Introduction
Liver cancer ranks fifth in frequency in the world
and is the third most common cause of lethal cancer (1). Liver cancer consists of
hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma
(ICC), with HCC as the most common. Regarding HCC therapy,
hepatectomy, percutaneous local therapy and transcatheter arterial
embolization (TAE) are common, but the recurrence rate with
conventional therapies for advanced HCC patients is still high
(2). Therefore, developing a novel
curative therapy or an effective adjuvant therapy for HCC is
important.
Recently, immunotherapy, which consists of a peptide
vaccine, protein vaccine, or DNA vaccine, has become a potentially
promising option for HCC (3,4).
Many tumor antigen-derived peptides recognized by cytotoxic
T-lymphocyte (CTL) have been identified (5). However, to date, vaccine therapy
using these peptides has not proven adequate antitumor efficacy in
clinical trials for advanced HCC patients (6–8).
In HCC, glypican-3 (GPC3) is overexpressed and is
not expressed in normal tissues except for the placenta and
embryonic liver (9). Hence, GPC3
is a novel target molecule in HCC patients. GPC3 is a member of the
heparan sulfate proteoglycan family and the glypican family
regulates cell growth and division through Wnt signaling,
Hedgehogs, fibroblast growth factors and bone morphogenetic
proteins (10–12). We previously identified
HLA-A*24:02-restricted GPC3298–306
(EYILSLEEL) and HLA-A*02:01-restricted GPC3144–152
(FVGEFFTDV) peptides and showed that both peptides can induce
GPC3-specific CTLs without an autoimmune response (13,14).
Clinical trials of a GPC3-derived peptide vaccine for HCC patients
are currently in progress. The phase I clinical trial of a
GPC3-derived peptide vaccine for advanced HCC showed safety as well
as immunological evidence and potential for improving overall
survival (15–17). The phase I clinical trial suggested
that the GPC3-derived peptide vaccine could be an attractive
approach for treatment of HCC, however, the effect of tumor
reduction was limited. Therefore, further studies are needed to
enhance the effect of GPC3-targeted immunotherapy and to establish
a GPC3-specific CTL-inducible mouse model. We previously conducted
a preclinical study of the GPC3-derived peptide vaccine using
HLA-A2.1 transgenic mice (18).
The treatment model experiment using HLA transgenic mice is
limited.
Mice with the C57BL/6 (B6) background have been
reported to spontaneously develop liver cancer (19,20).
Recently, the NASH mouse model (named STAM mice C57BL/6N-NASH),
which had a B6 background and spontaneously developed liver cancer,
was exploited by Stelic Institute & Co. In this mouse model,
the cancer incidence rate is high and cancer incident time is
short, thus, STAM mice C57BL/6N-NASH is an attractive model for
studying GPC3-targeted therapy for HCC. Therefore, identification
of a mouse major histocompatibility complex (MHC) class I epitope
peptide to induce GPC3-specific CTL was needed for establishment of
the appropriate mouse model.
Strategies to identify epitope peptides have
previously been reported (21–24).
A summary of our strategy follows. First, peptides binding MHC
class I epitope were predicted from antigen amino acid sequences
in silico by prediction software and the ability of the
predicted peptides to bind MHC class I was confirmed in
vitro by a binding assay. Then, the immunogenic potential of
the predicted peptides was examined by in vivo immunization
or in vitro stimulation. Lastly, whether peptides that have
immunogenic potential are presented by cells endogenously
expressing the antigen was confirmed. In summary, we identified
peptides with immunogenic potential that were presented by cells
endogenously expressing the antigen. We attempted to identify
H2-Kb or H2-Db restricted, GPC3-derived CTL
epitope peptides in C57BL/6 mice based on the above strategy.
Materials and methods
Mice
C57BL/6 (B6) mice were purchased from Charles River
Laboratories Japan, Inc. and STAM mice C57BL/6N-NASH were a gift
from this company. Mice were maintained under the institutional
guidelines set by the Animal Research Committee of the National
Cancer Center Hospital East. Mice were housed in specific
pathogen-free (SPF) conditions with a 12-h light cycle and food and
water at ad libitum. Six to eight-week-old female B6 mice
were used in all experiments and STAM mice C57BL/6N-NASH were
provided with a very high-fat rodent diet (rodent diet with 60%
kcal% fat, Research Diet Inc.). All animal procedures were
performed according to the guidelines for Animal Research Committee
of the National Cancer Center, Japan.
Cell lines and transfection
B6 thymoma RMA and RMA-S cell lines, which have
H2-Kb and -Db as MHC class I epitopes, were
maintained in our laboratory. RMA-S is an antigen
processing-defective cell line and the cells cannot present
endogenous antigens with MHC class I epitopes (25). To obtain RMA transiently expressing
murine GPC3 (RMA-GPC3-puro), RMA (GPC3-negative) was transfected
with pCAGGS-mGPC3-internal ribosomal entry site
(IRES)-puromycin-resistant (puro-R) using Lipofectamine 2000
reagent (Invitrogen Corp., Carlsbad, CA, USA) according to the
manufacturer’s protocols. As negative control, RMA, which was
transfected with pCAGGS-IRES-puro-R in a similar way, was named
RMA-puro. Expression of murine GPC3 (mGPC3) in RMA-GPC3-puro or
RMA-puro was confirmed by reverse transcription polymerase chain
reaction (RT-PCR). All cells were cultured in RPMI-1640 (Gibco,
USA) supplemented with 10% fetal bovine serum (FBS) (Gibco).
