Introduction
Recent vigorous research of somatic stem cells,
embryonic stem (ES) cells and induced pluripotent stem (iPS) cells
clarified several novel key regulators associated with cellular
pluripotency, undifferentiated phenotype, self-renewal and stem
cell maintenance. In cancer research, especially in cancer stem
cell (CSC) research, some reports focused on the expression of
pluripotency-associated factors and indicated their correlation
with cancer cell aggressiveness (1), chemoradiotherapy resistance (2) and cancer stem-like cell properties
(3), but little is known about the
relationship between ES cell-related cell surface markers and
cancer. In this study, we focused on stage-specific embryonic
antigen 3 (SSEA-3) in colorectal cancer (CRC). SSEAs are
globoseries glycolipids and are known to consist of 3 species;
SSEA-1, 3 and 4. SSEA-1 and SSEA-4 were established through
immunization of animals with human embryonic carcinoma cells, and
SSEA-3 with 4- to 8-cell stage mouse embryos (4–6).
SSEA-4 is similar to SSEA-3 in terms of structure; they share the
same globoseries glycolipid but SSEA-4 has an additional terminal
sialic acid moiety (7). SSEA-1 and
SSEA-3/-4 are reportedly expressed on the surface of the murine
(8–10) and human (11,12)
pluripotent stem cells; inner cell mass of early embryos, ES cells
and iPS cells, respectively. Though expression of SSEAs are thought
to be attenuated during the process of differentiation, a recent
report revealed that a small group of SSEA-3 expressing cells;
multilineage differentiating stress enduring (Muse) cells, exist in
adult skin fibroblasts and bone marrow stroma and that they possess
stress tolerance and endogenous pluripotency (13). In cancer, expression of SSEA-1 is
related to the poor prognosis (14) and tumor-initiating capacity in
human glioblastoma (15), whereas
the reduced expression of SSEA-4 is correlated with advanced tumor
stages and poor tumor cell differentiation in ovarian cancer
(16). In contrast to SSEA-1 and
SSEA-4, there are only a few reports that have assessed SSEA-3
expression in cancer; in breast cancer, 77.5% of clinical breast
cancer samples express SSEA-3 with a broad positive ratio (17), and in non-small cell lung cancer,
SSEA-3 expression increases after exposure to multiple anti-cancer
drugs (2). To the best of our
knowledge, there are no reports that have clearly assessed SSEA-3
expression and its biological characteristics in
gastro-enterological cancer. Furthermore, there are no reports that
have assessed SSEA-3 expression in association with CSC. In
addition, human colorectal epithelial cells were shown to be SSEA-3
positive (17), implying that CRC
tissue also contains SSEA-3 expressing immature subsets.
In this study, we assessed the existence of
SSEA-3+ cells in the cell lines of CRC. Next, we
investigated the relationship between SSEA-3 expression and
representative colorectal CSC or Muse cell marker, tumorigenic
activity and sphere formation activity to assess for CSC-like
properties. Then, we assessed the cell proliferation activity of
SSEA-3+ cells to characterize them. Finally, we assessed
the distribution of SSEA-3+ cells to confirm the
existence of SSEA-3+ cells in clinical specimens.
Materials and methods
Patients and tissue samples
Surgically resected CRC samples were obtained from
10 patients after informed consent from Osaka University Medical
Hospital with approval of the research ethics board. All specimens
were embedded in Tissue-Tek O.C.T. compound (Sakura Finetek, Tokyo,
Japan), rapidly frozen by immersion in liquid nitrogen and stored
at −80°C.
Cell lines and culture
Human CRC cell line Caco-2, DLD-1, HCT116, HT-29 and
SW480 were obtained from American Type Culture Collection
(Manassas, VA) and Car-1 from Japanese Collection of Research
Bioresources (Osaka, Japan). Caco-2 and Car-1 were maintained in
Eagle’s minimum essential medium (Wako Pure Chemical Industries,
Tokyo, Japan), DLD-1, SW480 and NTERA-2 were maintained in
Dulbecco’s minimum essential medium (Wako Pure Chemical
Industries), and HCT116 and HT-29 were maintained in McCoy’s 5a
medium (Invitrogen, Carlsbad, CA, USA), respectively supplemented
with 10% fetal bovine serum (HyClone, Logan, UT, USA), penicillin
and streptomycin (Pen-Strep; Invitrogen) at 37°C in a humidified
atmosphere containing 5% CO2.
Immunofluorescent staining
The 7 μm thick frozen sections were obtained
by using cryostat and fixed by 4% paraformaldehyde (Wako Pure
Chemical Industries). After blocking, sections were incubated with
rat anti-SSEA-3 monoclonal antibody (MAB4303, Millipore, Billerica,
MA, USA) and Alexa Fluor 488 mouse anti-human E-cadherin (BD
Pharmingen, San Diego, CA, USA). Alexa Fluor 594 goat anti-rat
antibody (Invitrogen) was used as a secondary antibody. Rat IgM
isotype (BD Pharmingen) was used as the control. Sections were
mounted with ProLong Gold Antifade Reagent with DAPI (Invitrogen)
and viewed with a fluorescent microscope (BZ-9000, Keyence, Osaka,
Japan).
For immunocytochemistry, cells were cultured on
8-well culture slides (BD Falcon, Franklin Lakes, NJ) at the
concentration of 20,000 cells/well for 24 h, fixed with 4%
paraformaldehyde, then permealized with Triton X-100
(Sigma-Aldrich, St. Louis, MO, USA) and processed using the same
protocol as the frozen sections.
