Introduction
Colon cancer is one of the most common causes of
cancer-related mortality worldwide, it continues to be a major
health concern in the world (1).
Although recent data have elucidated some of the molecular
mechanisms of tumor pathogenesis and progression, the etiology of
colon cancer is largely unknown. The development, progression and
malignancy phenotypes of colon cancer may be resulted from a
complex set of genetic events that promotes cell proliferation,
survival and invasion.
Sphingosine kinase (SphK) is a conserved lipid
kinase that catalyzes the phosphorylation of sphingosine to form
sphingosine-1-phosphate (S1P). SphK is a critical regulator of
sphingolipid-mediated functions. Up to now, two mammalian isozymes,
SphK1 and SphK2, have been characterized. It is widely accepted
that SphK1 resides at the centre of a cell-survival rheostat that
balances the cellular levels of pro-apoptotic ceramide with
anti-apoptotic S1P (2,3). Evidence also showed that SphK1
enhances cancer cell migration by secretion of S1P in a protein
kinase C-α- and ERK1/2-dependent manner (4). SphK1 appeared upregulated in some
solid tumors, high level of SphK1 was associated with poor survival
of tumor patients (5) and
suppressing SphK1 reduced tumor growth in animal model (6). SphK1 was significantly elevated in
azoxymethane-induced murine colon cancer tissues, SphK1 knockout
mice subjected to azoxymethane had significantly less aberrant
formation of crypt foci and cancer development (7). Moreover, SphK1 inhibitor or shRNA
enhanced the cytotoxicity and chemosensitivity of colon cancer
cells (8,9). Studies also showed that the
expression of SphK1 was required for the proliferation of small
intestinal tumor cell and SphK1 was able to promote intestinal
adenoma progression (10).
Therefore, SphK1 may play an important role in the carcinogenesis
and metastasis of gut tumors.
Focal adhesion kinase (FAK) is a non-receptor
tyrosine kinase that acts as a primary regulator of focal adhesion
signaling to regulate cell survival, adhesion and migration, which
are key processes for a transformed cell to become invasive and
metastatic (11). Numerous studies
had reported FAK overexpression in various tumor cells, including
gastrointestinal tract cancer, and its expression correlated with
tumor malignancy (12). LPA
induced FAK redistribution and activation in confluent monolayer
colon cancer Caco-2 cells conferring a migratory phenotype
(13). Lunasin inhibited
metastasis of human colon cancer cells by suppressing FAK signaling
pathway and potentiated the effect of oxaliplatin in preventing the
outgrowth of metastasis (14). FAK
signaling pathway may play an important role in keeping the
malignancy phenotypes of colon cancer cells. However, the
regulating mechanism of SphK1 and FAK and the events following
SphK1 and FAK activation in colon cancer are still needed to be
explored.
Mechanistic studies suggest a relation of SphK1/S1P
to the regulation of FAK. It was reported that S1P induced a rapid
increase in tyrosine phosphorylation of FAK and stimulated motility
of human endothelial cells (15,16).
Evidence was presented that activation of FAK pathway was involved
in the cell proliferation induced by S1P in human prostate cancer
cells (17). It seems reasonable
to hypothesize that SphK1 may promote the progression and confer
malignancy phenotype of colon cancer through mediating the
expression of FAK. In the present study, the correlations of SphK1,
FAK and p-FAK expression with clinicopathologic parameters were
investigated in colon cancers and paired normal tissues. SphK1
inhibitor N,N-dimethylsphingosine (DMS), SphK1 DNA and SphK1 shRNA
transfection were exploited to confirm the effect of SphK1 on
malignancy phenotypes in colon cancer LOVO cells. In addition,
effects of SphK1 on FAK, p-FAK, intercellular adhesion molecule-1
(ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) were
determined to explore the molecular mechanisms.
Materials and methods
Tissue specimens
Sixty-six fresh colon cancer tissues and matched
normal tissues from patients were collected and fresh-frozen in
liquid nitrogen after surgical resections performed at the First
Affiliated Hospital of Guangxi Medical University (Guangxi, China)
from 2009 to 2010. For the 66 fresh tissues, one-half of the tissue
was snap-frozen immediately in liquid nitrogen and stored at −80°C
and the other half was formalin-fixed and paraffin-embedded. Each
case was reviewed by 2 experienced histopathologists who were
blinded to disease status. The patients had not received
chemotherapy or radiation therapy before tumor resection. The study
was approved by the Institutional Ethics Committee of Guangxi
Medical University under full consideration of the declaration on
human rights of Helsinki.
Immunohistochemistry staining
Formalin-fixed, paraffin-embedded tissue blocks were
serially sectioned at 4 μm. Sections were deparaffinized in
xylene and rehydrated before analysis. Endogenous peroxidase was
quenched with 3.0% hydrogen peroxide in methanol for 10 min,
antigen retrieval was performed by microwave for 15 min and tissue
sections were blocked with normal rabbit serum for 20 min. This was
followed by incubation overnight at 4°C with mouse monoclonal FAK
(Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), rabbit
polyclonal p-FAK (Abcam, Cambridge, UK) and mouse monoclonal SphK1
(Sigma-Aldrich, St Louis, MO, USA) primary antibody. Sections were
washed with PBS and incubated with second antibody at room
temperature for 30 min. After being washed with PBS, sections were
stained by a streptavidin-peroxidase detection system. Antibody
binding was visualized using diaminobenzidine as chromogen and
counterstained with hematoxylin. Incubation with PBS instead of the
primary antibody served as a negative control.
The degree of staining was estimated by
semiquantitative evaluation and categorized by the extent and
intensity of staining as follows: i) The extent of positive cells
was scored as: 0, positive-staining cells <5%; 1,
positive-staining cells 5–25%; 2, positive-staining cells 26–50%;
3, positive-staining cells 51–75%; 4, positive-staining cells
>75%. ii) The intensity of staining was scored as: 0,
achromatic; 1, light yellow; 2, yellow; 3, brown. The percentage of
positive tumor cells and staining intensity were multiplied to
produce a weighted score for each case. Cases with weighted scores
≤1 were defined as negative; otherwise as positive.
Cell culture and transfection
Human colon cancer LOVO cell line was obtained from
ATCC. Cells were cultivated in DMEM high glucose medium (Invitrogen
Co., Carlsbad, CA, USA) supplemented with 10% FBS (Gibco-BRL,
Rockville, MD, USA) and antibiotics (100 U/ml penicillin plus 50 mM
streptomycin, Invitrogen Co.) in an atmosphere of 5% CO2
at 37°C.
In order to develop a new model system to better
study effects of SphK1 on cell proliferation, apoptosis and
invasion, LOVO cells were transfected with SphK1 DNA (SphK1 group),
SphK1 shRNA (shRNA group) and SphK1 non-targeting shRNA control (NC
group). In brief, 24 h before transfection, cells were cultivated
in medium without antibiotics. Cells were seeded in 6-well
plates(2×105 cells/well). The shRNA, non-targeting shRNA
control (NC) or DNA plasmid targeting SphK1 (Shanghai GenePharma
Co., Ltd., Shanghai, China) were diluted in Opti-MEM®
(Gibco-BRL) Reduced Serum Medium without serum. Cells were
transfected with 40 nmol shRNA oligomer, NC or 0.8 μg of DNA
using Lipofectamine™ 2000 reagent (Life Technology Co., Carlsbad,
CA, USA) according to the manufacturer’s instructions. Cells were
harvested 48 h later for the following experiment.
