Introduction
Birt-Hogg-Dubé syndrome (BHDS) is an autosomal
dominantly inherited disease characterized by spontaneous
pneumothorax, hair folliculomas and renal tumors (1). The mutation responsible for BHDS has
been shown by positional cloning to occur in the BHD gene,
which has tumor suppressor activity (2,3). We
previously investigated hereditary renal carcinoma in the Nihon rat
model and identified a germline mutation in Bhd, the rat
homolog of BHD(4).
Heterozygous Nihon mutant rats develop renal carcinomas that are
mainly characterized by clear-cell histology, while homozygotes for
the mutation are embryonic lethal (5).
Human BHD is located on chromosome 17p11.2
and encodes folliculin, an evolutionarily conserved protein (∼67
kDa) that has no apparent functional motif (2). Although the function of folliculin
has not been fully elucidated, two folliculin-interacting proteins,
folliculin-interacting protein 1 (FNIP1) and its homolog
folliculin-interacting protein 2 (FNIP2/FNIPL), have been
identified (6–8). Baba et al demonstrated that 5′
AMP-activated protein kinase (AMPK) interacts with FNIP1 and
phosphorylates both FNIP1 and folliculin (6). They also reported the phosphorylation
of folliculin is regulated directly or indirectly by mTOR. These
findings suggest that folliculin is involved in energy and nutrient
sensing through the AMPK and mTOR signaling pathways.
In the present study, we found that an increase in
cyclin D1 protein levels was caused in HeLa cells by knockdown of
BHD. Recently, it was reported that cyclin D1 protein levels
were elevated in renal cell tumors of Bhd heterozygous
knockout mice and renal cysts of kidney-specific Bhd
knockout mice, and that the absence of folliculin resulted in an
increase in CCND1 (cyclin D1 gene) mRNA levels in a
BHD-deficient renal tumor cell line (UOK257) (9–11).
CCND1 is an oncogenic cell cycle regulating gene and its
aberrant activation may be implicated in the pathogenesis
associated with BHD deficiency (12). Here, we focus on the mechanism for
the regulation by folliculin of cyclin D1 expression.
Materials and methods
Cell lines, culture conditions
HeLa cells were cultured in Dulbecco’s modified
Eagle’s medium (DMEM; Sigma, St. Louis, MO, USA) containing 10%
fetal bovine serum (FBS) and antibiotics (penicillin/streptomycin).
Treatment (2 h) with wortmannin (final concentration 100 nM),
SB203580 (10 μM), U0126 (10 μM), rapamycin (20 nM) or
SP600125 (20 μM) was performed using dimethylsulfoxide
(DMSO) as the vehicle. Treatment with cycloheximide (10
μg/ml) was performed using ethanol as the vehicle.
Stable cell lines with an expression vector for
folliculin were established from the NR32 (Bhd-deficient
renal tumor cell line from the Nihon rat) (13). First, NR32 cells were transfected
with the plasmid and stable cells were selected in DMEM containing
10% FBS and G418 (final concentration 100 μg/ml); clones
were then selected after limiting dilution and cultured under the
same conditions.
Plasmid construction
Rat full length Bhd cDNA was cloned into
pcDNA3.1 vector (Invitrogen) (4).
The CCND1 promoter region (−1106 to +159) was amplified by
PCR from HEK293 cell genomic DNA using the primers PHD1F1
(5′-CCGCTAGCCTCACGCTCACGAATTCAGT-3′) and PHD1R1
(5′-CCAAGCTTATGGCTGGGGCTCTTCCT-3′) and subcloned into pGL4.10
vector (Promega, Madison, WI, USA) after digestion with NheI
and HindIII. Fragments of the 3′ untranslated region (3′UTR)
of human CCND1, 3′UTR1 (nt 1–1026) and 3′UTR2 (894–2253),
were amplified from HEK293 cell genomic DNA using the following
primers: CCND1UTR1F (5′-GGTCTAGAAGCAGAACATGGACCCCAAG-3′) and
CCND1UTR1R (5′-GGTCTAGAGGACTGAAAGTGCTTGGAAA-3′) for 3′UTR1;
CCND1UTR2F (5′-CCTCTAGAACCTGTTTATGAGATGCTGG-3′) and CCND1UTR2R
(5′-CGTCTAGAGCCAAAGCAGGCAGAACCT-3′) for 3′UTR2. Amplified products
were subcloned into the XbaI site (downstream of firefly
luciferase coding region) of a vector consisting of pGL4.10 and the
CCND1 promoter. A partial cDNA clone containing the distal
1.8 kb 3′UTR of human CCND1 (HIBBN77) was obtained from
American Type Culture Collection (Manassas, VA, USA). CCND1
3′UTR3 (2209–3171) was amplified from HIBBN77 using primers
CCND1UTR3F (5′-CCTCTAGACACAATACCTCATGCTTCAC-3′) and CCND1UTR3R
(5′-TTTCTAGACTTTCATGTTTGTCTTTTTG-3′) and subcloned as 3′UTR1 and
-2. A construct containing the full length 3′UTR of CCND1 was
generated from three partial constructs UTR1, -2 and -3 using
PstI and NcoI sites. Full coding sequence of human
HuR cDNA was amplified from HeLa cell cDNAs using the primers HuRF1
(5′-CGGAATTCGCCCGCATCCAGATTTTTGA-3′) and HuRR1
(5′-GGCTCGAGTTTGTGGGACTTGTTGGTTTT-3′) and subcloned into a modified
pCAG-GS vector (pCAG-CFLAG) after digestion with EcoRI and
XhoI to introduce a carboxy-terminal FLAG tag (14). A full length cDNA clone (pF1KB9904)
for hnRNP L (heterogeneous nuclear ribonucleoprotein L) was
obtained from Promega and subcloned into pCAG-CFLAG vector.
RNA interference (RNAi)
Transfection of siRNA was performed with
Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) at a final
concentration of 25 nM. Sequences of siRNAs were described
previously (8).
Antibodies
Antibodies against cyclin D1 (C-20 and H295), p27
(C-19), S6K (C-18), HuR (3A2) and hnRNP L (A-11) were purchased
from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies
against phospho-p70 S6K (Thr389), phospho-4E-BP1 (Thr37/46), 4E-BP
and acetyl CoA carboxylase were obtained from Cell Signaling
Technology (Danvers, MA, USA). Antibodies against histone H1 (AE4)
and AUF1 were obtained from Millipore (Billerica, MA, USA).
Anti-β-actin (AC15) was obtained from Sigma. The anti-folliculin
(C1) antibody has been described previously (4).