RT-PCR
Total ribonucleic acid was isolated from
RMA-GPC3-puro or RMA-puro homogenized with the TRIzol Reagent (Life
Technologies, Inc., Rockville, MD, USA) according to the
manufacturer’s protocols. The first-strand complementary
deoxyribonucleic acid (cDNA) was synthesized with a
PrimeScript® II 1st strand cDNA Synthesis kit (Takara
Bio Inc., Japan), then mGPC3 was amplified using a Takara PCR
Amplification kit (Takara Bio Inc.). The amplification protocol was
as follows: 150 sec at 94°C for initial denaturation, 35
amplification cycles at 58°C for 40 sec and 72°C for 40 sec,
followed by a final extension at 72°C for 5 min. The primer
sequences for mGPC3 were as follows: sense, 5′-ACGGGATGGTGAAA
GTGAAGA-3′ and antisense, 5′-GAAAGAGAAAAGAGGGA AACA-3′. The primer
sequences for β-actin were as follows: sense,
5′-GAGCAATGATCTTGATCTTCAT-3′ and antisense,
5′-TCCATCATGAACTGTGACGT-3′. PCR products were visualized by
ethidium bromide staining after separation on a 1% agarose gel.
After normalization using β-actin messenger ribonucleic acid (mRNA)
as a control, we compared the expression of mGPC3 mRNA.
Generation of bone marrow-derived
dendritic cells (BM-DCs) from BM cells
BM cells (4×106) from B6 mice were
cultured in RPMI-1640 containing FBS (10%), 2-mercaptoethanol
(2-ME, 50 μM) and murine granulocyte macrophage
colony-stimulating factor (mGM-CSF, 20 ng/ml) for 1 week.
Peptides
Eleven types of 9- to 10-mer peptides predicted to
bind with H2-Kb or H2-Db were selected from
mGPC3 amino acid sequences (accession code AAH36126) based on the
binding score as calculated by BIMAS software (BioInformatics and
Molecular Analysis Section, Center for Information Technology, NIH,
Bethesda, MD, USA) and 11 synthetic peptides (custom ordered) were
purchased from Scrum Inc. (Tables
I and II). The 11 amino acid
sequences were as follows: mGPC3-1, AMFKNNYPSL; mGPC3-2,
SLFPVIYTQM; mGPC3-3, LFPVIYTQM; mGPC3-4, KSFINFYSAL; mGPC3-5,
LTARLNMEQL; mGPC3-6, LGSDINVDDM; mGPC3-7, QYVQKNGGKL; mGPC3-8,
YVQKNGGKL; mGPC3-9, DTLCWNGQEL; mGPC3-10, RNGMKNQFNL; mGPC3-11,
MKNQFNLHEL. Each peptide was dissolved in dimethyl sulfoxide (DMSO)
(Wako Pure Chemical Industries, Japan) and each peptide’s density
was 10 mg/ml.
 | Table ISynthetic peptides predicted to bind
with H2-Kb. |
Table I
Synthetic peptides predicted to bind
with H2-Kb.
| Peptide sequence
(position) | Binding
scorea |
|---|
| mGPC3-1 | AMFKNNYPSL
(127–136) | 52.8 |
| mGPC3-2 | SLFPVIYTQM
(172–181) | 44 |
| mGPC3-3 | LFPVIYTQM
(173–181) | 66 |
| mGPC3-4 | KSFINFYSAL
(395–404) | 40 |
| Table IISynthetic peptides predicted to bind
with H2-Db. |
Table II
Synthetic peptides predicted to bind
with H2-Db.
| Peptide sequence
(position) | Binding
scorea |
|---|
| mGPC3-5 | LTARLNMEQL
(82–91) | 200 |
| mGPC3-1 | AMFKNNYPSL
(127–136) | 343.2 |
| mGPC3-6 | LGSDINVDDM
(156–165) | 260 |
| mGPC3-7 | QYVQKNGGKL
(331–340) | 720 |
| mGPC3-8 | YVQKNGGKL
(332–340) | 240 |
| mGPC3-9 | DTLCWNGQEL
(418–127) | 600 |
| mGPC3-10 | RNGMKNQFNL
(437–446) | 200 |
| mGPC3-11 | MKNQFNLHEL
(440–449) | 288 |
H2-Kb or H2-Db
binding assay
To evaluate the binding affinity of the predicted
peptides to H2-Kb or H2-Db molecules, an
in vitro cellular binding assay was performed as previously
reported (23,26). Briefly, after incubation of RMA-S
cells in culture medium at 26°C overnight, cells (1×106)
were washed with PBS and suspended in 100 μl
Opti-MEM® (Invitrogen) with or without 10 μg
peptide, followed by incubation at 26°C for 3 h and then at 37°C
for 3 h. After washing with PBS, H2-Kb or
H2-Db expression was measured with a BD FACSCanto™ II
flow cytometer (BD) using FITC-conjugated H2-Kb
(BioLegend Inc., AF6-88.5) or H2-Db (BioLegend Inc.,
KH95) specific monoclonal antibody and mean fluorescence intensity
(MFI) was recorded. Percent MFI increase was calculated as follows:
percent MFI increase = (MFI with the given peptide - MFI without
peptide)/(MFI without peptide) × 100.
Vaccination
The mixed peptide vaccine per mouse consisted of 5
μl mGPC3-1 to mGPC3-11 solution, 55 μl sodium
bicarbonate solution and 110 μl incomplete Freund’s adjuvant
(IFA). Single peptide vaccine per mouse consisted of 5 μl
peptide, 45 μl sodium bicarbonate solution and 50 μl
IFA. Each vaccine solution was emulsified. The mice were immunized
by intradermal injection at the base of the tail every 7 days for a
total of two vaccinations. Similarly, STAM mice C57BL/6N-NASH were
immunized seven times with the mGPC3-1 peptide vaccine.
Restimulation of splenocytes obtained
from immunized mice
Seven days after the last immunization, splenocytes
were collected and cluster of differentiation 8 (CD8) positive
splenocytes were isolated by positive selection with anti-CD8
microbeads (Miltenyi Biotec) according to the manufacturer’s
protocol. CD8-positive splenocytes were cocultured with BM-DCs
pulsed with each peptide as previously described (13). Seven days after coculture, the
detection of antigen-specific T cells producing interferon (IFN)-γ
was performed using the BD ELISPOT kit (BD Bioscience, San Jose,
CA, USA) according to the manufacturer’s protocols.