SphK1 activity assay
Cells were cultured in a culture dish. Cells were
transfected with SphK1 shRNA, NC, SphK1 DNA or treated with 50
μM DMS (Merck KGaA, Darmstadt, Germany) for 24 h (DMS
group). Cells treated with equal amount of 0.9% NaCl instead of
drugs served as the control group. Activation of SphK1 was measured
as described previously with slight modification (18). Cells were resuspended in ice-cold
0.1 M phosphate buffer [(pH 7.4) containing 20% glycerol, 1 mM
mercaptoethanol, 1 mM EDTA, phosphatase inhibitors 1 mM sodium
orthovanadate, 15 mM sodium fluoride], protease inhibitors (10
μg/ml leupeptin, aprotinin, trypsin, chymotrypsin and 1 mM
phenylmethylsulfonylfluoride) and 0.5 mM 4-deoxypyridoxine, were
then disrupted by three 5-sec pulses with a Fisher 550 sonic
dismembranator. Each sample containing 100 μg protein was
assayed for sphingosine kinase activity by incubation with
sphingosine and [γ-32P]-ATP (Beijing Free Biotech. Co.,
Beijing, China) for 30 min at 37°C. Products were separated on TLC
on Silica Gel G60 (Merck KGaA) using 1-butanol/methanol/acetic
acid/water (80:20:10:20) and visualized by autoradiography. The
radioactive spots corresponding to sphingosine phosphate were
scraped and counted in a scintillation counter.
Cells viability assay
Cell viability was determined by colorimetric assay
utilizing MTT (Sigma-Aldrich). In brief, non-transfected LOVO cells
(DMS group), cells of SphK1, shRNA and NC group were seeded in
96-well plate (5×103 per well) as different experimental
groups. Cells of DMS group were incubated with 25, 50 and 100
μM DMS for 0, 6, 12 and 24 h. LOVO cells treated with equal
amount of 0.9% NaCl instead of drugs served as the control group.
Then 20 μl MTT solution (5 mg/ml) was added to each well and
incubated at 37°C for 4 h. The solution was carefully removed,
followed by addition of 150 μl dimethyl sulfoxide (DMSO,
Invitrogen Co.) to each well to solubilize MTT. The absorbance
(A) of sample was measured at 490 nm and the cell viability
was expressed as A value of experimental cells/control cells
×100%. Triplicate measurements were done at each
time-concentration.
Flow cytometry analysis
The treatment of cells was the same as in the SphK1
activity assay. Cells were harvested and washed twice with PBS. The
cells were resuspended in 500 μl binding buffer at a density
of 1×106 cells/ml, 1 μl Annexin V-FITC (Nanjing
KeyGen Biotech. Co., Nanjing, China) was added to the sample and
incubated for 20 min at room temperature in the dark, and then 5
μl propidium iodine (PI) buffer was added and incubated for
5 min at 4°C in the dark. Finally, samples were evaluated by flow
cytometry and data were analyzed using CellQuest (Becton-Dickinson,
San Jose, CA, USA).
Semi-quantitative reverse transcription
polymerase chain reaction
The cell treatment was the same as in the SphK1
activity assay. Total-RNAs were extracted from tissue samples or
cells using TRIzol reagent (Invitrogen Co., San Diego, CA, USA).
First-strand cDNA was synthesized from 1 μg of total-RNA
using oligo-dT primer and Moloney murine leukemia virus reverse
transcriptase (Fermentas International Inc., Burlington, Canada)
according to the instructions from the manufacturers. Then cDNA was
amplified by using the following primers: SphK1, 5′-ATG CAC GAG GTG
GTG AAC G-3′ (sense), 5′-GGA GGC AGG TGT CTT GGA AC-3′ (antisense),
FAK: 5′-ACC TCA GCT AGT GAC GTA TGG-3′ (sense), 5′-CGG AGT CCC AGG
ACA CTG TG-3′ (antisense), β-actin: 5′-AGC CAT GTA CGT AGC CAT
CC-3′ (sense), 5′-CTC TCA GCT GTG GTG GTG AA-3′ (antisense). The
PCR products were analyzed on 2% agarose gels and visualized by
ethidium bromide staining. Quantitation of expression levels was
achieved after adjustment for the expression levels of β-actin by
densitometry. A 100-base pair DNA ladder was used as a molecular
weight marker on each gel.
Western blot analysis
The treatment of cells was the same as in the SphK1
activity assay. Tissue samples or cells of different groups were
lysed in lysis buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton
X-100, 1% sodium deoxycholate, 0.1% SDS, 2 mM sodium pyrophosphate,
25 mM β-glycerophosphate, 1 mM EDTA, 1 mM
Na3VO4, 0.5 μg/ml leupeptin and 1 mM
PMSF] and then the lysate was incubated on ice for 30 min and
centrifuged at 12000 rpm for 10 min. The supernatant was collected
and the content of total protein was evaluated by the Bradford
colorimetry. After denaturation, 40 μg of protein from each
sample was separated on a 12% SDS-PAGE and electroblotted onto
nitrocellulose membrane. The nitrocellulose membrane was blocked
with 5% non-fat milk in TBST [10 mM Tris (pH 7.4), 100 mM NaCl,
0.5% Tween 20] for 2 h at room temperature. Membrane was
subsequently incubated overnight at 4°C in 5% non-fat milk in TBST
containing either mouse monoclonal β-actin, mouse monoclonal FAK,
mouse monoclonal ICAM-1, rabbit polyclonal VCAM-1 (Santa Cruz
Biotechnology, Inc.) or rabbit polyclonal p-FAK (Abcam) antibody,
followed by 1 h incubation with peroxidase-conjugated secondary
antibody (Santa Cruz Biotechnology, Inc.). The protein signals were
visualized using Pierce enhanced chemiluminescence (ECL) reaction
Western Blotting Substrate (Pierce Co., Rockford, IL, USA) and
exposed to medical X-ray film.
In vitro cell migration and invasion
assay
The cell treatment was the same as in the SphK1
activity assay. Cell migration and invasion were evaluated in
24-well transwell chamber model. Briefly, the upper and lower
culture compartments of each well were separated by polycarbonate
membranes (8-μm pore size). To determine baseline migration
ability, 1.0×105 cells in 0.2 ml of complete medium
containing 5% FBS were placed into the upper compartment of
uncoated wells (Corning Costar Inc., Corning, NY, USA), and 0.6 ml
of complete medium containing 10% FBS were placed into the lower
compartment. In parallel, to assess the ability of the cells to
penetrate a collagen matrix (invasiveness), the experiment was
repeated using the upper compartment coated with 100 μl 5
μg/ml collagen matrix (Corning Costar Inc.). The transwell
chambers were incubated for 24 h at 37°C in 95% air and 5%
CO2. Cell penetration through the porous membrane into
the plate was detected by crystal violet staining, and then
observed and photographed at ×400 magnification on a light
microscope. Data were quantitated by cell count and expressed as
average of cell number from 4 random fields. The cell number
indicates the cell migration capability and invasiveness.
Enzyme-linked immunosorbent assay
(ELISA)
The cell treatment was the same as in the SphK1
activity assay. The cell culture supernatant was collected for
ICAM-1 and VCAM-1 analysis using ELISA detection kits (Life
Technology Co.) according to the instructions from the
manufacturers. In this experiment, blank wells (no sample or
HRP-conjugate reagent), standard wells and testing sample well were
set, 50 μl diluted standard was added to standard wells, 40
μl sample dilution and 10 μl culture supernatants
were added to test sample wells, mixing gently with shaking and
incubated for 30 min at 37°C. Discard liquid and washed each well
with diluted washing liquid for 5 times. Then 50 μl
HRP-conjugate reagent was added to each well, except the blank
wells. Mixing gently with shaking, incubated for 30 sec at 37°C.
Liquid was discarded followed by 5 washes. Chromogen solution A and
B (50 μl each) were added to the wells, gently mixing, and
incubated for 10 min at 37°C. Then, 50 μl stop solution was
added to each well to stop the reaction. After zero setting, the
A value was measured at 450 nm. According to standard
concentration and the corresponding A values, the standard
curve linear regression equation was harvested, then the sample
concentration was calculated according to the regression
equation.