Flow cytometry
Cells were dissociated with Accutase (Invitrogen),
blocked with FcR blocking reagent (BD Biosciences) and incubated
with antibodies as follows; anti-SSEA-3 (MAB4303, Millipore),
anti-CD105 (APC-conjugated, BioLegend, San Diego, CA, USA),
anti-rat IgM (APC-conjugated; Jackson ImmunoResearch Laboratories,
West Grove, PA, FITC-conjugated; BD Biosciences, San Jose, CA,
USA), anti-CD44, anti-CD24, anti-CD26 (conjugated form, BD
Biosciences) and AldeFluor (Stemcell Technologies, BC, Canada).
7-AAD (BD Biosciences) was used to eliminate dead cells. Cells were
analyzed and isolated by using FACSAriaII equipped with FACS Diva
software (BD Biosciences).
RNA preparation and quantitative
real-time PCR
Total RNA was isolated using TRIzol reagent
(Invitrogen). In all cases, 1 μg of total RNA was
reverse-transcribed with High Capacity RNA-to-cDNA Master Mix
(Applied Biosystems, Foster, CA, USA) following the manufacturer’s
instructions. For quantitative assessments, real-time RT-PCR
analysis was performed with the LightCycler TaqMan Master kit
(Roche Diagnostics, Tokyo) and the LightCycler 480 system (Roche
Applied Science, Indianapolis, IN, USA). The sequences of the
primers used were as follows: GAPDH (NM_002046.3) sense
primer 5′-AGCCACATCGCTCAGACAC-3′ and antisense primer
5′-GCCCAATACGACCAAATCC-3′; OCT3/4 (NM_203289.3) sense primer
5′-AATCCAGTCCCAGGACATCA-3′ and anti-sense primer
5′-TGGCTGAATACCTTCCCAAA-3′; NANOG (NM_024865.2) sense primer
5′-ATGCCTCACACGGAGAC TGT-3′ and antisense primer
5′-AGGGCTGTCCTGAATAA GCA-3′; SOX2 (NM_003106.2) sense primer
5′-CTCCGGGA CATGATCAGC-3′ and antisense primer 5′-CTGGGACAT
GTGAAGTCTGC-3′; c-MYC (NM_002467.4) sense primer
5′-CACCAGCAGCGACTCTGA-3′ and antisense primer
5′-GATCCAGACTCTGACCTTTTGC-3′.
Western blot analysis
Western blot analysis was performed as described
previously (18). The following
antibodies, at appropriate concentrations, were applied on
membranes after the transfer of a sodium dodecyl
sulfate-polyacrylamide gel (Bio-Rad Laboratories, Hercules, CA,
USA): mouse anti-Oct4 monoclonal antibody (MAB4305, Millipore),
anti-Nanog goat polyclonal antibody (R&D Systems, Minneapolis,
MN, USA), anti-Sox2 rabbit polyclonal antibody (MBL, Nagoya,
Japan), anti-cMyc, anti-p21Cip1/Waf1,
anti-p27Kip1 and anti-cyclin D1 mouse monoclonal
antibodies, anti-cyclin A2 and anti-cyclin B1 rabbit monoclonal
antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA),
anti-p16 mouse monoclonal antibody (BD Pharmingen), and anti-cyclin
E mouse monoclonal antibody (Calbiochem, La Jolla, CA, USA). Equal
loading of the protein samples was confirmed by parallel western
blots for actin with anti-actin rabbit polyclonal antibody
(Sigma-Aldrich).
Cell cycle analysis
To synchronize cell cycle, double-thymidine block
(DTB) method (19,20) was used. On 10 cm dishes,
1×106 cells of HCT116 were cultured in media containing
2.5 mM thymidine (Sigma-Aldrich) for 18 h, placed in thymidine-free
media for 12 h, and followed by an additional 18 h in thymidine
containing media. Between the exchange of media, dishes were washed
twice with PBS (Wako Pure Chemical Industries). Cell cycle was
analyzed at 0, 4, 8, 16 and 24 h after release of block with
PI/RNase Staining Buffer (BD Biosciences) on FACSAriaII and data
analysis was performed using FlowJo software (Tree Star, Ashland,
OR, USA).
Sphere formation assay
Single cells of isolated SSEA-3+ and
SSEA-3− in HCT116 were seeded in 96-well ultra-low
attachment plates (Corning, New York, NY, USA), cultured in mTeSR1
(Stem Cell Technologies) media without serum. Sphere formation was
assessed 28 days after seeding. In each of the 48 wells, the number
of formed spheres was calculated. This study was performed as
triplicated study.
Cell proliferation assay
Isolated HCT116 SSEA-3− and
SSEA-3+ cells were put into 96-well plates at a density
of 2×103 cells in 100 μl medium per well,
cultured for 24 h in McCoy’s 5A medium supplemented with 10% FBS,
and then cultured for an additional 24, 48, 72 and 96 h. Cell
proliferation was assessed with Cell Counting kit-8 incorporating
WST-8 (Dojindo Molecular Technologies, Kumamoto, Japan) following
the manufacturer’s instructions with a plate reader (Model 680XR;
Bio-Rad Laboratories).
Xenotransplantation
To assess tumorigenic properties, each of the 5,000
and 1,000 SSEA-3− and SSEA-3+ cells were
inoculated into dorsal flanks of 8-week-old NOD/SCID mice (CREA,
Tokyo) with a mixture of Matrigel matrix (BD Biosciences). Tumor
growth was monitored every three or four days by measurement with
calipers and the volume of the subcutaneous tumor was calculated by
the following formula; v = l ×
w2/2, where v = volume, 1 =
length and w = width (21).
