Introduction
Angiogenesis is an essential component of tumour
progression and is regulated by a number of hormones, cytokines,
growth factors and low molecular mediators (1). One of these molecules, vascular
endothelial cell growth factor (VEGF), has a particularly important
role. In the solid tumour microenvironment, it is conceivable that
focal hypoxia functions as an essential trigger for pathological
angiogenesis by the upregulation of VEGF gene expression (2). The principal transcription factor
that regulates VEGF expression is hypoxia inducible factor
(HIF)-1α, which is stabilised and accumulates under hypoxic
conditions as a result of decreased ubiquitination and degradation.
HIF-1α dimerises with HIF-1β and translocates to the nucleus, where
they bind to a hypoxia response element (HRE) on the VEGF gene and
activate its transcription (3).
On the other hand, hepatocyte growth factor (HGF) is
one of the molecules that induces tumour angiogenesis (4). HGF was identified originally as a
potent hepatotrophic factor responsible for vigorous regeneration
of the liver (5). It is now
recognised as a multipotent cytokine that mediates tumour-stromal
interaction with mitogenic, motogenic and morphogenic activities
(6,7). Moreover, HGF exerts potent angiogenic
activity in vascular endothelial cells (8). HGF is a natural ligand for the c-Met
proto-oncogene product of receptor tyrosine kinase (7,9). In
fact, HGF and c-Met are upregulated in a number of human cancers,
such as colorectal, gastric, oesophageal, breast and lung cancers
(10–12). The upregulation of c-Met inversely
correlates with the survival of patients with these types of cancer
(13–15).
HGF/c-Met signalling activates multiple signal
transduction pathways, including phosphatidylinositol 3-kinase
(PI3K)/Akt, mitogen-activated protein kinase (MAPK) and signal
transducer and activator of transcription 3 (STAT3) (16–18).
The synchronous activation of several signalling pathways is
essential for the various biological abilities of HGF (7). In terms of induced angiogenesis, HGF
stimulates endothelial cells directly through the c-Met receptor
and indirectly by facilitating the expression of other angiogenic
factors, represented by VEGF (19,20).
In the tumour-stromal interaction, HGF is mainly produced by
stromal cells and acts on tumour cells (21,22).
Therefore, we hypothesised that stroma-derived HGF induces VEGF
expression in tumour cells, resulting in CT26 tumour
angiogenesis.
We previously demonstrated that NK4, a competitive
antagonist for HGF, potently suppressed murine CT26 tumour growth
via inhibiting angiogenesis rather than HGF antagonism (23). NK4 inhibits the angiogenic
responses induced by VEGF and basic fibroblast growth factor
(bFGF), as well as those of HGF (24). However, the molecular mechanisms by
which HGF/c-Met signalling regulates VEGF expression in tumour
cells in the hypoxic tumour environment are not yet completely
understood. To provide further insight into the molecular
mechanisms underlying the angiogenic effect induced by the
cooperation between HGF and VEGF, we examined the effect of HGF
with/without anti-HGF antibody, NK4, or the inhibitors of kinases
downstream of HGF/c-Met signalling, on VEGF and HIF-1α expression
in CT26 cells.
Materials and methods
Reagents
Recombinant mouse HGF and polyclonal anti-mouse HGF
antibody were purchased from R&D Systems (Minneapolis, MN,
USA). Human recombinant NK4 was purified from the conditioned
medium of Chinese hamster ovary cells transfected with human NK4
cDNA (25). The PI3K inhibitor,
LY294002, and the MAPK inhibitor, PD98059, were purchased from
Promega Corp. (Madison, WI, USA). The STAT3 inhibitor, Stattic (a
non-peptidic small molecule shown to selectively inhibit the
function of the STAT3 SH2 domain, resulting in the inhibition of
STAT3 activation and dimerisation) was obtained from
Calbiochem-Merck Co. (Darmstadt, Germany).
Cell lines and culture conditions
CT26 is an undifferentiated colon adenocarcinoma
cell line originally derived from intrarectal injections of
N-nitroso-N-methylethylamine in a female BALB/c mouse. Cells were
maintained in RPMI-1640 (Nacalai Tesque, Inc., Kyoto, Japan)
supplemented with 100 IU/ml penicillin, 100 μg/ml
streptomycin (Sigma, Welwyn Garden City, UK) and 10%
heat-inactivated fetal bovine serum (FBS; JRH Biosciences, Inc.,
Lenexa, KS, USA) at 37°C in a humidified atmosphere containing 5%
CO2. Hypoxic condition (1% O2, 5%
CO2, 94% N2) was achieved using a Wakenyaku
9000E incubator (Wakenyaku Co., Ltd., Kyoto, Japan).
The genetic modification of CT26 to produce NK4 has
been described previously (23).
The transfectant expressing the highest amount of NK4 was
designated as CT26-NK4. Cells transfected with the
neomycin-resistance gene (pSVneo) alone were used as the control
(CT26-NEO).
Animal experiments
Female BALB/c mice (8–10 weeks old) were purchased
from the Shimizu Laboratory Animal Center (Kyoto, Japan). To
generate tumours, 5×105 CT26-NEO or CT26-NK4 cells in
0.1 ml phosphate-buffered saline (PBS) were inoculated
subcutaneously into syngeneic BALB/c mice in the right lower flank
(n=7 for each group). Considering the difference in tumour growth
between CT26-NEO and CT26-NK4, the mice were sacrificed under
general anaesthesia when the tumour diameter was 10 mm. After
general perfusion of the mice with PBS, tumours were removed and
homogenised by a sonic homogeniser. VEGF concentration in the
homogenates was quantitatively analysed by ELISA (R&D Systems).
The results were normalised to tumour weight. The animal experiment
was approved by the Animal Ethics Committee of the Kyoto
Prefectural University of Medicine and was carried out in
accordance with the ‘Guidelines for the welfare and use of animals
in cancer research’ (26).
Determination of VEGF expression
The CT26-NEO and CT26-NK4 cells were plated in
24-well culture plates at 1×105 cells/well in RPMI with
10% FBS. After an overnight incubation, the culture medium was
replaced and the cells were incubated under serum-starved
conditions (RPMI with 0.1% BSA) for 12 h, followed by treatment
with 10 ng/ml mouse recombinant HGF for 24 h under normoxic and
hypoxic conditions. To examine the effect of HGF/c-Met signalling,
HGF with/without 20 mg/ml NK4 or 5 mg/ml anti-HGF antibody were
added. Moreover, to examine the activities of the respective kinase
inhibitors, cultured cells were pre-treated with 50 mM PI3K
inhibitor (LY294002), 20 mM MAPK inhibitor (PD98059), or 20 mM
STAT3 inhibitor (Stattic) for 60 min prior to the addition of HGF.
The culture supernatants were collected and VEGF concentrations
were determined by ELISA.
Quantification of HIF-1α transcriptional
activity
The DNA binding activity of HIF-1α was evaluated
using the HIF-1α transcription factor assay kit (Cayman Chemical,
Ann Arbor, MI, USA) according to the manufacturer’s instructions.