Statistical analysis
The significance between SphK1, FAK, p-FAK and
clinicopathologic characteristics of the patients was assessed with
the χ2 test. The correlation between SphK1, FAK and
p-FAK was calculated by the method of Pearson’s correlation
coefficient. Data are presented as mean ± standard deviation (SD)
and Student’s t-test were used to compare continuous variables
among groups. A p-value of <0.05 was considered significant.
Results
Immunohistochemistry analysis of SphK1,
FAK and p-FAK in colon cancer and matched normal colonic
tissues
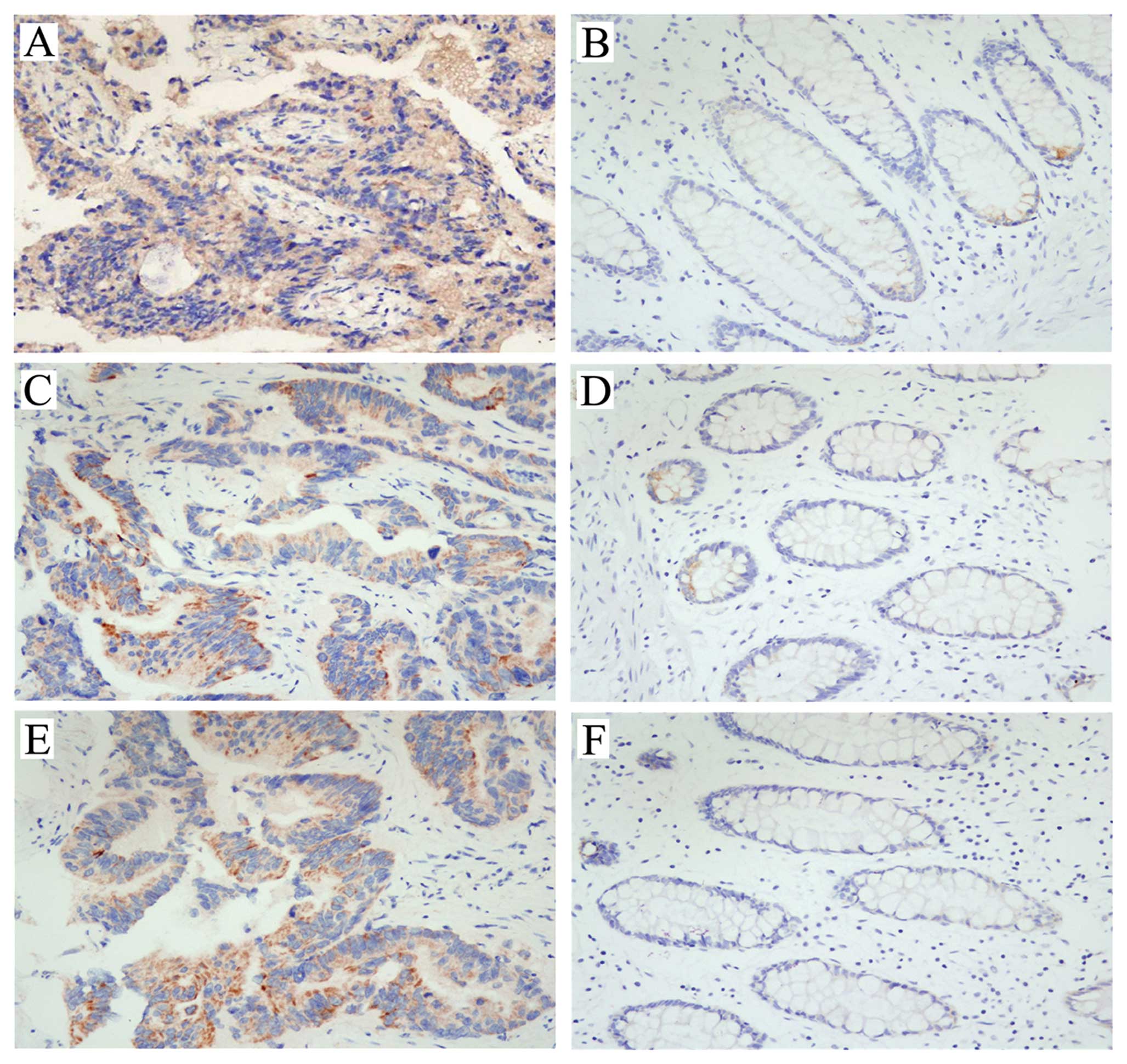
Immunohistochemistry staining for SphK1, FAK and
p-FAK was revealed as yellow or brown color and present in the
cytoplasm of colon cancer cells, and the staining of cancerous
tissue was significantly stronger than that of matched normal
tissue (Fig. 1). As shown in
Table II, in 66 cancerous tissues,
48 (72.73%) showed positive staining for SphK1, 51 (77.27%) showed
positive staining for FAK and 39 (59.09%) showed positive staining
for p-FAK. In 66 matched normal tissues, 23 (34.85%) showed
positive staining for SphK1, 15 (22.73%) showed positive staining
for FAK and 11 (16.67%) showed positive staining for p-FAK, the
positive staining rates were significantly lower than those of
cancerous tissues (p<0.01). Moreover, there was a close
correlation between the expression of SphK1 and FAK or p-FAK
(Table I). The co-expression of
SphK1, FAK and p-FAK significantly associated with histological
grade, Dukes’ stage, lymph node metastasis and distant metastasis,
but no correlation was found between SphK1, FAK, p-FAK expression
and age or gender (Table II).
 | Table II.Clinicopathologic characteristics and
their association with SphK1, FAK and p-FAK expression. |
Table II.
Clinicopathologic characteristics and
their association with SphK1, FAK and p-FAK expression.
| | SphK1
| FAK
| p-FAK
|
|---|
| Clinicopathologic
characteristics | N | Positive (%) | p-value | Positive (%) | p-value | Positive (%) | p-value |
|---|
| Gender | | | | | | | |
| Male | 37 | 28 (75.68) | 0.244 | 28 (75.68) | 0.727 | 22 (59.46) | 0.945 |
| Female | 29 | 20 (68.97) | | 23 (79.31) | | 17 (58.62) | |
| Age | | | | | | | |
| ≤50 | 27 | 21 (77.78) | 0.588 | 21 (77.78) | 0.935 | 14 (51.85) | 0.320 |
| >50 | 39 | 27 (69.23) | | 30 (76.92) | | 25 (64.10) | |
| Histological
grading | | | | | | | |
| Well
differentiated | 14 | 10 (71.43) | 0.049 | 8 (57.14) | 0.063 | 4 (28.57) | 0.014 |
| Moderately
differentiated | 36 | 23 (63.89) | | 28 (77.78) | | 22 (61.11) | |
| Poorly
differentiated | 16 | 15 (93.75) | | 15 (93.75) | | 13 (81.25) | |
| Dukes’ stage | | | | | | | |
| A and B | 34 | 21 (61.76) | 0.039 | 22 (64.71) | 0.012 | 15 (44.12) | 0.011 |
| C and D | 32 | 27 (84.38) | | 29 (90.63) | | 24 (75.00) | |
| Lymph node
metastasis | | | | | | | |
| Positive | 24 | 21 (87.50) | 0.042 | 22 (91.67) | 0.035 | 18 (75.00) | 0.047 |
| Negative | 42 | 27 (64.29) | | 29 (69.05) | | 21 (50.00) | |
| Distance
metastasis | | | | | | | |
| Positive | 20 | 18 (90.00) | 0.038 | 19 (95.00) | 0.023 | 16 (80.00) | 0.023 |
| Negative | 46 | 30 (65.22) | | 32 (69.57) | | 23 (50.00) | |
| Colon cancer
tissues | 66 | 48 (72.73) | 0.000 | 51(77.27) | 0.000 | 39(59.09) | 0.000 |
| Normal colon
tissues | 66 | 23 (34.85) | | 15(22.73) | | 11(16.67) | |
| Table I.The correlation between the protein
expression of FAK, p-FAK and SphK1 in colon cancer tissues. |
Table I.