Statistical analysis
The relationships among gene expressions, cell
counts, and tumor volume were analyzed using Student’s t-tests. All
tests were analyzed with JMP 9 software (SAS Institute, Cary, NC,
USA). A value of P<0.05 was taken as statistically
significant.
Results
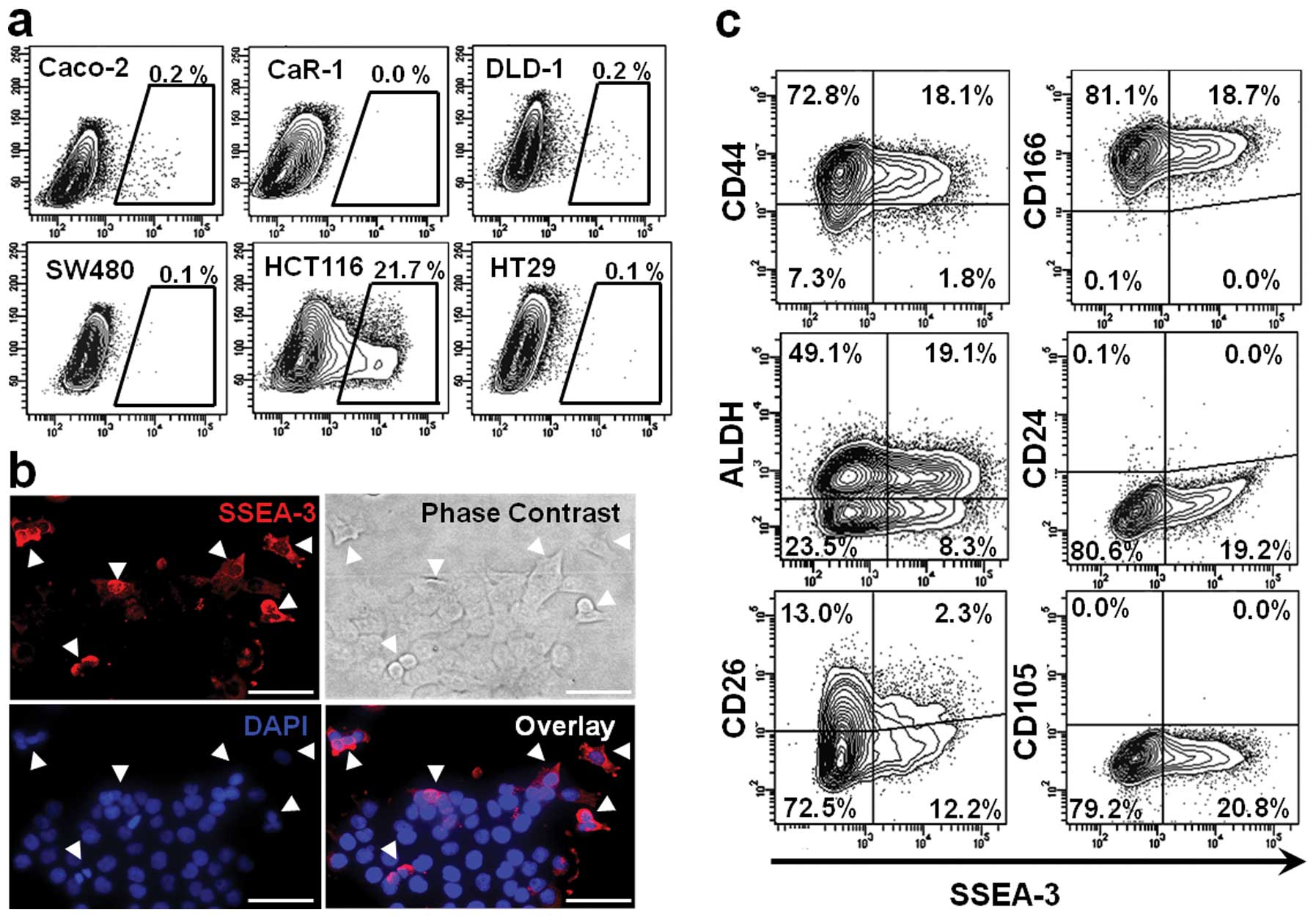
SSEA-3 expression in CRC cell lines
To assess SSEA-3 expression in CRC, flow-cytometric
analysis was performed on 6 CRC cell lines. It indicated that a
small number of SSEA-3+ population existed in most of
the CRC cell lines; 0.1 to 0.2% in Caco-2, DLD-1, HT-29 and SW480.
Among those tested, HCT116 had the highest expression level; it had
as much as 21.7±8.3% SSEA-3+ population. In CaR-1 cells,
expression of SSEA-3 could not be identified in this triplicate
study (Fig. 1a). To confirm SSEA-3
expression in HCT116 cells, immunofluorescent staining of SSEA-3
was performed. Consistent with the results of flow cytometric
analysis, a small number of HCT116 cells expressed SSEA-3 (Fig. 1b). In this study, HCT116 was used
for the following analyses because it had abundant
SSEA-3+ cells and it was easy to isolate
SSEA-3− and SSEA-3+ populations.