Cells were plated in 100-mm culture dishes and allowed to grow
until subconfluent. Cells were starved for 12 h, pre-treated with
the respective kinase inhibitors (50 mM LY294002, 20 mM PD098059
and 20 mM Stattic) and then treated with 10 ng/ml HGF under
normoxic and hypoxic conditions for 8 h.
Nuclear extracts were prepared and incubated in
96-well plates coated with immobilised double-stranded
oligonucleotides containing the HIF-1α response element
(5′-ACGTG-3′). The HIF-1α transcription factor complex was detected
by the addition of a specific primary antibody directed against
HIF-1α, visualised by an anti-IgG horseradish peroxidase
(HRP)-conjugate and quantified by measuring the absorbance at 450
nm. The DNA binding activity of HIF-1α was expressed relative to
the value of the control (CT26-NEO without HGF treatment under
normoxic conditions). The experiments were repeated 2 or 3 times
and similar results were obtained.
Western blot analysis
Aliquots of nuclear extracts (50 mg) were
fractionated by 7% sodium dodecyl sulphate (SDS)-polyacrylamide gel
electrophoresis and transferred onto a polyvinylidene difluoride
(PVDF) membrane. The membrane was blocked with 5% non-fat dried
milk in Tris-buffered saline containing 0.05% Tween-20, for 1 h at
room temperature, followed by overnight incubation at 4°C with
anti-HIF-1α rabbit polyclonal antibody (NB100-497) (Novus
Biologicals, Littleton, CO, USA) at a 1:500 dilution.
HRP-conjugated goat anti-rabbit secondary antibody was used at a
1:2,000 dilution. The signal was developed with a chemiluminescence
reagent (ECL plus; GE Healthcare, Piscataway, NJ, USA) and the band
images were detected with Versa Doc-4000 (Bio-Rad Laboratories,
Hercules, CA, USA).
Real-time reverse
transcription-polymerase chain reaction (RT-PCR)
Subconfluent cells in 100-mm culture dishes were
starved for 12 h and treated with/without 10 ng/ml mouse
recombinant HGF for 6 h under normoxic and hypoxic conditions. To
examine the activities of the respective kinase inhibitors, the
cells were pre-treated with 50 mM LY294002, 20 mM PD98059, or 20 mM
Stattic for 60 min before the addition of HGF.
Total RNA was isolated from the cultured cells using
an acid guanidinium thiocyanate-phenol (AGTP) solution (Isogen;
Nippon Gene, Tokyo, Japan) according to the manufacturer’s
instructions. The PCR primers used were as follows: VEGF-A,
5′-CTGGATATGTTTGACTGCTGTGGA-3′ (sense) and
5′-GTTTCTGGAAGTGAGCCAATGTG-3′ (antisense); HIF-1α,
5′-AGCAGGAATTGGAACATTATTGCAG-3′ (sense) and
5′-TGTGGTAATCCACTCTCATCCATTG-3′ (antisense). For normalization, the
18S ribosomal protein was used as the housekeeping gene: Rps18,
5′-TTCTGGCCAACGGTCTAG ACAAC-3′ (sense) and
5′-CCAGTGGTCTTGGTGTGCTGA-3′ (antisense). Real-time RT-PCR was then
performed using a LightCycler 1.5 (Roche, Basel, Switzerland) and
SYBR-Green I (Qiagen, Inc., Valencia, CA). The RT-PCR protocol
consisted of a 50°C reverse transcription step for 20 min; a 95°C
PCR initial activation step for 15 min; followed by 40 cycles of a
94°C denaturation for 15 sec, 60°C annealing for 30 sec and 72°C
extension for 30 sec. In order to confirm specific amplification,
the PCR products were subjected to melting curve analysis. The
results from real-time RT-PCR analysis were normalised to Rps18 and
expressed relative to the value of the control (CT26-NEO without
HGF treatment under normoxic conditions). Relative expression
levels were calculated using the ΔΔCt method. Experiments were
repeated 2 or 3 times and similar results were obtained.
Statistical analysis
Statistical evaluation was performed with the
two-tailed Student’s t-test, unless mentioned otherwise in the
text. Differences with P-values <0.05 were considered
statistically significant.
Results
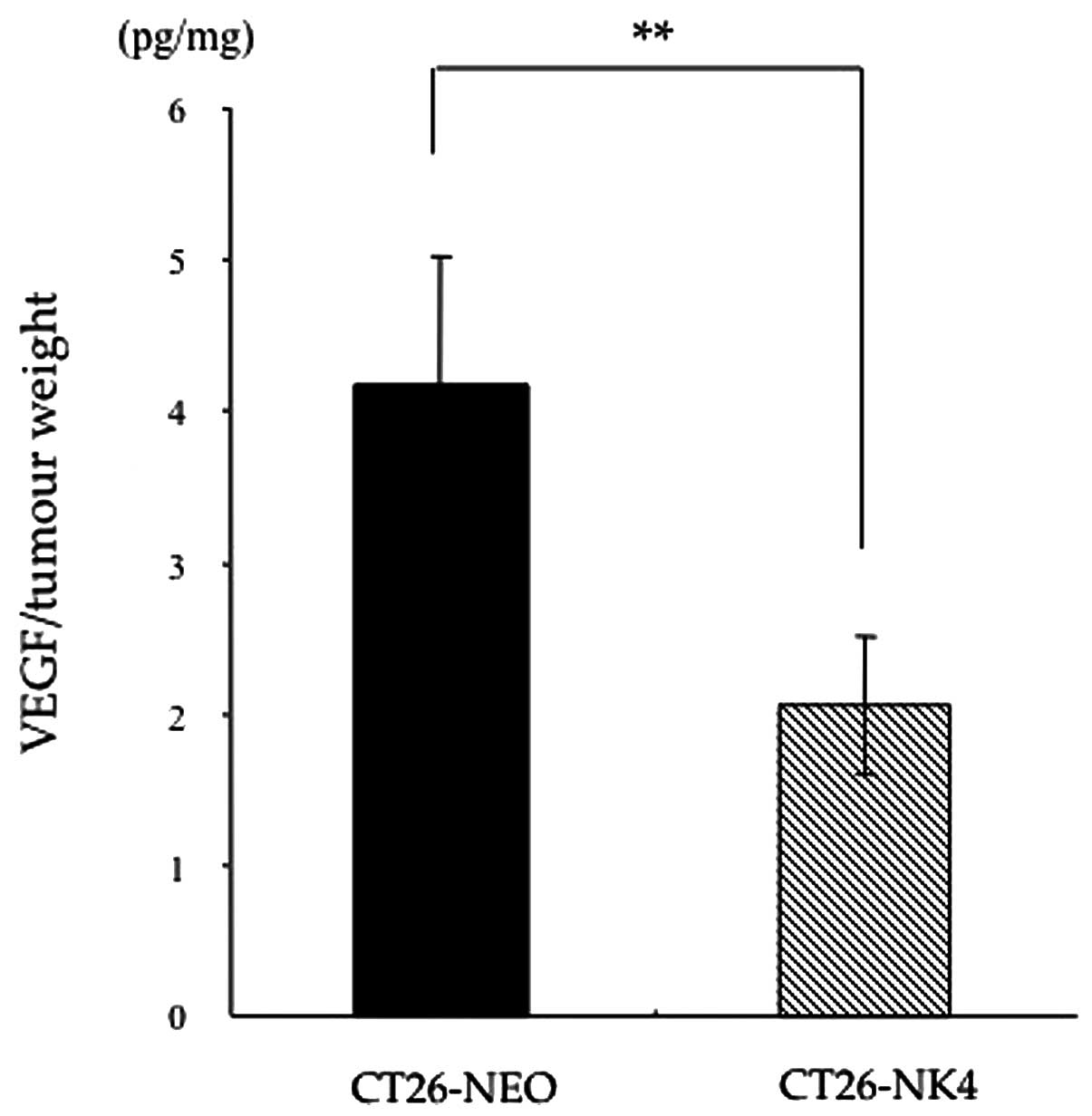
VEGF expression is reduced in homografts
of CT26-NK4
To confirm the hypothesis that the blockade of
HGF/c-Met signalling would reduce VEGF expression, resulting in
anti-angiogenesis, we first compared VEGF expression in vivo
between CT26-NEO and CT26-NK4 subcutaneous tumours. CT26
transfectants were inoculated into mice and the VEGF concentrations
in the homografts were determined. The VEGF concentration in the
CT26-NK4 homografts, relative to the respective tumour weights, was
significantly lower than that in the CT26-NEO homografts (CT26-NK4
2.1±0.5 pg/mg, CT26-NEO 4.2±0.9 pg/mg; P<0.01) (Fig. 1).
