Introduction
Recent studies have shown that mediators of
inflammation, such as prostaglandins (PGs), play an important role
in tumorigenesis (1,2). Cyclooxygenase-2 (COX-2) is the key
enzyme involved, as it triggers PG synthesis. The increased
expression of COX-2 and the production of PGs are involved in the
genesis of various human cancers, including carcinoma of the liver,
colon, stomach, breast and lung (3–7). The
knockdown of COX-2 gene expression suppresses skin carcinogenesis
(8), and the targeted expression
of COX-2 promotes colon cancer cell growth (9) and enhances skin tumorigenesis
(8). Accumulating evidence has
indicated that prostaglandin E2 (PGE2)
promotes liver cancer cell growth (10,11);
however, the exact mechanisms through which PGE2
regulates liver cancer development are currently unknown.
PGE2 signaling stimulates its
G-protein-coupled plasma membrane receptors [E prostanoid (EP)1–4],
which activate multiple signal transduction pathways leading to
downstream responses. The EP1 receptor mainly couples to Gq protein
and upregulates the level of intracellular Ca2+; EP2 and
EP4 receptors couple to Gs protein, activate adenylate cyclase (AC)
and increase the production of intracellular cyclic AMP (cAMP);
however, the EP3 receptor couples to Gi protein, inactivates AC and
decreases the formation of intracellular cAMP (12). Thus, the specific target of
PGE2 in regulating cancer cell growth through EP
receptors has not yet been well illustrated.
The EP3 receptor has multiple isoforms generated
through alternative mRNA splicing in the carboxyl tail of the EP3
receptor gene. Thus far, 11 mRNA splice variants of the human EP3
receptor have been identified (13–15).
Evidence of different signal transduction pathways and the
regulation of gene expression among different EP3 receptor isoforms
has also been demonstrated in a number of studies (16–18).
The FUSE-binding proteins (FBPs) are a family of 3
regulatory proteins, termed FBP1, FBP2 and FBP3 (19). FBP1 was initially characterized as
a protein targeting the far upstream element, a positive
cis-element of the human c-myc gene (19). In liver, renal and cervix carcinoma
cell lines, FBP1 plays a role in tumorigenesis by regulating
c-myc transcript and protein levels (19–22).
FBP1 knockdown suppresses cell proliferation (20,23),
increases sensitivity to apoptotic stimuli (23) and affects the maintenance of
morphology in human hepatocellular carcinoma cells (20). Consistent with these observations,
FBP1 knockdown has been shown to impair liver tumor formation in a
mouse xenograft transplantation model (23). The overexpression of FBP1 promotes
the proliferation of liver cancer cells (20,22,24).
FBP1 overexpression significantly correlates with the proliferation
and motility of human non-small cell lung cancer cells (25). Thus, FBP1 plays a role in malignant
cell transformation. These findings strongly suggest the importance
of FBP1 in the development and progression of human cancers.
Our previous studies demonstrated that EP3 receptor
agonist upregulated FBP1 protein expression and promoted the
proliferation of liver cancer cells (unpublished data). Thus, we
hypothesized that PGE2 may promote liver cancer cell
growth through the upregulation of FBP1 expression via the EP3
receptor pathway; the molecular mechanisms involved have not yet
been reported.
Our present results revealed that EP3 receptor
activated by PGE2 couples to Gs protein and activates
cAMP-protein kinase A (PKA), downregulating the level of JTV1
protein, consequently inhibiting the ubiquitination of FBP1 and
increasing FBP1 protein expression, thus promoting liver cancer
cell growth.
Materials and methods
Antibodies and reagents
PGE2, the EP3 receptor agonist,
sulprostone, and the Cyclic AMP EIA kit were purchased from Cayman
Chemical Co. (Ann Arbor, MI, USA). The cell proliferation assay
reagent, WST-1, was purchased from Dojindo Laboratories (Kumamoto,
Japan). The human transforming growth factor-β1 (TGF-β1)
was purchased from R&D Systems (Minneapolis, MN, USA). The EP3
receptor selective antagonist, L-798106, the PKA inhibitor, H89,
the AC inhibitor, SQ22536, the cAMP analog, db-cAMP, and the Gi
inhibitor, pertussis toxin (PTX), were obtained from Sigma-Aldrich
(St. Louis, MO, USA). Lipofectamine™ 2000 and siRNA were purchased
from Invitrogen (Carlsbad, CA, USA). Anti-EP3 (AV34104) and
anti-β-actin antibodies were obtained from Sigma-Aldrich. Anti-FBP1
antibody (sc-11098) and protein A/G (sc-2003) were purchased from
Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-JTV1 antibody
(10424-1-AP) was purchased from Proteintech (Chicago, IL, USA).
Anti-ubiquitin antibody (ab19247) was purchased from Abcam
(Cambridge, UK). Anti-p-Smad2 (BS4172) and anti-Smad2 (BS1425)
antibodies were obtained from Bioworld Technology Inc. (St. Louis
Park, MN, USA).
Cell lines and culture
CCLP1 human liver cancer cells from the Department
of Transplantation Pathology, University of Pittsburgh Medical
Center (UPMC; Pittsburgh, PA, USA) were cultured in Dulbecco’s
modified Eagle’s medium (DMEM), supplemented with 10% FBS, 2 mM
L-glutamine, and 50 μg/ml gentamicin at 37°C in 5%
CO2.
Cell proliferation
Cell growth was determined using the cell
proliferation reagent, WST-1, a tetrazolium salt that is cleaved by
mitochondrial dehydrogenases in viable cells. Briefly, 100
μl of cell suspension (containing 0.5–2×104
cells) were plated in each well of 96-well plates. Cells were
cultured overnight. The cells were then incubated with different
treatments at the indicated concentrations and time periods. Cell
proliferation reagent, WST-1 (10 μl), was subsequently added
to each well. The incubation was continued from 30 min to 4 h at
37°C, and absorbance at 450 nm was measured using an automatic
ELISA plate reader.
Overexpression of EP3-4 plasmid in CCLP1
cells
The CCLP1 cells were exposed to the mixture of
Lipofectamine 2000 and EP3-4 plasmid or pcDNA3.1 control vector for
4 h. Following the removal of the transfection mixtures, fresh DMEM
with 10% fetal bovine serum was added. On the second day, the
medium was changed, and the cells were incubated with medium
containing 300 μg/ml G418 sulfate. Subsequent cultures of
selected CCLP1 cells were routinely grown in the presence of
selective pressure. Western blot analysis was performed in the
selected cells permanently transfected with EP3-4 or control
plasmids. The selected cells with the successful increase in EP3-4
expression were subsequently used for further experiments.
RNA interference
Cells were transfected with either EP3-4 siRNA or
with the negative RNA duplex as the control using Lipofectamine
2000. The depletion of EP3-4 was confirmed by western blot
analysis.
Preparation of whole cell lysate
At the end of each treatment, cellular extracts were
prepared in radio immunoprecipitation assay (RIPA) buffer
consisting of 50 mM Tris (pH 7.4), 150 mM NaCl, 1% NP-40, 0.25%
sodium deoxycholate, in the presence of protease inhibitors and
phosphatase inhibitors as follows: 2 mM sodium pyrophosphate, 1 mM
sodium orthovanadate, 1 mM sodium fluoride, 1 mM EDTA, 0.5
μg/ml leupeptin and 1 mM phenyl-methylsulfonyl fluoride
(PMSF). After sonication, the whole cell lysate was collected by
centrifugation at 10,000 rpm at 4°C for 10 min using a
microcentrifuge to remove cell debris. The samples were stored at
−80°C until use. The protein concentrations in the cell extracts
were determined by the Bio-Rad protein assay.
Western blot analysis
Equal amounts of protein (20 μg) or protein
purified by immunoprecipitation were separated by 12% SDS-PAGE and
electrotransferred onto nitrocellulose membranes for western blot
analysis. Membranes were blocked with 5% defatted milk in TBST (10
mM Tris, pH 7.4, 0.1% Tween-20, and 100 mM NaCl) for 1 h at room
temperature. Blotted proteins were probed with the primary
antibodies overnight at 4°C in TBST containing 1% defatted milk.
The membranes were then washed and incubated with horseradish
peroxidase-conjugated secondary antibodies in TBST for 1 h. Signals
were generated by enhanced chemiluminescent reagent (ECL, Amersham)
according to the manufacturer’s instructions and visualized by
exposing with the Bio-Rad system. Quantification was performed
using ImageJ software. The results are expressed as the fold change
vs. the control.
Immunoprecipitation
Cellular extracts (100 μg protein) were
incubated overnight at 4°C in RIPA buffer with antibody against
FBP1 (2 μg). Protein A/G-agarose beads were then added. The
mixture was gently vortexed and incubated for 2 h at 4°C. The beads
were recovered by centrifugation at 10,000 × g and gently washed 3
times with RIPA buffer. SDS sample loading buffer for SDS-PAGE was
added, and the mixture was incubated at 100°C for 5 min. The beads
were centrifugated, and the supernatants were applied to 12%
SDS-PAGE.
cAMP production
To measure cAMP production, the cells cultured in
6-well plates were serum-starved overnight. The cells were exposed
to sulprostone, PGE2 and the vehicle. After a 10-min
incubation, the cells were collected and resuspended in 0.1 M HCl,
then a 50-μl centrifuged sample was analyzed for cAMP
production according to the manufacturer’s instructions.
Statistical analysis
The values are expressed as the means ± SD. Data
were analyzed by one-way analysis of variance followed by the
Student’s t-test. A value of p<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of EP3-4 receptor overexpression
in CCLP1 cells
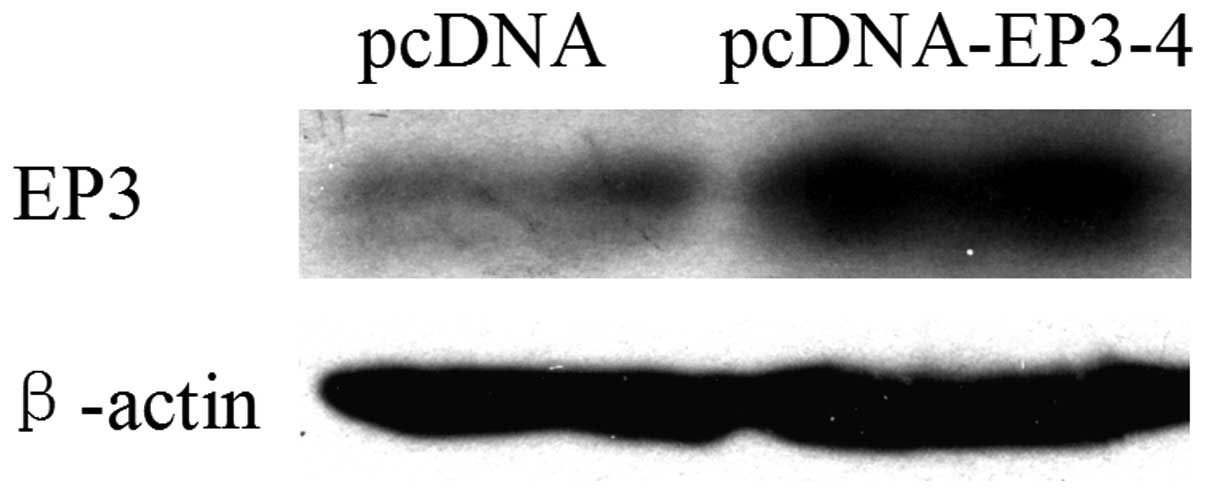
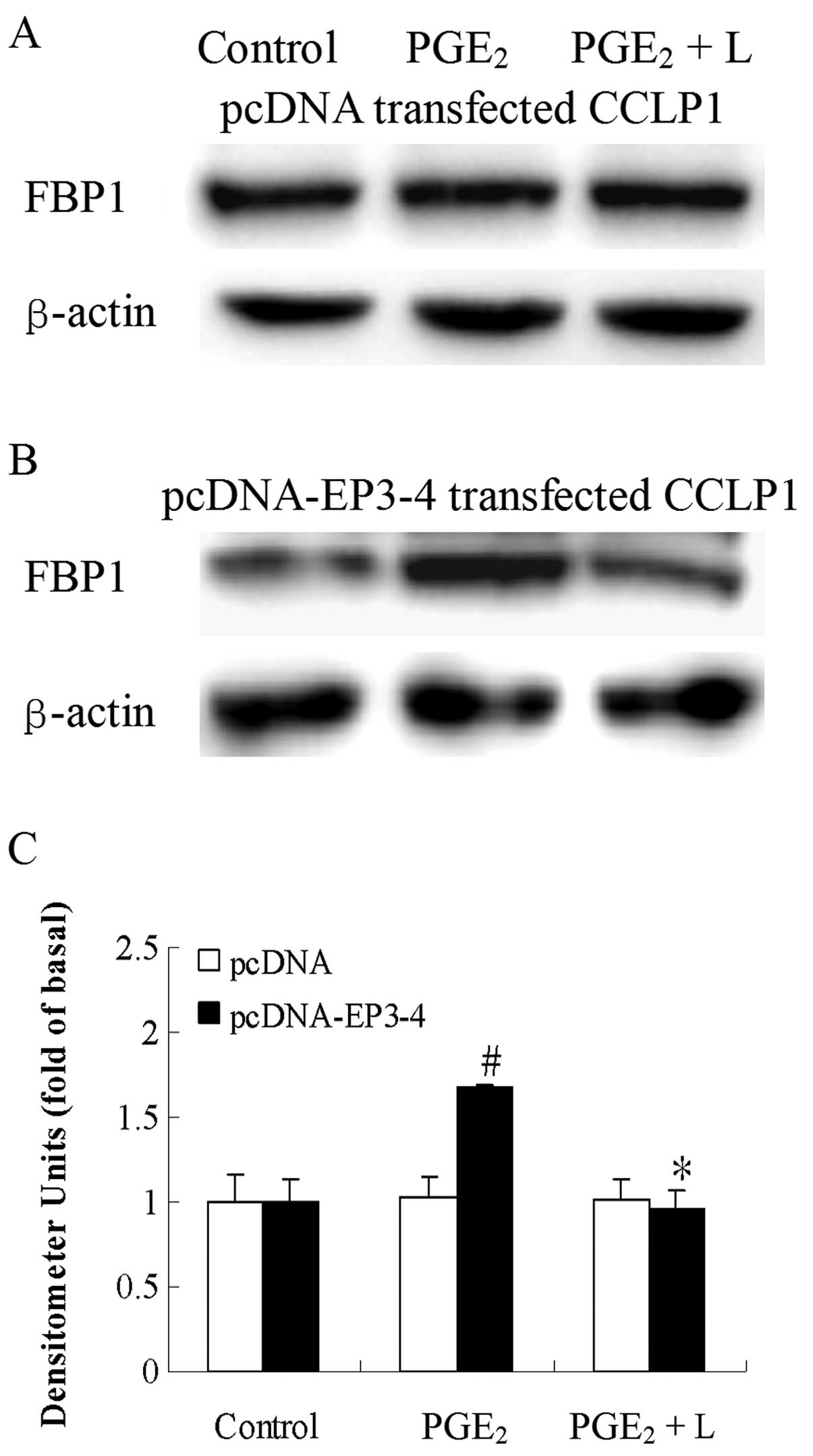
As shown in Fig. 1,
CCLP1 cells were stably transfected with the EP3-4 expression
plasmid or the pcDNA3.1 control plasmid. The western blot analysis
results showed that the EP3-4 receptor was overexpressed in the
EP3-4-pcDNA3.1-transfected CCLP1 cells.