Western blot analysis
Protein concentration was determined by the
DC-protein assay (Bio-Rad, Hercules, CA, USA). Equal amounts of
proteins were subjected to SDS-polyacrylamide gel electrophoresis
and transferred to immobilon-P membranes (Millipore). The membranes
were blocked in 1% skim milk in Tris-buffered saline containing
0.05% Tween-20 and then the appropriate antibodies were applied.
Rabbit antibodies were detected using the Envision system (Dako,
Glostrup, Denmark) as previously described (15). Mouse antibodies were detected using
anti-mouse immunoglobulin and streptavidin-biotinylated horseradish
peroxidase (GE Healthcare, Buckinghamshire, UK). ECL reagents (GE
Healthcare) were used for detection.
RNA isolation, cDNA synthesis and
quantitative RT-PCR (qRT-PCR)
Total RNA for mRNA analysis was extracted from
cultured cells using FastPure RNA kit (Takara Bio, Ohtsu, Japan).
cDNA was synthesized using SuperScript II Reverse Transcriptase
with random primers (Invitrogen). qRT-PCR was performed using Power
SYBR-Green PCR Master mix (Applied Biosystems, Life Technologies,
Carlsbad, CA) and the following thermocycler conditions: 95°C for
10 min, and 40 cycles of 95°C for 15 sec and 60°C for 1 min. All
values were normalized against β-actin expression levels. Primer
sequences were as follows: SBHCD1F1 (5′-TCGGTGTCCTACTTCAAATG-3′)
and SBHCD1R1 (5′-TTCTGTTCCTCGCAGACCTC-3′) for CCND1;
SBHCD3F1 (5′-GCCCTCTGTGCTACAGATT-3′) and SBHCD3R1
(5′-CAGTCCACTTCAGTGCCAGT-3′) for CCND3;
(5′-CCATTTGCCATGGTTATAAGG-3′) and SBHCER1
(5′-CTTTGCTCGGGCTTTGTCCA-3′) for CCNE1; SBHVEGFAF1
(5′-CATGAACTTTCTGCTGTCTTGG-3′) and SBHVEGFAR1
(5′-ATGATTCTGCCCTCCTCCTT-3′) for VEGFA; SBHCDC25AF1
(5′-CAAGCGTGTCATTGTTGTGTT-3′) and SBHCDC25AR1
(5′-CCCTTCAGGACATACAGCTCA-3′) for CDC25A; SBHBAF1
(5′-CGCGAGAAGATGACCCAGA-3′) and SBHBAR1
(5′-GAGTCCATCACGATGCCAGT-3′) for β-actin.
Reporter assay
After 24 h of siRNA treatment, HeLa cells were
transfected with the various constructs using Fugene 6 (Roche,
Mannheim, Germany). One microgram of each reporter construct was
co-transfected with 20 ng renilla luciferase expressing plasmid
(pGL4.73, Promega). At 24 h after transfection, the cells were
lysed for the analyses. PicaGene dual SeaPangy luminescence kit
(Toyo B-net, Tokyo, Japan) was used to measure firefly and renilla
luciferase activities.
miRNA analysis
Total RNA for microRNA analysis was isolated using
the miRNeasy mini kit (Qiagen, Valencia, CA). cDNA was synthesized
using the NCode VILO miRNA cDNA Synthesis kit (Invitrogen). qRT-PCR
was performed using Express SYBR-GreenER qPCR SuperMixes and
Two-Step qRT-PCR kits (Invitrogen) and the following thermocycler
conditions: 50°C for 2 min, 95°C for 10 min and 40 cycles of 95°C
for 15 sec and 60°C for 1 min. All values were normalized against
U6 expression levels. The primer sequences for U6 and miR-16
amplification were described previously (16). Other primer sequences can be
obtained from the NCode miRNA Database provided by Invitrogen
(http://escience.invitrogen.com/ncode/).
Cell fractionation
Cytoplasmic and nuclear extracts from HeLa cells
were obtained using NE-PER nuclear and cytoplasmic extraction
reagent (Thermo Scientific, Waltham, MA, USA) with protease
inhibitors (aprotinin, leupeptin, pepstatin A) following the
manufacturer’s instructions.
Results
Increase in cyclin D1 levels by BHD
knockdown
siRNA mediated suppression of BHD expression
in HeLa cells caused an increase in expression of cyclin D1 protein
(Fig. 1A). Upregulation of cyclin
D1 levels was observed in BHD suppressed cells even under
serum starvation conditions (Fig.
1B). We examined cyclin D1 protein levels in rat renal tumor
cell lines that had been stably transduced with an expression
vector for folliculin or an empty vector as a control; the cell
lines were derived from NR32, a Bhd deficient renal tumor
cell line from the Nihon rat. Cyclin D1 levels in the cell lines
with restoration of Bhd activity (W2 and W15 in Fig. 1C) were lower than in Bhd
deficient cells (E7 and E19) in both serum starvation and serum
added conditions. These results suggest that the downstream pathway
of folliculin negatively regulates cyclin D1 expression.