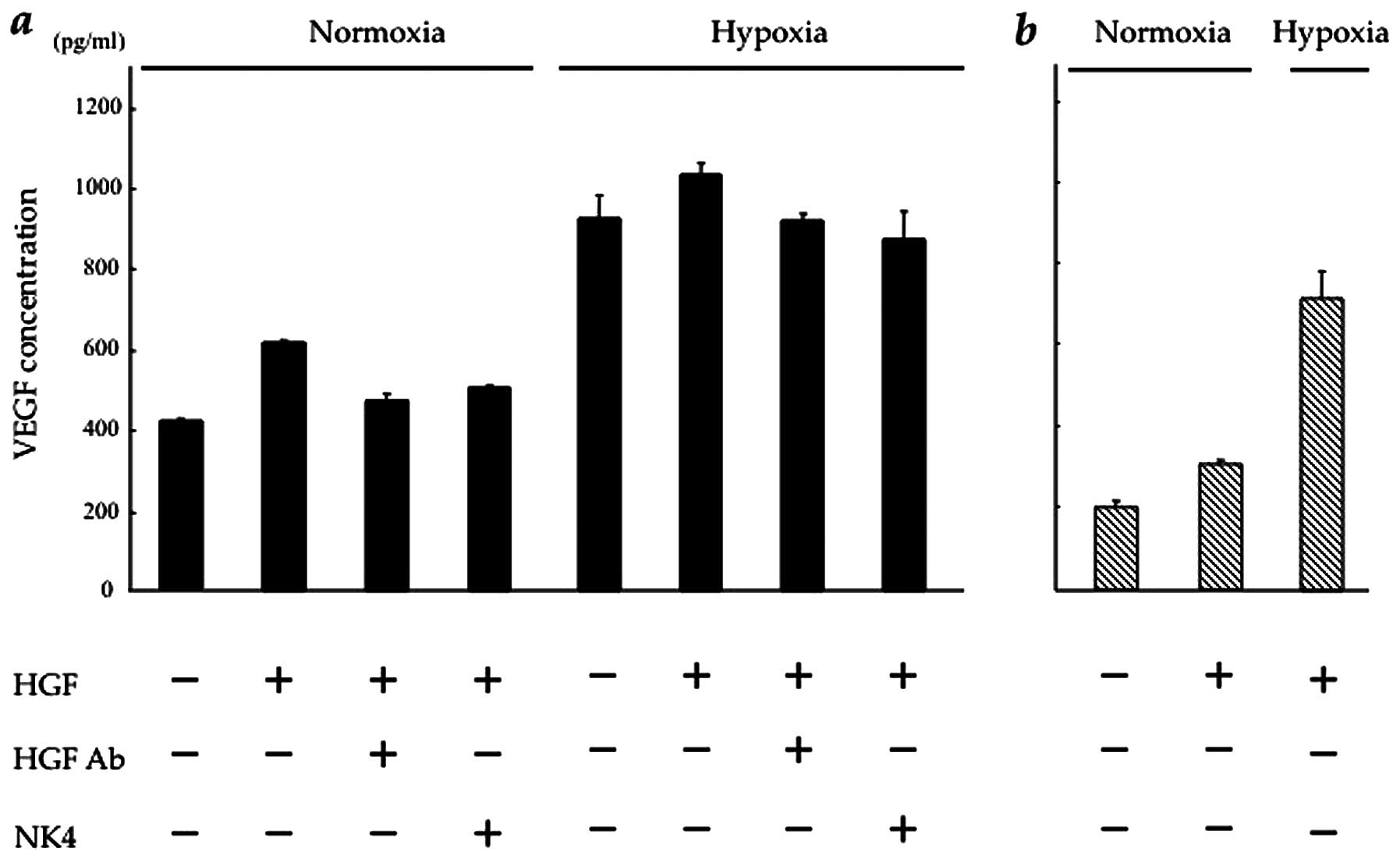
Blockade of HGF/c-Met signalling inhibits
HGF-induced VEGF expression in CT26 cells
We then investigated the effect of HGF on VEGF
expression in vitro in CT26-NEO and CT26-NK4 cells under
normoxic and hypoxic conditions. In our preliminary experiment,
VEGF expression in both transfectants under hypoxic conditions was
markedly higher than that under normoxic conditions (data not
shown). As regards the effect of HGF, VEGF expression in the
transfectants was increased in a HGF-dose-dependent manner.
However, VEGF expression in response to HGF reached a plateau at 10
ng/ml HGF in CT26-NEO and at 2.5 ng/ml HGF in CT26-NK4 cells.
Under normoxic conditions, in CT26-NEO cells, VEGF
expression was 1.6-fold higher under HGF (10 ng/ml) stimulation,
but was blocked by the addition of NK4 (20 mg/ml) or anti-HGF
antibody (5 mg/ml). Under hypoxic conditions, in CT26-NEO cells,
the baseline VEGF expression was significantly increased (by
2.4-fold), but a similar block was observed in the presence of HGF
with/without NK4 or anti-HGF antibody (Fig. 2a). These results indicate that the
alteration in VEGF expression in CT26 cells involves the HGF/c-Met
signalling pathway.