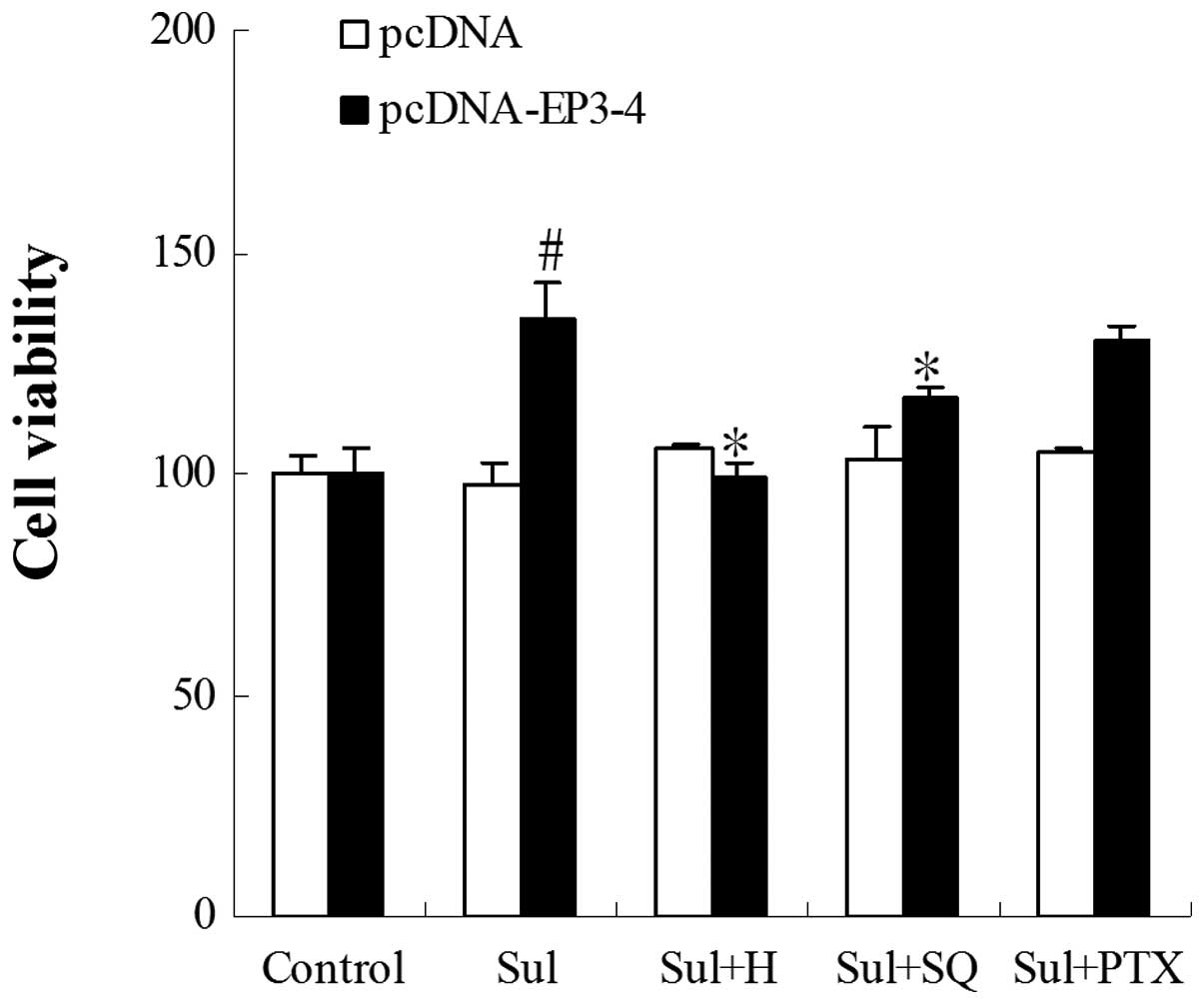
Effect of EP3 receptor activation on the
growth of CCLP1 cells
The EP3-4- and control plasmid pcDNA3.1-transfected
CCLP1 cells were examined for their response to treatment with the
EP3 agonist, sulprostone. To determine the proliferation of the
cells, the cells were treated with 10 μM of EP3 receptor
agonist (sulprostone), 10 μM PKA inhibitor (H89), 50
μM AC inhibitor (SQ22536) and 50 nm Gi subunit inhibitor
(PTX) (Fig. 2). The treatment of
EP3-4-transfected CCLP1 cells with sulprostone for 24 h induced a
35.17% increase in cell growth. The treatment of these cells with
sulprostone + H89 and sulprostone + SQ22536, dereased the growth
rate by 26.5 and 13.5% compared to treatment with sulprostone
alone. However, PTX had no effect on the cell growth induced by
sulprostone. The empty pcDNA3.1-transfected cells showed no
response.
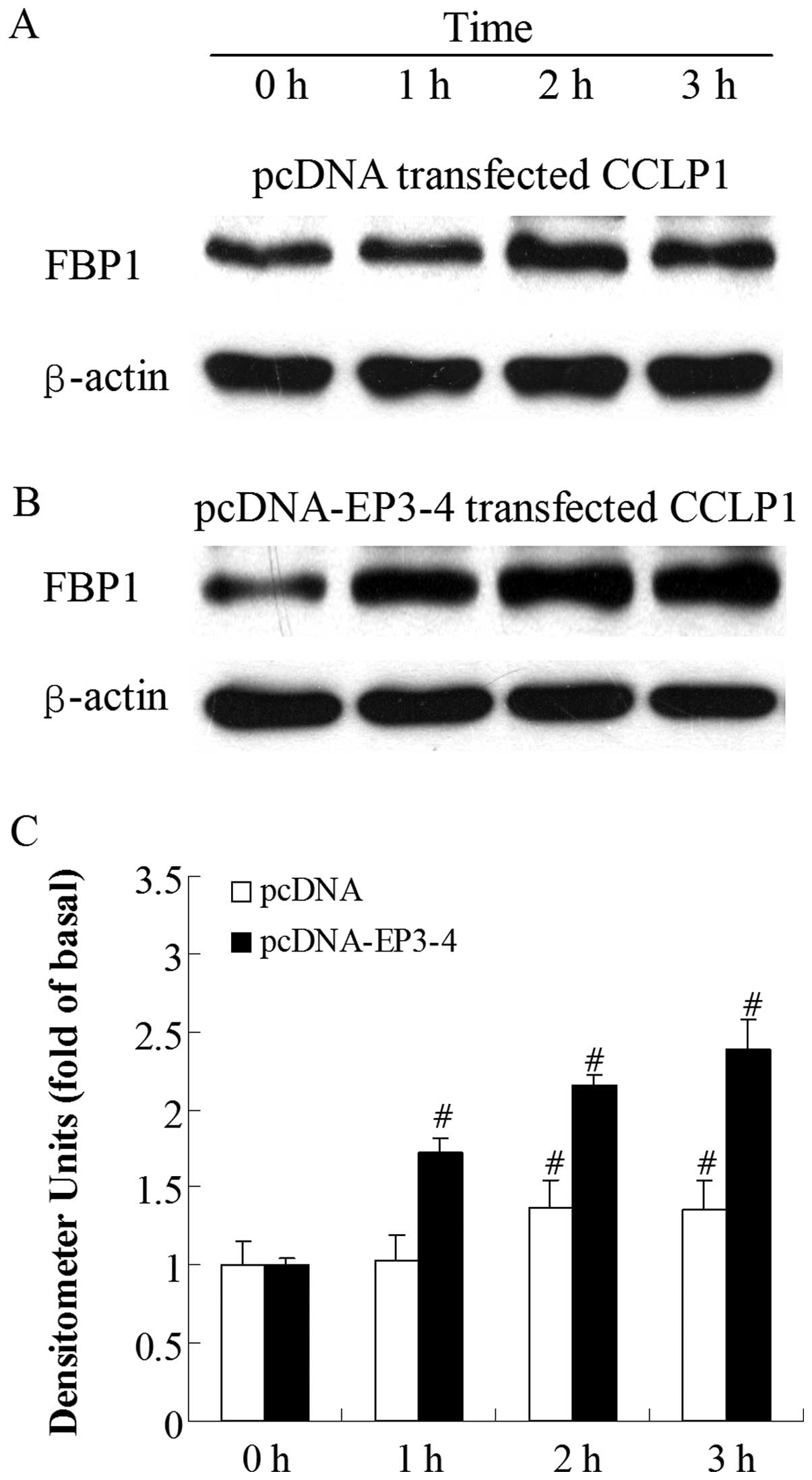
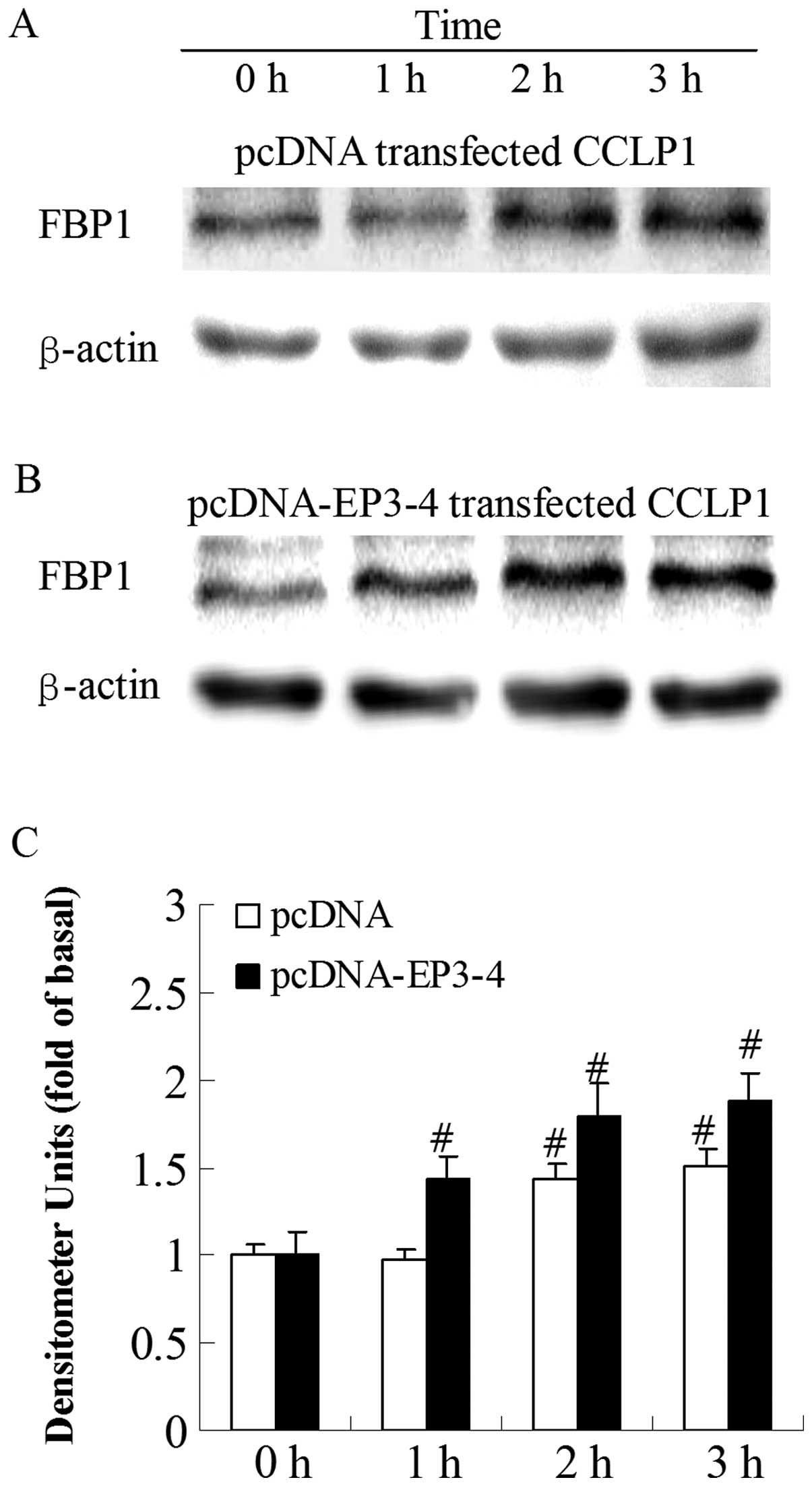
Sulprostone and PGE2 induce an
increase in FBP1 protein expression in CCLP1 cells
To investigate which molecule was regulated by
PGE2 viathe EP3 receptor, we examined the effects of the
EP3 receptor agonist, sulprostone, and PGE2 on the level
of FBP1 protein. By contrast, at the 1-h time-point, the treatment
of the empty pcDNA3.1-transfected cells with 10 μM
sulprostone had no effect on FBP1 protein levels and the increase
in FBP1 protein expression was only observed at 2 h (Fig. 3A). Fig. 3B shows that the treatment of the
EP3-4-transfected cells with 10 μM sulprostone induced an
increase in FBP1 levels in a time-dependent manner. The level of
FBP1 protein was rapidly increased within 1 h (1.72-fold increase
compared to 0 h). A similar pattern of increased FBP1 protein
expression was observed when the cells were treated with 10
μM PGE2, with a 1.43-fold increase at 1 h
compared to 0 h in the EP3-4-transfected cells (Fig. 4B). PGE2 had no effect on
the FBP1 protein at 1 h in the empty pcDNA3.1-transfected cells
(Fig. 4A).