Correlation of SSEA-3 and stem cell
markers
To clarify the correlation between SSEA-3 and
representative colorectal CSC markers, HCT116 cells were
double-stained with CD44, CD166, ALDH, CD24 and CD26 (22). In CD44+ fraction 19.9%
of cells were SSEA-3 positive. In CD166+ fraction 18.7%,
in ALDH+ fraction 28.0%, in CD24+ fraction 0.0% and in
CD26+ fraction 15.0% of cells were SSEA-3 positive. These findings
indicated that there was no apparent correlation between the
representative CSC markers and SSEA-3 expression (Fig. 1c). Next, correlation between the
expression of SSEA-3 and CD105 [a specific mesenchymal/Muse cell
marker (23)] was assessed. In
contrast to the abundant expression of SSEA-3, expression of CD105
could not be identified in HCT116 cells, implying that properties
of SSEA-3+ cancer cells may be somewhat different from
those of Muse cells (Fig. 1c).
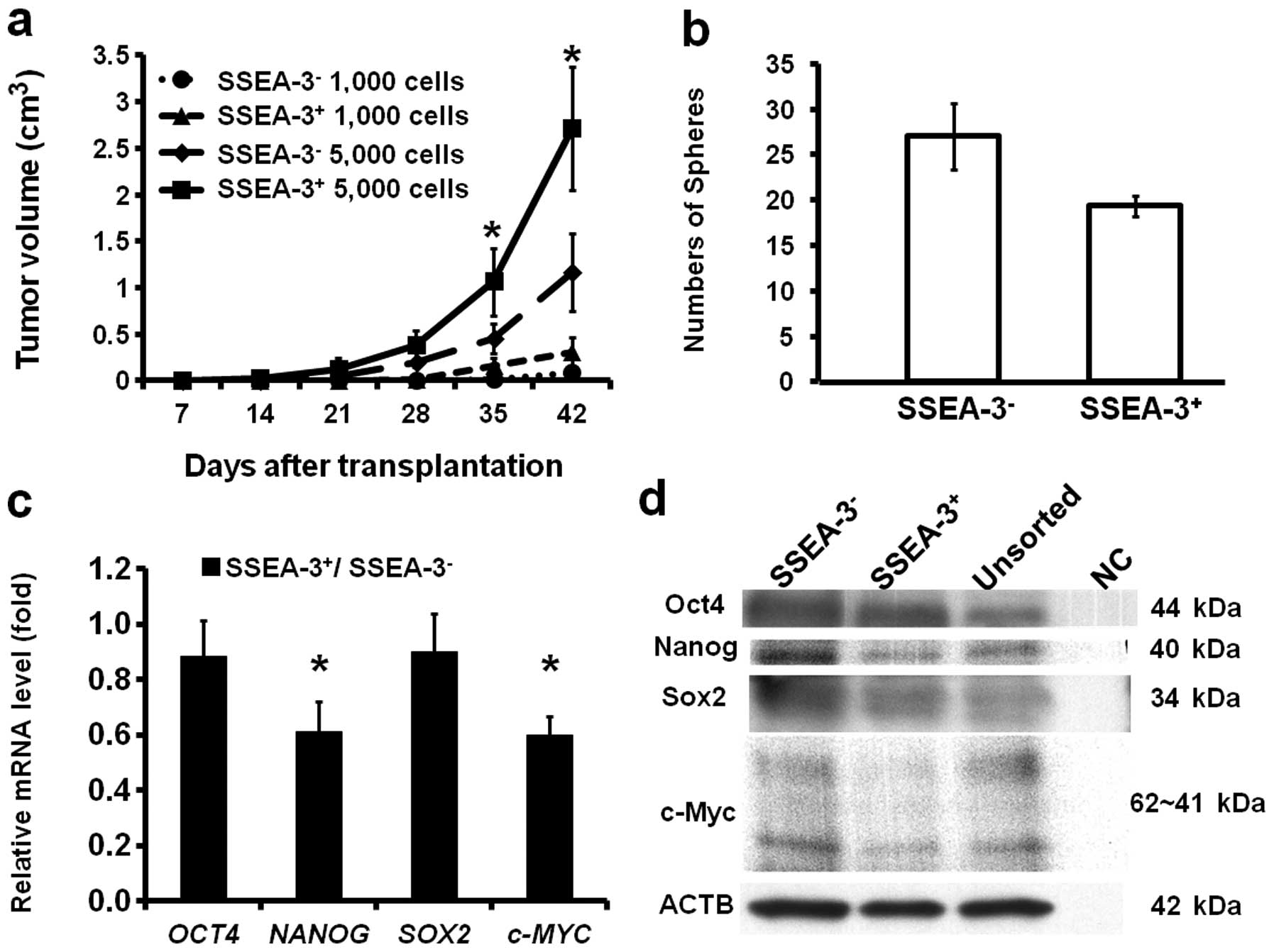
Correlation of SSEA-3 and tumorigenicity
and sphere formation ability
To confirm if SSEA-3 expression status actually does
not correlate with CSC characteristics, the tumorigenic activity of
SSEA-3− and SSEA-3+ cells were assessed. The
freshly isolated 1,000 and 5,000 SSEA-3− and
SSEA-3+ HCT116 cells were transplanted subcutaneously
into NOD/SCID mice. Notably, though SSEA-3 expression was not
correlated with representative colorectal CSC markers,
SSEA-3+ cells revealed higher tumorigenic activity
compared to that of SSEA-3− cells in both 1,000 and
5,000 cell inoculations (Fig. 2a).
To assess self-renewal ability, sphere formation assays were
performed three times. This revealed that SSEA-3+ cells
actually did form spheres, but the number of formed spheres was
significantly smaller (P<0.05) compared to that of
SSEA-3− cells (Fig.
2b).
