigen in all 5 patients after vaccination,
while GPC-3 or MAGE-1 antigens were moderate in their capacity to
induce T cell responses. AFP-specific IFN-γ-producing cells peaked
10 weeks after the first vaccination in 2 patients, and 18 weeks in
2 patients.
Discussion
HCC is one of the major malignancies in Asian
countries including China, Korea and Japan (1). Screenings based on imaging studies,
such as ultrasonography and CT, and serum tumor markers have
improved HCC detection in high-risk patients at a relatively early
stage. Such patients may have some benefits by curative treatments
for inhibition of local recurrence in the liver; however, the
surrounding non-tumor liver tissues exhibit a high carcinogenic
potential, such as liver cirrhosis and chronic hepatitis. The high
rate of intrahepatic recurrence is a key feature correlated with
poor prognosis, and its prevention is an issue for urgent
investigation (5).
HCC is a potentially ideal tumor for targeting by
immune-based therapies (24–26).
However, the observation of tumor progress in HCC despite the
presence of tumor-specific immune responses suggests that
development of HCC leads to a number of immune suppressor
mechanisms, including increase of regulatory T cells (27), myeloid-derived suppressor cells
(28), and impairment of
antigen-presenting cells. DCs are the most potent
antigen-presenting cells effective to induce appropriate adaptive
immune responses (7,8). However, DC function is suppressed in
patients with HCC (29,30), and may lead to a failure of the
induction and maintenance of antitumor immunity. Therefore, these
observations provide a rationale for activating DC in vitro
and infusing them into patients to overcome tumor-related
immunosuppression to induce sufficient anti-tumor immunity. A
series of clinical trials using DC-based vaccines demonstrated
evidence of safety and immune activity; however, clinical benefits
have shown to be limited (11–20).
Therefore, clinical trials with a well established DC vaccination
protocol are highly recommended in the field of DC-based
immunotherapy.
We investigated the safety and efficacy of the
autologous DC-based tumor vaccine charged with
HCC-specific/associated recombinant antigens in 5 patients with
advanced HCC. No technical hardships were encountered with blood
procurement or the subsequent generation of DC vaccine. No severe
treatment-related complications were noted (Table IV), and antigen-specific immunity
was induced in all patients (Fig.
4). A clinical response, defined as stable disease (SD) was
achieved in one patient (Fig. 3).
These results indicate that DC vaccine used in this study is well
tolerated and able to induce anti-tumor immunity in patients with
HCC that may be associated with clinical benefits.
Our DC vaccine protocol for the treatment of the
patients with HCC comprises major modifications from the previous
studies in several points. First, we used mature DCs which were
antigen-charged and stimulated with a cytokine mixture, poly I:C,
and OK432 (Fig. 2 and Table II). Immature DCs have been used in
several clinical trials (11–18).
Evidence suggests that mature DCs are better in inducing clinical
impact in DC-based cancer immunotherapy (31). Recently, Nakamoto et
al(20) demonstrated that
infusion of mature DCs, but not immature DCs, during the TACE
procedures prolonged recurrence-free survival. Antigen uptake assay
was not exactly preceded because of shortage of PBMC. However,
based on another set of experiments which were performed using DC
derived from HCC patients, the result of antigen uptake capacity of
DC vaccine was always >70% evaluated by FITC-dextran uptake
assay (data not shown). Second, topical application of imiquimod, a
TLR7 ligand, was also used to enhance anti-tumor immunity in
synergy with DC vaccine (32).
Aldara™ Cream (5% imiquimod) is a new type of treatment in the
category of medicines known as immune response modifiers and is
indicated for the treatment of condyloma acuminate. In this study,
we demonstrated the feasibility and safety of DC vaccine designed
to have synergistic effects with imiquimod in HCC patients. Third,
we used a novel approach for the delivery of tumor antigens into
DCs. CTP has a strong membrane transduction potential (21), and was very efficient for the
delivery of antigens into the cytoplasm of DCs. DC vaccine pulsed
with CTP-conjugated antigens elicited a robust Th1-mediated
immunity and antigen-specific CTL responses when compared with
antigen alone, which is probably attributable to the CTP
technology. The feasibility was confirmed in this clinical trial.