In CT26-NK4 cells, the baseline VEGF expression
decreased and responded weakly to HGF under normoxic conditions.
However, the expression level was lower than that of the control.
Under hypoxic conditions, VEGF expression in CT26-NK4 cells
significantly increased, but was also lower than that in CT26-NEO
cells under hypoxic conditions (Fig.
2b). These results indicate that VEGF expression in CT26 cells
is regulated by HGF/c-Met signalling. On the other hand, the
exogenous expression of NK4 potently reduced VEGF expression in
CT26 cells, even under hypoxic conditions.
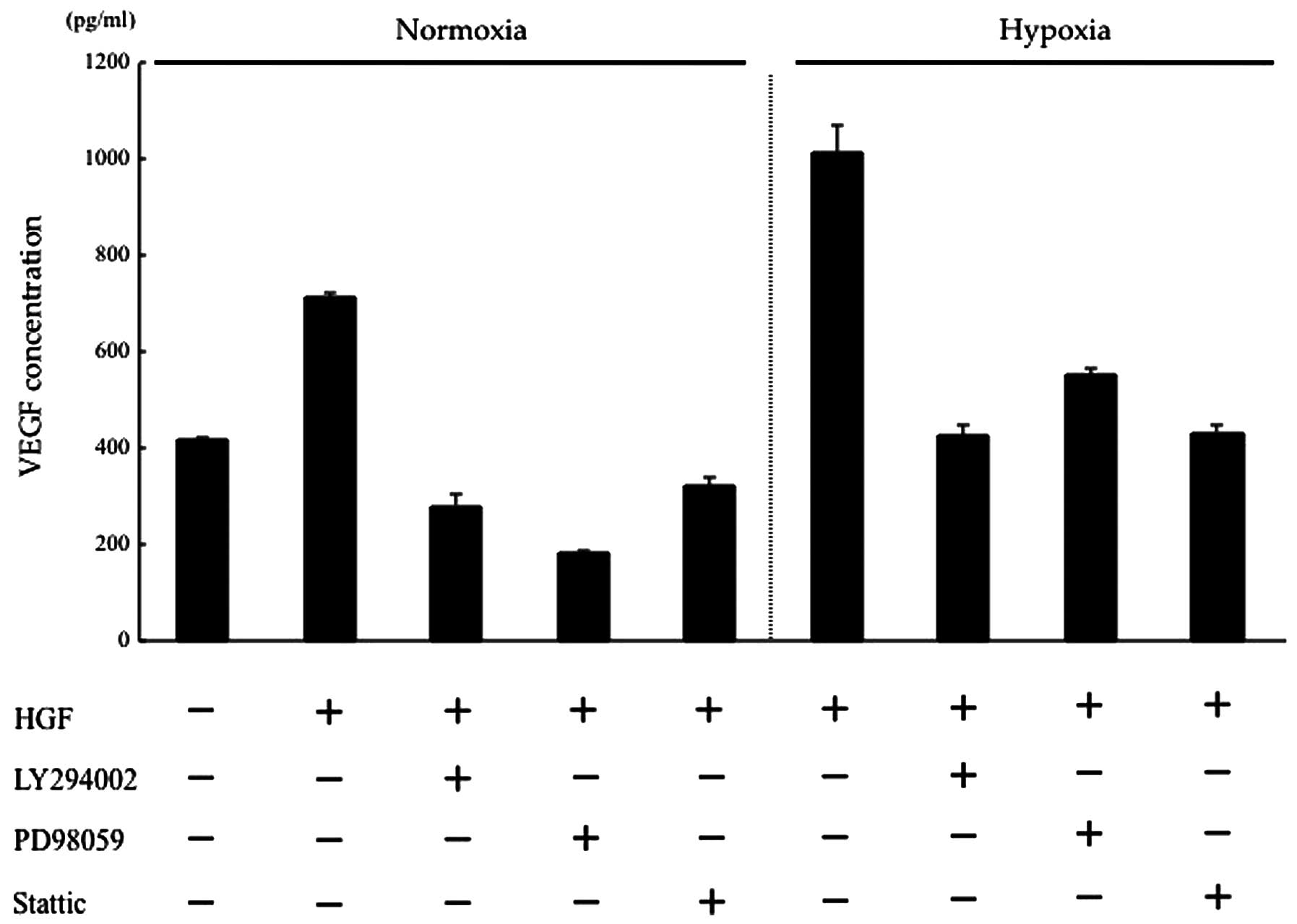
HGF regulates VEGF expression of CT26
cells via the PI3K/Akt, MAPK and STAT3 pathways
HGF/c-Met binding activates several intracellular
signalling pathways, including PI3K/Akt, MAPK and STAT3 (4,27).
Previously, we showed that the phosphorylation of PI3K, Akt,
extracellular-signal-regulated kinase 1/2 (ERK1/2) and STAT3 in the
CT26 transfectants activated by HGF was inhibited by the addition
of anti-HGF antibody or NK4 (28).
In this study, we determined which of these pathways may be
involved in regulating VEGF expression in CT26 cells, using kinase
inhibitors of the PI3K/Akt, MAPK and STAT3 pathways. Under normoxic
conditions, HGF-induced VEGF expression was suppressed to a level
lower than that of the control by the respective kinase inhibitors.
Under hypoxic conditions, VEGF expression, which was increased by
HGF, was suppressed to a level almost equal to that of the control
by the respective kinase inhibitors. These results suggest that in
intracellular HGF/c-Met signalling, the PI3K/Akt, MAPK, STAT3
pathways are involved in the changes in VEGF expression induced by
hypoxia (Fig. 3).