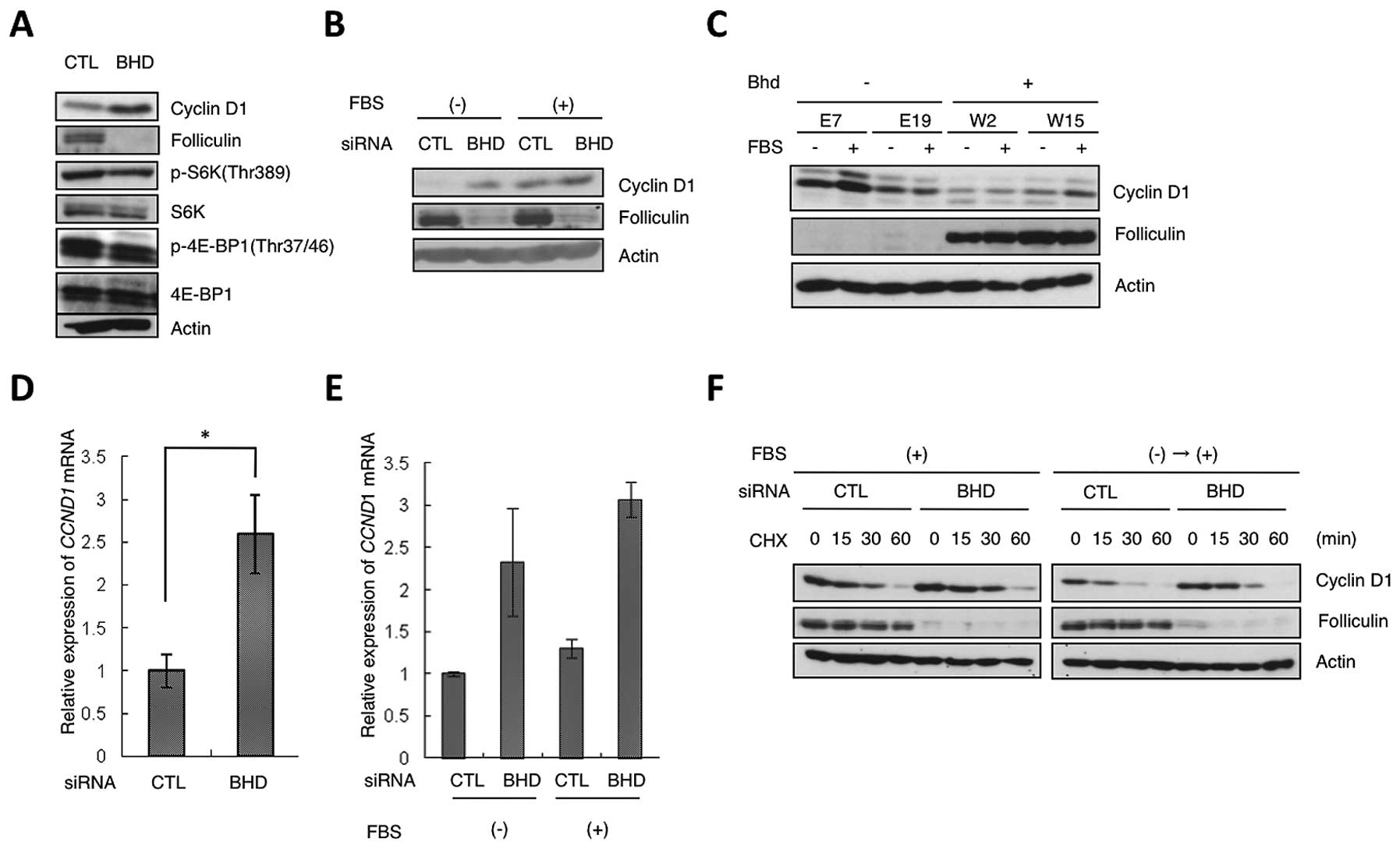 | Figure 1Regulation of cyclin D1 expression by
folliculin. (A) Upregulation of cyclin D1 protein expression in
HeLa cells by BHD knockdown. HeLa cells were transfected
with control (CTL) or BHD siRNA (BHD). Lysates were analyzed
by western blot analysis with the indicated antibodies. β-actin was
used as the control. (B) Increased expression of cyclin D1 protein
in BHD knockdown cells during serum starvation. HeLa cells
were transfected as in (A). During the final 24 h of culture, the
cells were starved of serum and then cultured in the presence (+)
or absence (−) of 10% serum (FBS) for 2 h. Western blot analysis
was performed with the indicated antibodies. (C) Expression of
cyclin D1 in Bhd-deficient (E7, E19) and Bhd-restored
(W2, W15) cells. FBS (−), serum starvation for 24 h; (+), with
serum for 24 h. Lysates were analyzed by western blot analysis with
the indicated antibodies. (D) Relative change in CCND1 mRNA
levels induced by BHD knockdown. CCND1 mRNA
expression in control HeLa (CTL) and BHD knockdown cells
(BHD) was analyzed by qRT-PCR. Error bars represent SD; *P<0.05.
Three independent experiments were performed in triplicate. (E)
qRT-PCR of the relative expression levels of CCND1 under
serum starvation conditions. As in (B), during the last 24 h of the
siRNA treatment, the cells were starved of serum and then grown for
2 h in the presence (+) or absence (−) of 10% serum (FBS). Error
bars represent SD. Three independent experiments were performed in
triplicate. (F) Stability of cyclin D1 protein. HeLa cells were
transfected with control (CTL) or BHD siRNA (BHD) and
cultured under different culture conditions: FBS(+), with serum;
FBS(−)→(+), cells were arrested by serum starvation for 24 h, and
then grown for 2 h in DMEM containing 10% FBS. Cycloheximide (CHX)
was added for the indicated time; lysates were analyzed by western
blot analysis with the indicated antibodies. |
To determine whether the induced increase in cyclin
D1 levels after BHD knockdown was a result of upregulation
of mRNA expression, we performed qRT-PCR. This analysis showed that
CCND1 mRNA levels in BHD knockdown cells were also
higher than those in control cells (Fig. 1D and E). We also examined the
half-life of cyclin D1 using cycloheximide to ascertain whether
cyclin D1 turnover was downregulated by BHD knockdown in
HeLa cells; no significant difference was present (Fig. 1F). These results suggest that
folliculin regulates cyclin D1 expression at transcriptional or
posttranscriptional level but not via protein turnover.