Establishment of GPC3-1-specific CTL
line
The GPC3-1-specific CTL line was established as
previously described (27).
Splenocytes (1×104) derived from B6 mice immunized with
the GPC3-1 peptide vaccine were cocultured with B6-derived and
irradiated (35 Gy) splenocytes (5×104) in RPMI-1640
contained with FBS (10%), sodium pyruvate (1 mM, Gibco), MEM
non-essential amino acid solution (1X, Gibco) and 2-ME (50
μM). Seven days later, recombinant interleukin-2 (rIL-2, 50
U/ml, Nipro, Osaka, Japan) was added to the culture medium.
IFN-γ enzyme-linked immunospot (ELISPOT)
analysis
IFN-γ ELISPOT assay was performed according to the
manufacturer’s protocols. Briefly, restimulated CD8-positive
splenocytes (5×104) as target cells were added to the
plate and then BM-DCs (5×104) pulsed with each peptide
(10 μg/ml) as effector cells or non-pulsed BM-DCs
(5×104) as control and target cells were added to the
plate, which was then incubated for 20 h at 37°C, 5%
CO2. Using the GPC3-1-reactive CTL line
(1×105) as effector cells, RMA-S (5×104)
pulsed with each peptide (10 μg/ml) as target cells and
non-pulsed RMA-S as control and target cells (5×104),
the plate was incubated for 20 h at 37°C, 5% CO2. Using
the mGPC3-1-reactive CTL line (1×105) as effector cells,
RMA-GPC3-puro as target cells (5×105) and RMA-puro
(5×105) as control and target cells, the plate was
incubated for 48 h at 37°C, 5% CO2. The number of spots
was automatically counted using the Eliphoto system (Minerva Tech,
Tokyo, Japan).
Cytotoxicity assay
Cytotoxic activity against target cells was analyzed
using the Terascan VPC system (Minerva Tech) as previously
described (28). Target cells were
incubated with calcein AM (Dojindo, Kumamoto, Japan) solution for
30 min at 37°C and labeled. Then the labeled cells were incubated
with effector cells for 4 h. Fluorescence intensity was measured
before and after the culture and specific cytotoxic activity was
evaluated using the following formula: % cytotoxicity = {1-
[(average fluorescence of the sample wells - average fluorescence
of the maximal release control wells) - (average fluorescence of
the minimal release control wells - average fluorescence of the
maximal release control wells)]} × 100%.
Statistical analysis
Statistical analyses were performed with a
Mann-Whitney U test (n=3). Significant differences were defined as
*p<0.05 or R2 >0.5.
Results
Evaluation of selected peptide-binding
affinity to H2-Kb or H2-Db
The selected 11 peptides derived from mGPC3 by the
BIMAS software were evaluated by an in vitro binding assay
to determine each peptide’s binding affinity to H2-Kb or
H2-Db. The peptide with the highest binding affinity for
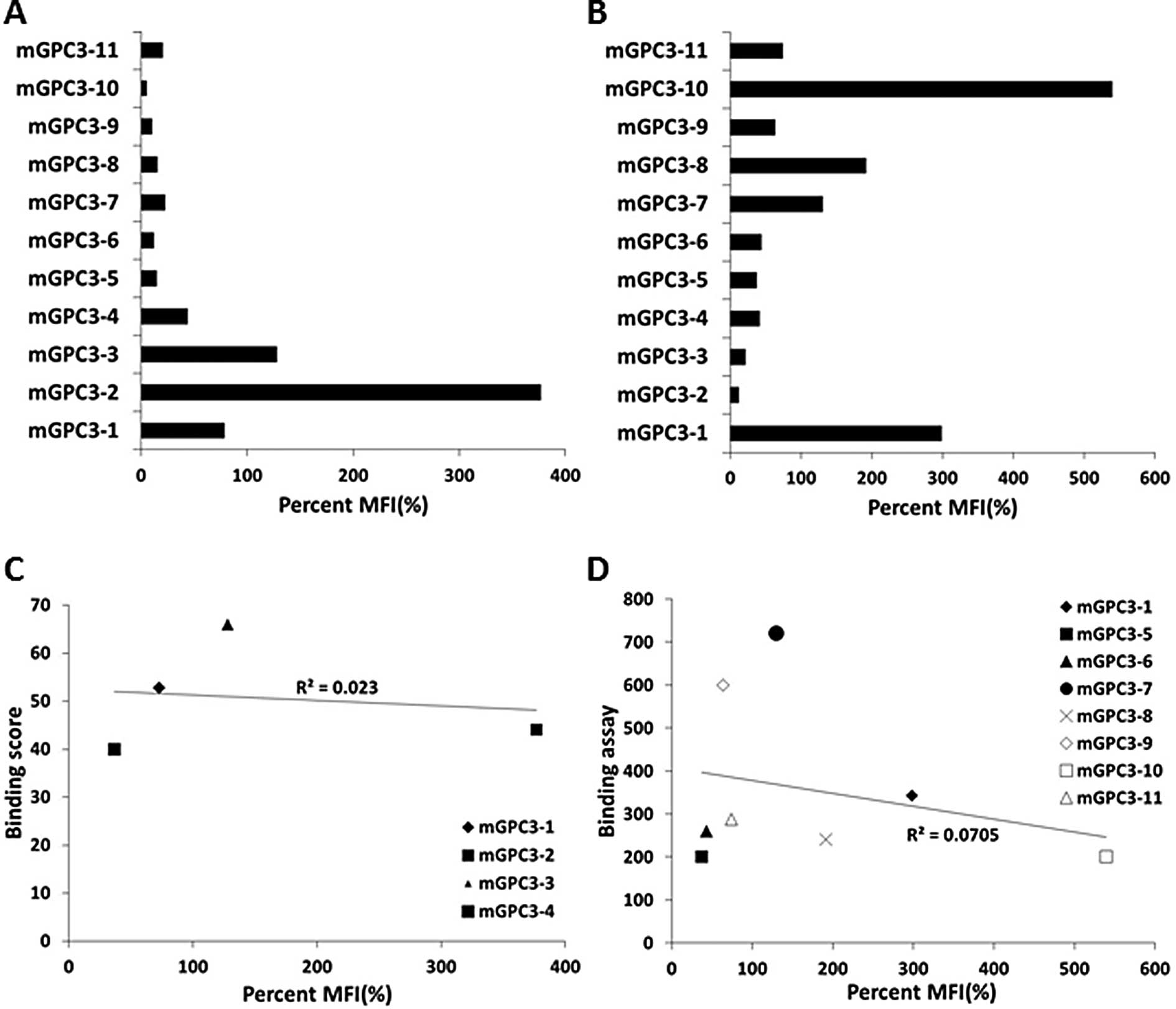
H2-Kb was mGPC3-2 (percent MFI, 376.6%), followed by the
mGPC3-3 peptide (128.0%) and the mGPC3-1 peptide (72.7%) (Fig. 1A). That for H2-Db was
mGPC3-10 peptide (539.1%) followed by the mGPC3-1 peptide (298.2%)
and the mGPC3-8 peptide (191.1%) (Fig.