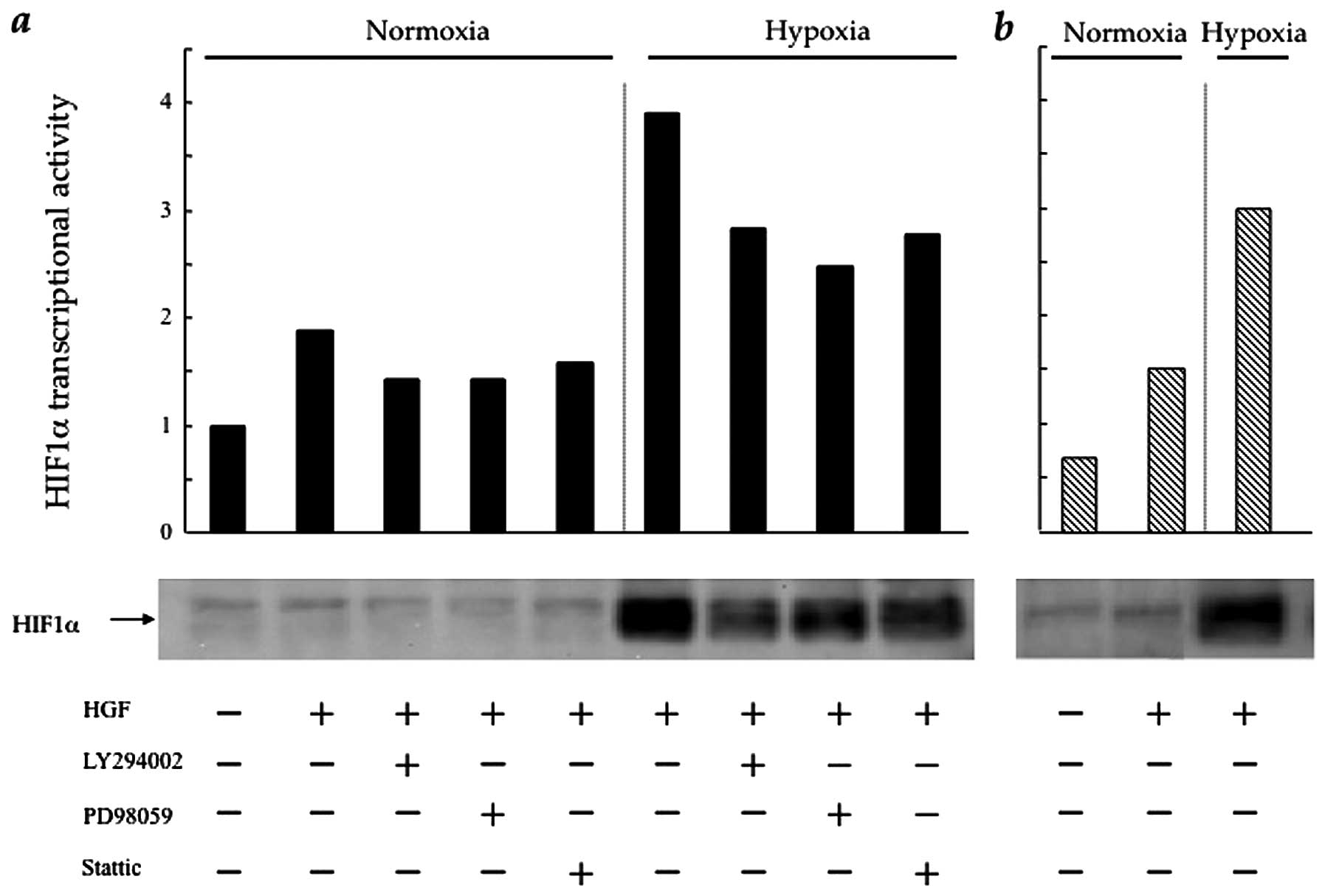
HGF regulates HIF-1α protein synthesis
and transcriptional activity in CT26 cells via the PI3K/Akt, MAPK
and STAT3 pathways
HIF-1α is the main transcriptional factor of VEGF
under hypoxic conditions (3).
Therefore, we investigated the influence of intracellular HGF/c-Met
signalling pathways upon HIF-1α expression and HIF-1α
transcriptional activity in the CT26 transfectants. In CT26-NEO
cells, HIF-1α expression, which increased slightly in the presence
of HGF under normoxic conditions, was stimulated significantly in
response to hypoxia. The PI3K, MAPK and STAT3 inhibitors
significantly, although incompletely, blocked HIF-1α expression
under hypoxic, as well as normoxic conditions. HIF-1α
transcriptional activity showed a similar trend under normoxic and
hypoxic conditions (Fig. 4a). In
CT26-NK4 cells, HIF-1α expression and HIF-1α transcriptional
activity only weakly responded to HGF (Fig. 4b). It was not surprising that
HIF-1α expression and HIF-1α transcriptional activity were
upregulated by hypoxia; however, these results suggest a partial
involvement of intracellular HGF/c-Met signalling pathways in
regulating HIF-1α expression and HIF-1α transcriptional
activity.
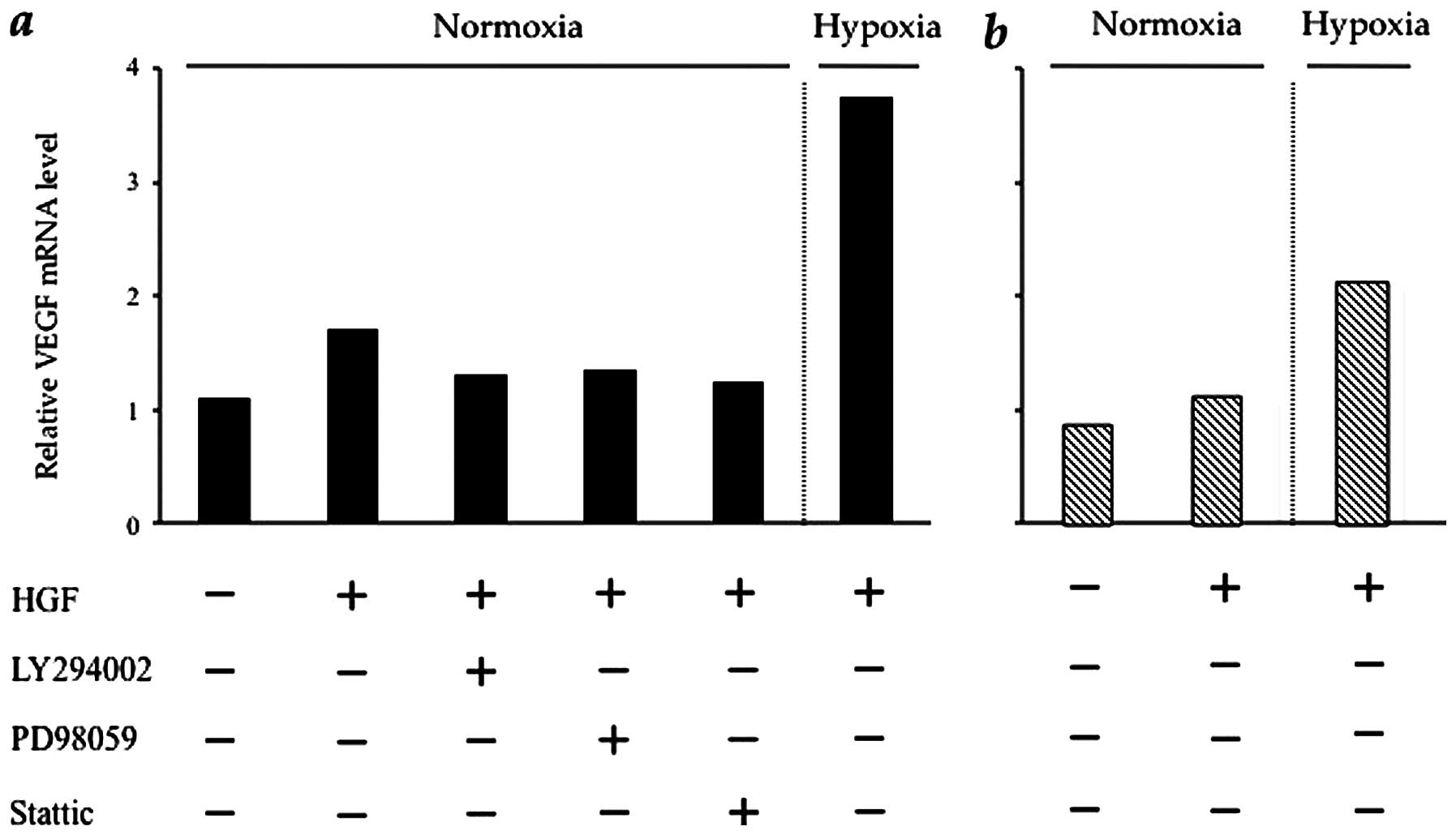
HGF regulates VEGF mRNA in CT26 cells via
the PI3K/Akt, MAPK and STAT3 pathways
Based on the result presented in the previous
section, we investigated the influence of intracellular HGF/c-Met
signalling pathways upon VEGF and HIF-1α mRNA expression. In
CT26-NEO cells, the VEGF mRNA expression level was increased by HGF
and, in particular, under hypoxic conditions (by approximately
4-fold) in CT26-NEO cells. HGF induction of VEGF mRNA was prevented
by the respective kinase inhibitors (Fig. 5a). The effect of HGF on VEGF mRNA
expression in CT26-NK4 cells was minimal. VEGF mRNA expression in
CT26-NK4 cells was increased by hypoxia (by approximately 2-fold),
but was lower than that in CT26-NEO cells under hypoxic conditions
(Fig. 5b). These results
correspond to those obtained for HIF-1α expression and HIF-1α
transcriptional activity.