BHD knockdown does not stimulate CCND1
promoter activity
To determine whether folliculin regulates the
transcription of CCND1, we carried out a reporter analysis
by transfecting HeLa cells with a luciferase expressing vector
carrying the CCND1 promoter region (−1106 to +159) (15). Although the CCND1 mRNA
levels increased, luciferase activity was not elevated but rather
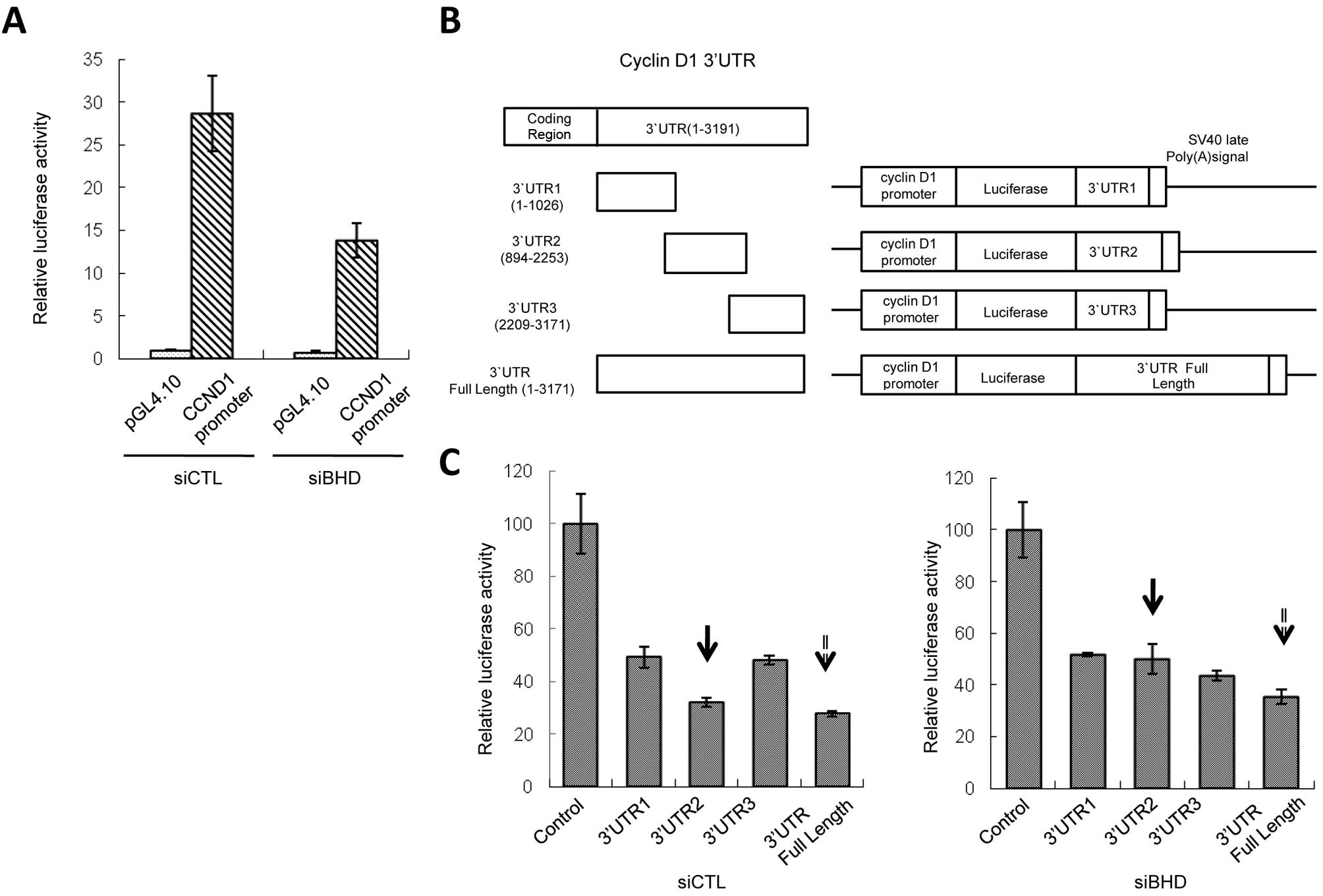
suppressed in BHD knockdown cells (Fig. 2A). Therefore, we concluded that
folliculin-mediated control of the level of CCND1 mRNA is
likely not to be via transcriptional mechanisms through known
cis-acting elements.
Regulation of CCND1 mRNA level through
3′UTR by folliculin
Next, we tested whether the expression of
CCND1 was regulated by 3′UTR of mRNA. Three constructs
carrying different 3′UTR fragments (3′UTR1-3) of CCND1
(Fig. 2B) exhibited inhibition of
reporter activity, possibly due to the existence of negative
regulatory elements. Knockdown of BHD showed no effect on
reporter activity of the constructs containing 3′UTR1 or 3′UTR3.
However, for the third construct, 3′UTR2, BHD knockdown
increased reporter activity (Fig.
2C, arrows with solid line) and this phenomenon was not
cancelled by rapamycin treatment (data not shown). Full length
CCND1 3′UTR also gave similar results, although to a lesser
extent than 3′UTR2 (Fig. 2C,
arrows with dotted line). We also tested the effects of 3′UTR2
inserted in reverse orientation or upstream of the promoter region.
Neither of these constructs elevated luciferase activity in
BHD knockdown cells compared to control cells, indicating
that the function of 3′UTR2 is position and orientation dependent
(data not shown). These results imply that the 3′UTR2 sequence in