The correlation between the protein
expression of FAK, p-FAK and SphK1 in colon cancer tissues.
| SphK1 expression
| | |
|---|
| Expression | Positive (%) | Negative (%) | Total | Correlation | p-value |
|---|
| FAK | | | | | |
| Positive (%) | 43 (65.16) | 8 (12.12) | 51 (77.27) | 0.480 | 0.000 |
| Negative (%) | 5 (7.58) | 10 (15.15) | 15 (22.73) | | |
| p-FAK | | | | | |
| Positive (%) | 36 (54.55) | 3 (4.55) | 39 (59.09) | 0.499 | 0.000 |
| Negative (%) | 12 (18.18) | 15 (22.72) | 27 (40.91) | | |
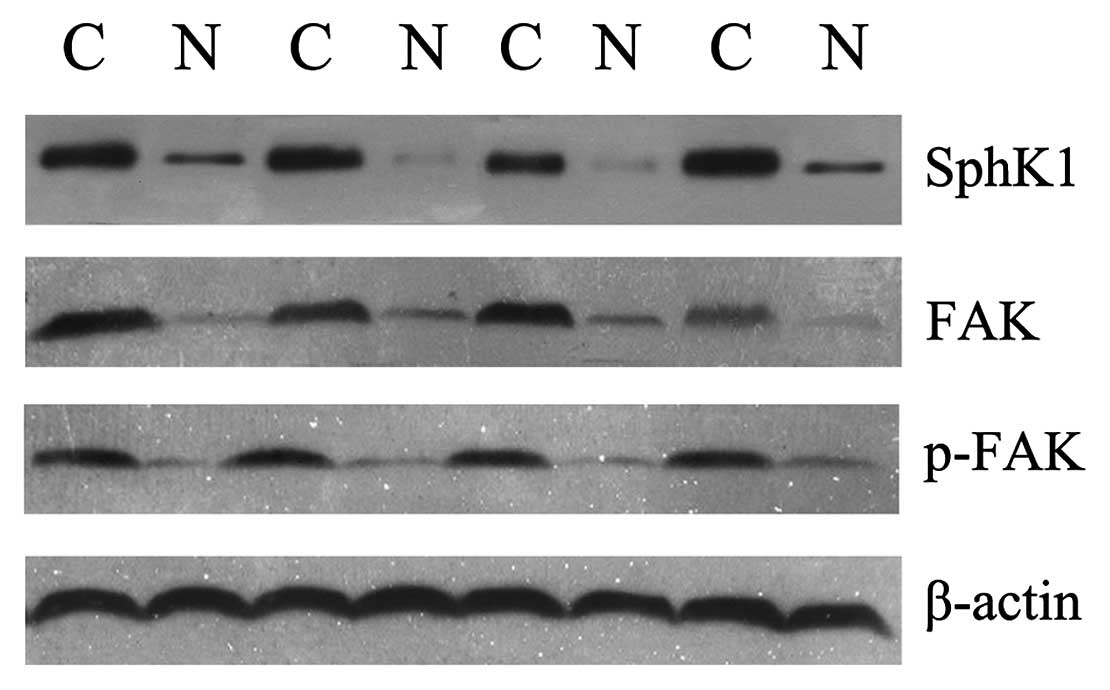
Western blot detection of SphK1, FAK and
p-FAK in colon cancer and matched normal colonic tissues
The expression of SphK1, FAK and p-FAK was also
detected in these 66 colon cancer specimens and matched normal
colonic tissues using western blot analysis. The density of SphK1,
FAK and p-FAK (Tyr 397) expression in cancerous tissues was much
stronger than that in matched normal tissues (Fig. 2). The western blot analysis results
fit very well with those obtained by immunohistochemistry.
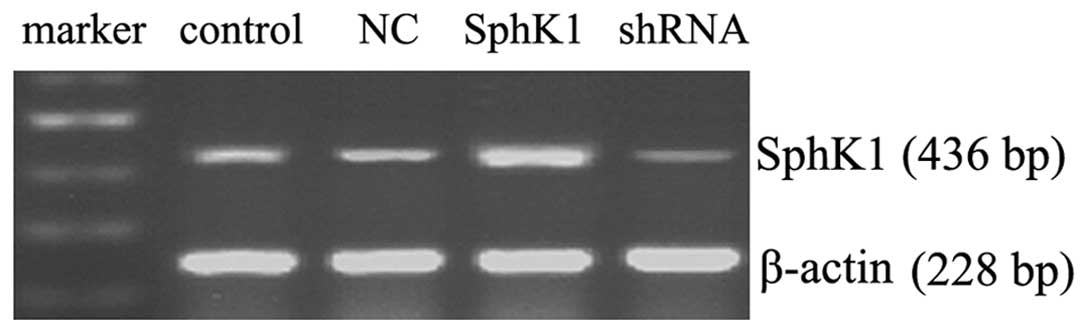
Activity and mRNA expression of
SphK1
Fig. 3 shows that
SphK1 DNA transfection dramatically enhanced the mRNA expression of
SphK1, shRNA knockdown significantly decreased the mRNA expression
of SphK1 and NC transfection had no significant effect on the mRNA
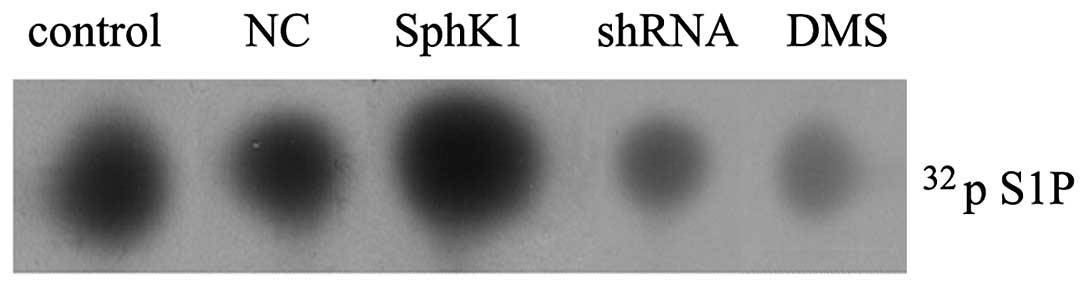
expression of SphK1. As shown in Fig.
4, LOVO cells possessed a basal activity of SphK1 (26.16
pmol/mg/min), SphK1 DNA transfection significantly enhanced the
activity of SphK1 (65.19 pmol/mg/min), SphK1 shRNA knockdown
significantly suppressed the activity of SphK1 (6.28 pmol/mg/min)
and NC had no significant effect on the activity of SphK1 (25.46
pmol/mg/min). Moreover, in cells treated with 50 μM DMS for
24 h, the SphK1 activity reduced to 5.31 pmol/mg/min.