Correlation of SSEA-3 and
pluripotency-associated genes
To resolve the reason why SSEA-3+ cells
reveal high tumorigenic activity, expressions of
pluripotency-associated genes; OCT4, NANOG,
SOX2 and c-MYC, which play key roles in iPS cell
induction (10) and are
overexpressed in Muse cells (23),
were analyzed on freshly isolated SSEA-3− and
SSEA-3+ HCT116 cells by qRT-PCR. The results indicated
that the expression of NANOG and c-MYC in
SSEA-3+ cell population was significantly lower than
that of SSEA-3−, while the expression of OCT4 and
SOX2 were not significantly different when stratified by
SSEA-3 levels (Fig. 2c), both of
which were consistent with the results from the western blot
analysis (Fig. 2d). These results
indicated that SSEA-3+ cancer cells had clearly distinct
properties from Muse cells (23).
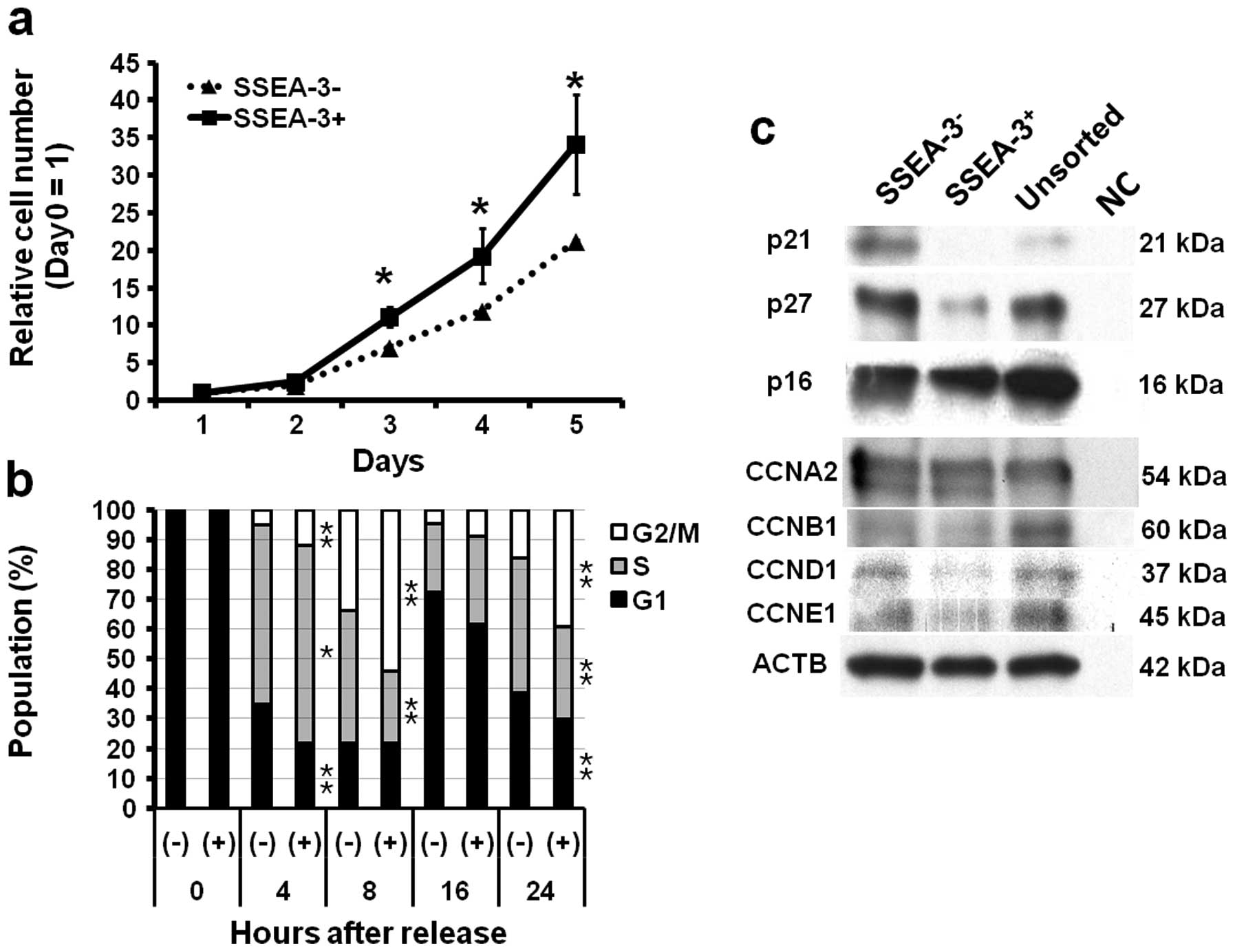
Correlation of SSEA-3 and proliferation
ability and cell cycle
To elucidate the background of the difference of
tumorigenic ability, cell proliferation ability of
SSEA-3− and SSEA-3+ cells were assessed. As
compared to SSEA-3− cells, SSEA-3+ cells
showed significantly higher proliferative activity in vitro
(P<0.05 at 3, 4 and 5 days after seeding) (Fig. 3a). To clarify the background of
high proliferation ability of SSEA-3+ cells observed
in vivo and in vitro, the cell cycle status of
SSEA-3− and SSEA-3+ cells were assessed,
because SSEA-3+ cells in ES cells were reported to show
rapid cell cycles (24). Cell
cycle analysis using HCT116 cells synchronized with a
double-thymidine block method revealed faster entry from G1 to S
and from S to G2/M phase in SSEA-3+ cells (Fig. 2c); there were significant increases
of S phase (P<0.05 at 4 h) and G2/M phase (P<0.05 at 4, 8,
and 24 h) population and significant decrease of G1 phase
(P<0.05 at 4 and 24 h) and S phase (P<0.05 at 8 and 24 h)
population in SSEA-3+ cells as compared to
SSEA-3− cells (Fig.