the CCND1 mRNA influences the level of expression level and
that the regulation of CCND1 by folliculin may be associated
with microRNA(s) or RNA binding protein(s) (RBPs) that binds to the
3′UTR2.
CCND1 3′UTR-related microRNAs and BHD
knockdown
It was reported previously that the 3′UTR of
CCND1 is a target of several microRNAs or RBPs and that
these transacting factors regulate CCND1 mRNA stability
and/or protein expression (16,18,19).
We hypothesized that BHD knockdown alters the regulatory
pathway that controls microRNA or RBP binding to CCND1
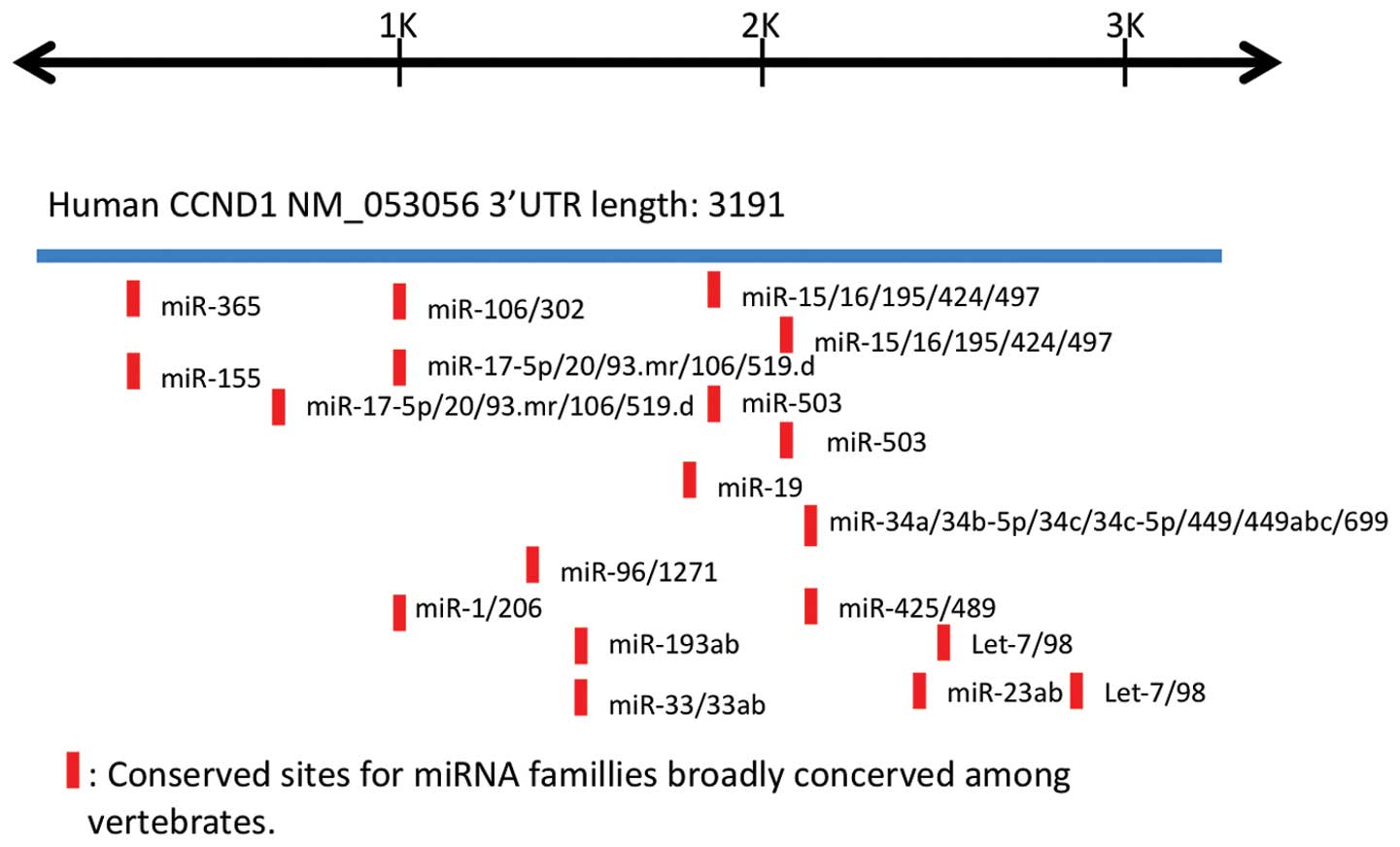
3′UTR. We searched for putative microRNA binding sites on
CCND1 3′UTR using the publicly accessible database
TargetScan (http://www.targetscan.org) and
evaluated the results by comparison with those described in a
previous report (Fig. 3) (16). Several of the potential microRNA
targets were found to be conserved among vertebrates. We carried
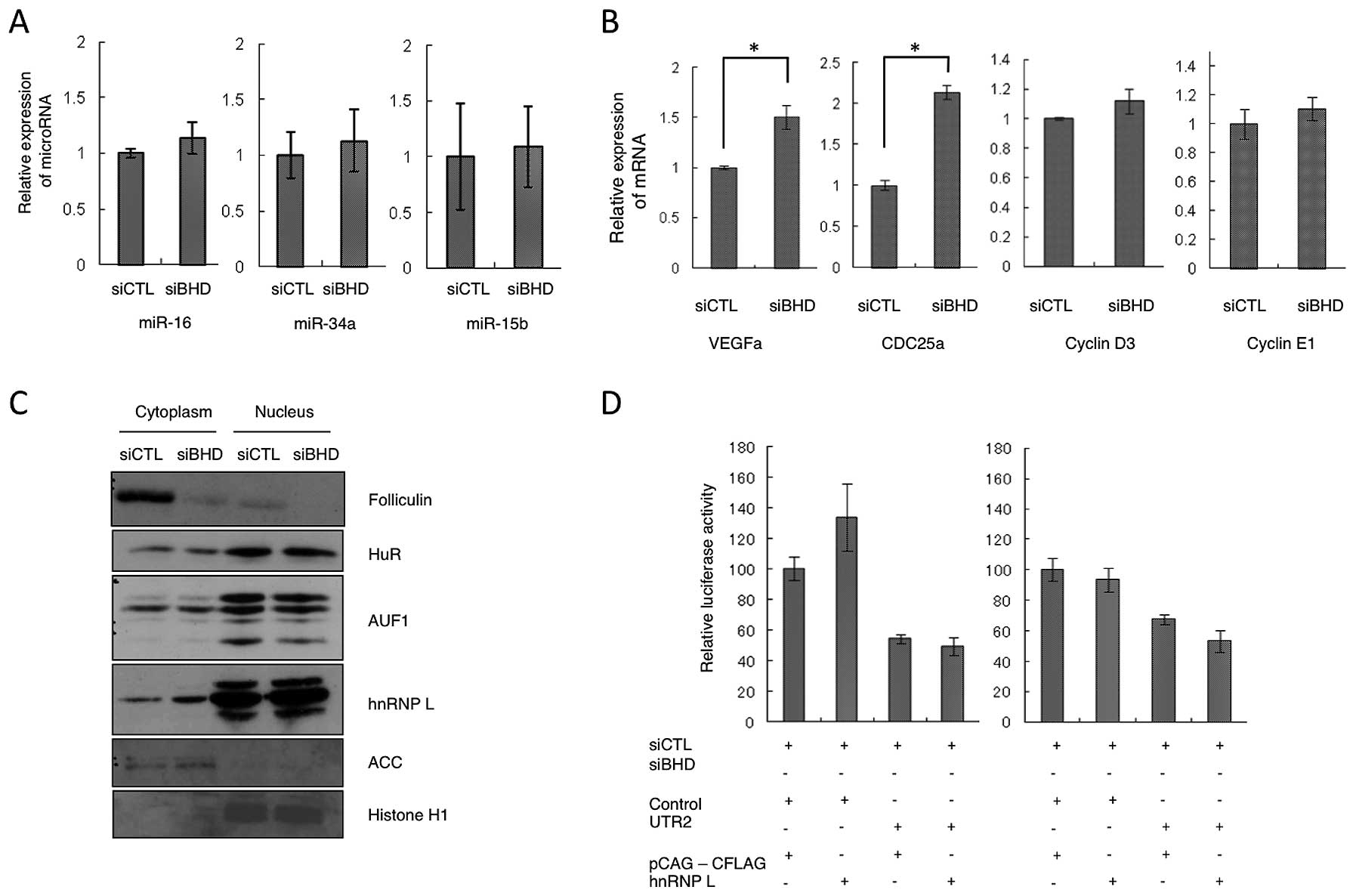
out a qRT-PCR on 22 candidate microRNAs (Table I), but did not identify any clear
change in their expression levels following BHD knockdown
(Fig. 4A, and data not shown). We
also investigated the mRNA levels of miR-16 target genes (16,19–21).
miR-16 is believed to be a major microRNA that binds to the
CCND1 3′UTR. The analysis showed that BHD knockdown
increased the relative level of expression of VEGFA,
CDC25A mRNA, but not of CCND3 or CCNE1
(Fig. 4B). From these
observations, we conclude that the microRNAs listed here do not
play a major role in cyclin D1 regulation by folliculin.
 | Table IList of microRNAs predicted to target
cyclin D1 by TargetScan. |
Table I
List of microRNAs predicted to target
cyclin D1 by TargetScan.