Effects of blocking EP3 receptor on
PGE2-induced increase in FBP1 protein expression in
CCLP1 cells
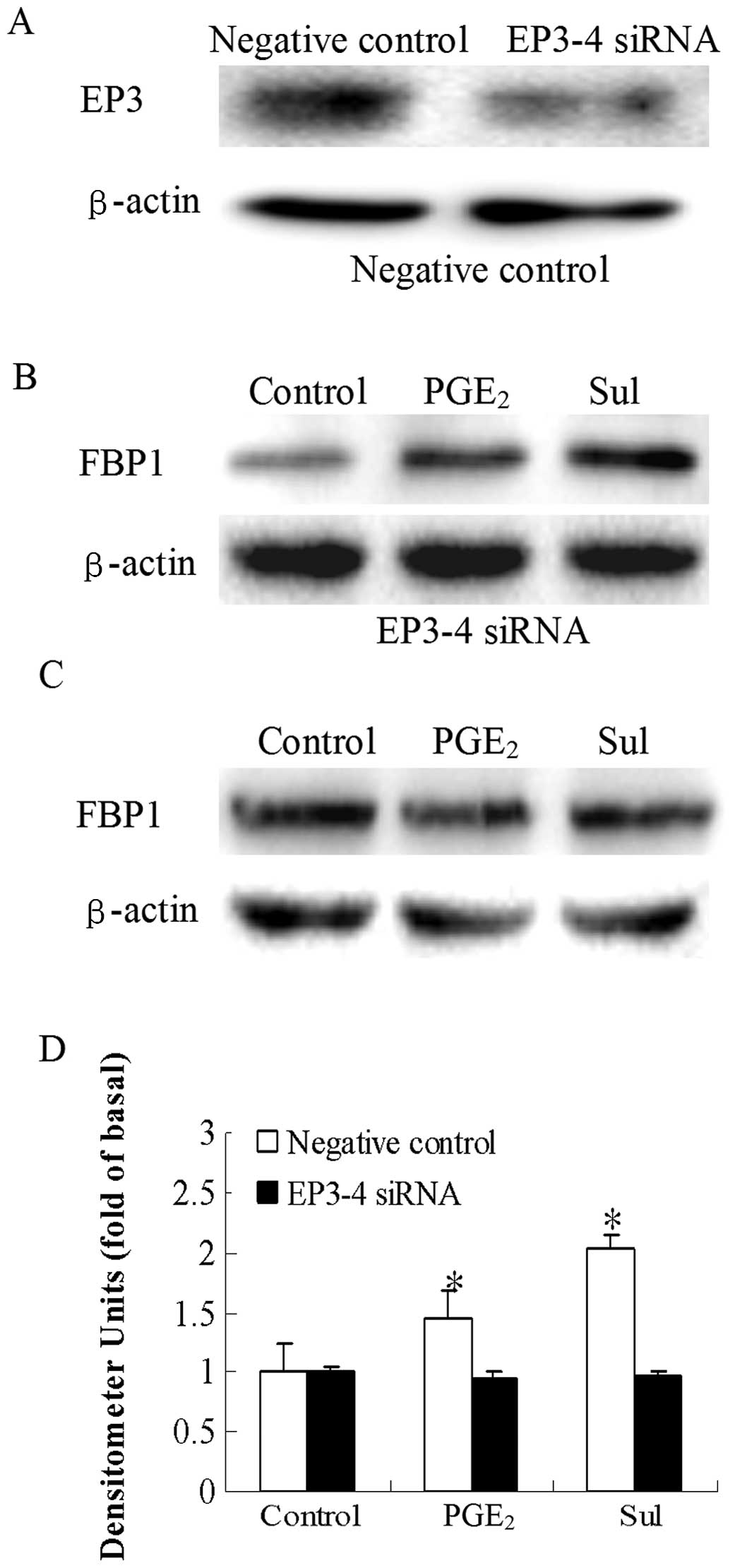
We then examined the direct effects of the EP3
receptor antagonist, L-798106, and EP3-4 siRNA on the
PGE2-induced increase in FBP1 protein expression. In the
EP3-4-transfected cells, treatment with 10 μM
PGE2 and 10 μM L-798106 resulted in a 43%
decrease in FBP1 protein expression induced by PGE2
(Fig. 5B). L-798106 had no effect
on the empty pcDNA3.1-transfected cells (Fig. 5A). Consistent with these results,
the PGE2 and sulprostone-induced increase in FBP1
protein expression was also blocked by the siRNA suppression of the
EP3-4 receptor in the EP3-4-transfected cells (Fig. 6C). In the negative
siRNA-transfected cells, the levels of FBP1 protein in the
PGE2 and sulprostone groups were 1.46- and 2.02-fold
higher compared to those in the the control group (Fig. 6B). The effect of the siRNA
suppression of the EP3-4 receptor in the EP3-4-transfected cells
was detected by western blot analysis (Fig. 6A). These findings demonstrate the
key role of the EP3 receptor in the regulation of FBP1 protein
expression by PGE2.
Effects of EP3 receptor activation on the
cytoplasmic cAMP production in CCLP1 cells
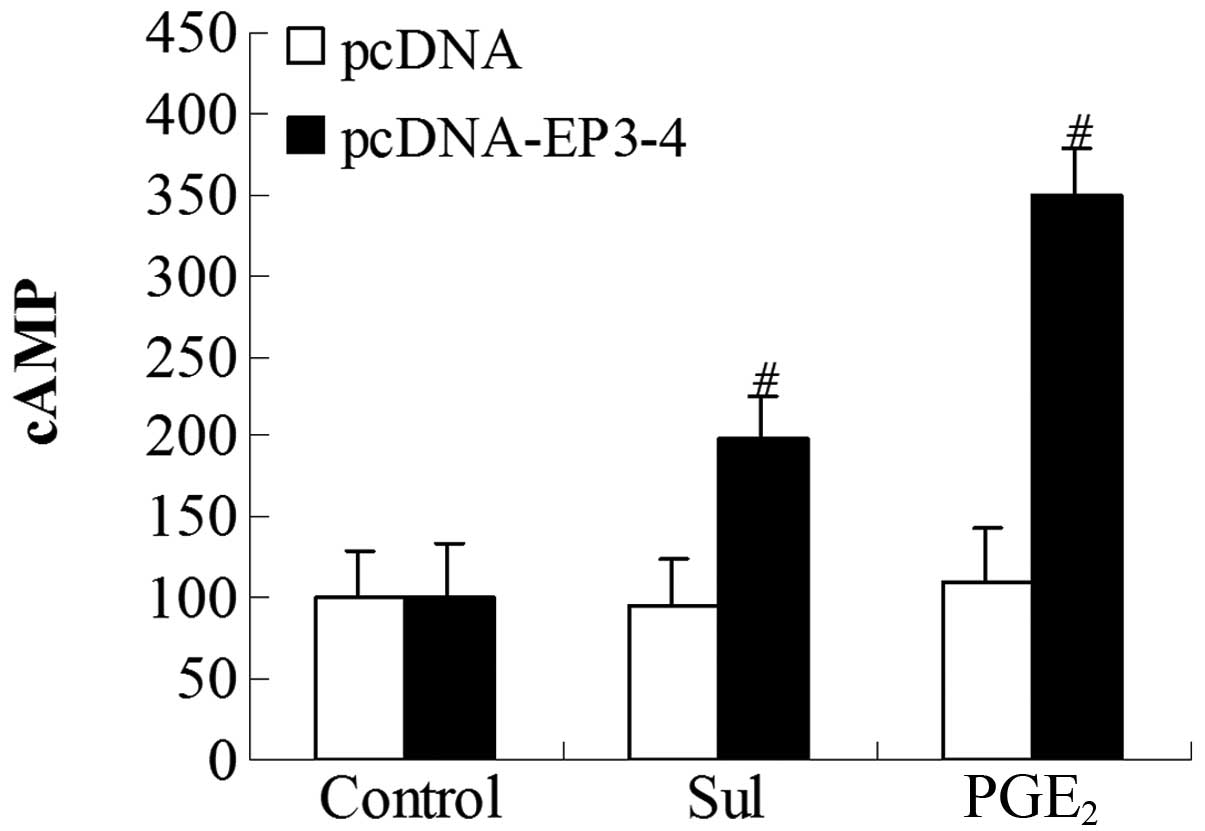
To further investigate whether the EP3 receptor
couples to the Gs subunit, we examined the cytoplasmic cAMP
production induced by PGE2 and sulprostone. The
EP3-4-transfected cells and the empty pcDNA3.1-transfected cells
were treated with 10 μM PGE2 and 10 μM
sulprostone. In the EP3-4-transfected cells, the levels of cAMP
induced by PGE2 and sulprostone were relatively
increased by 248.56 and 99.42%, respectively. PGE2 and
sulprostone had no effect on the empty pcDNA3.1-transfected cells
(Fig. 7).
Effects of the AC activator, forskolin,
cAMP analog, db-cAMP, EP3 agonist, sulprostone, and PGE2
on PKA inhibitor H89-induced Smad2 phosphorylation in CCLP1
cells
Since PKA downregulates TGF-β activity, we examined
the effects of the AC activator, forskolin, the cAMP analog,
db-cAMP, the EP3 agonist, sulprostone, and PGE2 on PKA
inhibitor H89-induced Smad2 phosphorylation. Fig. 8B shows that in the
EP3-4-transfected cells, 10 μM H89 treatment induced the
rapid phosphorylation of Smad2 (1.49-fold compared to the control).
In addition, treatment with 10 μM forskolin, 100 μM
db-cAMP, 10 μM sulprostone and 10 μM PGE2
reduced the Smad2 phosphorylation induced by H89 by 49, 26, 32 and
38%, respectively. As shown in Fig.
8A, these reagents had no effect on the empty
pcDNA3.1-transfected cells.