3b). Western blotting showed the decrease of
p21Cip1/Waf1 and p27Kip1 (hereafter
designated p21 and p27, respectively) in SSEA-3+ cells,
which are consistent with the results of cell cycle analyses, while
cyclins showed no apparent difference between SSEA-3−
and SSEA-3+ cells (Fig.
3c).
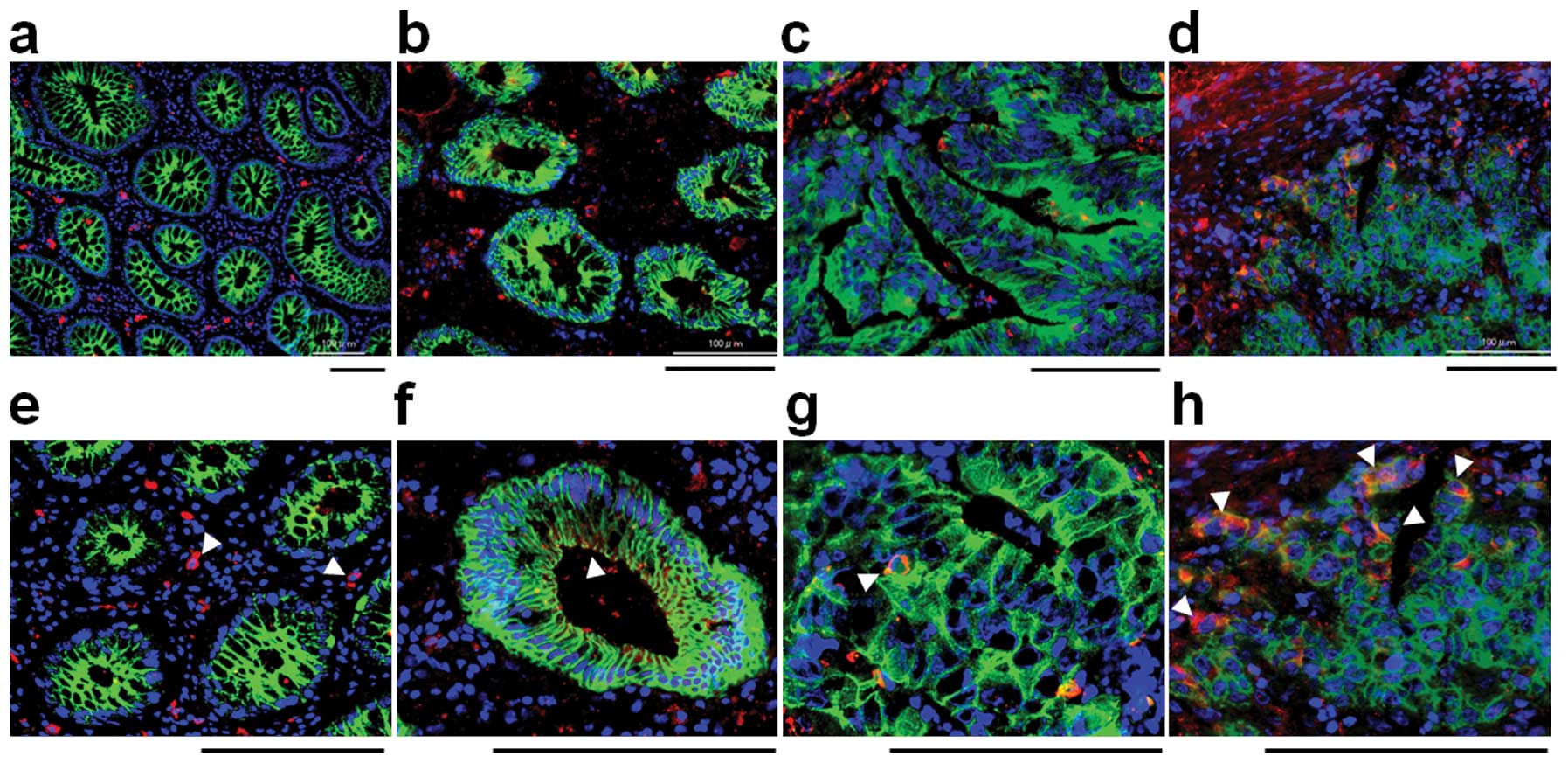
SSEA-3 expression in colorectal normal
and cancer tissues
To investigate SSEA-3 expression in normal
colorectal epithelia and colorectal cancer, frozen specimens, which
contain both normal and cancerous areas in the same section were
stained with SSEA-3 (2 cases of well differentiated, 6 cases of
moderately differentiated and 2 cases of poorly differentiated
adenocarcinoma). In normal areas, small numbers of stromal cells
were strongly positive for SSEA-3, but epithelial cells were not
apparently positive for SSEA-3 (Fig.
4a). In cancer areas, SSEA-3+ cancer cells were
identified in 50% (1 out of 2 cases) of well differentiated, in
66.7% (4 out of 6 cases) of moderately differentiated and in 100%
(2 out of 2) of poorly differentiated adenocarcinomas (Fig. 4b–d). The positive ratio and the
expression level of SSEA-3 had a tendency to be high in poorly
differentiated adenocarcinomas but low in those that were well
differentiated (Fig. 4d).