HGF regulates HIF-1α mRNA in CT26 cells
via the STAT3 pathway
The HIF-1α mRNA expression level under hypoxic
conditions was equal to or less than the control CT26-NEO cells. Of
note, HGF induction of HIF-1α mRNA was not inhibited by LY294002 or
PD98059, but only by Stattic (Fig.
5c). HGF consistently had no effect on CT26-NK4 cells and the
effect of hypoxia was similar to that observed in CT26-NEO cells
(Fig. 5d). Taken together, the
results of real-time PCR and the quantification of HIF-1α
transcriptional activity demonstrated that the PI3K/Akt and MAPK
pathways regulate HIF-1α translation, whereas the STAT3 pathway
regulates HIF-1α mRNA expression.
Discussion
In the present study, we demonstrate that the
HGF/c-Met signalling pathway regulates VEGF expression in CT26
tumour cells. VEGF, which is produced by various cancer cells, acts
on endothelial cells and promotes tumour growth and angiogenesis
(29). Given that CT26 cells
strongly express VEGF, it is hypothesised that VEGF participates in
CT26 angiogenesis and subsequent tumour progression. We previously
revealed that the production of tumour microvessels in CT26 tumours
was significantly inhibited by NK4 gene transfer (23). NK4 is a potent angiogenic inhibitor
and its action is independent of HGF antagonism. In in vivo
tumours in particular, HGF seems to be the predominant mechanism of
angiogenesis (30,31). Kuba et al(24) reported that NK4 exerted a potent
anti-angiogenic effect, not only by the blockade of HGF/c-Met
signalling, but also by the interruption of intracellular
signalling of other growth factors, such as VEGF or bFGF. However,
the mechanism we propose in this study is distinct from that
previously reported.
In our study, we focused on the alteration of VEGF
expression in CT26-NK4 tumours. It has now become apparent that no
single factor alone can induce structural and functional
neovascularisation. HGF stimulates VEGF production in
non-endothelial cells (32,33)
and increases the expression of VEGF receptors and c-Met in
endothelial cells (19). Sulpice
et al(34) reported that
cross-talk between VEGF and HGF signalling pathways promoted
neovascularisation by enhancing intracellular signalling in
endothelial cells. Therefore, we hypothesised that NK4 may
contribute to the inhibition of tumour growth by decreasing VEGF
expression through the HGF/c-Met signalling pathway. Our hypothesis
was then proven by the fact that VEGF expression in CT26-NK4
homografts was reduced as compared to the control homografts. In
addition, this result was supported by the in vitro
experiment under normoxic and hypoxic conditions.
The effect of hypoxia on malignant progression is
mediated by a series of hypoxia-induced proteomic and genomic
changes, activating angiogenesis, anaerobic metabolism and other
processes that enable tumour cells to survive or escape their
oxygen-deficient environment (35). The transcription factor, HIF-1α, is
a major regulator of tumour cell adaptation to hypoxic stress.
Therefore, we assessed which intracellular signalling pathway may
be involved in VEGF expression under hypoxic conditions, using the
PI3K inhibitor, LY294002, the MAPK inhibitor, PD98059 and the STAT3
inhibitor, Stattic. Our data revealed that the respective kinase
inhibitors suppressed the HGF induction of HIF-1α, subsequent
HIF-1α transcriptional activity, VEGF mRNA expression, and VEGF
expression, even under hypoxic conditions, indicating that HGF
induces HIF-1α expression and subsequent processes to promote the
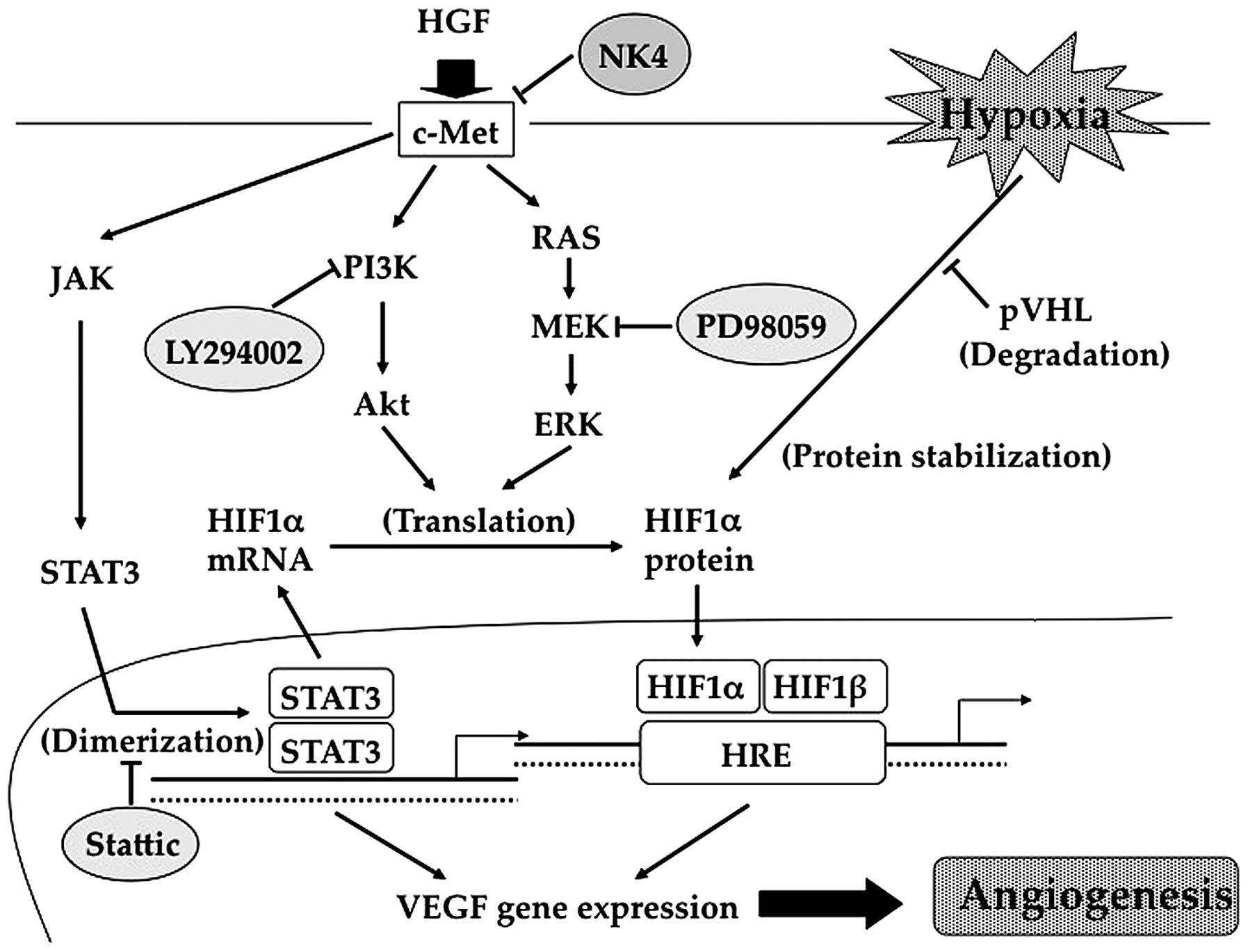
expression of VEGF via PI3K, MAPK and STAT3 activation (Fig. 6). However, our results demonstrated
that the mechanisms involved are more complex. In this study, the
HGF induction of HIF-1α mRNA was not inhibited by the PI3K and MAPK
inhibitors, but only by the STAT3 inhibitor. Certain studies have
reported that growth factors induce HIF-1-mediated VEGF expression,
which is dependent on the MAPK and PI3K pathways (36,37).
Therefore, the PI3K/Akt and MAPK pathways regulate VEGF expression
via HIF-1α translation. On the other hand, the ability of STAT3 to
activate the VEGF gene as a direct transcriptional activator has
previously been demonstrated (38,39).
A recent study reported that STAT3 signalling also enhanced the
transcriptional activation of the HIF-1α promoter and contributed
to the upregulation of HIF-1α mRNA (40). Our findings are consistent with