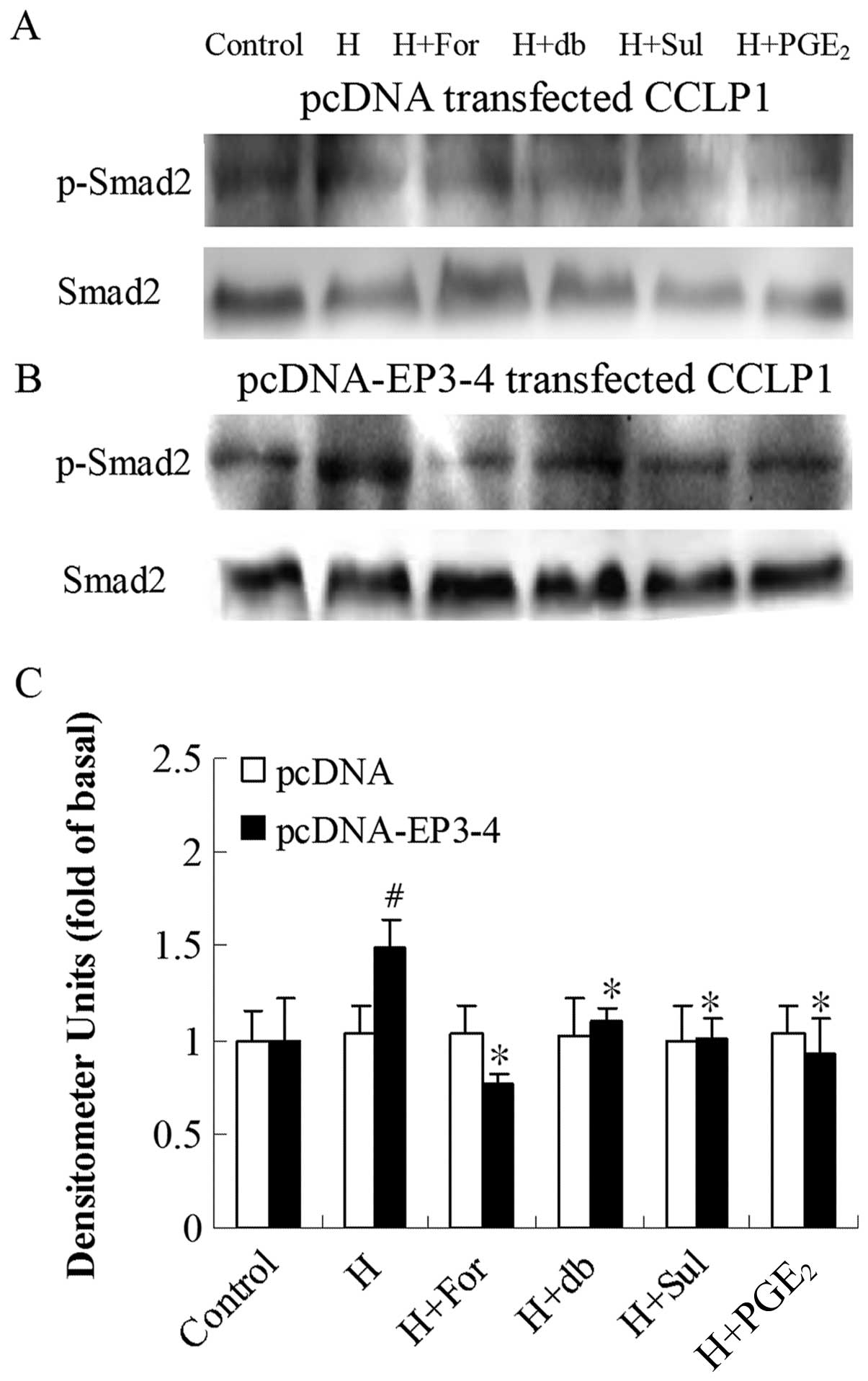 | Figure 8Effects of the AC activator,
forskolin, cAMP analog, db-cAMP, EP3 agonist, sulprostone and
PGE2 on PKA inhibitor H89-induced Smad2 phosphorylation.
(A) The empty pcDNA3.1-transfected CCLP1 cells and (B) the
EP3-4-transfected CCLP1 cells (B) were treated with 10 μM
forskolin, 100 μM db-cAMP, 10 μM sulprostone and 10
μM PGE2 for 1 h prior to stimulation with 10
μM H89 for 1 h. The cell lysates were obtained for western
blot analysis with polyclonal antibodies against p-Smad2 and Smad2.
(C) Statistical plots of data from experiments are shown. The data
represent an average of 3 independent experiments.
#p<0.05 comparison of H treatment with control.
*p<0.05 comparison of H + For, H + db, H + Sul and H
+ PGE2 treatments with H treatment. H, H89; For,
forskolin; db, db-cAMP; Sul, sulprostone; pcDNA, pcDNA3.1. |
Effects of the AC activator, forskolin,
cAMP analog, db-cAMP, EP3 agonist, sulprostone, and PGE2
on TGF-β1-induced Smad2 phosphorylation in CCLP1
cells
To further document that PGE2-EP3-Gs-PKA
inhibits TGF-β activity, we examined the effects of the AC
activator ,forskolin, the cAMP analog, db-cAMP, the EP3 agonist,
sulprostone, and PGE2 on TGF-β1-induced Smad2
phosphorylation. Fig. 9B shows
that in the EP3-4-transfected cells, treatment with 2 ng/ml
TGF-β1 induced an increase in the phosphorylation of
Smad2 (2.26-fold compared to the control). In addition, treatment
with forskolin, db-cAMP, sulprostone and PGE2 reduced
the Smad2 phosphorylation induced by TGF-β1 by 24, 40,
40 and 30%, respectively. As shown in Fig. 9A, these reagents had no effect on
the empty pcDNA3.1-transfected cells.
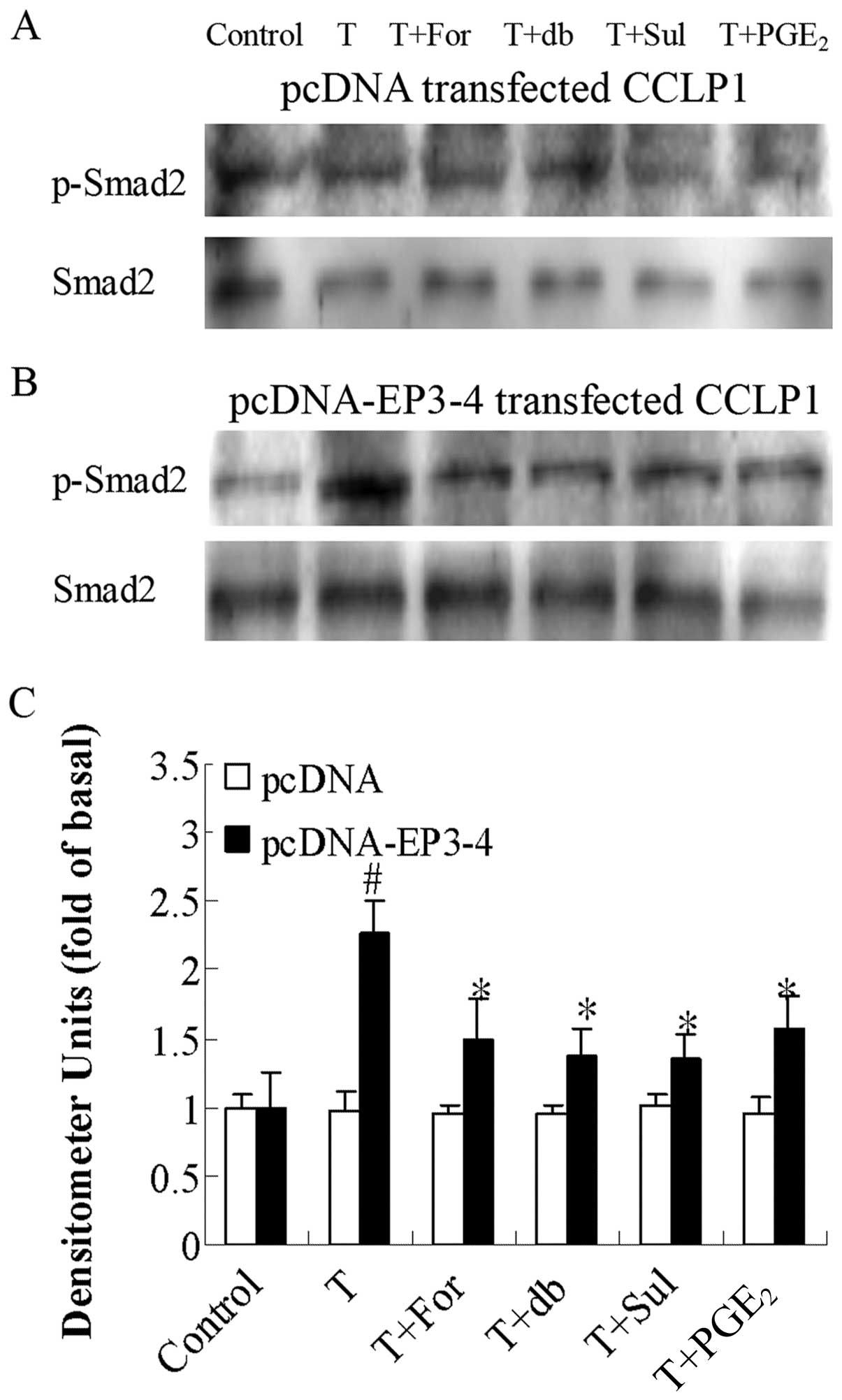 | Figure 9Effects of the AC activator,
forskolin, the cAMP analog, db-cAMP, the EP3 agonist, sulprostone,
and PGE2 on TGF-β1-induced Smad2
phosphorylation. (A) The empty pcDNA3.1-transfected CCLP1 cells and
(B) the EP3-4-transfected CCLP1 cells (B) were treated with 10
μM forskolin, 100 μM db-cAMP, 10 μM
sulprostone and 10 μM PGE2 for 1 h prior to
stimulation with 2 ng/ml TGF-β1 for 1 h. The cell
lysates were obtained for western blot analysis with polyclonal
antibodies against p-Smad2 and Smad2. (C) Statistical plots of data
from experiments are shown. The data represent an average of 3
independent experiments. #p<0.05 comparison of T
treatment with control, *p<0.05 comparison of T +
For, T + db, T + Sul and T + PGE2 treatments with T
treatment. T, TGF-β1; For, forskolin; db, db-cAMP; Sul,
sulprostone; pcDNA, pcDNA3.1. |
Effects of the AC activator, forskolin,
cAMP analog, db-cAMP, EP3 agonist, sulprostone, and PGE2
on TGF-β1-induced FBP1 and JTV1 protein expression in
CCLP1 cells
We then examined the effects of the AC activator,
forskolin, the cAMP analog, db-cAMP, the EP3 agonist, sulprostone,
and PGE2 on TGF-β1-induced FBP1 and JTV1
protein expression. Fig. 10B
shows that in the EP3-4-transfected cells, the level of FBP1
protein induced by TGF-β1 was decreased by 37% of the
control. The levels of FBP1 protein in the TGF-β1 +
forskolin, TGF-β1 + db-cAMP, TGF-β1 +
sulprostone and TGF-β1 + PGE2 groups were
decreased by 1.90-, 1.56-, 1.84-and 1.62-fold, respectively
compared to the TGF-β1 group. The level of JTV1 protein
induced by TGF-β1 was 1.48-fold of the control. The
levels of JTV1 protein in the TGF-β1 + forskolin,
TGF-β1 + db-cAMP, TGF-β1 + sulprostone and
TGF-β1 + PGE2 groups were decreased by 54,
45, 44 and 45%, respectively compared to the TGF-β1
group. As shown in Fig. 10A,
these reagents had no effect on the empty pcDNA3.1-transfected
cells. These results indicate that the PGE2-EP3-Gs-PKA
inhibition of TGF-β1 regulates the protein expression of
FBP1 and JTV1.
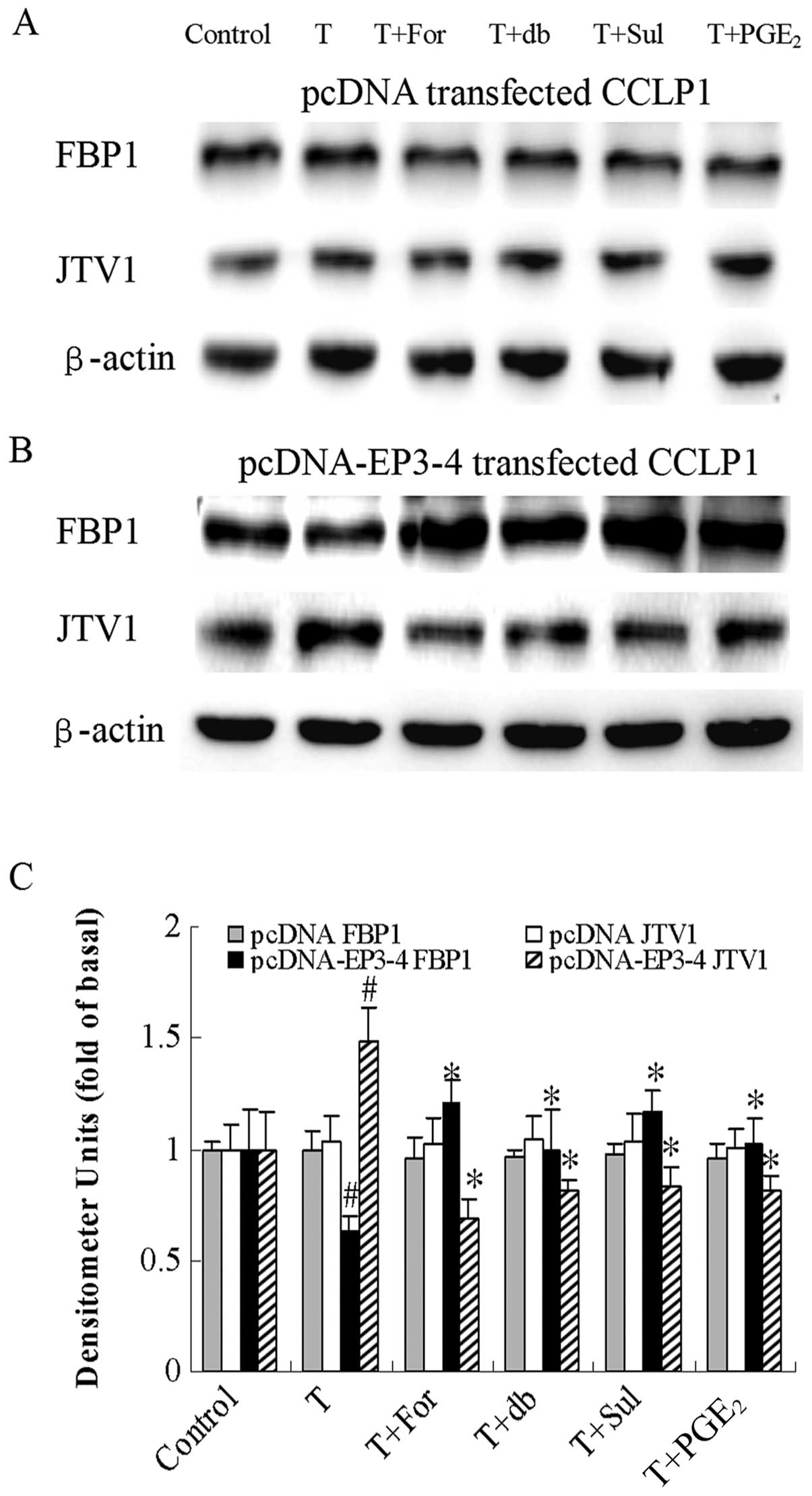 | Figure 10Effects of the AC activator,
forskolin, the cAMP analog, db-cAMP, the EP3 agonist, sulprostone
and PGE2 on TGF-β1-induced FBP1 and JTV1
protein expression. (A) The empty pcDNA3.1-transfected CCLP1 cells
and (B) the EP3-4-transfected CCLP1 cells were treated with 10
μM forskolin, 100 μM db-cAMP, 10 μM
sulprostone and 10 μM PGE2 for 1 h prior to
stimulation with 2 ng/ml TGF-β1 for 1 h. The cell
lysates were obtained for western blot analysis with antibodies
against FBP1, JTV1 and β-actin. (C) Statistical plots of data from
experiments are shown. The data represent an average of 3
independent experiments. #p<0.05 comparison of T
treatment with control. *p<0.05 comparison of T +
For, T + db, T + Sul and T + PGE2 treatments with T
treatment. T, TGF-β1; For, forskolin; db, db-cAMP; Sul,
sulprostone; pcDNA, pcDNA3.1. |
Effects of the AC activator, forskolin,
cAMP analog, db-cAMP, EP3 agonist, sulprostone, and PGE2
on TGF-β1-induced binding of JTV1 with FBP1 and the
ubiquitination of FBP1 in CCLP1 cells
As shown in Fig.