1B). These results show that all 11 peptides could bind
H2-Kb or H2-Db, although the binding score
calculated by the BIMAS software did not correlate with the actual
binding affinity (Fig. 1C and
D).
Induction of CTL response against
mGPC3-derived peptides in B6 mice
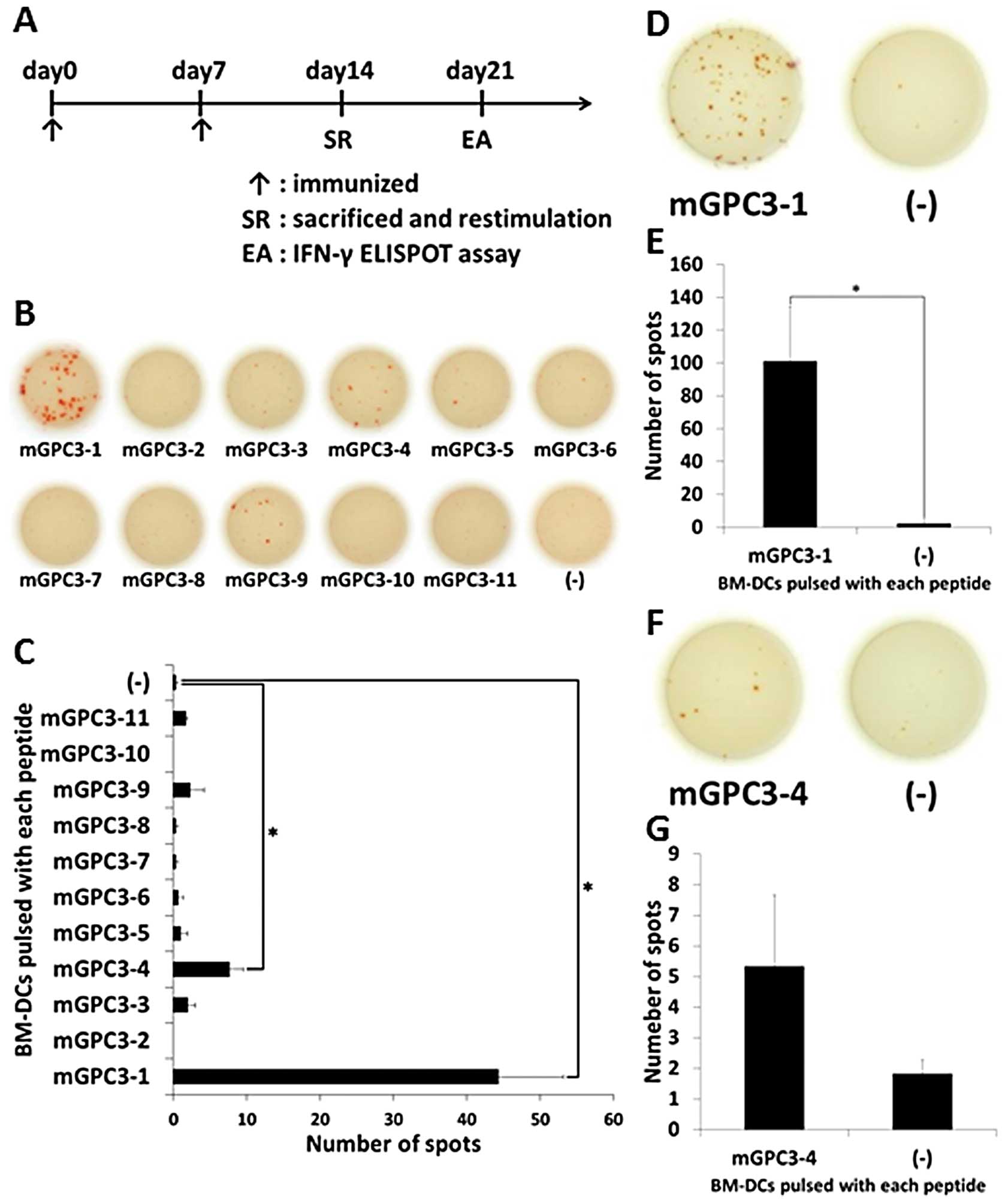
The vaccine schedule was performed as follows
(Fig. 2A): At days 0 and 7,
peptide vaccine was given. At day 14, primed mice were sacrificed
and CD8-positive splenocytes were collected. CD8-positive
splenocytes were restimulated with BM-DCs pulsed with each peptide.
At day 21, the peptide’s immunogenic potential was evaluated by
IFN-γ ELISPOT assay.
The mixed peptide vaccination was performed to
evaluate immunogenic potential of the 11 peptides and IFN-γ ELISPOT
assays were performed using BM-DCs pulsed with each peptide and
non-pulsed BM-DCs as target cells. The CD8-positive splenocytes
from mice primed with the mixed vaccine released more IFN-γ to
BM-DCs pulsed with mGPC3-1 peptide (average number of spots,
44.3±15.3) and mGPC3-4 peptide (average number of spots, 7.6±3.2)
than to non-pulsed BM-DCs (average number of spots, 0.3±0.5). These
results suggest that the mGPC3-1 and mGPC3-4 peptides had
immunogenic potential and were able to induce peptide-specific CTLs
in B6 mice primed by the mixed vaccine system (Fig. 2B and C).