miR-15a, miR-15b,
miR-16, miR 17, miR-19a, miR-19b,
miR-20a, miR-20b, miR-34a, miR-93, miR-106a, miR-106b,
miR-155, miR-195, miR-302a, miR-302b, miR-302c,
miR-302d, miR-424, miR-497, miR-503, miR-519d |
RNA binding proteins and BHD
knockdown
We analyzed three RBPs, HuR, AUF1 and hnRNP L, that
have previously been reported to bind to 3′UTR and to control mRNA
stability. HuR and AUF1 have been identified as binding to the AU
rich element (ARE) on the CCND1 3′UTR. HuR stabilizes
CCND1 mRNA, whereas AUF1 causes its destabilization
(18). hnRNP L has been reported
to bind to the CA rich element of VEGFA and to increase mRNA
stability (22). First we
performed a cell fractionation analysis and found that cytoplasmic
hnRNP L levels were increased by BHD knockdown (Fig. 4C), whereas those of HuR and AUF1
were unchanged in both the cytoplasm and nucleus. To determine
whether these RBPs could bind CCND1 3′UTR and regulate mRNA
expression in a folliculin-dependent manner, a reporter assay using
constructs expressing hnRNP L and HuR was performed. We found that
luciferase activities from the 3′UTR2-containing reporter were not
elevated following co-transfection of hnRNP L or HuR, regardless of
the folliculin status (Fig. 4D and
data not shown). Accordingly, although possible regulation of hnRNP
L localization was suggested, the folliculin-dependent association
between 3′UTR2 and RBPs could not be identified in our
experiments.
BHD knockdown elevates rapamycin
sensitivity of CCND1 mRNA expression
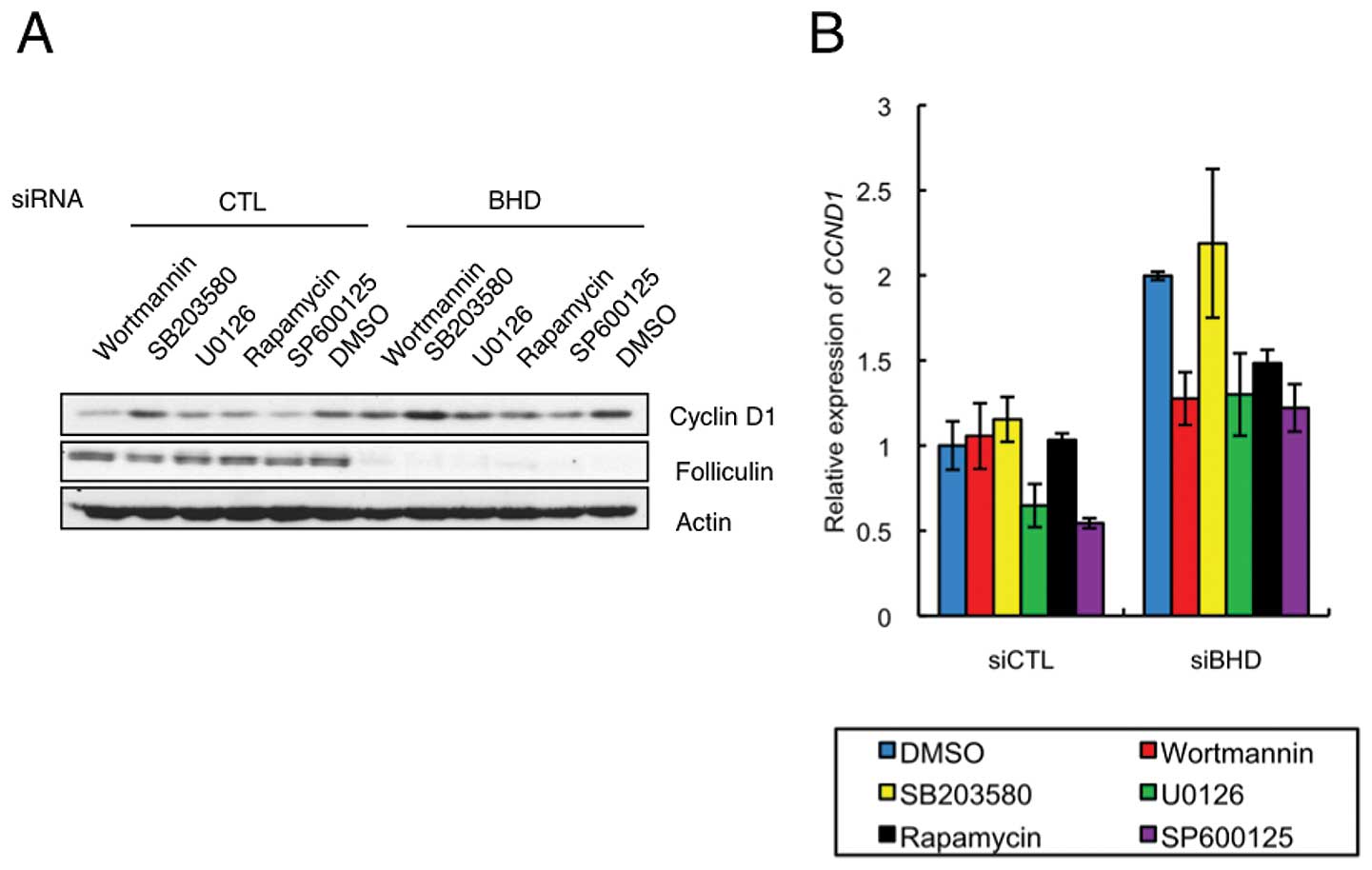
To explore the signaling pathway modulated after
BHD knockdown, we investigated the effects of several signal
inhibitors on cyclin D1 expression. This analysis showed that 2-h
treatment of wortmannin, U0126, rapamycin or SP600125 inhibited
cyclin D1 protein levels in control and BHD knockdown cells,
whereas SB203580 was ineffective in both cell types (Fig. 5A). Each of the positive inhibitors
had a similar effect in control and BHD knockdown cells
relative to the vehicle control. Interestingly, the relative levels
of CCND1 mRNA in drug treated BHD knockdown cells
differed from that in control cells (Fig. 5B). Treatment with wortmannin or
rapamycin significantly decreased the relative levels of
CCND1 mRNA in a BHD knockdown-specific manner
(Fig. 5B). These results suggest
that the regulatory mechanism for CCND1 mRNA expression was
more sensitive to inhibition of the mTORC1 pathway in BHD
knockdown cells.