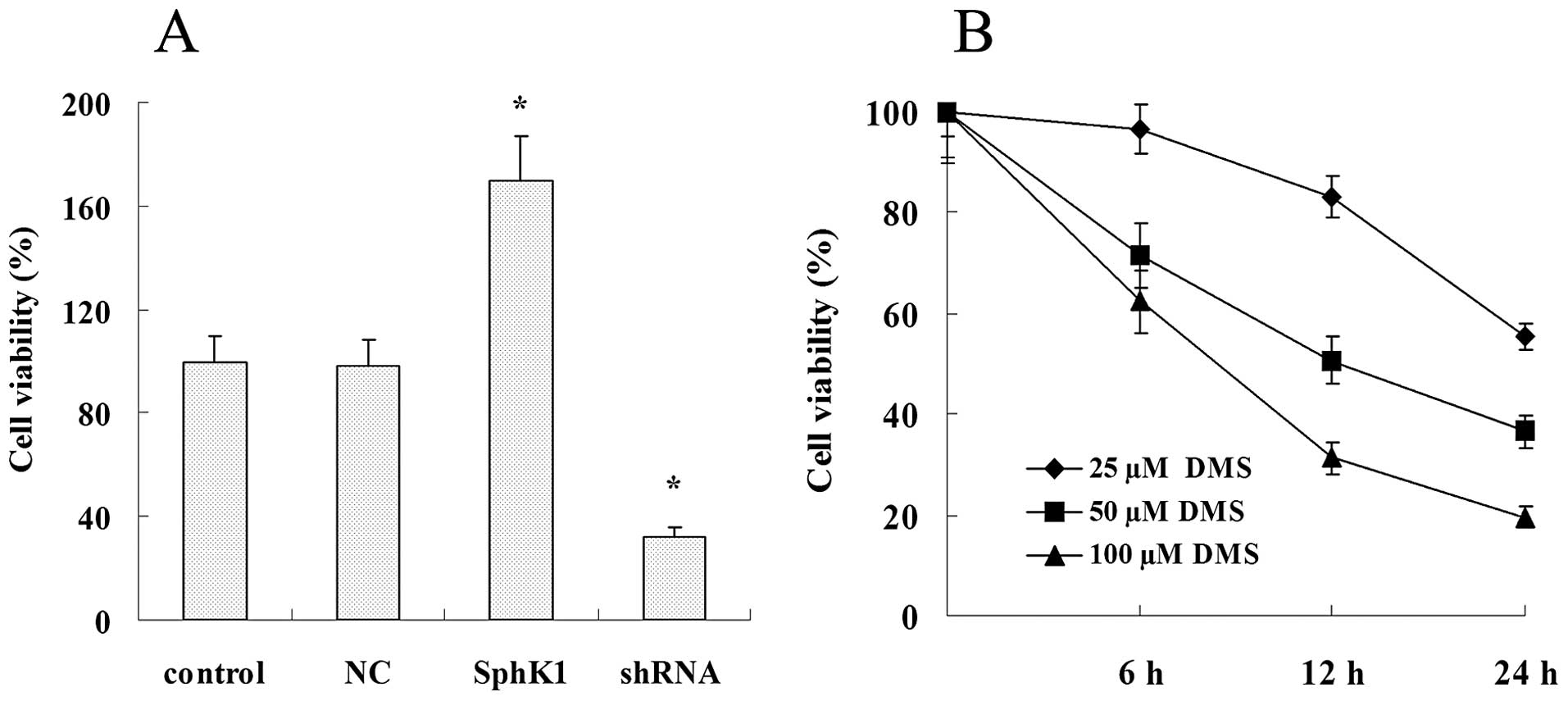
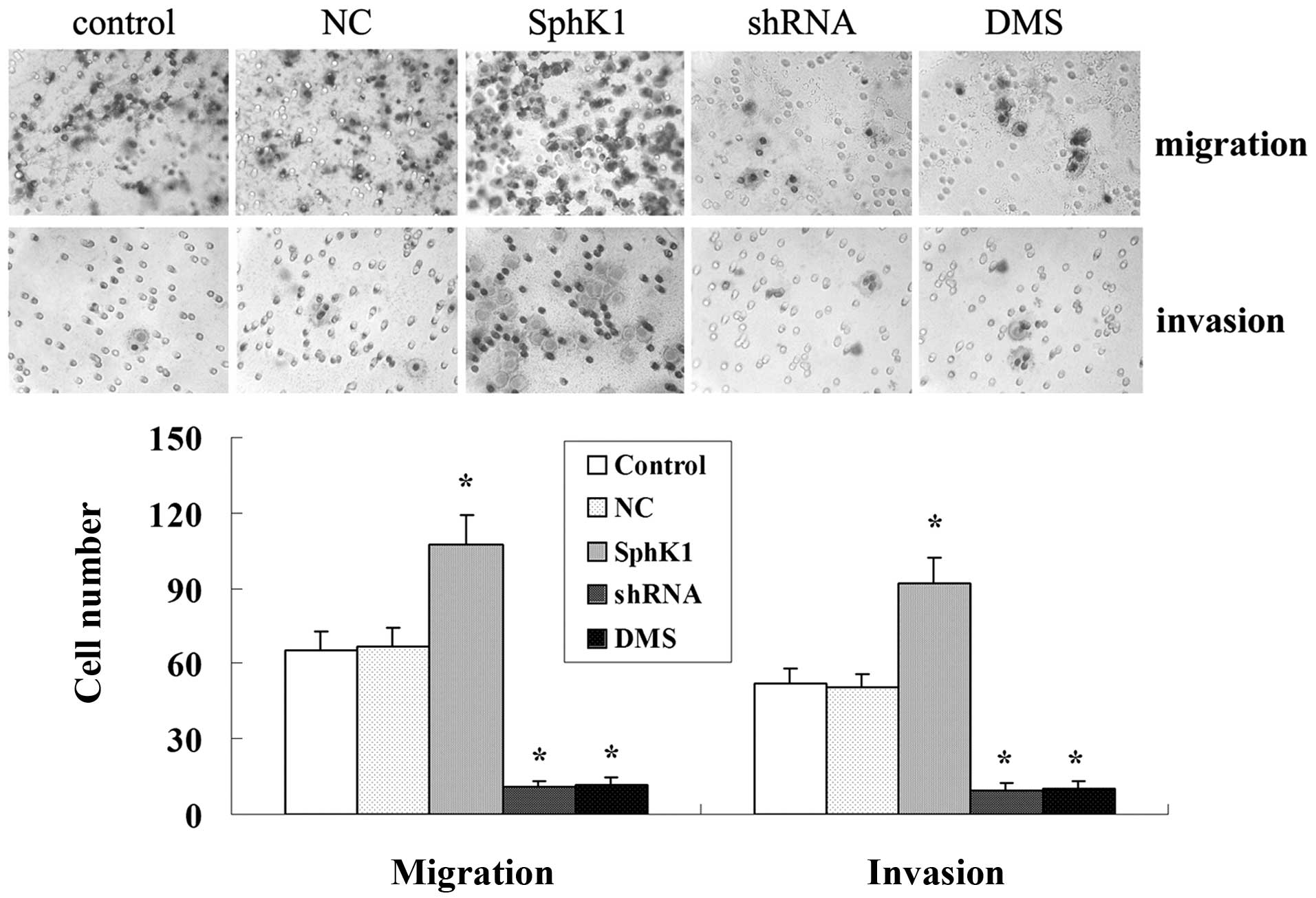
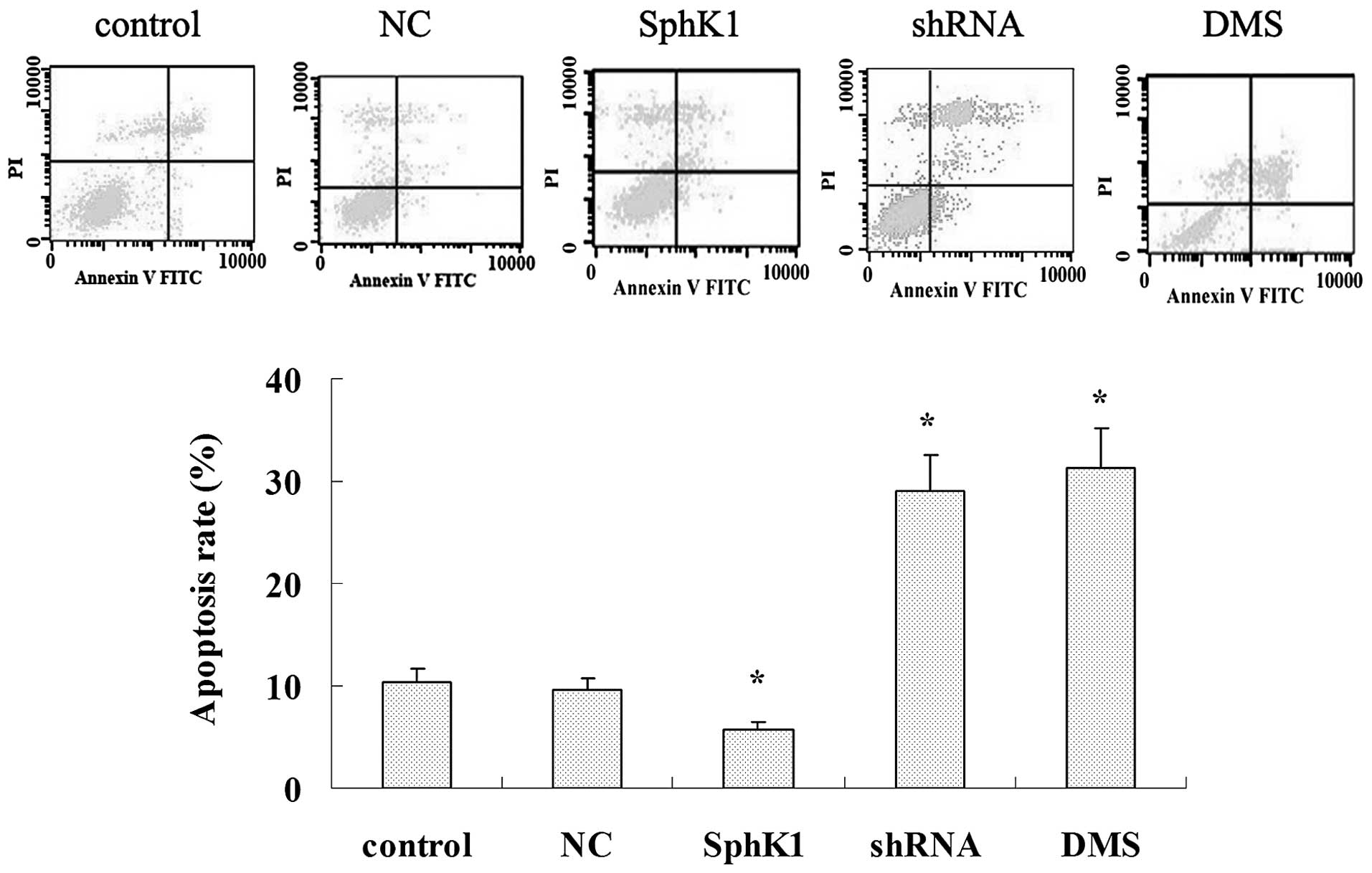
SphK1 enhances tumor cell proliferation,
migration and invasiveness while suppresses cell apoptosis
Overexpression of SphK1 dramatically enhanced the
cell proliferation, migration and invasiveness (Figs. 5A and 7), and significantly suppressed cell
apoptosis (Fig. 6). Compared with
control group, SphK1 shRNA significantly suppressed the cell
proliferation, cell migration and invasiveness (Figs. 5A and 7), while significantly promoted cell
apoptosis (Fig. 6). DMS suppressed