Next, to confirm whether the peptides are
CTL-inducible peptides, a single peptide vaccine was given and
IFN-γ ELISPOT assays were performed using BM-DCs pulsed with either
peptide and non-pulsed BM-DCs as target cells. The CD8-positive
cells from mice immunized with mGPC3-1 peptide released more IFN-γ
to BM-DCs pulsed with mGPC3-1 peptide (average number of spots,
101.0±33.2) than to non-pulsed BM-DCs (average number of spots,
2.1±3.7) (Fig. 2D and E). The
CD8-positive cells from mice immunized with mGPC3-4 peptide
released more IFN-γ to BM-DCs pulsed with mGPC3-4 peptide (average
number of spots, 5.3±4.0) than to non-pulsed BM-DCs (average number
of spots, 1.8±0.7), but no significant differences were observed
(Fig. 2F and G). These results
suggest that mGPC3-1 peptide is more efficient for inducing CTLs
than the mGPC3-4 peptide in a single peptide vaccine system.
Taken together, the above results suggest that
mGPC3-1 peptide is the most efficient peptide for inducing CTLs
among the 11 peptides.
mGPC3-1 peptide-specific CTL line
recognition of target cells endogenously expressing mGPC3
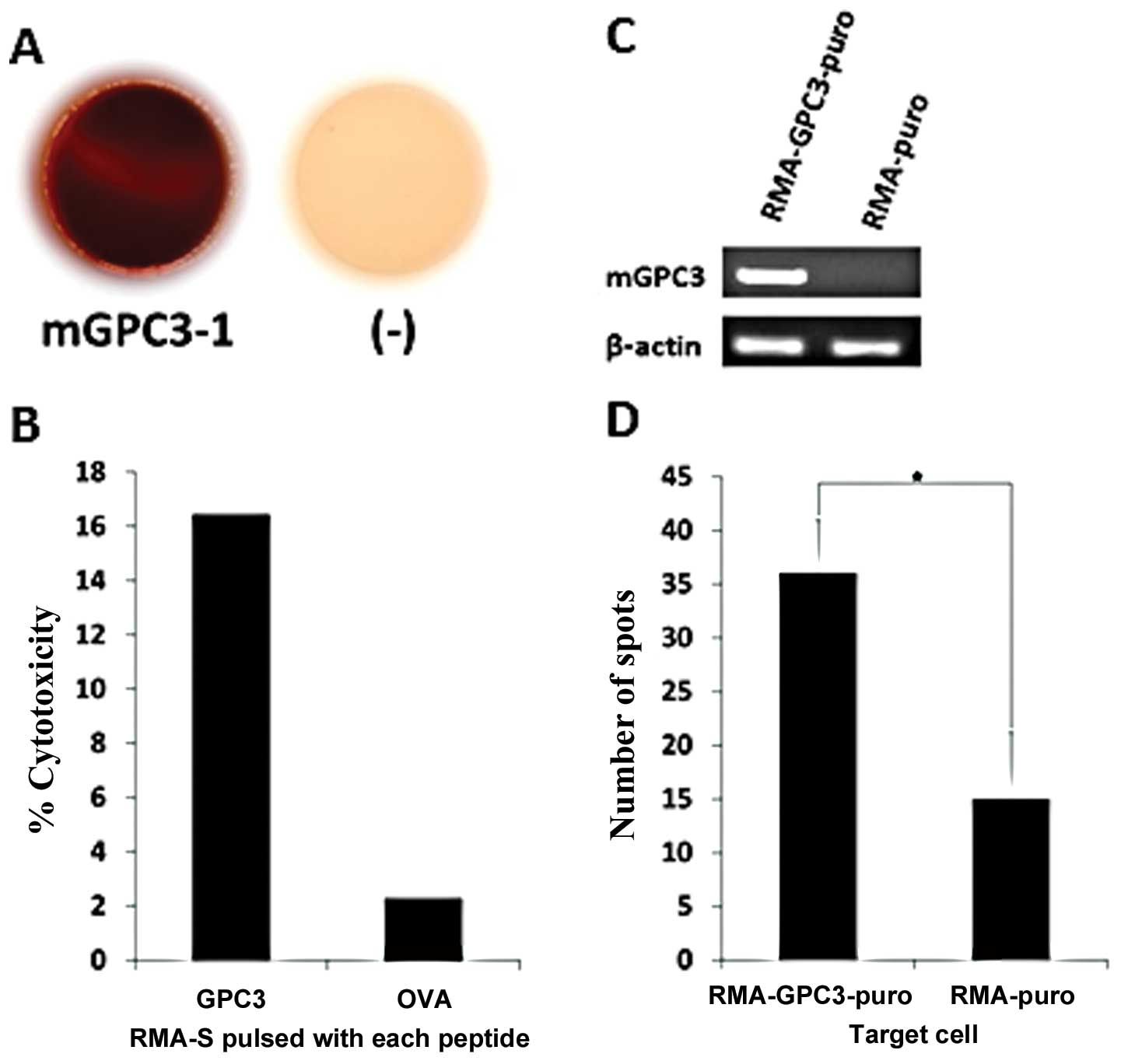
To further investigate the ability of mGPC3-1
peptide-specific CTLs induced by peptide vaccination, we
established a CTL line from immunized mice according to the above
described protocol. IFN-γ ELISPOT assays were performed using RMA-S
pulsed with mGPC3-1 peptide and non-pulsed RMA-S to confirm whether
the CTL line had mGPC3-1 peptide specificity. The CTL line clearly
released more IFN-γ to RMA-S pulsed with mGPC3-1 peptide than to
non-pulsed RMA-S, which suggests that the CTL line is the mGPC3-1
peptide-specific CTL (Fig.