Discussion
Although BHD is regarded as a tumor
suppressor gene, its function is not clearly understood and its
contribution to cell cycle regulation, including the suppression of
cyclin D1, has yet to be determined. In this study, we found an
increase in cyclin D1 protein and mRNA levels in BHD
knockdown cells. Based on the results of our experiments, we
suggest that the increase in CCND1 mRNA and protein levels
induced by BHD suppression is mediated, at least in part, by
a 3′UTR (3′UTR2)-related mechanism.
The CCND1 promoter region (-1106 to +159)
used in this study was reported to contain core positive regulatory
elements that respond to cell cycle promoting signals (17). Although we did not identify any
element in the CCND1 promoter region that responded to
BHD downregulation, the existence of such cis-acting
elements in other regions of the CCND1 gene cannot be
excluded.
One of the mechanisms for regulation of mRNA levels
is microRNA-mediated degradation (23). There are a number of microRNA
binding sites in the CCND1 3′UTR2 and many of these are
conserved between humans and rat (14). For example, miR-16 has been
implicated in the regulation of cell cycle related genes including
cyclin D1 (16,19–21).
The increase in CCND1 mRNA levels after knockdown of
BHD could be explained as the consequence of downregulation
by a microRNA-mediated mechanism. To date, we have not identified
any expression change in microRNAs nor of miR-16 target genes in
BHD knockdown cells. However, as there are additional
microRNA binding sites to those analyzed in 3′UTR2, the potential
contribution of other microRNAs remains to be determined. Possibly,
comprehensive microRNA analysis will identify candidates for
3′UTR2-associated miRNAs downstream of folliculin.
The CCND1 3′UTR has been reported to have
seven AREs composed of an AUUUA pentamer; six of these seven motifs
are present in 3′UTR2 (24). Many
RBPs have been found to selectively recognize and bind to the ARE
of mRNAs and to modulate the translation and/or stability of the
mRNAs (25). We tested the effects
of three RBPs (HuR, AUF1, hnRNP L) on 3′UTR2-mediated regulation,
but did not find a relationship between these RBPs and folliculin.
Other RBPs may bind CCND1 mRNA 3′UTR2 to regulate
folliculin-mediated mRNA translation/stability. Interestingly,
cytoplasmic hnRNP L levels were increased by BHD knockdown
in our study. hnRNP L has been reported to bind to CA repeats/CA
rich elements in the 3′UTR of VEGFA, BCL-2 and
GLUT1, and to increase the stability of their mRNAs
(22,26,27).
Notably, the CCND1 3′UTR2 also has CA repeats. Jafarifar
et al reported that hypoxia induces translocation of nuclear
hnRNP L to the cytoplasm, resulting in a marked increase in hnRNP L
binding to VEGFA mRNA and in the stability of VEGFA
mRNA (22). Although our reporter
analysis did not detect any specific effects, it is possible that
in CCND1 3′UTRs other than 3′UTR2, some cooperative elements
are required for the efficient binding and regulation by hnRNP L.
Singh et al(28) suggested
that folliculin may be a component of an RBP complex on the basis
of observation that the Drosophila folliculin protein binds
to RBP9, a homologue of human Hu protein, and shows weak homology
to Pumilio, another RBP (29).
Thus, the possibility of direct binding and regulation of
CCND1 mRNA 3′UTR2 by a folliculin-RBP complex still
remains.
We performed a search for signaling molecules
involved in the upregulation of cyclin D1 expression using various
signal inhibitors. This analysis showed that rapamycin and
wortmannin suppressed CCND1 mRNA levels in BHD
knockdown cells but not in control cells. These results suggest
that the mTORC1 pathway may contribute to upregulation of
CCND1 expression caused by BHD knockdown. Rapamycin
has been reported to affect CCND1 mRNA and protein stability
and thereby decrease levels of cyclin D1 protein (30). Recent studies have also reported
that rapamycin regulates CCND1 transcription and mRNA
stability in an AKT-dependent manner; under conditions of
relatively quiescent AKT activity, treatment of cells with
rapamycin results in the upregulation of CCND1 transcription
and mRNA stability, whereas in cells containing active AKT,
rapamycin these changes are repressed (31,32).
This phenomenon may be related to the rapamycin sensitivity of
BHD knockdown cells found in this study. We demonstrated
that S6K1 phosphorylation and total mTOR levels are decreased by
BHD knockdown in HeLa cells, and this also may lead to an
increased sensitivity to the effects of rapamycin and wortmannin
(8). Cyclin D1 protein and mRNA
levels were decreased by rapamycin treatment in BHD
knockdown cells, but this treatment did not affect the upregulated
reporter activity by 3′UTR2 in these cells. Thus, although mTORC1
signaling is required, it may be not a direct cause of the
CCND1 mRNA increase caused by BHD downregulation.
In recent studies, it has been reported that cyclin
D1 protein levels were elevated in renal cell tumors of Bhd
heterozygous knockout mice and the absence of folliculin resulted
in an increase in CCND1 mRNA levels in UOK257 cells
(10,11). Analysis of the folliculin-mediated
mechanism regulating cyclin D1 expression may provide important
clues to understanding the pathogenesis of BHDS and other
BHD mutation-associated diseases.
Acknowledgements
We thank Dr K. Kajino, Dr S. Matsuoka,
Dr D. Zhang, Dr Y. Ito, Dr H. Kawano, Dr X. Piao, Dr L. Wang, Mr.
M. Abe and Mr. T. Takagaki in the Department of Pathology and
Oncology for helpful discussion and technical supports. We also
thank Mrs. T. Ikegami in the Division of Molecular and Biochemical
Research, Biochemical Research Center (Juntendo University Graduate
School of Medicine) for expert support. We appreciate Dr K.
Okimoto, Dr I. Matsumoto and Dr M. Kouchi (Dainippon-Sumitomo
Pharma) for providing NR32. This study was supported by the grants
from the Japan Society for the Promotion of Science (JSPS), from
the Ministry of Education, Culture, Sports and Technology of Japan,
and from the Ministry of Health, Labour and Welfare of Japan.
References
|
1.
|
Birt AR, Hogg GR and Dubé WJ: Hereditary
multiple fibrofolliculomas with trichodiscomas and acrochordons.
Arch Dermatol. 113:1674–1677. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Nickerson ML, Warren MB, Toro JR, et al:
Mutations in a novel gene lead to kidney tumors, lung wall defects,
and benign tumors of the hair follicle in patients with the
Birt-Hogg-Dubé syndrome. Cancer Cell. 2:157–164. 2002.PubMed/NCBI
|
|
3.
|
Vocke CD, Yang Y, Palvovich CP, et al:
High frequency of somatic frameshift BHD mutations in
Birt-Hogg-Dubé-associated renal tumors. J Natl Cancer Inst.