cell proliferation in a time- and dose-dependent manner (Fig. 5B), DMS significantly promoted cell
apoptosis and suppressed cell migration and invasiveness (Figs. 6 and 7). NC transfection had no significant
effect on cell biological behavior.
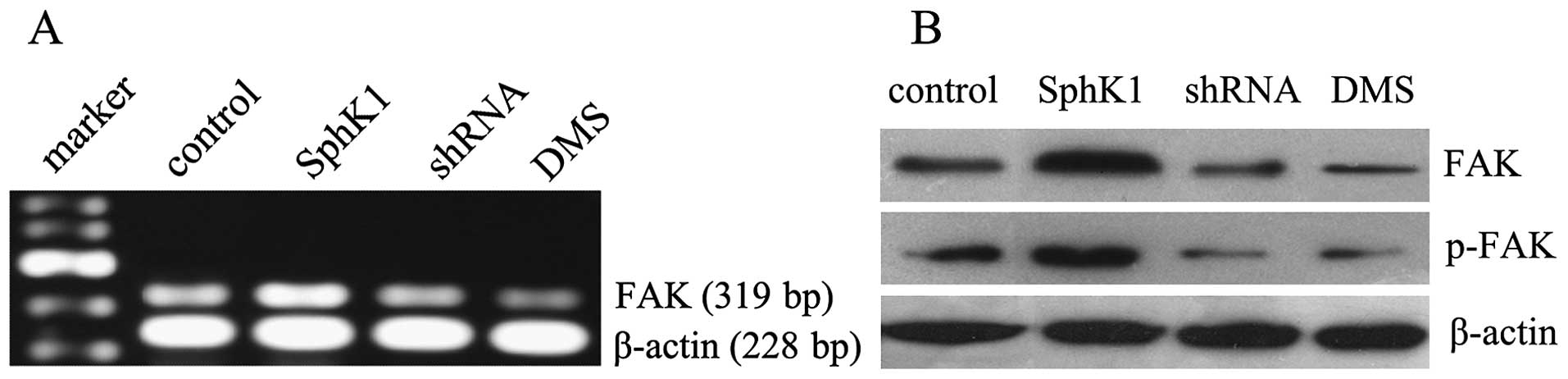
SphK1 enhances the expression of FAK and
p-FAK in LOVO cells
Overexpression of SphK1 not only enhanced the mRNA
expression of FAK, but also promoted the protein expression of FAK
and p-FAK in tumor cells. In contrast, suppression of SphK1 by
shRNA and DMS reduced the mRNA and protein expression of FAK
(Fig. 8). These results suggest
that SphK1 may activate the FAK pathway in tumor cells.