Discussion
In this study, we directly revealed the existence of
SSEA-3+ population in gastroenterological cancer, as far
as we know, for the first time. We have also clarified the cellular
properties of SSEA-3+ cells; highly tumorigenic and high
proliferative ability.
Though we could not identify a significant
correlation between SSEA-3 expression and representative colorectal
CSC markers (CD44, CD166, ALDH, CD24 and CD26), we revealed that
SSEA-3+ cells in the HCT116 CRC cell line possess high
tumorigenic ability, which is one of the representative properties
of CSCs, in immunodeficient mice. In sphere formation assay,
SSEA-3+ cells showed sufficient potency of sphere
formation ability, but it was significantly lower than that of
SSEA-3− cells. These results indicate that
SSEA-3+ cells in CRC retain immature phenotypes, but
have diminished stem cell properties, including self-renewal
capacity and pluripotency. In the CRC cell line, we have identified
abundant SSEA-3+ expression in HCT116 cells which
possess unique properties; they do not differentiate, even with
forced differentiation process and contain mainly CSCs (25). This report also supports our
findings that SSEA-3+ cells in CRC possess immature
phenotypes. A recent study indicated that colorectal CSCs involve
at least three characteristic sub-populations; long-term tumor
initiating cells (LT-TIC), tumor transient amplifying cells (T-TAC)
and delayed contributing TIC (DC-TIC) (26). On this basis, we hypothesized that
SSEA-3+ cells play a role as T-TAC in cancer
progression. To resolve these questions, we assessed the
proliferation activity and cell cycle status in SSEA-3+
cells, the data indicated that SSEA-3+ cells had high
proliferative activity. In the study of ES cells,
SSEA-3+ cells reportedly showed a faster cell cycle
(24), and we further detected the
decreased expression of cyclin-dependent kinase inhibitors p21 and
p27 in SSEA-3+ cells. p27 is not only a cell cycle
inhibitory factor of G1/S transition, but also a
differentiation-promoting factor (27), and decreased expression of p27 in
SSEA-3+ population has been reported in teratocarcinoma
(28). p21 negatively regulates
not only G1/S transition, but also G2/M transition (29), and its relation to SSEA-3
expression has not been reported. These findings also supported our
hypothesis.
SSEA-3 is known as a specific marker for Muse cells,
and Muse cells co-express CD105 (23). In this study, we could not identify
CD105 expression in SSEA-3 expressing CRC cells, which indicates
that SSEA-3+ cells in CRC differ from Muse cells in
marker expression. In addition, though Muse cells overexpress iPS
related genes (Oct3/4, NANOG, SOX2 and c-Myc) (23), the expression pattern of
iPS-related genes in SSEA-3+ CRC cells was clearly
different from that of Muse cells, suggesting that cellular
characteristics of SSEA-3+ cells in CRC are also
different from Muse cells. Moreover, the immunofluorescent finding
that no SSEA-3+ cells were detected in normal colorectal
epithelia also supports our findings.
In this study, unfortunately, we could not show a
correlation between SSEA-3 and clinical outcome because
immunohistochemical analysis for SSEA-3 on formalin-fixed paraffin
embedded (FFPE) samples was very difficult. SSEA-3 is a glycolipid
and can be lost in formalin, ethanol or methanol, meaning that FFPE
samples are not accurate. Though Chang et al showed the
existence of SSEA-3+ cells in normal human colorectal
tissues using FFPE samples (17),
we could not obtain reproducible immunohistochemical staining of
SSEA-3 in FFPE sections. For the same reason, qRT-PCR analysis and
western blot analysis are not routine methodologies for SSEA-3. For
further studies, it is necessary to identify a novel marker that
co-expresses on SSEA-3+ cells in CRC cells.
In the study of CSCs, especially in solid tumors,
tumorigenic properties have been considered to reveal CSC
properties. Based on this study, we imply that the assessment of
tumorigenicity is not sufficient to isolate CSC subsets, LT-TIC,
T-TAC and DC-TIC. To isolate and identify CSC subsets, it may be
essential to establish a new approach with a combination of
repeated serial transplantation assays.
Acknowledgements
We thank Dr Mari Dezawa and Dr Masaaki
Kitada at Tohoku University and Dr Eiichi Morii at Osaka University
for a valuable discussion on immunohistochemical sections. This
study was supported by a Grant-in-Aid for Cancer Research from the
Ministry of Education, Science, Sports and Culture Technology,
Japan, to H.Y. (grant no. 21390360).
References
|
1.
|
Ben-Porath I, Thomson MW, Carey VJ, et al:
An embryonic stem cell-like gene expression signature in poorly
differentiated aggressive human tumors. Nat Genet. 40:499–507.
2008.
|
|
2.
|
Levina V, Marrangoni AM, DeMarco R,
Gorelik E and Lokshin AE: Drug-selected human lung cancer stem
cells: cytokine network, tumorigenic and metastatic properties.
PLoS One. 3:E30772008.
|
|
3.
|
Rajasekhar VK, Studer L, Gerald W, Socci
ND and Scher HI: Tumour-initiating stem-like cells in human
prostate cancer exhibit increased NF-kappaB signalling. Nat Commun.
2:1622011.
|
|
4.
|
Wright AJ and Andrews PW: Surface marker
antigens in the characterization of human embryonic stem cells.