11B, in the EP3-4-transfected cells, TGF-β1
treatment induced the binding of JTV1 with FBP1 (1.47-fold of the
control) and the ubiquitination of FBP1 (1.76-fold of the control).
In addition, the binding of FBP1 with JTV1 in the TGF-β1
+ forskolin, TGF-β1 + db-cAMP, TGF-β1 +
sulprostone and TGF-β1 + PGE2 groups was
deceased by 59, 48, 59 and 63%, respectively compared to the
TGF-β1 group. The ubiquitination of FBP1 in the
TGF-β1 + forskolin, TGF-β1 + db-cAMP,
TGF-β1 + sulprostone and TGF-β1 +
PGE2 groups was deceased by 41, 36, 57 and 33%,
respectively compared to the TGF-β1 group. As shown in
Fig. 11A, these reagents had no
effect on the empty pcDNA3.1-transfected cells.
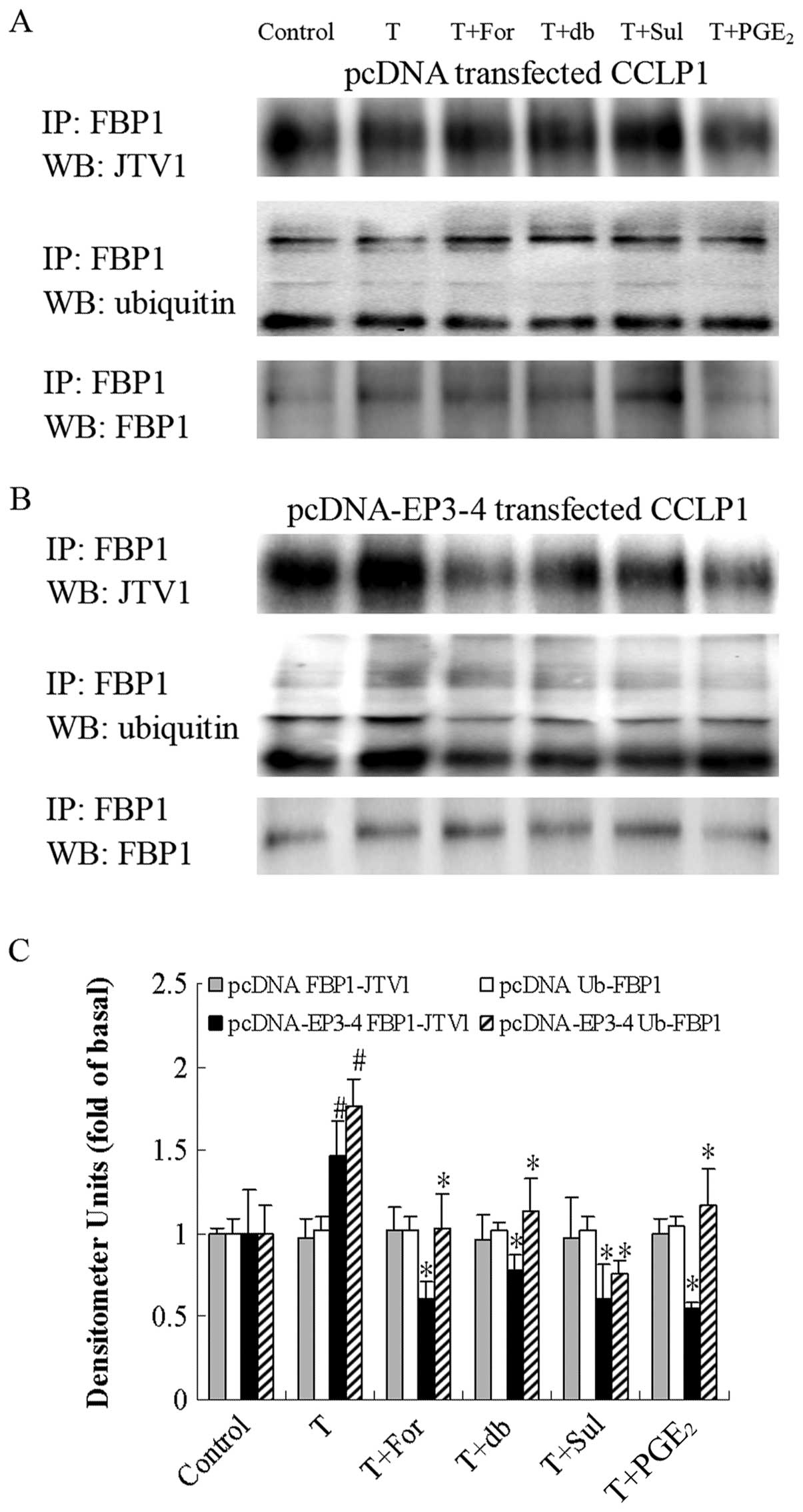 | Figure 11Effects of the AC activator
forskolin, the cAMP analog db-cAMP, the EP3 agonist, sulprostone,
and PGE2 on the TGF-β1-induced binding of
JTV1 with FBP1 and the ubiquitination of FBP1. (A) The empty
pcDNA3.1-transfected CCLP1 cells and (B) the EP3-4-transfected
CCLP1 cells were treated with 10 μM forskolin, 100 μM
db-cAMP, 10 μM sulprostone and 10 μM PGE2
for 1 h prior to stimulation with 2 ng/ml TGF-β1 for 1
h. The cell lysates were obtained for immunoprecipitation with
polyclonal antibody against FBP1. The precipitated pellets were
then separated by gel electrophoresis on 12% Tris-glycine gels,
followed by western blot analysis with polyclonal antibodies
against JTV1 and ubiquitin. (C) Statistical plots of data from
experiments are shown. The data represent an average of 3
independent experiments. #p<0.05 comparison of T
treatment with control, *p<0.05 comparison of T +
For, T + db, T + Sul and T + PGE2 treatments with T
treatment. T, TGF-β1; For, forskolin; db, db-cAMP; Sul,
sulprostone; pcDNA, pcDNA3.1. |
Effects of the PKA inhibitor, H89, and AC
inhibitor, SQ22536, on sulprostone and PGE2-induced FBP1
and JTV1 protein expression in CCLP1 cells
The findings presented above suggested that the
activation of cAMP-PKA induced by PGE2 via the EP3
receptor suppressed TGF-β, regulating the binding of JTV1 with FBP1
and the ubiquitination of FBP1, and thus regulating FBP1 protein
expression. To further evaluate this hypothesis, we examined
whether the inhibition of cAMP-PKA would alter the levels of JTV1
and FBP1 protein induced by PGE2 via the EP3 receptor.
The cells were treated with sulprostone and PGE2 in the
presence or absence of the PKA inhibitor, H89, and the AC
inhibitor, SQ22536, to determine the levels of JTV1 and FBP1
protein. As shown in Fig. 12B, in
the EP3-4-transfected cells, sulprostone increased the FBP1 protein
levels by 82% compared to the control. Treatment with H89 and
SQ22536 followed by sulprostone decreased the FBP1 protein levels
by 42 and 47%, respectively compared to treatment with sulprostone
alone. Sulprostone decreased the JTV1 protein levels by 40%
compared to the control. Treatment with H89 and SQ22536 followed by
sulprostone increased the JTV1 protein levels by 36 and 66%,
respectively compared to treatment with sulprostone alone.
Consistent with these results, as shown in in Fig. 13B, in the EP3-4-transfected cells,
PGE2 increased the FBP1 protein levels by 73% compared
to the control. Treatment with H89 and SQ22536 followed by
PGE2 decreased the FBP1 protein levels by 22 and 27%,
respectively compared to treatment with sulprostone alone.
PGE2 decreased the JTV1 protein levels by 20% compared
to the control. Treatment with H89 and SQ22536 followed by
PGE2 increased the JTV1 protein levels by 61 and 94%,
respectively compared to treatment with PGE2 alone. H89
and SQ22536 had no effect on the empty pcDNA3.1-transfected cells
(Figs. 12A and 13A).
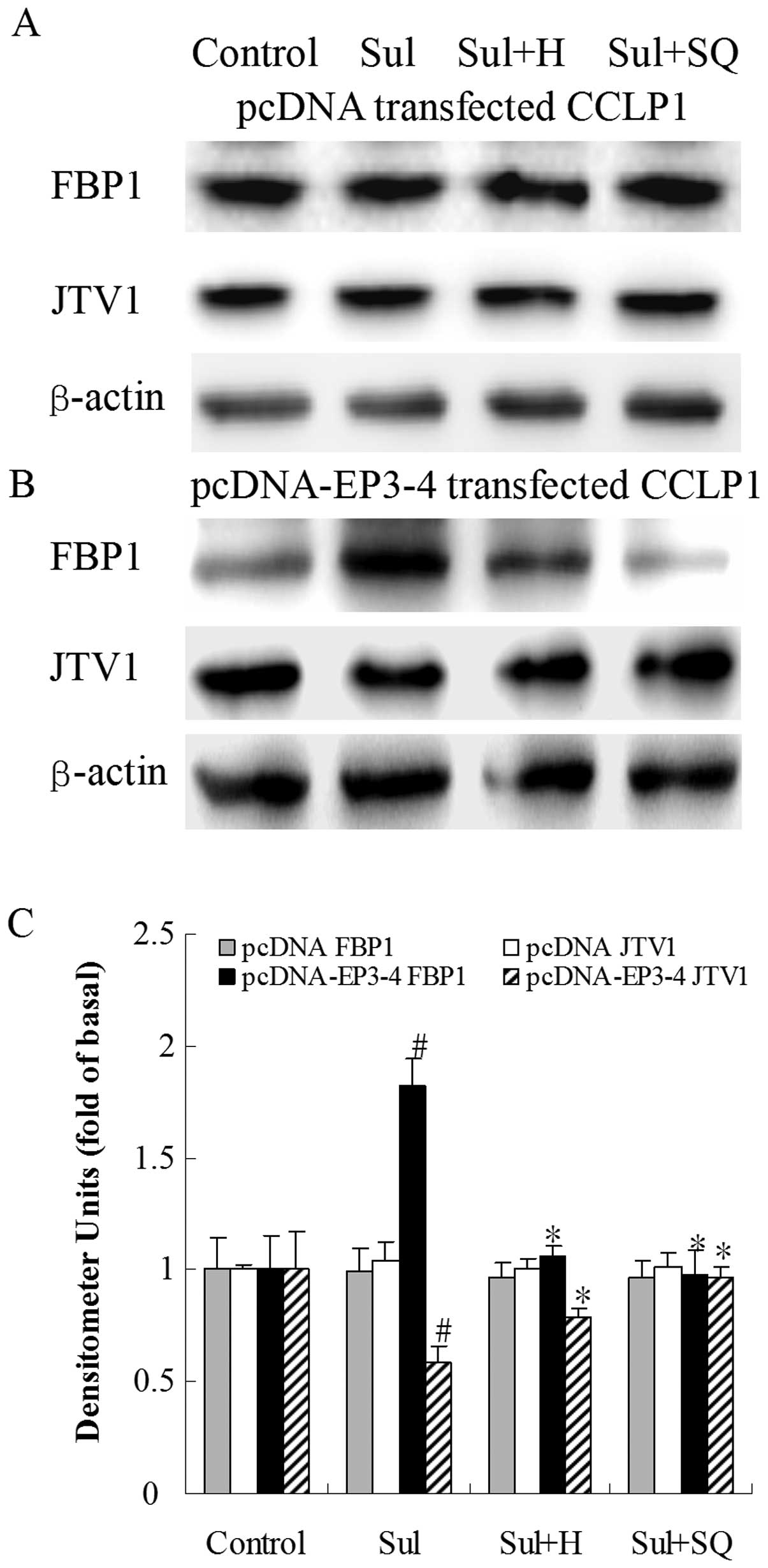 | Figure 12Effects of the PKA inhibitor, H89,
and the AC inhibitor, SQ22536, on sulprostone-induced FBP1 and JTV1
protein expression. (A) The empty pcDNA3.1-transfected CCLP1 cells
and (B) the EP3-4-transfected CCLP1 cells were treated with 10
μM sulprostone in the presence or absence of 10 μM of
the PKA inhibitor, H89, and 50μM of the AC inhibitor,
SQ22536. The cell lysates were obtained for western blot analysis
with antibodies against FBP1, JTV1 and β-actin. (C) Statistical
plots of data from experiments are shown. The data represent an
average of 3 independent experiments. #p<0.05
comparison of Sul treatment with control, *p<0.05
comparison of Sul + H and Sul + SQ treatments with Sul treatment.