SphK1 is required for the secretion and
expression of ICAM-1 and VCAM-1 in LOVO cells
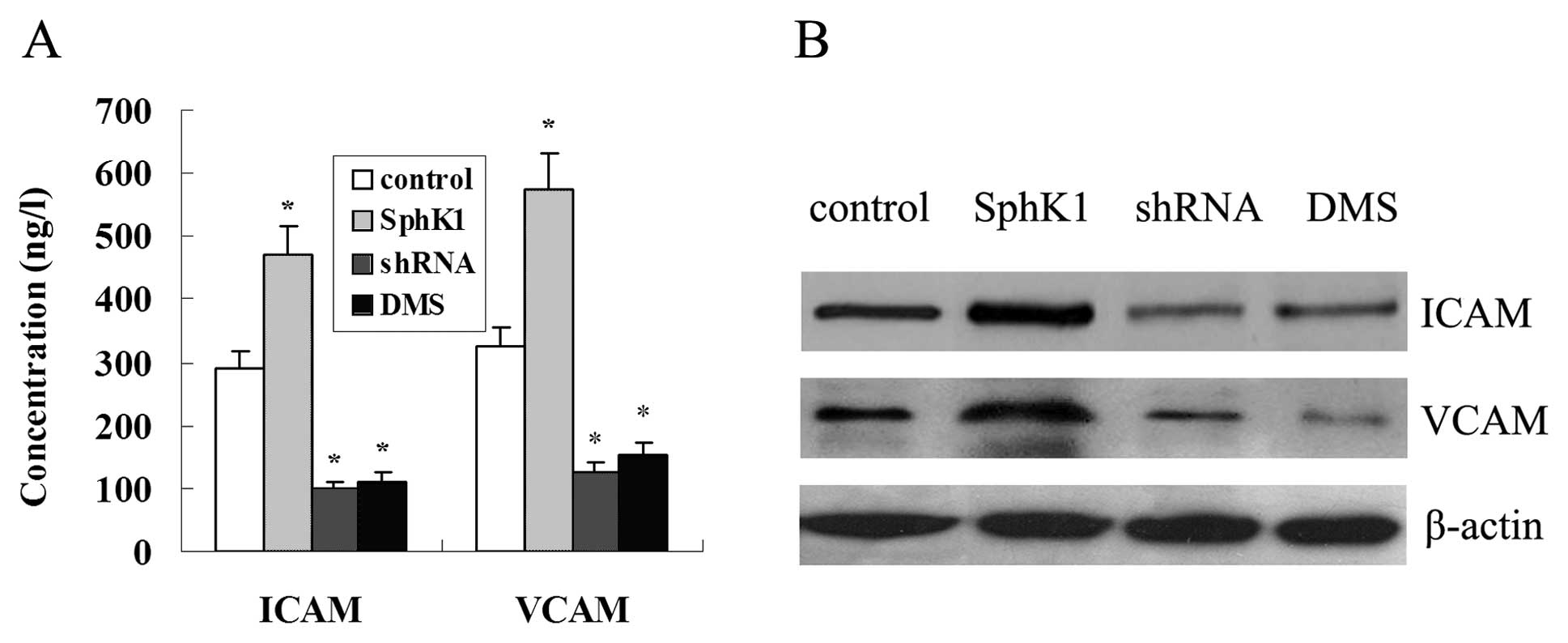
Fig. 9A shows that
overexpression of SphK1 promoted the secretion of ICAM-1 and VCAM-1
in LOVO cells, in contrast, the secretion of ICAM-1 and VCAM-1 was
reduced with the suppression of SphK1 by DMS and shRNA. In
addition, overexpression of SphK1 enhanced the protein expression
of ICAM-1 and VCAM-1, suppression of SphK1 significantly reduced
the protein expression of ICAM-1 and VCAM-1 (Fig. 9B).
Discussion
Previous evidence indicated that SphK1 was an
oncogenic enzyme and its activation was closely associated with
angiogenesis, lymphangiogenesis, anti-apoptosis, transformation,
proliferation and survival of tumor cells (19–21).
Studies also showed the expression of SphK1 was enhanced in colon
cancer (7). The present research
confirmed this finding and demonstrated that SphK1 was
overexpressed in colon cancer tissue but was absent or weakly
expressed in normal colonic tissue. Moreover, the expression of
SphK1 has been found to correlate with the Dukes’ stage,
histological grading, lymph node metastasis and distant metastasis.
These results indicated that SphK1 may contribute to colon
carcinogenesis and potentially also to metastasis of the malignancy
phenotype. However, the molecular mechanisms of the tumor promoting
activity of SphK1 remain to be determined.
Elevated FAK expression and activity is associated
with malignancy in a variety of cancer cells, indicating FAK plays
a critical role in tumor progression and metastasis (22,23).
Blocking phosphorylation of FAK not only inhibited the migration of
pancreatic cancer cells but also reduced tumor growth, invasion and
metastases in pancreatic cancer murine model (24). In the present investigation,
enhanced expression of FAK and p-FAK (Tyr397) in colon cancer
tissues was observed. The expression of FAK and p-FAK has been
found to correlate with the Dukes’ stage, lymph node metastasis and
distant metastasis. These results also supported the role of FAK in
progress of colon cancer (25).
Moreover, the results of immunohistochemistry and western blot
detection confirmed that the expression of SphK1 was closely
related with the expression of FAK and p-FAK, which indicated that
activation of FAK pathway may be regulated by SphK1 in the
progression of colon cancer.
The co-expression of SphK1, FAK and p-FAK correlates
with Dukes’ stage, lymph node and distant metastasis of colon
cancer supports the hypothesis that overexpression of SphK1 may
promote the progression and confer malignancy phenotypes of colon
cancer through FAK pathway activation. To confirm this hypothesis,
the activity and expression of SphK1 was regulated in human colon
cancer LOVO cells, results show that the expression of FAK and
p-FAK were closely correlated with the activity and expression of
SphK1, and suppressing SphK1 and FAK greatly inhibited the
proliferation, migration and invasiveness of colon cancer cells.
These results suggest the role of FAK pathway in SphK1-mediated
malignancy phenotypes of colon cancer cells. However, further
research should concentrate in filling the gap between SphK1 and
FAK pathway and in exploring the events following FAK
activation.
Cell adhesion molecules are involved in a variety of
pathologies including carcinogenesis, particularly VCAM-1 and
ICAM-1 play major roles in the initiation of tumor progression
(26). Moreover, some reports
indicated VCAM-1 and VCAM-1 were promising targets for the
prevention and inhibition of tumor metastasis (27,28).
In patients with colorectal cancer, studies also show the serum
levels of ICAM-1 and VCAM-1 were significantly higher than those of
healthy controls and there was a significant association between
the serum levels of these molecules, disease stage and the presence
of both lymph node and distant metastases (29,30).
In the present study LOVO cells showed that the levels of ICAM-1
and VCAM-1 were upregulated with the overexpression of SphK1 and
downregulated with the suppression of SphK1. These results
indicated ICAM-1 and VCAM-1 were involved in SphK1 promoting the
progression and conferring malignancy phenotypes of colon cancer
cells. However, the exact mechanism of SphK1 regulating FAK, ICAM-1
and VCAM-1 need further investigation.
There is evidence that S1P is a potent stimulator of
VCAM-1 and ICAM-1 expression (31)
and CCN6 enhanced the migration of chondrosarcoma cells by
increasing ICAM-1 expression at least partly through the FAK signal
pathway (32). In endothelial
cells FAK was essential for induction of ICAM-1 and sVCAM-1-induced
cell migration and was almost completely blocked by adenovirus
containing FAK-related non-kinase (33). In human colon cancer,
gastrin-releasing peptide was found to induce ICAM-1 via FAK and
promoted tumor cell motility and attachment to the extracellular
matrix (34). Collectively, these
results indicate that the secretion and expression of ICAM-1 and
VCAM-1 may be regulated by the FAK pathway. However, further
studies in colon cancer cell lines and tissues are needed to
validate this.
In summary, our results suggest that overexpression
of SphK1 may be one of the mechanisms by which colon cancer gain
malignancy phenotypes during malignant transformation. Furthermore,
SphK1 may regulate VCAM-1 and ICAM-1 in a FAK pathway-dependent
manner, which ultimately contributes to the tumor progression and
malignancy phenotypes in colon cancer. SphK1 has potential as a
therapeutic target in the antineoplastic treatment of colon
cancer.
Acknowledgements
This study was supported by the
National Natural Science Foundation of China (no. 30760275) and
Natural Science Foundation from Guangxi Autonomous Region of China
(no. 2011GXNSFA018182).
References
|
1.
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar
|
|
2.
|
Pitson SM: Regulation of sphingosine
kinase and sphingolipid signaling. Trends Biochem Sci. 36:97–107.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Gamble JR, Sun WY, Li X, Hahn CN, Pitson
SM, Vadas MA and Bonder CS: Sphingosine kinase-1 associates with
integrin {alpha}V{beta}3 to mediate endothelial cell survival. Am J
Pathol. 175:2217–2225. 2009.PubMed/NCBI
|
|
4.
|
Bergelin N, Blom T, Heikkilä J, et al:
Sphingosine kinase as an oncogene: autocrine sphingosine
1-phosphate modulates ML-1 thyroid carcinoma cell migration by a
mechanism dependent on protein kinase C-alpha and ERK1/2.