Stem Cell Res. 3:3–11. 2009.
|
|
5.
|
Solter D and Knowles BB: Monoclonal
antibody defining a stage-specific mouse embryonic antigen
(SSEA-1). Proc Natl Acad Sci USA. 75:5565–5569. 1978.
|
|
6.
|
Shevinsky LH, Knowles BB, Damjanov I and
Solter D: Monoclonal antibody to murine embryos defines a
stage-specific embryonic antigen expressed on mouse embryos and
human teratocarcinoma cells. Cell. 30:697–705. 1982.
|
|
7.
|
Kannagi R, Cochran NA, Ishigami F, et al:
Stage-specific embryonic antigens (SSEA-3 and -4) are epitopes of a
unique globo-series ganglioside isolated from human teratocarcinoma
cells. EMBO J. 2:2355–2361. 1983.
|
|
8.
|
Solter D, Shevinsky L, Knowles BB and
Strickland S: The induction of antigenic changes in a
teratocarcinoma stem cell line (F9) by retinoic acid. Dev Biol.
70:515–521. 1979.
|
|
9.
|
Evans MJ and Kaufman MH: Establishment in
culture of pluri-potential cells from mouse embryos. Nature.
292:154–156. 1981.
|
|
10.
|
Takahashi K and Yamanaka S: Induction of
pluripotent stem cells from mouse embryonic and adult fibroblast
cultures by defined factors. Cell. 126:663–676. 2006.
|
|
11.
|
Thomson JA, Itskovitz-Eldor J, Shapiro SS,
et al: Embryonic stem cell lines derived from human blastocysts.
Science. 282:1145–1147. 1998.
|
|
12.
|
Takahashi K, Tanabe K, Ohnuki M, et al:
Induction of pluripotent stem cells from adult human fibroblasts by
defined factors. Cell. 131:861–872. 2007.
|
|
13.
|
Kuroda Y, Kitada M, Wakao S, et al: Unique
multipotent cells in adult human mesenchymal cell populations. Proc
Natl Acad Sci USA. 107:8639–8643. 2010.
|
|
14.
|
Kannagi R: Carbohydrate-mediated cell
adhesion involved in hematogenous metastasis of cancer. Glycoconj
J. 14:577–584. 1997.
|
|
15.
|
Son MJ, Woolard K, Nam DH, Lee J and Fine
HA: SSEA-1 is an enrichment marker for tumor-initiating cells in
human glioblastoma. Cell Stem Cell. 4:440–452. 2009.
|
|
16.
|
Ye F, Li Y, Hu Y, Zhou C and Chen H:
Stage-specific embryonic antigen 4 expression in epithelial ovarian
carcinoma. Int J Gynecol Cancer. 20:958–964. 2010.
|
|
17.
|
Chang WW, Lee CH, Lee P, et al: Expression
of Globo H and SSEA3 in breast cancer stem cells and the
involvement of fucosyl transferases 1 and 2 in Globo H synthesis.
Proc Natl Acad Sci USA. 105:11667–11672. 2008.
|
|
18.
|
Yamamoto H, Kondo M, Nakamori S, et al:
JTE-522, a cyclooxygenase-2 inhibitor, is an effective
chemopreventive agent against rat experimental liver fibrosis1.
Gastroenterology. 125:556–571. 2003.
|
|
19.
|
Bello LJ: Studies on gene activity in
synchronized culture of mammalian cells. Biochim Biophys Acta.
179:204–213. 1969.
|
|
20.
|
Wilker EW, van Vugt MA, Artim SA, et al:
14-3-3sigma controls mitotic translation to facilitate cytokinesis.
Nature. 446:329–332. 2007.
|
|
21.
|
Ware JL and DeLong ER: Influence of tumour
size on human prostate tumour metastasis in athymic nude mice. Br J
Cancer. 51:419–423. 1985.
|
|
22.
|
Todaro M, Francipane MG, Medema JP and
Stassi G: Colon cancer stem cells: promise of targeted therapy.
Gastroenterology. 138:2151–2162. 2010.
|
|
23.
|
Wakao S, Kitada M, Kuroda Y, et al:
Multilineage-differentiating stress-enduring (Muse) cells are a
primary source of induced pluripotent stem cells in human
fibroblasts. Proc Natl Acad Sci USA. 108:9875–9880. 2011.
|
|
24.
|
Stewart MH, Bosse M, Chadwick K, Menendez
P, Bendall SC and Bhatia M: Clonal isolation of hESCs reveals
heterogeneity within the pluripotent stem cell compartment. Nat
Methods. 3:807–815. 2006.
|
|
25.
|
Yeung TM, Gandhi SC, Wilding JL, Muschel R
and Bodmer WF: Cancer stem cells from colorectal cancer-derived
cell lines. Proc Natl Acad Sci USA. 107:3722–3727. 2010.
|
|
26.
|
Dieter SM, Ball CR, Hoffmann CM, et al:
Distinct types of tumor-initiating cells form human colon cancer
tumors and metastases. Cell Stem Cell. 9:357–365. 2011.
|
|
27.
|
Wander SA, Zhao D and Slingerland JM: p27:
a barometer of signaling deregulation and potential predictor of
response to targeted therapies. Clin Cancer Res. 17:12–18.
2011.
|
|
28.
|
Baldassarre G, Barone MV, Belletti B, et
al: Key role of the cyclin-dependent kinase inhibitor p27kip1 for
embryonal carcinoma cell survival and differentiation. Oncogene.
18:6241–6251. 1999.
|
|
29.
|
Niculescu AB III, Chen X, Smeets M, Hengst
L, Prives C and Reed SI: Effects of p21(Cip1/Waf1) at both the G1/S
and the G2/M cell cycle transitions: pRb is a critical determinant
in blocking DNA replication and in preventing endoreduplication.
Mol Cell Biol. 18:629–643. 1998.
|