Sul, sulprostone; H, H89; SQ, SQ22536; pcDNA, pcDNA3.1. |
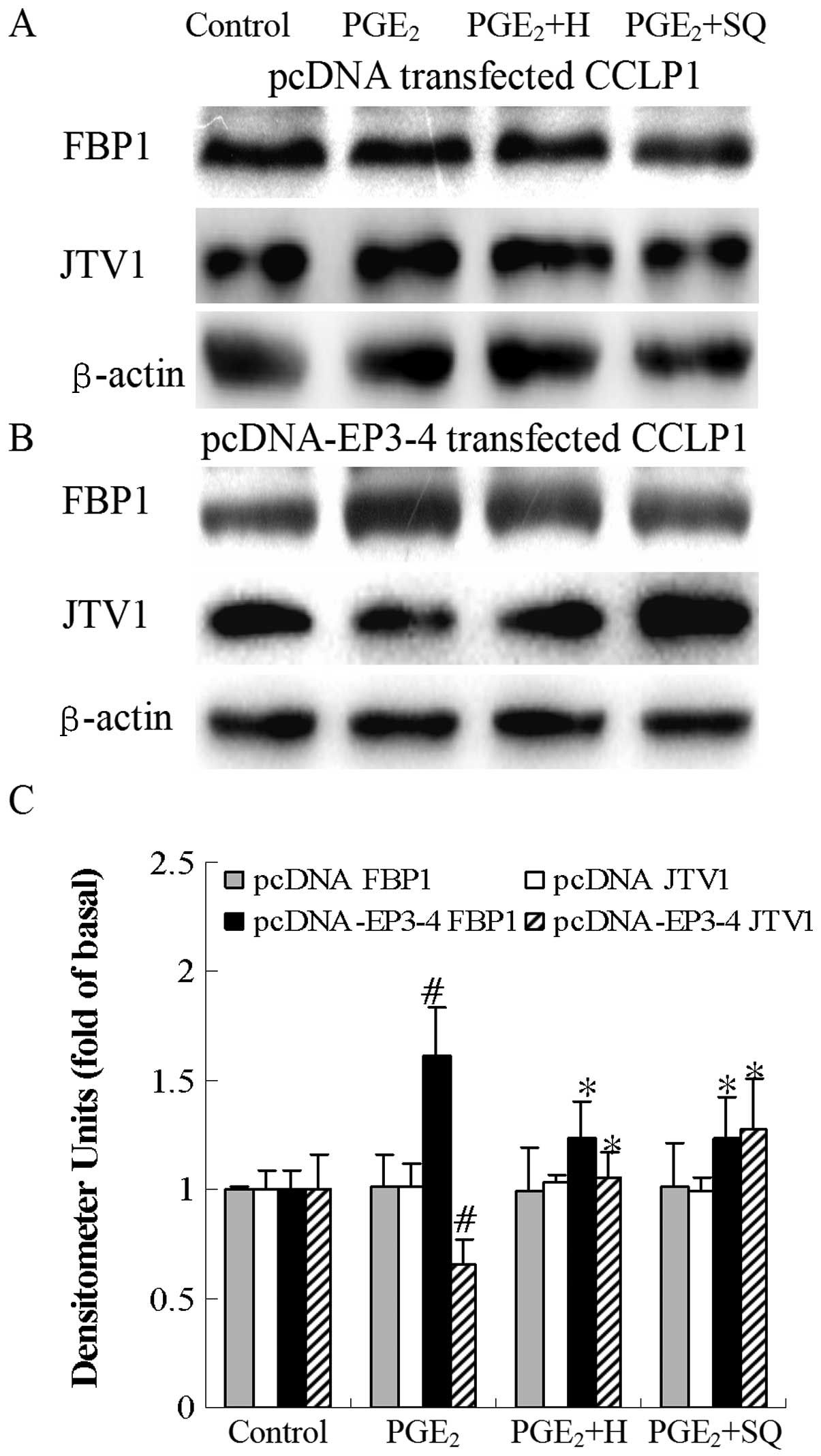 | Figure 13Effects of the PKA inhibitor, H89,
and the AC inhibitor, SQ22536, on PGE2-mediated FBP1 and
JTV1 protein expression. (A) The empty pcDNA3.1-transfected CCLP1
cells and (B) the EP3-4-transfected CCLP1 cells were treated with
10 μM PGE2 in the presence or absence of 10
μM of the PKA inhibitor, H89, and 50 μM of the AC
inhibitor, SQ22536. The cell lysates were obtained for western blot
analysis with antibodies against FBP1, JTV1 and β-actin. (C)
Statistical plots of data from experiments are shown. The data
represent an average of 3 independent experiments.
#p<0.05 comparison of PGE2 with control.
*p<0.05 comparison of PGE2 + H and
PGE2 + SQ treatments with PGE2 treatment. H,
H89; SQ, SQ22536; pcDNA, pcDNA3.1. |
Effects of the PKA inhibitor, H89, and AC
inhibitor, SQ22536, on the sulprostone and PGE2-mediated
binding of JTV1 with FBP1 and the ubiquitination of FBP1 in CCLP1
cells
The cells were treated with sulprostone and
PGE2 in the presence or absence of the PKA inhibitor,
H89, and the AC inhibitor, SQ22536, to determine the binding of
JTV1 with FBP1 and the ubiquitination of FBP1. As shown in Fig. 14B, in the EP3-4-transfected cells,
sulprostone decreased the binding of JTV1 with FBP1 by 64% compared
to the control. The binding of JTV1 with FBP1 following treatment
with H89 and SQ22536 followed by sulprostone was increased by
3.31-and 3.39-fold, respectively compared to treatment with
sulprostone. Sulprostone decreased the ubiquitination of FBP1 by
31% compared to the control. The ubiquitination of FBP1 following
treatment with H89 and SQ22536 followed by sulprostone was
increased by 1.77- and 1.79-fold, respectively compared to
treatment with sulprostone. Consistent with these results, as in
shown in Fig. 15B,
PGE2 decreased the binding of JTV1 with FBP1 by 44%
compared to the control. The binding of JTV1 with FBP1 following
treatment with H89 and SQ22536 followed by PGE2 was
increased by 2.21- and 2.34-fold, respectively compared to
treatment with PGE2. PGE2 decreased the
ubiquitination of FBP1 by 30% compared to the control. In the
EP3-4-transfected cells, the ubiquitination of FBP1 following
treatment with H89 and SQ22536 followed by PGE2 was
increased by 1.53- and 1.46-fold, respectively compared to
treatment with PGE2. H89 and SQ22536 had no effect on
the empty pcDNA3.1-transfected cells (Figs. 14A and 15A).
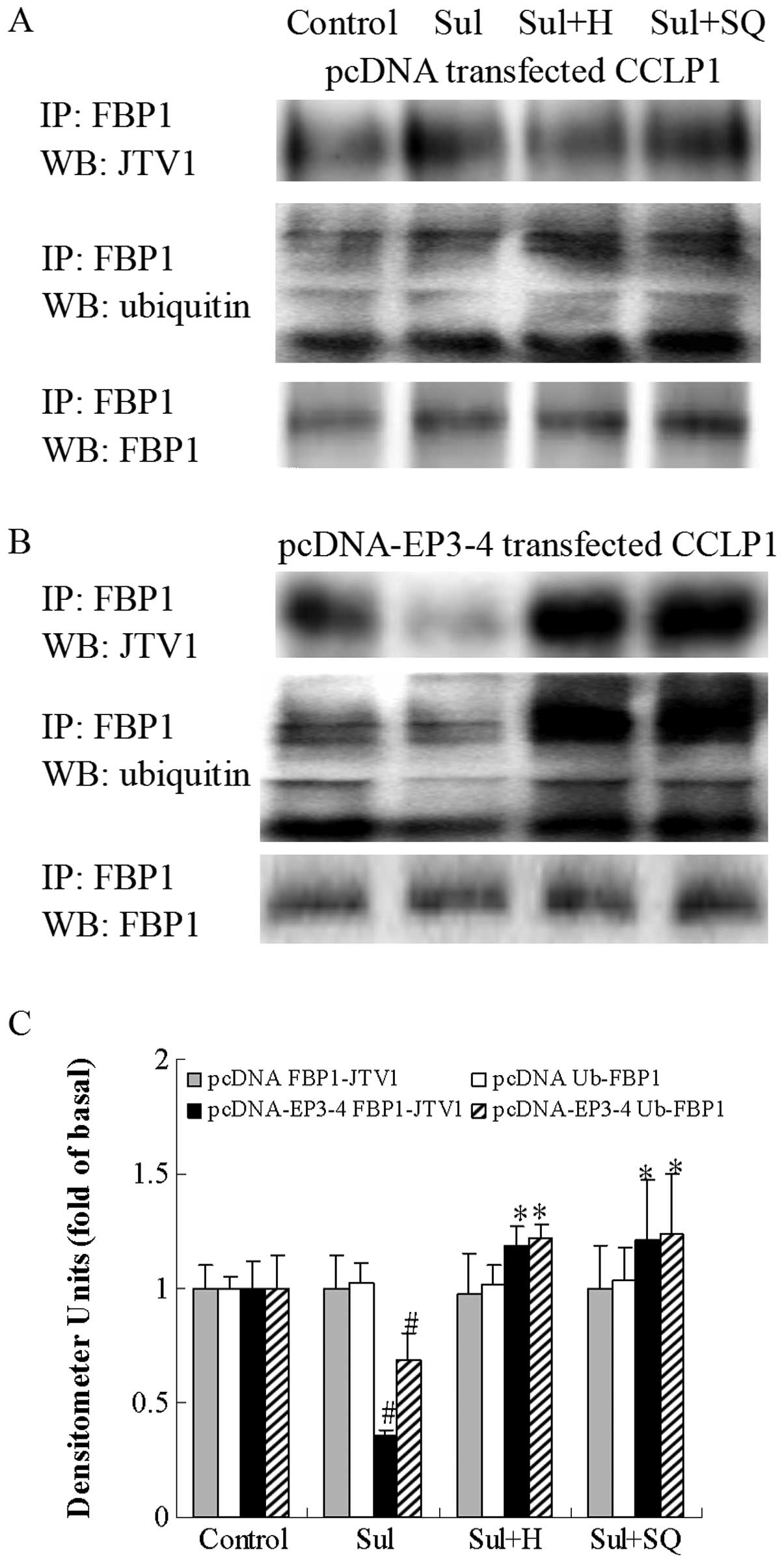 | Figure 14Effects of the PKA inhibitor, H89,
and the AC inhibitor, SQ22536, on the sulprostone-induced binding
of JTV1 with FBP1 and the ubiquitination of FBP1. (A) The empty
pcDNA3.1-transfected CCLP1 cells and (B) the EP3-4-transfected
CCLP1 cells were treated with 10 μM sulprostone in the
presence or absence of 10 μM of the PKA inhibitor, H89, and
50 μM of the AC inhibitor, SQ22536. The cell lysates were
obtained for immunoprecipitation with polyclonal antibody against
FBP1. The precipitated pellets were then separated by gel
electrophoresis on 12% Tris-glycine gels, followed by western blot
analysis with polyclonal antibodies against JTV1 and ubiquitin. (C)
Statistical plots of data from experiments are shown. The data
represent an average of 3 independent experiments.