these reports. Accordingly, we hypothesised, as shown in Fig. 6, that STAT3 is required not only
for the direct activation of the VEGF promoter, but also for
HIF-1α-mediated VEGF expression. Therefore, the increase in HIF-1α
mRNA expression induced by HGF was due to STAT3 activation. STAT3
has been reported to influence HIF-1α protein stability under
hypoxic conditions (41), in
addition to HIF-1α protein synthesis induced by oncogenic growth
signalling (42). Such phenomena
are also evident in our results. Taken together, these data
indicate that HGF/c-Met signalling promotes VEGF expression by a
complex mechanism involving the PI3K/Akt, MAPK and STAT3 pathways
(Fig. 6). Conversely, NK4
suppresses VEGF expression in CT26 tumour cells by inhibiting the
activation of intracellular signalling pathways downstream of
HGF/c-Met.
In conclusion, the present study provides new and
important information concerning the mechanisms by which NK4 exerts
its anti-angiogenic effects. These mechanisms partly involve the
suppression of VEGF expression in CT26 tumour cells by blocking the
activation of the HGF/c-Met signalling pathway. Furthermore, a
detailed analysis of the involvement of intracellular HGF/c-Met
signalling in VEGF expression showed that the PI3K/Akt and MAPK
pathways regulated HIF-1α translational activity, whereas the STAT3
pathway regulated HIF-1α transcriptional activity and directly
affected VEGF transcriptional activity. These data therefore
suggest that HGF/c-Met signalling may be a promising target for the
future development of anti-angiogenic strategies to improve
response rate and survival in cancer patients.
References
|
1.
|
Carmeliet P and Jain RK: Angiogenesis in
cancer and other diseases. Nature. 407:249–257. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Shweiki D, Itin A, Soffer D and Keshet E:
Vascular endothelial growth factor induced by hypoxia may mediate
hypoxia-initiated angiogenesis. Nature. 359:843–845. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Liao D and Johnson RS: Hypoxia: a key
regulator of angiogenesis in cancer. Cancer Metastasis Rev.
26:281–290. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Trusolino L and Comoglio PM:
Scatter-factor and semaphorin receptors: cell signalling for
invasive growth. Nat Rev Cancer. 2:289–300. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Nakamura T, Nishizawa T, Hagiya M, et al:
Molecular cloning and expression of human hepatocyte growth factor.
Nature. 342:440–443. 1989. View
Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Jiang WG and Hiscox S: Hepatocyte growth
factor/scatter factor, a cytokine playing multiple and converse
roles. Histol Histopathol. 12:537–555. 1997.PubMed/NCBI
|
|
7.
|
Matsumoto K and Nakamura T: Emerging
multipotent aspects of hepatocyte growth factor. J Biochem.
119:591–600. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Bussolino F, Di Renzo MF, Ziche M, et al:
Hepatocyte growth factor is a potent angiogenic factor which
stimulates endothelial cell motility and growth. J Cell Biol.
119:629–641. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Bottaro DP, Rubin JS, Faletto DL, et al:
Identification of the hepatocyte growth factor receptor as the
c-met proto-oncogene product. Science. 251:802–804. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Birchmeier C, Birchmeier W, Gherardi E and
Vande Woude GF: Met, metastasis, motility and more. Nat Rev Mol
Cell Biol. 4:915–925. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Comoglio PM, Giordano S and Trusolino L:
Drug development of MET inhibitors: targeting oncogene addiction
and expedience. Nat Rev Drug Discov. 7:504–516. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Maulik G, Shrikhande A, Kijima T, Ma PC,
Morrison PT and Salgia R: Role of the hepatocyte growth factor
receptor, c-Met, in oncogenesis and potential for therapeutic
inhibition. Cytokine Growth Factor Rev. 13:41–59. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Di Renzo MF, Olivero M, Giacomini A, et
al: Overexpression and amplification of the met/HGF receptor gene
during the progression of colorectal cancer. Clin Cancer Res.
1:147–154. 1995.PubMed/NCBI
|
|
14.
|
Ghoussoub RA, Dillon DA, D’Aquila T, Rimm
EB, Fearon ER and Rimm DL: Expression of c-met is a strong
independent prognostic factor in breast carcinoma. Cancer.