3A).
Subsequently, a cytotoxicity assay was performed to
confirm whether the mGPC3-1-specific CTLs could kill RMA-S pulsed
with mGPC3-1 peptide. The CTLs killed RMA-S pulsed with the mGPC3-1
peptide (16.4%) better than non-pulsed RMA-S (2.2%), suggesting
that the mGPC3-1-specific CTL line could specifically recognize and
kill RMA-S pulsed with the mGPC3-1 peptide (Fig. 3B).
Finally, we examined whether the CTL line could
recognize RMA GPC3-puro endogenously expressing mGPC3. Expression
of mGPC3 in RMA-GPC3-puro and RMA-puro was confirmed by RT-PCR. The
results showed that RMA-GPC3-puro expressed mGPC3 and RMA-puro did
not express mGPC3 (Fig. 3C). IFN-γ
ELISPOT assays were performed using RMA-GPC3-puro and RMA-puro as
target cells to investigate whether the CTL line could recognize
RMA-GPC3-puro expressing endogenous mGPC3. The CTL line released
more IFN-γ to RMA-GPC3-puro (average number of spots, 32.2±5.0)
than to RMA-puro (average number of spots, 18.2±6.2). This result
suggests that the mGPC3-1 peptide is an endogenously presented
peptide (Fig. 3D).
CTL response against the mGPC3-derived
peptides induced in STAM mice
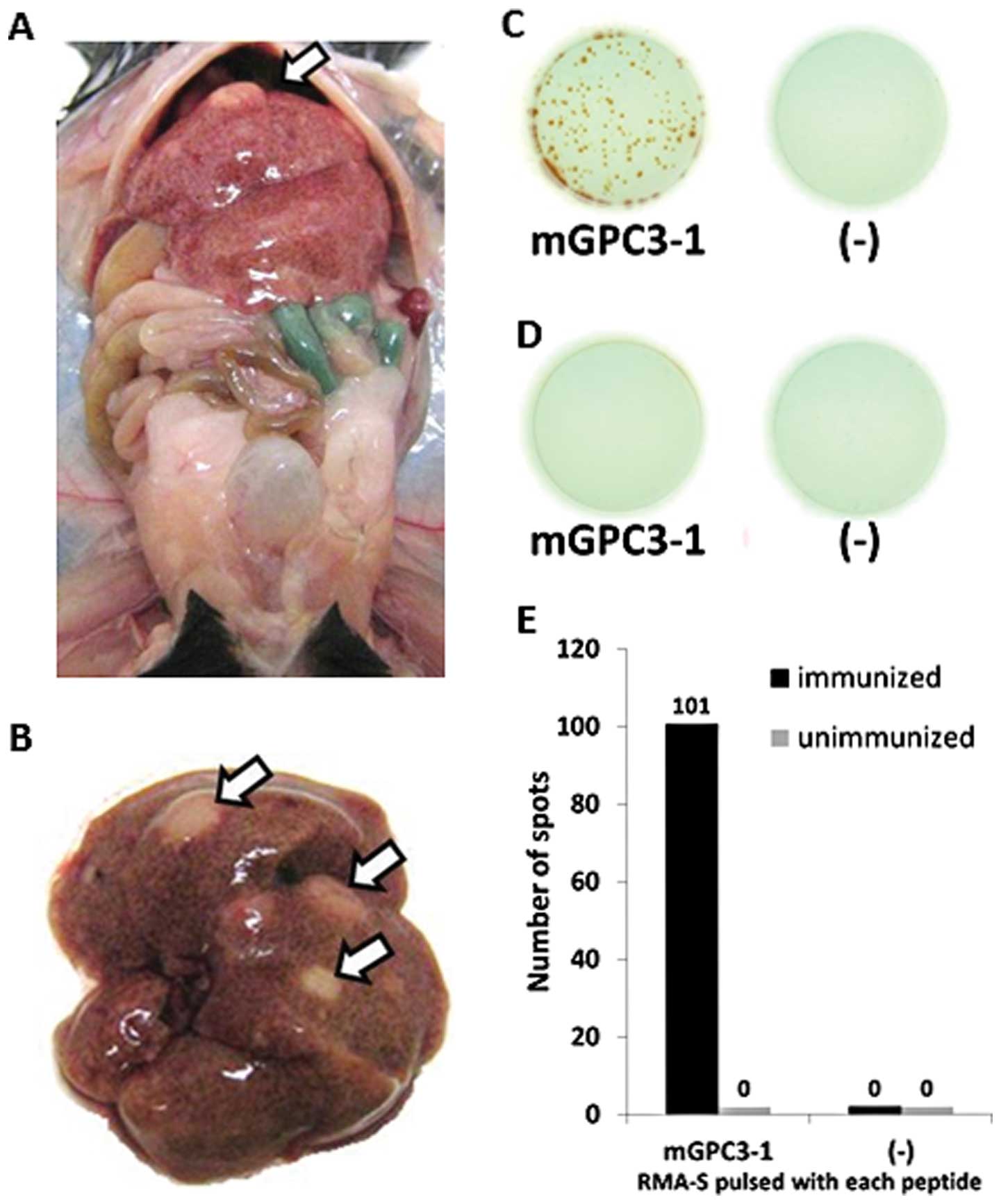
Previously, the NASH mouse model (named STAM mice
C57BL/6N-NASH) was exploited by Stelic Institute & Co. and STAM
mice with a B6 background spontaneously developed liver cancer. We
observed that liver cancer developed in 18-week-old STAM mice
(Fig. 4A and B). Furthermore, to
verify whether mGPC3-1 peptide-specific CTLs were induced in STAM
mice C57BL/6N-NASH, a mGPC3-1 peptide vaccine was given and an
IFN-γ ELISPOT assay was performed using RMA-S pulsed with mGPC3-1
peptide or non-pulsed RMA-S. The CD8-positive cells derived from
immunized mice released IFN-γ only to pulsed RMA-S (average number
of spots, 100±74.3), not to non-pulsed RMA-S (average number of
spots, 0.0±0.0) (Fig. 4C and E).