Finally, we used 3 different recombinant proteins as a source of
HCC antigens for the generation of DC vaccine. Because any single
antigen is ubiquitously expressed in HCC, we selected AFP, GPC-3
and MAGE-1 as target antigens for DC vaccine through the analysis
of the tissue array of a tumor tissues obtained from 412 patients
with HCC in Korea (data not shown). AFP has been studied as a
possible candidate antigen for anti-HCC immunotherapy. T-cell
responses to AFP-CTL epitope peptides were strongly induced in
patients with HCC (33,34). In addition, the overexpression of
GPC-3 specifically in human HCC has been reported (35), and DC expressing GPC-3 induced
protective immunity against highly meta-static cancer (36). Furthermore, the MAGE-1 was reported
to be expressed in 30% to 78% in HCC tissue samples (37,38).
An advantage of this approach is that recombinant proteins were
used for equipping DC vaccine to overcome the HLA restriction of
epitope peptides. In this study, AFP-specific T cell response was
significantly induced in all 5 patients after DC vaccination, but
those against GPC-3 and MAGE-1 were moderate even after DC
vaccination (Fig. 4). Moderate
responses to GPC-3 and MAGE-1 in the vaccine remain to be further
characterized, but are likely, at least in part, attributable to
the limited immunogenicity of each antigens in vivo. The
recombinant protein CTP-GPC-3 does not have trans-membrane and
cytoplasmic domains, latter of which contains immunogenic epitopes
(39).
We could not investigate the expression pattern of
each tumor antigen in HCC nodules for the limitations of biopsy
samples. Therefore, we were not able to analyze correlation between
TAA expression and TAA-specific T cell response after vaccination.
Further studies are necessary in this regard. However, the results
of the present study confirmed the feasibility, safety and immune
activity of recombinant tumor antigen-pulsed DC vaccine for
therapeutic use in HCC patients. Genome profiling studies of HCC
have revealed that HCC is a very heterogeneous tumor (40). Furthermore, HCC demonstrates
multicentric carcinogenesis and develops at different time points.
These data indicate that the identification of many more target
antigens and their optimization is necessary to evoke better
clinical responses.
In conclusion, we conducted a phase I/II clinical
trial using DC vaccine in 5 patients with advanced HCC and liver
cirrhosis. DC vaccine was well tolerated in all patients and
induced anti-tumor immune responses in vaccine, but clinical
response was detected only in 1 patient (patient 3) with advanced
HCC and liver cirrhosis. The tumor-load of this patient was
relatively smaller compared to those of other 4 patients (Table I). Including our study, most of
DC-based immunotherapies have been studied in patients with
advanced stage disease, resulting in poor clinical responses.
Future trials in less advanced disease may accompany better
clinical responses. DC-based tumor immunotherapy will be a good
indication as an adjuvant setting to radical therapy, such as
surgical resection or RFA, to prevent tumor recurrences in patients
with HCC.
Acknowledgements
We thank Ms. Sawa Yamamoto and Ms.
Sakiko Sugawasa for their excellent technical assistance. This work
was supported in part by the Japanese Ministry of Education,
Culture, Sports, Science and Technology (JSPS KAKENHI 21790669) to
M.A and Korean Ministry of Health and Welfare Bio New Drug Grants
(A110054).
References
|
1.
|
HB El-SeragHepatocellular carcinomaN Engl
J Med36511181127201110.1056/NEJMra100168321992124
|
|
2.
|
I IkaiM KudoS AriiReport of the 18th
follow-up survey of primary liver cancer in JapanHepatol
Res4010431059201010.1111/j.1872-034X.2010.00731.x
|
|
3.
|
R LencioniLoco-regional treatment of
hepatocellular
carcinomaHepatology52762773201010.1002/hep.2372520564355
|
|
4.
|
Y OkuwakiT NakazawaA ShibuyaIntrahepatic
distant recurrence after radiofrequency ablation for a single small
hepatocellular carcinoma: risk factors and patternsJ
Gastroenterol437178200810.1007/s00535-007-2123-z
|
|
5.
|
N IzumiPrediction and prevention of
intrahepatic recurrence of hepatocellular carcinomaHepatol
Res42226232201210.1111/j.1872-034X.2011.00922.x22181559
|
|
6.