97:931–935. 2005.PubMed/NCBI
|
|
4.
|
Okimoto K, Sakurai J, Kobayashi T, et al:
A germ-line insertion in the Birt-Hogg-Dubé (BHD) gene gives rise
to the Nihon rat model of inherited renal cancer. Proc Natl Acad
Sci USA. 101:2023–2027. 2004.
|
|
5.
|
Okimoto K, Kouchi M, Matsumoto I, Sakurai
J, Kobayashi T and Hino O: Natural history of the Nihon rat model
of BHD. Curr Mol Med. 4:887–893. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Baba M, Hong SB, Sharma N, et al:
Folliculin encoded by the BHD gene interacts with a binding
protein, FNIP1, and AMPK, and is involved in AMPK and mTOR
signaling. Proc Natl Acad Sci USA. 103:15552–15557. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Hasumi H, Baba M, Hong SB, et al:
Identification and characterization of a novel
folliculin-interacting protein FNIP2. Gene. 415:60–67. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Takagi Y, Kobayashi T, Shiono M, et al:
Interaction of folliculin (Birt-Hogg-Dubé product) with a novel
Fnip1-like (FnipL/Fnip2) protein. Oncogene. 27:5339–5347. 2008.
|
|
9.
|
Baba M, Furihata M, Hong SB, et al:
Kidney-targeted Birt-Hogg-Dubé gene inactivation in a mouse model:
Erk1/2 and Akt-mTOR activation, cell hyperproloferation, and
polycystic kidneys. J Natl Cancer Inst. 100:140–154. 2008.
|
|
10.
|
Hasumi Y, Baba M, Ajima R, et al:
Homozygous loss of BHD causes early embryonic lethality and kidney
tumor development with activation of mTORC1 and mTORC2. Proc Natl
Acad Sci USA. 106:18722–18727. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Preston RS, Philip A, Classens T, et al:
Absence of Birt-Hogg-Dubé gene product is associated with increased
hypoxia-inducible factor transcriptional activity and a loss of
metabolic flexibility. Oncogene. 30:1159–1173. 2010.
|
|
12.
|
Sherr CJ and Roberts JM: CDK inhibitors:
positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Matsumoto I, Kouchi M, Okimoto K, et al:
Establishment and characterization of renal carcinoma cell lines
from a Bhd gene mutant (Nihon) rat. Tumour Biol. 30:249–256. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Niwa H, Yamamura K and Miyazaki J:
Efficient selection for high-expression transfectants with a novel
eukaryotic vector. Gene. 108:193–199. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Fukuda T, Tani Y, Kobayashi T, Hirayama Y
and Hino O: A new western blotting method using polymer
immunocomplexes: detection of Tsc1 and Tsc2 expression in various
cultured cell lines. Anal Biochem. 285:274–276. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Chen RW, Bemis LT, Amato CM, et al:
Truncation in CCND1 mRNA alters miR-16-1 regulation in mantle cell
lymphoma. Blood. 112:822–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Tetsu O and McCormick F: β-catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999.
|
|
18.
|
Lal A, Mazan-Mamczarz K, Kawai T, Yang X,
Martindale JL and Gorospe M: Concurrent versus individual binding
of HuR and AUF1 to common labile target mRNAs. EMBO J.
23:3092–3102. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Liu Q, Fu H, Sun F, et al: miR-16 family
induces cell cycle arrest by regulating multiple cell cycle genes.
Nucleic Acids Res. 36:5391–5404. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Hua Z, Lv Q, Ye W, et al: miRNA-directed
regulation of VEGF and other angiogenic factors under hypoxia. PLoS
One. 1:e1162006. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Pothof J, Verkaik NS, van Ijcken W, et al:
MicroRNA-mediated gene silencing modulates the UV-induced DNA
damage response. EMBO J. 28:2090–2099. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Jafarifar F, Yao P, Eswarappa SM and Fox
PL: Repression of VEGFA by CA-rich element-binding microRNAs is
modulated by hnRNP L. EMBO J. 30:1324–1334. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Guo H, Ingolia NT, Weissman JS and Bartel
DP: Mammalian microRNAs predominantly act to decrease target mRNA
levels. Nature. 466:835–840. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Deshpande A, Pastore A, Deshpande AJ, et
al: 3′UTR mediated regulation of cyclin D1 proto-oncogene. Cell
Cycle. 32:3592–3600. 2009.
|
|
25.
|
Barrreau C, Paillard L and Osborne HB:
AU-rich elements and associated factors: are there unifying
principles? Nucleic Acids Res. 33:7138–7150. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Hamilton BJ, Nichols RC, Tsukamoto H,
Boado RJ, Pardridge WM and Rigby WF: hnRNP A2 and hnRNP L bind the
3′UTR of glucose transporter 1 mRNA and exist as a complex in vivo.
Biochem Biophys Res Commun. 261:646–651. 1999.
|
|
27.
|
Lee DH, Lim MH, Youn DY, Jung SE, Ahn YS,
Tsujimoto Y and Lee JH: hnRNP L binds to CA repeats in the 3′UTR of
bcl-2 mRNA. Biochem Biophys Res Commun. 382:583–587.
2009.PubMed/NCBI
|
|
28.
|
Singh SR, Zhen W, Wang H, et al: The
Drosophila homolog of the human tumor supressor gene BHD
interacts with the JAK-STAT and Dpp signaling pathways in
regulating male germline stem cell maintenance. Oncogene.
25:5933–5941. 2006.
|
|
29.
|
Park SJ, Yang ES, Kim-Ha J and Kim YJ:
Down regulation of extramacrochaetae mRNA by drosophila neural RNA
binding protein Rbp9 which is homologous to human Hu proteins.
Nucleic Acids Res. 26:2989–2994. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Hashemolhosseini S, Nagamine Y, Morley S,
Desrivières S, Mercep L and Ferrari S: Rapamycin inhibition of G1
to S transition is mediated by effect on cyclin D1 mRNA and protein
stability. J Biol Chem. 273:14424–14429. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Manderosian M, Sharma A, Funk AP,
Vartanian R, Mastri J, Jo OD and Gera JF: Tristetraprolin regulates
cyclin D1 and c-Myc mRNA stability in response to rapamycin in an
Akt-dependant manner via p38 MAPK signaling. Oncogene.
25:6277–6290. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Vartanian R, Mastri J, Martin J, et al:
AP-1 regulates cyclin D1 and c-MYC transcription in an
AKT-dependent mannner in response to mTOR inhibition: role of
AIP4/Itch-mediated JUNB degradation. Mol Cancer Res. 9:115–130.
2011. View Article : Google Scholar : PubMed/NCBI
|