However, the CD8-positive cells derived from unimmunized mice did
not release IFN-γ to either pulsed (average number of spots, 0±0.0
or non-pulsed (average number of spots, 0.0±0.0) RMA-S (Fig. 4D and E). These results suggest that
peptide-specific mGPC3-1 could be induced in STAM mice
C57BL/6N-NASH immunized with the mGPC3-1 peptide vaccine but could
not be induced in un-immunized STAM mice C57BL/6N-NASH.
Discussion
HCC is the most common liver cancer and the
recurrence rate for treated HCC patients is high, thus
establishment of an effective preventative method, such as a
vaccination to prevent the occurrence and recurrence of HCC, is
needed. GPC3 is overexpressed in HCC and is not expressed in normal
tissue except for the placenta and embryonic liver. Clinical trials
of a GPC3-derived peptide vaccine for HCC have been performed and a
phase I clinical trial has shown the safety and immunological and
clinical potential of the vaccine (15,16).
Moreover, to study the preventive effect as a potential of the
GPC3-derived peptide vaccine, we attempted to establish a mouse
model to induce GPC3-specific CTLs by the peptide vaccine.
First, mGPC3-derived peptides binding to
H2-Kb or H2-Db were determined in
silico using BIMAS software. Moreover, a binding assay was
performed in vitro and showed that all peptides predicted by
the BIMAS software could bind H2-Kb and
H2-Db. However, the BIMAS score did not correlate with
the actual binding affinity.
Peptides that can bind to MHC class I are not always
able to induce peptide-specific CTLs (21,29).
Therefore, to investigate actual CTL-inducible peptides among the
11 selected peptides, a mixed peptide vaccine and single peptide
vaccine were given to mice. These results (Fig. 2) suggested that mGPC3-1 could
induce peptide-specific CTLs. In addition, antigen-derived and
CTL-inducible peptides are not necessarily presented by cancer
cells endogenously expressing the antigen (23,30).
Hence, we confirmed whether the mGPC3-1 peptide-specific CTL line
could recognize RMA-GPC3-puro endogenously expressing mGPC3
(Fig. 3D). Furthermore, confirming
whether the mGPC3-1 peptide-specific CTL line killed cancer cells
presenting the mGPC3-1 peptide is important, thus a cytotoxicity
assay was performed (Fig. 3B).
Mice with a B6 background that spontaneously develop
liver cancer have been reported (19,20).
These mice enable investigations as to whether a peptide vaccine
for GPC3 has a preventive capability. Recently, the STAM mice
C57BL/6N-NASH was established as a non-alcoholic-steatohepatitis
(NASH) mouse model by Stelic Institute & Co. STAM mice
C57BL/6N-NASH are drug-treated B6 mice and liver cancer occurs
spontaneously and early in NASH mice. Therefore, this mouse is an
attractive model for studying the preventive effects of a cancer
vaccine. We showed that mGPC3-1 peptide-specific CTL could be
induced in STAM mice C57BL/6N-NASH (Fig. 4E). Simultaneously, we established a
liver cancer cell line derived from STAM mice C57BL/6N-NASH and
observed the cancer cell line expressed mGPC3 (data not shown).
However, the GPC3 peptide vaccine did not prevent
the occurrence of liver cancer in STAM mice C57BL/6N-NASH (data not
shown). Therefore, further research to develop strong GPC3-specific
immunotherapies or combinational approaches in an appropriate mouse
model is needed. Identification of an H2-Kb or
H2-Db restricted, GPC3-derived peptide is the first
step. The established cell line from STAM mice C57BL/6N-NASH, which
show GPC3 expression, may help us to develop a new mouse model
system for a GPC3-targeted therapy.
In conclusion, mGPC3-1127–136 AMFKNNYPSL
was identified as an H2-Kb or H2-Db
restricted, GPC3-derived CTL most-inducible epitope peptide and
mGPC3-1 peptide-specific CTL can kill RMA-S pulsed with the mGPC3-1
peptide. Furthermore, we established an mGPC3-1-specific
CTL-inducible model in B6 mice using an mGPC3-1 peptide
vaccine.
Acknowledgements
D.N., H.K. and Y.S. would like to
thank the Foundation for Promotion of Cancer Research (Japan) for
the Third-Term Comprehensive Control Research for Cancer for
awarding them a research resident fellowship. This study was
supported in part by Health and Labor Science Research Grants for
Clinical Research and Third Term Comprehensive Control Research for
Cancer from the Ministry of Health, Labor and Welfare, Japan and
the National Cancer Center Research and Development Fund.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Yamamoto J, Okada S, Shimada K, et al:
Treatment strategy for small hepatocellular carcinoma: comparison
of long-term results after percutaneous ethanol injection therapy
and surgical resection. Hepatology. 34:707–713. 2001. View Article : Google Scholar
|
|
3
|
Greten TF, Manns MP and Korangy F:
Immunotherapy of hepatocellular carcinoma. J Hepatol. 45:868–878.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Motomura Y, Senju S, Nakatsura T, et al:
Embryonic stem cell-derived dendritic cells expressing glypican-3,
a recently identified oncofetal antigen, induce protective immunity
against highly metastatic mouse melanoma, B16-F10. Cancer Res.
66:2414–2422. 2006. View Article : Google Scholar
|
|
5
|
Mizukoshi E, Nakamoto Y, Arai K, et al:
Comparative analysis of various tumor-associated antigen-specific
T-cell responses in patients with hepatocellular carcinoma.
Hepatology. 53:1206–1216. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Butterfield LH, Ribas A, Dissette VB, et
al: A phase I/II trial testing immunization of hepatocellular
carcinoma patients with dendritic cells pulsed with four
alpha-fetoprotein peptides. Clin Cancer Res. 12:2817–2825. 2006.