82:1513–1520. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Miller CT, Lin L, Casper AM, et al:
Genomic amplification of MET with boundaries within fragile site
FRA7G and upregulation of MET pathways in esophageal
adenocarcinoma. Oncogene. 25:409–418. 2006.PubMed/NCBI
|
|
16.
|
Ponzetto C, Bardelli A, Maina F, et al: A
novel recognition motif for phosphatidylinositol 3-kinase binding
mediates its association with the hepatocyte growth factor/scatter
factor receptor. Mol Cell Biol. 13:4600–4608. 1993.
|
|
17.
|
Xiao GH, Jeffers M, Bellacosa A, et al:
Anti-apoptotic signaling by hepatocyte growth factor/Met via the
phosphatidylinositol 3-kinase/Akt and mitogen-activated protein
kinase pathways. Proc Natl Acad Sci USA. 98:247–252. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Boccaccio C, Andò M, Tamagnone L, et al:
Induction of epithelial tubules by growth factor HGF depends on the
STAT pathway. Nature. 391:285–288. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Gerritsen ME, Tomlinson JE, Zlot C, Ziman
M and Hwang S: Using gene expression profiling to identify the
molecular basis of the synergistic actions of hepatocyte growth
factor and vascular endothelial growth factor in human endothelial
cells. Br J Pharmacol. 140:595–610. 2003. View Article : Google Scholar
|
|
20.
|
Dong G, Chen Z, Li ZY, Yeh NT, Bancroft CC
and Van Waes C: Hepatocyte growth factor/scatter factor-induced
activation of MEK and PI3K signal pathways contributes to
expression of proangiogenic cytokines interleukin-8 and vascular
endothelial growth factor in head and neck squamous cell carcinoma.
Cancer Res. 61:5911–5918. 2001.
|
|
21.
|
Rosen EM, Goldberg ID, Kacinski BM,
Buckholz T and Vinter DW: Smooth muscle releases an epithelial cell
scatter factor which binds to heparin. In Vitro Cell Dev Biol.
25:163–173. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Stoker M, Gherardi E, Perryman M and Gray
J: Scatter factor is a fibroblast-derived modulator of epithelial
cell mobility. Nature. 327:239–242. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Kubota T, Fujiwara H, Amaike H, et al:
Reduced HGF expression in subcutaneous CT26 tumor genetically
modified to secrete NK4 and its possible relation with antitumor
effects. Cancer Sci. 95:321–327. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Kuba K, Matsumoto K, Date K, Shimura H,
Tanaka M and Nakamura T: HGF/NK4, a four-kringle antagonist of
hepatocyte growth factor, is an angiogenesis inhibitor that
suppresses tumor growth and metastasis in mice. Cancer Res.
60:6737–6743. 2000.PubMed/NCBI
|
|
25.
|
Date K, Matsumoto K, Shimura H, Tanaka M
and Nakamura T: HGF/NK4 is a specific antagonist for pleiotrophic
actions of hepatocyte growth factor. FEBS Lett. 420:1–6. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Workman P, Aboagye EO, Balkwill F, et al:
Committee of the National Cancer Research Institute: Guidelines for
the welfare and use of animals in cancer research. Br J Cancer.
102:1555–1577. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Furge KA, Zhang YW and Vande Woude GF: Met
receptor tyrosine kinase: enhanced signaling through adapter
proteins. Oncogene. 19:5582–5589. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Kubota T, Taiyoh H, Matsumura A, et al:
NK4, an HGF antagonist, prevents hematogenous pulmonary metastasis
by inhibiting adhesion of CT26 cells to endothelial cells. Clin Exp
Metastasis. 26:447–456. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Brown LF, Berse B, Jackman RW, et al:
Expression of vascular permeability factor (vascular endothelial
growth factor) and its receptors in adenocarcinomas of the
gastrointestinal tract. Cancer Res. 53:4727–4735. 1993.PubMed/NCBI
|
|
30.
|
Tomioka D, Maehara N, Kuba K, et al:
Inhibition of growth, invasion, and metastasis of human pancreatic
carcinoma cells by NK4 in an orthotopic mouse model. Cancer Res.
61:7518–7524. 2001.PubMed/NCBI
|
|
31.
|
Saimura M, Nagai E, Mizumoto K, et al:
Tumor suppression through angiogenesis inhibition by SUIT-2
pancreatic cancer cells genetically engineered to secrete NK4. Clin
Cancer Res. 8:3243–3249. 2002.PubMed/NCBI
|
|
32.
|
Van Belle E, Witzenbichler B, Chen D, et
al: Potentiated angiogenic effect of scatter factor/hepatocyte
growth factor via induction of vascular endothelial growth factor:
the case for paracrine amplification of angiogenesis. Circulation.
97:381–390. 1998.PubMed/NCBI
|
|
33.
|
Xin X, Yang S, Ingle G, et al: Hepatocyte
growth factor enhances vascular endothelial growth factor-induced
angiogenesis in vitro and in vivo. Am J Pathol. 158:1111–1120.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Sulpice E, Ding S, Muscatelli-Groux B, et
al: Cross-talk between the VEGF-A and HGF signalling pathways in
endothelial cells. Biol Cell. 101:525–539. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Vaupel P: The role of hypoxia-induced
factors in tumor progression. Oncologist. 9(Suppl 5): 10–17. 2004.
View Article : Google Scholar
|
|
36.
|
Fukuda R, Hirota K, Fan F, Jung YD, Ellis
LM and Semenza GL: Insulin-like growth factor 1 induces
hypoxia-inducible factor 1-mediated vascular endothelial growth
factor expression, which is dependent on MAP kinase and
phosphatidylinositol 3-kinase signaling in colon cancer cells. J
Biol Chem. 277:38205–38211. 2002. View Article : Google Scholar
|
|
37.
|
Burroughs KD, Oh J, Barrett JC and
DiAugustine RP: Phosphatidylinositol 3-kinase and mek1/2 are
necessary for insulin-like growth factor-I-induced vascular
endothelial growth factor synthesis in prostate epithelial cells: a
role for hypoxia-inducible factor-1? Mol Cancer Res. 1:312–322.
2003.
|
|
38.
|
Niu G, Wright KL, Huang M, et al:
Constitutive Stat3 activity up-regulates VEGF expression and tumor
angiogenesis. Oncogene. 21:2000–2008. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Wei LH, Kuo ML, Chen CA, et al:
Interleukin-6 promotes cervical tumor growth by VEGF-dependent
angiogenesis via a STAT3 pathway. Oncogene. 22:1517–1527. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Niu G, Briggs J, Deng J, et al: Signal
transducer and activator of transcription 3 is required for
hypoxia-inducible factor-1alpha RNA expression in both tumor cells
and tumor-associated myeloid cells. Mol Cancer Res. 6:1099–1105.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Jung JE, Kim HS, Lee CS, et al: STAT3
inhibits the degradation of HIF-1alpha by pVHL-mediated
ubiquitination. Exp Mol Med. 40:479–485. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Xu Q, Briggs J, Park S, et al: Targeting
Stat3 blocks both HIF-1 and VEGF expression induced by multiple
oncogenic growth signaling pathways. Oncogene. 24:5552–5560. 2005.
View Article : Google Scholar : PubMed/NCBI
|