|
F KorangyB HöchstMP MannsTF GretenImmune
responses in hepatocellular carcinomaDig
Dis28150154201010.1159/00028207920460904
|
|
7.
|
J BanchereauRM SteinmanDendritic cells and
the control of immunityNature392245252199810.1038/32588
|
|
8.
|
M OnjiSM AkbarDendritic Cells in
Clinics2nd editionSpringerTokyo2008
|
|
9.
|
J BanchereauAK PaluckaDendritic cells as
therapeutic vaccines against cancerNat Rev
Immunol5296306200510.1038/nri159215803149
|
|
10.
|
FO NestleA FarkasC
ConradDendritic-cell-based therapeutic vaccination against
cancerCurr Opin
Immunol17163169200510.1016/j.coi.2005.02.00315766676
|
|
11.
|
A LadhamsC SchmidtG SingTreatment of
nonresectable hepatocellular carcinoma with autologous tumor-pulsed
dendritic cellsJ Gastroenterol
Hepatol17889896200210.1046/j.1440-1746.2002.02817.x12164965
|
|
12.
|
Y IwashitaK TaharaS GotoA phase I study of
autologous dendritic cell-based immunotherapy for patients with
unresectable primary liver cancerCancer Immunol
Immunother52155161200312649744
|
|
13.
|
A StiftJ FriedlP DubskyDendritic
cell-based vaccination in solid cancerJ Clin
Oncol21135142200310.1200/JCO.2003.02.13512506182
|
|
14.
|
T KumagiSM AkbarN HoriikeAdministration of
dendritic cells in cancer nodules in hepatocellular carcinomaOncol
Rep14969973200516142359
|
|
15.
|
KH ChiSJ LiuCP LiCombination of conformal
radiotherapy and intratumoral injection of adoptive dendritic cell
immunotherapy in refractory hepatomaJ
Immunother28129135200510.1097/01.cji.0000154248.74383.5e15725956
|
|
16.
|
WC LeeHC WangCF HungPF HuangCR LiaMF
ChenVaccination of advanced hepatocellular carcinoma patients with
tumor lysate-pulsed dendritic cells: a clinical trialJ
Immunother28496504200510.1097/01.cji.0000171291.72039.e216113606
|
|
17.
|
LH ButterfieldA RibasVB DissetteA phase
I/II trial testing immunization of hepatocellular carcinoma
patients with dendritic cells pulsed with four alpha-fetoprotein
peptidesClin Cancer
Res1228172825200610.1158/1078-0432.CCR-05-2856
|
|
18.
|
Y NakamotoE MizukoshiH TsujiCombined
therapy of transcatheter hepatic arterial embolization with
intratumoral dendritic cell infusion for hepatocellular carcinoma:
clinical safetyClin Exp
Immunol147296305200710.1111/j.1365-2249.2006.03290.x
|
|
19.
|
DH PalmerRS MidgleyN MirzaA phase II study
of adoptive immunotherapy using dendritic cells pulsed with tumor
lysate in patients with hepatocellular
carcinomaHepatology49124132200910.1002/hep.2262618980227
|
|
20.
|
Y NakamotoE MizukoshiM KitaharaProlonged
recurrence-free survival following OK432-stimulated dendritic cell
transfer into hepatocellular carcinoma during transarterial
embolizationClin Exp
Immunol163165177201110.1111/j.1365-2249.2010.04246.x
|
|
21.
|
D KimC JeonJH KimCytoplasmic transduction
peptide (CTP): new approach for the delivery of biomolecules into
cytoplasm in vitro and in vivoExp Cell
Res31212771288200610.1016/j.yexcr.2005.12.02916466653
|
|
22.
|
JH KimY LeeYS BaePhase I/II study of
immunotherapy using autologous tumor lysate-pulsed dendritic cells
in patients with metastatic renal cell carcinomaClin
Immunol125257267200710.1016/j.clim.2007.07.01417916447
|
|
23.
|
P TherasseSG ArbuckEA EisenhauerNew
guidelines to evaluate the response to treatment in solid tumors
European Organization for Research and Treatment of Cancer,
National Cancer Institute of the United States, National Cancer
Institute of CanadaJ Natl Cancer
Inst92205216200010.1093/jnci/92.3.205
|
|
24.