View Article : Google Scholar
|
|
7
|
Greten TF, Forner A, Korangy F, et al: A
phase II open label trial evaluating safety and efficacy of a
telomerase peptide vaccination in patients with advanced
hepatocellular carcinoma. BMC Cancer. 10:2092010. View Article : Google Scholar
|
|
8
|
Butterfield LH, Ribas A, Meng WS, et al:
T-cell responses to HLA-A*0201 immunodominant peptides
derived from alpha-fetoprotein in patients with hepatocellular
cancer. Clin Cancer Res. 9:5902–5908. 2003.PubMed/NCBI
|
|
9
|
Nakatsura T, Yoshitake Y, Senju S, et al:
Glypican-3, over-expressed specifically in human hepatocellular
carcinoma, is a novel tumor marker. Biochem Biophys Res Commun.
306:16–25. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Capurro MI, Xiang YY, Lobe C and Filmus J:
Glypican-3 promotes the growth of hepatocellular carcinoma by
stimulating canonical Wnt signaling. Cancer Res. 65:6245–6254.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Filmus J: Glypicans in growth control and
cancer. Glycobiology. 11:R19–R23. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Filmus J, Capurro M and Rast J: Glypicans.
Genome Biol. 9:2242008. View Article : Google Scholar
|
|
13
|
Komori H, Nakatsura T, Senju S, et al:
Identification of HLA-A2-or HLA-A24-restricted CTL epitopes
possibly useful for glypican-3-specific immunotherapy of
hepatocellular carcinoma. Clin Cancer Res. 12:2689–2697. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nakatsura T, Komori H, Kubo T, et al:
Mouse homologue of a novel human oncofetal antigen, glypican-3,
evokes T-cell-mediated tumor rejection without autoimmune reactions
in mice. Clin Cancer Res. 10:8630–8640. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sawada Y, Sakai M, Yoshikawa T, Ofuji K
and Nakatsura T: A glypican-3-derived peptide vaccine against
hepatocellular carcinoma. Oncoimmunology. 1:1448–1450. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sawada Y, Yoshikawa T, Nobuoka D, et al:
Phase I trial of a glypican-3-derived peptide vaccine for advanced
hepatocellular carcinoma: immunologic evidence and potential for
improving overall survival. Clin Cancer Res. 18:3686–3696. 2012.
View Article : Google Scholar
|
|
17
|
Nobuoka D, Yoshikawa T, Takahashi M, et
al: Intratumoral peptide injection enhances tumor cell antigenicity
recognized by cytotoxic T lymphocytes: a potential option for
improvement in antigen-specific cancer immunotherapy. Cancer
Immunol Immunother. Nov 11–2012.(Epub ahead of print).
|
|
18
|
Motomura Y, Ikuta Y, Kuronuma T, et al:
HLA-A2 and -A24-restricted glypican-3-derived peptide vaccine
induces specific CTLs: preclinical study using mice. Int J Oncol.
32:985–990. 2008.PubMed/NCBI
|
|
19
|
Koike K, Moriya K, Iino S, et al:
High-level expression of hepatitis B virus HBx gene and
hepatocarcinogenesis in transgenic mice. Hepatology. 19:810–819.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Moriya K, Yotsuyanagi H, Shintani Y, et
al: Hepatitis C virus core protein induces hepatic steatosis in
transgenic mice. J Gen Virol. 78:1527–1531. 1997.PubMed/NCBI
|
|
21
|
Ikuta Y, Hayashida Y, Hirata S, et al:
Identification of the H2-Kd-restricted cytotoxic T
lymphocyte epitopes of a tumor-associated antigen, SPARC, which can
stimulate antitumor immunity without causing autoimmune disease in
mice. Cancer Sci. 100:132–137. 2009.
|
|
22
|
Wu X, Xu X, Gu R, et al: Prediction of HLA
class I-restricted T-cell epitopes of islet autoantigen combined
with binding and dissociation assays. Autoimmunity. 45:176–185.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakatsugawa M, Horie K, Yoshikawa T, et
al: Identification of an HLA-A*0201-restricted cytotoxic
T lymphocyte epitope from the lung carcinoma antigen, Lengsin. Int
J Oncol. 39:1041–1049. 2011.
|
|
24
|
Hofmann UB: Identification and
characterization of survivin-derived H-2Kb-restricted CTL epitopes.
Eur J Immunol. 39:1419–1424. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou X, Glas R, Momburg F, Hammerling GJ,
Jondal M and Ljunggren HG: TAP2-defective RMA-S cells present
Sendai virus antigen to cytotoxic T lymphocytes. Eur J Immunol.
23:1796–1801. 1993. View Article : Google Scholar
|
|
26
|
Stuber G, Leder GH, Storkus WT, et al:
Identification of wild-type and mutant p53 peptides binding to
HLA-A2 assessed by a peptide loading-deficient cell line assay and
a novel major histocompatibility complex class I peptide binding
assay. Eur J Immunol. 24:765–768. 1994. View Article : Google Scholar
|
|
27
|
Tsukahara T, Kawaguchi S, Torigoe T, et
al: HLA-A*0201-restricted CTL epitope of a novel
osteosarcoma antigen, papillomavirus binding factor. J Transl Med.
7:442009.
|
|
28
|
Yoshikawa T, Nakatsugawa M, Suzuki S, et
al: HLA-A2-restricted glypican-3 peptide-specific CTL clones
induced by peptide vaccine show high avidity and antigen-specific
killing activity against tumor cells. Cancer Sci. 102:918–925.
2011. View Article : Google Scholar
|
|
29
|
Yamazoe S, Tanaka H, Iwauchi T, et al:
Identification of HLA-A*0201- and
A*2402-restricted epitopes of mucin 5AC expressed in
advanced pancreatic cancer. Pancreas. 40:896–904. 2011.
|
|
30
|
Guo Y, Zhu Y and Sun S: Identification and
functional studies of HLA-A0201 restricted CTL epitopes in the X
protein of hepatitis B virus. Acta Virologica. 55:107–115. 2011.
View Article : Google Scholar : PubMed/NCBI
|