#p<0.05 comparison of Sul treatment with control,
*p<0.05 comparison of Sul + H and Sul + SQ treatments
with Sul treatment. Sul, sulprostone; H, H89; SQ, SQ22536; pcDNA,
pcDNA3.1; IP, immunoprecipitationl; WB, western blot analysis. |
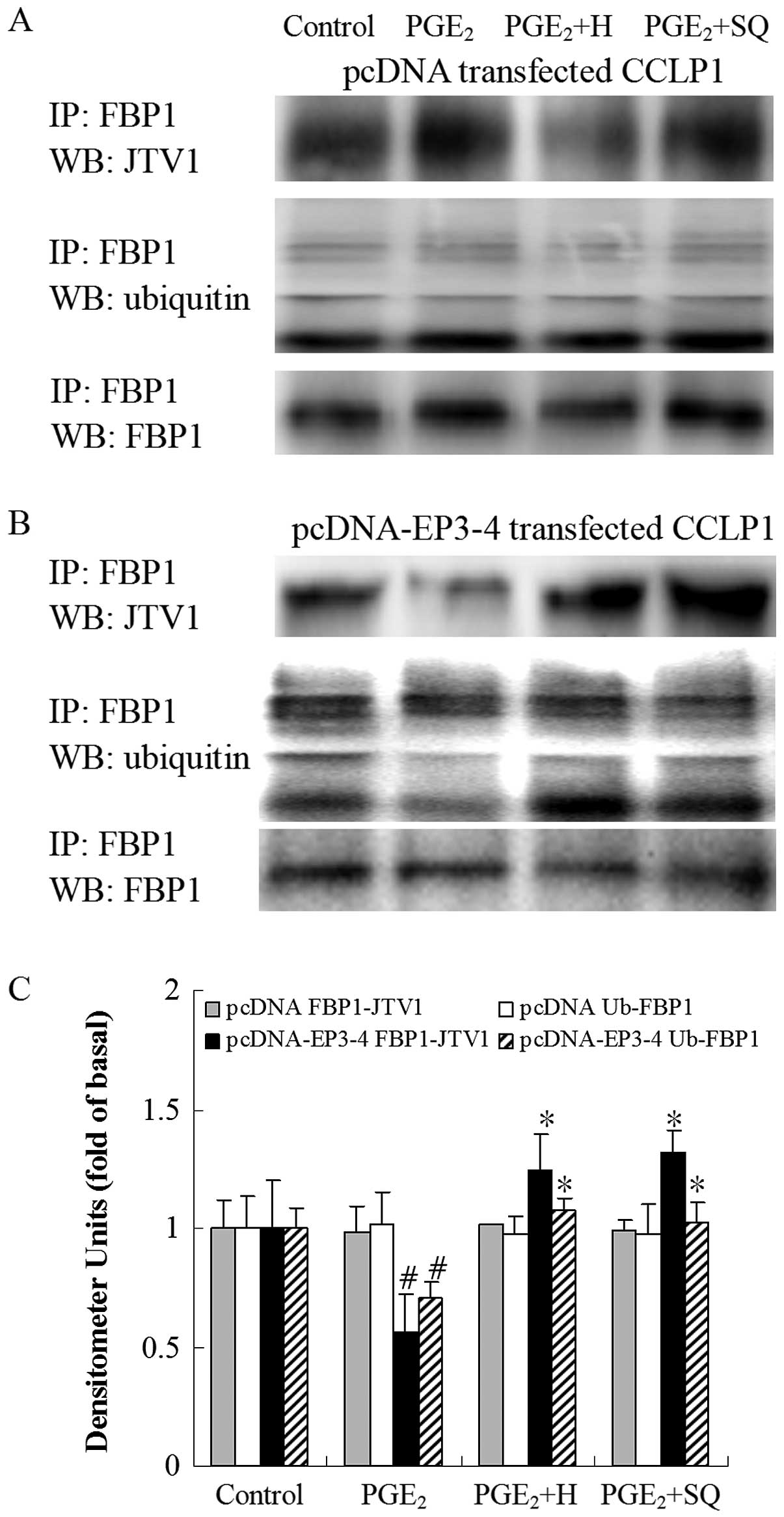 | Figure 15Effects of the PKA inhibitor, H89,
and the AC inhibitor, SQ22536, on the PGE2-mediated
binding of JTV1 with FBP1 and the ubiquitination of FBP1. (A) The
empty pcDNA3.1-transfected CCLP1 cells and (B) the
EP3-4-transfected CCLP1 cells were treated with 10 μM
PGE2 in the presence or absence of 10 μM of the
PKA inhibitor, H89, and 50 μM of the AC inhibitor, SQ22536.
The cell lysates were obtained for immunoprecipitation with
polyclonal antibody against FBP1. The precipitated pellets were
then separated by gel electrophoresis on 12% Tris-glycine gels,
followed by western blot analysis with polyclonal antibodies
against JTV1 and ubiquitin. (C) Statistical plots of data from
experiments are shown. The data represent an average of 3
independent experiments. #p<0.05 comparison of
PGE2 treatment with control. *p<0.05
comparison of PGE2 + H and PGE2 + SQ
treatments with PGE2 treatment. H, H89; SQ, SQ22536;
pcDNA, pcDNA3.1; IP, immunoprecipitationl; WB, western blot
analysis. |
Discussion
FBPs preferentially bind to single-stranded DNA and
to RNA sequences, and are known to act as transcription factors,
but have been postulated to regulate transcript stability (19). The FBPs are therefore likely to be
multifunctional. FBP1, as the family progenitor, is involved in
regulation of multiple physiological functions, such as gene
expression and tissue differentiation (26–28).
FBP1 binds through its 4 K-homology domains to FUSE of the
c-myc promoter, leading to the upregulation of
c-myc(29,30). Moreover, inhibition or loss of FBP1
function abrogates c-myc expression and arrests cellular
proliferation (26,31). FBP1 is developmentally regulated in
the mouse and chicken embryonic brain (32) and has been identified as a Parkin
substrate (33). FBP1 is present
in undifferentiated cells and is downregulated following
differentiation (26,34,35).
FBP1 is critical for cancer cell growth.
Our findings suggest that PGE2 and the
EP3 receptor agonist, sulprostone, upregulate the level of FBP1
protein and promote liver cancer cell growth. The EP3 receptor
inhibitor, L-798106, and EP3-4 siRNA suppressed the increased FBP1
protein expression induced by PGE2 and sulprostone.
These results demonstrate that PGE2 upregulates FBP1
protein via the EP3 receptor. More significantly, this study
provides important experimental evidence and mechanisms for
PGE2/EP3/FBP1 signaling pathways in liver cancer
cells.
According to previous reports, the EP3 receptor
couples to the Gi subunit and decreases cytoplasmic cAMP (12). However, in this study, we showed
that the Gi subunit inhibitor, PTX, exhibited no significant
effect. By contrast, the inhibitor of the Gs subunit pathway
suppressed the proliferation of liver cancer cells induced by EP3
receptor activation. Therefore, we hypothesized that the EP3
receptor might couple to the Gs subunit, not the Gi subunit. If the
EP3 receptor couples to the Gs subunit, it may increase cytoplasmic
cAMP production.
The observations that cytoplasmic cAMP was
increased by PGE2 and the EP3 agonist support our
hypothesis. These results indicate that the EP3 receptor may couple
to the Gs subunit and upregulate cAMP, which is not consistent with
previous data on the EP3 receptor. Moreover, studies supporting our
results of the EP3 receptor clarify that the EP3 receptor couples
to the Gs subunit, activates AC, increases cAMP, and promotes tumor
growth, angiogenesis and metastasis (36–38).
The G protein consists of α, β and γ subunits and is divided into
Gs, Gi and Gq, etc. Different types of G proteins mediate various
signaling pathways; the Gs protein activates AC, upregulates cAMP
production and induces PKA activation; the Gi protein inactivates
AC, downregulates cAMP production; and the Gq protein induces the
increase in Ca2+ and the activation of PKC (12,39).
In this study, the EP3 receptor activated by PGE2
coupled to the Gs protein, GDP of Gα subunit of Gs exchanged with
GTP followed by the dissociation of Gα and Gβγ, leading to AC
activation and an increased in cAMP production.
It has been indicated that cAMP activates PKA in
adipose cells (40). Our data
showed that the PKA inhibitor, H89, and the AC inhibitor, SQ22536,
suppressed the increase in FBP1 protein expression, as well as the
decrease in JTV1 protein expression, blocked the suppression of the
binding of JTV1 with FBP1 and the decreased ubiquitination of FBP1
induced by PGE2 and sulprostone, which demonstrates that
cAMP-PKA is involved in the signaling pathway mediated by the EP3
receptor.
The activation of PKA has been shown to decrease
TGF-β activity in osteoblasts (41). TGF-βs are multifunctional cytokines
that regulate cell proliferation, differentiation, apoptosis,
migration and extracellular matrix production (42–46).
The TGF-β receptor is composed of a heteromeric complex of
transmembrane serine/threonine kinases, the type I, II and III
receptors (TβRI, TβRII and TβRIII). Following ligand binding to
TβRII, TβRI is recruited to the complex, allowing for the
constitutively active TβRII kinase to transphosphorylate and
activate the TβRI kinase, which in turn phosphorylates Smad2 and
Smad3. Phosphorylated Smad2/3 then binds with Smad4 and
translocates to the nucleus, regulating gene transcription.
The present study shows that TGF-βs affect tumor
growth and function as tumor suppressors. Transgenic mice
overexpressing TGF-β can resist tumorigenesis (47,48),
the deletion of TβRII and the destruction of Smad3 and Smad4 genes
may enhance tumorigenesis (49–54),
and the Smad2, Smad4 and TβRII genes mutate or disappear in a
number of human tumors (42,44–46).
Since the TGF-β/Smad pathway can suppress the growth and metastasis
of tumors (55–57), the blocking of TGF-β/Smad
transduction maybe promote tumor growth (58,59).
Our data demonstrate the role of forskolin,
db-cAMP, PGE2 and sulprostone in TGF-β-induced Smad2
phosphorylation. These results illustrate that PGE2
facilitates cell growth by inhibiting TGF-β activity through the
EP3-Gs-cAMP-PKA pathway.
TβRIII is also termed β-glycan, lacks a distinct
intracellular signaling motif and may control the stability of the
ligand binding capacity of TβRII and has complex effects on signal
generation through TβRI. Perhaps PKA activation enhances TβRIII
promoter activity and increases the mRNA and protein expression of
TβRIII, inhibiting TGF-β activity (41); the mechanisms of the
PGE2 regulation of TβRIII through PKA require further
investigation.
It is worth noting that the suppression of cancer
cell differentiation induced by TGF-β is related with JTV1
(60). JTV1, another FBP partner,
also termed aminoacyl tRNA synthetase complex-interacting
multifunctional protein 2 (AIMP2/p38), is a structural subunit of a
multi-aminoacyl-tRNA synthetase (ARS) complex (61,62).
In response to signals, individual subunits of the ARS complex may
be released to participate in a variety of cellular processes,
including transcription (63),
translational silencing (64),
angiogenesis (65) and apoptosis
(61,66). For example, following DNA damage,
JTV1 is liberated from the ARS complex, phosphorylated in a
JNK2-dependent pathway and translocated into the nucleus where it
has been suggested to bind and sequester p53 from Mdm2-dependent
ubiquitination (66). JTV1 has
also been shown to be a substrate of the E3 ligase Parkin (67). The accumulation of JTV1 as a result
of Parkin mutation has been speculated to contribute to the
characteristic dopaminergic cell death observed in individuals with
Parkinson’s disease (67). The
increasing level of JTV1 protein may inhibit the proliferation of
cancer cells (62). TGF-β induces
the increase in JTV1 protein levels and promotes its translocation
to the nucleus during lung differentiation (60). In the nucleus, JTV1 binds with FBP1
for the ubiquitination and degradation of FBP1 (62). The knockdown of JTV1 increases the
levels of FBP1 and c-myc in fetal lungs and intestines
(63).
Treatment with forskolin, db-cAMP, PGE2
and sulprostone suppressed the increase in JTV1 protein levels, the
binding of JTV1 with FBP1 and the ubiquitination of FBP1 induced by
TGF-β. Thus, PGE2 downregulates JTV1, decreases the
binding of FBP1 with JTV1 and reduces the ubiquitination and
degradation of FBP1 by TGF-β in liver cancer cells.
In conclusion, in this study, a novel hypothesis is
established that the EP3 receptor activated by PGE2
couples to the Gs protein and activates cAMP-PKA, which inhibits
the activity of TGF-β. Moreover, the suppression of TGF-β reduces
the level of JTV1 protein, suppresses the binding of JTV1 with FBP1
and the ubiquitination of FBP1, leading to the upregulation of FBP1
protein, stimulating tumor cell growth. This study provides further
insight into the mechanisms by which PGE2 promotes liver
cancer cell growth. Our data may thus aid in the prevention and
treatment of malignant diseases by novel therapeutic
strategies.
Acknowledgements
This study was supported by the
National Natural Science Foundation of China (30871015, 81172003)
and a Project Funded by the Priority Academic Program Development
of Jiangsu Higher Education Institutions (PAPD).
References
|
1
|
LeBlanc MM, Giguère S, Lester GD, Brauer K
and Paccamonti DL: Relationship between infection, inflammation and
premature parturition in mares with experimentally induced
placentitis. Equine Vet J. (Suppl)41:8–14. 2012. View Article : Google Scholar
|
|
2
|
Menter DG and Dubois RN: Prostaglandins in
cancer cell adhesion, migration, and invasion. Int J Cell Biol.
2012:7234192012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Granado-Serrano AB, Martín MÁ, Bravo L,
Goya L and Ramos S: Quercetin attenuates TNF-induced inflammation
in hepatic cells by inhibiting the NF-κB pathway. Nutr Cancer.
4:588–598. 2012.PubMed/NCBI
|
|
4
|
Sasaki Y, Kamei D, Ishikawa Y, et al:
Microsomal prostaglandin E synthase-1 is involved in multiple steps
of colon carcinogenesis. Oncogene. 24:2943–2952. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thiel A, Narko K, Heinonen M, et al:
Inhibition of cyclooxygenase-2 causes regression of gastric
adenomas in trefoil factor 1 deficient mice. Int J Cancer.
131:1032–1041. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hoellen F, Kelling K, Dittmer C, Diedrich
K, Friedrich M and Thill M: Impact of cyclooxygenase-2 in breast
cancer. Anticancer Res. 12:4359–4367. 2011.PubMed/NCBI
|
|
7
|
Nadda N, Vaish V, Setia S and Sanyal SN:
Angiostatic role of the selective cyclooxygenase-2 inhibitor
etoricoxib (MK0663) in experimental lung cancer. Biomed
Pharmacother. 66:474–483. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Smith KA, Tong X, Abu-Yousif AO, Mikulec
CC, Gottardi CJ, Fischer SM and Pelling JC: UVB radiation-induced
β-catenin signaling is enhanced by COX-2 expression in
keratinocytes. Mol Carcinog. 51:734–745. 2012.
|
|
9
|
Phutthaphadoong S, Yamada Y, Hirata A, et
al: Chemopreventive effect of fermented brown rice and rice bran
(FBRA) on the inflammation-related colorectal carcinogenesis in
ApcMin/+ mice. Oncol Rep. 1:53–59.