|
LH ButterfieldImmunotherapeutic strategies
for hepatocellular carcinomaGastroenterology127Suppl
1S232S241200410.1053/j.gastro.2004.09.03815508089
|
|
25.
|
TF GretenMP MannsF KorangyImmunotherapy of
HCCRev Recent Clin Trials33139200810.2174/157488708783330549
|
|
26.
|
P MatarL AlanizV RozadosImmunotherapy for
liver tumors: present status and future prospectsJ Biomed
Sci1630200910.1186/1423-0127-16-3019272130
|
|
27.
|
LA OrmandyT HillemannH WedemeyerMP MannsTF
GretenF KorangyIncreased populations of regulatory T cells in
peripheral blood of patients with hepatocellular carcinomaCancer
Res6524572464200510.1158/0008-5472.CAN-04-323215781662
|
|
28.
|
B HoechstLA OrmandyM BallmaierA new
population of myeloid-derived suppressor cells in hepatocellular
carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T
cellsGastroenterology135234243200818485901
|
|
29.
|
T NinomiyaSM AkbarT MasumotoN HoriikeM
OnjiDendritic cells with immature phenotype and defective function
in the peripheral blood from patients with hepatocellular
carcinomaJ
Hepatol31323331199910.1016/S0168-8278(99)80231-110453947
|
|
30.
|
S KakumuS ItoT IshikawaDecreased function
of peripheral blood dendritic cells in patients with hepatocellular
carcinoma with hepatitis B and C virus infectionJ Gastroenterol
Hepatol15431436200010.1046/j.1440-1746.2000.02161.x10824889
|
|
31.
|
D McIlroyM GregoireOptimizing dendritic
cell-based anticancer immunotherapy: maturation state does have
clinical impactCancer Immunol
Immunother52583591200310.1007/s00262-003-0414-712827310
|
|
32.
|
RM PrinsN CraftKW BruhnThe TLR-7 agonist,
imiquimod, enhances dendritic cell survival and promotes tumor
antigen-specific T cell priming: relation to central nervous system
antitumor immunityJ
Immunol176157164200610.4049/jimmunol.176.1.157
|
|
33.
|
LH ButterfieldA RibasWS MengT-cell
responses to HLA-A*0201 immunodominant peptides derived from
alpha-fetoprotein in patients with hepatocellular cancerClin Cancer
Res9590259082003
|
|
34.
|
E MizukoshiY NakamotoK AraiComparative
analysis of various tumor-associated antigen-specific T-cell
responses in patients with hepatocellular
carcinomaHepatology5312061216201110.1002/hep.2414921480325
|
|
35.
|
T NakatsuraY YoshitakeS SenjuGlypican-3,
over-expressed specifically in human hepatocellular carcinoma, is a
novel tumor markerBiochem Biophys Res
Commun3061625200310.1016/S0006-291X(03)00908-212788060
|
|
36.
|
Y MotomuraS SenjuT NakatsuraEmbryonic stem
cell-derived dendritic cells expressing glypican-3, a recently
identified oncofetal antigen, induce protective immunity against
highly metastatic mouse melanoma, B16-F10Cancer
Res6624142422200610.1158/0008-5472.CAN-05-2090
|
|
37.
|
K SuzukiS TsujitaniI KonishiY YamaguchiY
HirookaN KaibaraExpression of MAGE genes and survival in patients
with hepatocellular carcinomaInt J Oncol1512271232199910568832
|
|
38.
|
K KariyamaT HigashiY KobayashiExpression
of MAGE-1 and -3 genes and gene products in human hepatocellular
carcinomaBr J
Cancer8110801087199910.1038/sj.bjc.669081010576668
|
|
39.
|
J O’BeirneF FarzanehPM HarrisonGeneration
of functional CD8+ T cells by human dendritic cells
expressing glypican-3 epitopesJ Exp Clin Cancer Res29482010
|
|
40.
|
JS LeeSS ThorgeirssonGenome-scale
profiling of gene expression in hepatocellular carcinoma:
classification, survival prediction, and identification of
therapeutic targetsGastroenterology127Suppl
1S51S55200410.1053/j.gastro.2004.09.01515508103
|