Endocrinology. 150:2055–2063. 2009. View Article : Google Scholar
|
|
5.
|
Li W, Yu CP, Xia JT, et al: Sphingosine
kinase 1 is associated with gastric cancer progression and poor
survival of patients. Clin Cancer Res. 15:1393–1399. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Pchejetski D, Doumerc N, Golzio M, et al:
Chemosensitizing effects of sphingosine kinase-1 inhibition in
prostate cancer cell and animal models. Mol Cancer Ther.
7:1836–1845. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Kawamori T, Kaneshiro T, Okumura M, et al:
Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J.
23:405–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Nemoto S, Nakamura M, Osawa Y, et al:
Sphingosine kinase isoforms regulate oxaliplatin sensitivity of
human colon cancer cells through ceramide accumulation and Akt
activation. J Biol Chem. 284:10422–10432. 2009. View Article : Google Scholar
|
|
9.
|
Wang H, Maurer BJ, Liu YY, et al:
N-(4-Hydroxyphenyl) retinamide increases dihydroceramide and
synergizes with dimethylsphingosine to enhance cancer cell killing.
Mol Cancer Ther. 7:2967–2976. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Kohno M, Momoi M, Oo ML, et al:
Intracellular role for sphingosine kinase 1 in intestinal adenoma
cell proliferation. Mol Cell Biol. 26:7211–7223. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Schwock J, Dhani N and Hedley DW:
Targeting focal adhesion kinase signaling in tumor growth and
metastasis. Expert Opin Ther Targets. 14:77–94. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Hao HF, Naomoto Y, Bao XH, et al: Progress
in researches about focal adhesion kinase in gastrointestinal
tract. World J Gastroenterol. 15:5916–5923. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Leve F, Marcondes TG, Bastos LG, Rabello
SV, Tanaka MN and Morgado-Díaz JA: Lysophosphatidic acid induces a
migratory phenotype through a crosstalk between RhoA-Rock and
Src-FAK signalling in colon cancer cells. Eur J Pharmacol.
671:7–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Dia VP and Gonzalez de Mejia E: Lunasin
potentiates the effect of oxaliplatin preventing outgrowth of colon
cancer metastasis, binds to α5β1 integrin and suppresses
FAK/ERK/NF-κB signaling. Cancer Lett. 313:167–180. 2011.PubMed/NCBI
|
|
15.
|
Zhao J, Singleton PA, Brown ME, Dudek SM
and Garcia JG: Phosphotyrosine protein dynamics in cell membrane
rafts of sphingosine-1-phosphate-stimulated human endothelium: role
in barrier enhancement. Cell Signal. 21:1945–1960. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Lee OH, Lee DJ, Kim YM, Kim YS, Kwon HJ,
Kim KW and Kwon YG: Sphingosine 1-phosphate stimulates tyrosine
phosphorylation of focal adhesion kinase and chemotactic motility
of endothelial cells via the G(i) protein-linked phospholipase C
pathway. Biochem Biophys Res Commun. 268:47–53. 2000. View Article : Google Scholar
|
|
17.
|
Gibbs TC, Rubio MV, Zhang Z, Xie Y, Kipp
KR and Meier KE: Signal transduction responses to lysophosphatidic
acid and sphingosine 1-phosphate in human prostate cancer cells.
Prostate. 69:1493–1506. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Pettus BJ, Bielawski J, Porcelli AM, et
al: The sphingosine kinase 1/sphingosine-1-phosphate pathway
mediates COX-2 induction and PGE2 production in response to
TNF-alpha. FASEB J. 17:1411–1421. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Cuvillier O, Ader I, Bouquerel P, Brizuela
L, Malavaud B, Mazerolles C and Rischmann P: Activation of
sphingosine kinase-1 in cancer: implications for therapeutic
targeting. Curr Mol Pharmacol. 3:53–65. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Nagahashi M, Ramachandran S, Kim EY, et
al: Sphingosine-1-phosphate produced by sphingosine kinase 1
promotes breast cancer progression by stimulating angiogenesis and
lymphangiogenesis. Cancer Res. 72:726–735. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Maceyka M, Harikumar KB, Milstien S and
Spiegel S: Sphingosine-1-phosphate signaling and its role in
disease. Trends Cell Biol. 22:50–60. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Hao H, Naomoto Y, Bao X, Watanabe N, et
al: Focal adhesion kinase as potential target for cancer therapy
(Review). Oncol Rep. 22:973–979. 2009.PubMed/NCBI
|
|
23.
|
Lechertier T and Hodivala-Dilke K: Focal
adhesion kinase and tumour angiogenesis. J Pathol. 226:404–412.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Stokes JB, Adair SJ, Slack-Davis JK, et
al: Inhibition of focal adhesion kinase by PF-562,271 inhibits the
growth and metastasis of pancreatic cancer concomitant with
altering the tumor microenvironment. Mol Cancer Ther. 10:2135–2145.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Yu HG, Tong SL, Ding YM, et al: Enhanced
expression of cholecystokinin-2 receptor promotes the progression
of colon cancer through activation of focal adhesion kinase. Int J
Cancer. 119:2724–2732. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Kobayashi H, Boelte KC and Lin PC:
Endothelial cell adhesion molecules and cancer progression. Curr
Med Chem. 14:377–386. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Shah N, Cabanillas F, McIntyre B, Feng L,
et al: Prognostic value of serum CD44, intercellular adhesion
molecule-1 and vascular cell adhesion molecule-1 levels in patients
with indolent non-Hodgkin lymphomas. Leuk Lymphoma. 53:50–56. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Lu X, Mu E, Wei Y, Riethdorf S, et al:
VCAM-1 promotes osteolytic expansion of indolent bone
micrometastasis of breast cancer by engaging α4β1-positive
osteoclast progenitors. Cancer Cell. 20:701–714. 2011.PubMed/NCBI
|
|
29.
|
Dymicka-Piekarska V, Guzinska-Ustymowicz
K, Kuklinski A and Kemona H: Prognostic significance of adhesion
molecules (sICAM-1, sVCAM-1) and VEGF in colorectal cancer
patients. Thromb Res. 129:e47–e50. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Alexiou D, Karayiannakis AJ, Syrigos KN,
Zbar A, Kremmyda A, Bramis I and Tsigris C: Serum levels of
E-selectin, ICAM-1 and VCAM-1 in colorectal cancer patients:
correlations with clinicopathological features, patient survival
and tumour surgery. Eur J Cancer. 37:2392–2397. 2001. View Article : Google Scholar
|
|
31.
|
Kase H, Hattori Y, Jojima T, et al:
Globular adiponectin induces adhesion molecule expression through
the sphingosine kinase pathway in vascular endothelial cells. Life
Sci. 81:939–943. 2007. View Article : Google Scholar
|
|
32.
|
Fong YC, Lin CY, Su YC, et al: CCN6
enhances ICAM-1 expression and cell motility in human
chondrosarcoma cells. J Cell Physiol. 227:223–232. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Petzold T, Orr AW, Hahn C, Jhaveri KA,
Parsons JT and Schwartz MA: Focal adhesion kinase modulates
activation of NF-kappaB by flow in endothelial cells. Am J Physiol
Cell Physiol. 297:C814–C822. 2009. View Article : Google Scholar
|
|
34.
|
Taglia L, Matusiak D, Matkowskyj KA and
Benya RV: Gastrin-releasing peptide mediates its morphogenic
properties in human colon cancer by upregulating intracellular
adhesion protein-1 (ICAM-1) via focal adhesion kinase. Am J Physiol
Gastrointest Liver Physiol. 292:G182–G190. 2007. View Article : Google Scholar
|