2010.PubMed/NCBI
|
|
10
|
Bai XM, Jiang H, Ding JX, et al:
Prostaglandin E2 upregulates survivin expression via the EP1
receptor in hepatocellular carcinoma cells. Life Sci. 86:214–223.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang L, Jiang L, Sun QY, Peng T, Lou KX,
Liu NB and Leng J: Prostaglandin E2 enhances mitogenactivated
protein kinase/Erk pathway in human cholangiocarcinoma cells:
involvement of EP1 receptor, calcium and EGF receptors signaling.
Mol Cell Biochem. 305:19–26. 2007. View Article : Google Scholar
|
|
12
|
Wu T: Cyclooxygenase-2 and prostaglandin
signaling in cholangiocarcinoma. Biochim Biophys Acta.
1775:135–150. 2005.
|
|
13
|
Kotelevets L, Foudi N, Louedec L,
Couvelard A, Chastre E and Norel X: A new mRNA splice variant
coding for the human EP3-I receptor isoform. Prostaglandins Leukot
Essent Fatty Acids. 77:195–201. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Regan JW, Bailey TJ, Donello JE, et al:
Molecular cloning and expression of human EP3 receptors: evidence
of three variants with differing carboxyl termini. Br J Pharmacol.
112:377–385. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schmid A, Thierauch KH, Schleuning WD and
Dinter H: Splice variants of the human EP3 receptor for
prostaglandin E2. Eur J Biochem. 228:23–30. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kotani M, Tanaka I, Ogawa Y, et al:
Molecular cloning and expression of multiple isoforms of human
prostaglandin E receptor EP3 subtype generated by alternative
messenger RNA splicing: multiple second messenger systems and
tissue-specific distributions. Mol Pharmacol. 48:869–879. 1995.
|
|
17
|
Kotani M, Tanaka I, Ogawa Y, et al:
Multiple signal transduction pathways through two prostaglandin E
receptor EP3 subtype isoforms expressed in human uterus. J Clin
Endocrinol Metab. 85:4315–4322. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Israel DD and Regan JW: EP(3) prostanoid
receptor isoforms utilize distinct mechanisms to regulate ERK 1/2
activation. Biochim Biophys Acta. 4:238–245. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rydziel S, Delany AM and Canalis E:
AU-rich elements in the collagenase 3 mRNA mediate stabilization of
the transcript by cortisol in osteoblasts. J Biol Chem.
279:5397–5404. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Malz M, Weber A, Singer S, et al:
Overexpression of far upstream element binding proteins: a
mechanism regulating proliferation and migration in liver cancer
cells. Hepatology. 50:1130–1139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Weber A, Kristiansen I, Johannsen M, et
al: The FUSE binding proteins FBP1 and FBP3 are potential c-myc
regulators in renal, but not in prostate and bladder cancer. BMC
Cancer. 8:3692008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chung HJ, Liu J, Dundr M, Nie Z, Sanford S
and Levens D: FBPs are calibrated molecular tools to adjust gene
expression. Mol Cell Biol. 26:6584–6597. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rabenhorst U, Beinoraviciute-Kellner R,
Brezniceanu ML, et al: Overexpression of the far upstream element
binding protein 1 in hepatocellular carcinoma is required for tumor
growth. Hepatology. 4:1121–1129. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Andersen SS: Spindle assembly and the art
of regulating micro-tubule dynamics by MAPs and Stathmin/Op18.
Trends Cell Biol. 10:261–267. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Singer S, Malz M, Herpel E, Warth A,
Bissinger M, Keith M, Muley T, Meister M, Hoffmann H, Penzel R,
Gdynia G, Ehemann V, Schnabel PA, Kuner R, Huber P, Schirmacher P
and Breuhahn K: Coordinated expression of stathmin family members
by far upstream sequence element-binding protein-1 increases
motility in non-small cell lung cancer. Cancer Res. 6:2234–2243.
2009. View Article : Google Scholar
|
|
26
|
He LS, Liu JH, Collins I, et al: Loss of
FBP function arrests cellular proliferation and extinguishes c-myc
expression. EMBO J. 5:1034–1044. 2000.PubMed/NCBI
|
|
27
|
Liu J, Kouzine F, Nie Z, et al: The
FUSE/FBP/FIR/TFIIH system is a molecular machine programming a
pulse of c-myc expression. EMBO J. 10:2119–2130. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Avigan MI, Strober B and Levens D: A far
upstream element stimulates c-myc expression in undifferentiated
leukemia cells. J Biol Chem. 30:18538–18545. 1990.PubMed/NCBI
|
|
29
|
Hsiao HH, Nath A, Lin CY, et al:
Quantitative characterization of the interactions among c-myc
transcriptional regulators FUSE, FBP, and FIR. Biochemistry.
49:4620–4634. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wierstra I and Alves J: The c-myc
promoter: still MysterY and challenge. Adv Cancer Res. 99:113–333.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jang M, Park BC, Kang S, et al: Far
upstream element-binding protein-1, a novel caspase ubstrate, acts
as a cross-talker between apoptosis and the c-myc oncogene.
Oncogene. 28:1529–1536. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang X, Avigan M and Norgren RB:
FUSE-binding protein is developmentally regulated and is highly
expressed in mouse and chicken embryonic brain. Neurosci Lett.
252:191–194. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ko HS, Kim SW, Sriram SR, Dawson VL and
Dawson TM: Identification of far upstream element-binding protein-1
as an authentic Parkin substrate. J Biol Chem. 281:16193–16196.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Duncan RD, Bazar L, Michelotti G, et al: A
sequence-specific, single-strand binding protein activates the far
upstream element of c-myc and defines a new DNA-binding motif.
Genes. 8:465–480. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bazar L, Harris V, Sunitha I, Hartmann D
and Avigan MI: A transactivator of c-myc is coordinately regulated
with the protooncogene during cellular growth. Oncogene.
10:2229–2238. 1995.PubMed/NCBI
|
|
36
|
Yamaki T, Endoh K, Miyahara M, et al:
Prostaglandin E2 activates Src signaling in lung adenocarcinoma
cell via EP3. Cancer Lett. 214:115–120. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yutaka S, Mami T, Nobuo T, et al:
Prostaglandin E receptor EP3 deficiency modifies tumor outcome in
mouse two-stage skin carcinogenesis. Carcinogenesis. 26:2116–2122.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Finetti F, Solito R, Morbidelli L,
Giachetti A, Ziche M and Donnini S: Prostaglandin E2
regulates angiogenesis via activation of fibroblast growth factor
receptor-1. J Biol Chem. 283:2139–2146. 2008.
|
|
39
|
Gutierrez DV, Mark MD, Masseck O, et al:
Optogenetic control of motor coordination by
Gi/o protein-coupled vertebrate rhodopsin in
cerebellar Purkinje cells. J Biol Chem. 286:25848–25858. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Deng J, Liu S, Zou L, Xu C, Geng B and Xu
G: Lipolysis response to endoplasmic reticulum stress in adipose
cells. J Biol Chem. 287:6240–6249. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
McCarthy TL, Pham TH, Knoll BI and
Centrella M: Prostaglandin E2 increases transforming
growth factor-β type III receptor expression through CCAAT
enhancer-binding protein δ in osteoblasts. Mol Endocrinol.
11:2713–2724. 2007.PubMed/NCBI
|
|
42
|
Massague J, Blain SW and Lo RS: TGFbeta
signaling in growth control, cancer, and heritable disorders. Cell.
103:295–309. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shi Y and Massague J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Akhurst RJ and Derynck R: TGF-beta
signaling in cancer - a double-edged sword. Trends Cell Biol.
11:S44–S51. 2001.PubMed/NCBI
|
|
45
|
Derynck R, Akhurst RJ and Balmain A:
TGF-beta signaling in tumor suppression and cancer progression. Nat
Genet. 29:117–129. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wakefield LM and Roberts AB: Learning
together: clinical skills teaching for medical and nursing
students. Curr Opin Genet. 12:22–29. 2002.
|
|
47
|
Pierce DF Jr, Johnson MD, et al:
Inhibition of mammary duct development but not alveolar outgrowth
during pregnancy in transgenic mice expressing active TGF-beta 1.
Genes Dev. 7:2308–2317. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cui W, Fowlis DJ, Bryson S, et al:
TGFbeta1 inhibits the formation of benign skin tumors, but enhances
progression to invasive spindle carcinomas in transgenic mice.
Cell. 86:531–542. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bottinger EP, Jakubczak JL, Haines DC,
Bagnall K and Wakefield LM: Transgenic mice overexpressing a
dominant-negative mutant type II transforming growth factor β
receptor show enhanced tumorigenesis in the mammary gland and lung
in response to the carcinogen 7,12-dimethylbenz-[a]-anthracene.
Cancer Res. 57:5564–5570. 1997.
|
|
50
|
Gorska AE, Joseph H, Derynck R, Moses HL
and Serra R: Dominant-negative interference of the transforming
growth factor beta type II receptor in mammary gland epithelium
results in alveolar hyperplasia and differentiation in virgin mice.
Cell Growth Differ. 9:229–238. 1998.
|
|
51
|
Engle SJ, Hoying JB, Boivin GP, Ormsby I,
Gartside PS and Doetschman T: Transforming growth factor beta1
suppresses nonmetastatic colon cancer at an early stage of
tumorigenesis. Cancer Res. 59:3379–3386. 1999.PubMed/NCBI
|
|
52
|
Zhu Y, Richardson JA, Parada LF and Graff
JM: Smad3 mutant mice develop metastatic colorectal cancer. Cell.
94:703–714. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xu X, Brodie SG, Yang X, et al: Haploid
loss of the tumor suppressor Smad4/Dpc4 initiates gastric polyposis
and cancer in mice. Oncogene. 19:1868–1874. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tang B, Bottinger EP, Jakowlew SB, et al:
Transforming growth factor-beta1 is a new form of tumor suppressor
with true haploid insufficiency. Nat Med. 4:802–807. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Han C, Demetris AJ, Liu Y, Shelhamer JH
and Wu T: Transforming growth factor-β (TGF-β) activates cytosolic
phospholipase A2α (cPLA2α)-mediated prostaglandin
E2 (PGE)2/EP1 and peroxisome proliferator-activated
receptor-γ (PPAR-γ)/Smad signaling pathways in human liver cancer
cells. A novel mechanism for subversion of TGF-β-induced
mitoinhibition. J Biol Chem. 43:44344–44354. 2004.
|
|
56
|
Markowitz SD, Itzkowitz SH and Berger BM:
The effectiveness of colonoscopy in reducing mortality from
colorectal cancer. Ann Intern Med. 150:816–817. 2009. View Article : Google Scholar
|
|
57
|
Chowdhury S, Howell GM, Rajput A, et al:
Identification of a novel TGFβ/PKA signaling transduceome in
mediating control of cell survival and metastasis in colon cancer.
PLoS One. 5:e193352011.
|
|
58
|
Liu X, Sun S and Ostrom R: Fibrotic lung
fibroblasts show blunted inhibition by cAMP due to deficient cAMP
response element-binding protein phosphorylation. J Pharmacol Exp
Ther. 2:678–687. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Schiller M, Verrecchia F and Mauviel A:
Cyclic adenosine 3′,5′,-monophosphate-elevating agents inhibit
transforming growth factor-beta-induced SMAD¾-dependent
transcription via a protein kinase A-dependent mechanism. Oncogene.
22:8881–8890. 2003.
|
|
60
|
Kim MJ, Park BJ, Kang YS, Kim HJ, Park JH
and Kang JW: Downregulation of FUSE-binding protein and c-myc by
tRNA synthetase cofactor p38 is required for lung cell
differentiation. Nat Genet. 34:330–336. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kim JY, Kang YS, Lee JW, Kim HJ, Ahn YH,
Park H, Ko YG and Kim S: p38 is essential for the assembly and
stability of macromolecular tRNA synthetase complex: implications
for its physiological significance. Proc Natl Acad Sci USA.
99:7912–7916. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Liu J, Chung HJ, Vogt M, et al: JTV1
co-activates FBP to induce USP29 transcription and stabilize p53 in
response to oxidative stress. EMBO J. 30:846–858. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sampath P, Mazumder B, Seshadri V, et al:
Noncanonical function of glutamyl-prolyl-tRNA synthetase:
genespecific silencing of translation. Cell. 119:195–208. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Park SG, Kang YS, Ahn YH, et al:
Dose-dependent biphasic activity of tRNA synthetase-associating
factor, p43, in angiogenesis. J Biol Chem. 277:45243–45248. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Han JM, Park BJ, Park SG, et al:
AIMP2/p38, the scaffold for the multi-tRNA synthetase complex,
responds to genotoxic stresses via p53. Proc Natl Acad Sci USA.
105:11206–11211. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Corti O, Hampe C, Koutnikova H, et al: p38
subunit of the aminoacyl-tRNA synthetase complex is a Parkin
substrate: linking protein biosynthesis and neurodegeneration. Hum
Mol Genet. 12:1427–1437. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ko HS, Coelln R, Sriram SR, et al:
Accumulation of the authentic parkin substrate aminoacyl-tRNA
synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell
death. J Neurosci. 25:7968–7978. 2005. View Article : Google Scholar : PubMed/NCBI
|