Introduction
Prostate cancer is the most commonly diagnosed male
malignancy and the second cause of malignant male death worldwide
after lung cancer (1). Infectious
agents, physical trauma, hormones and a break of immune tolerance
to prostate antigen are considered as the cause of prostate cancer
(2). There are several therapeutic
options such as radical prostatectomy, radiation and hormonal
therapy for prostate cancer (3).
However, these protocols have a limitation for metastatic and
hormone refractory prostate cancer. Hormonal therapy in the form of
medical or surgical castration can induce significant long-term
remissions, but androgen-independent patients ultimately develop
metastatic prostate cancer resulting in death due to widespread
metastases (3–5). Chemotherapy is also effective but
long-term use is not feasible due to its toxicity (6). Therefore, the development of
alternative therapeutics are required for prostate cancer.
Imiquimod is a low-molecular-weight compound
belonging to the imidazoquinolines family. It was first identified
as a compound that has anti-viral activity in guinea pigs infected
with herpes simplex virus, and has been successfully used for the
treatment of genital warts caused by human papilloma virus in the
clinic (7,8). In addition, recent studies have
attracted considerable interest owing to their profound antitumoral
activities (9–11). Imiquimod exerts antitumor effect by
activating immune response to suppress tumor growth in a variety of
transplantable tumors (9), it also
has direct proapoptotic activity against various tumor cell
populations in vitro and in vivo(12,13).
The 5% imiquimod cream that is commercially available has been
successfully used for the treatment of several cancers including
basal cell carcinomas and melanoma (11,14–16).
Antitumor effects of the imidazoquinolines family
have been demonstrated in urogenital cancers including bladder
cancer and renal cell carcinoma (17–21).
Treatment with an imidazoquinoline (3M–011) downregulated c-Myc
expression in bladder cancer cells and reduced its transcriptional
activity (21). It also
significantly suppressed in vivo tumor growth in a mouse
model for orthotopic bladder cancer (21). Imiquimod also induced apoptosis and
cytokines production in various bladder cancer cell lines and
effectively inhibited in vivo tumor growth (17). An imidazoquinoline also enhanced
in vivo apoptosis and increased lymphocytic infiltration and
proinflammatory cytokine production in a mouse model of renal cell
carcinoma (22). However, the
effect of imidazoquinolines on the growth of prostate cancer has
not been studied. Therefore, in this study, we evaluated in
vitro and in vivo antitumor effect of an
imidazoquinoline, imiquimod, against prostate cancer.
Materials and methods
Mice
Specific pathogen-free (SPF) C57BL/6 mice were
purchased from Koatech (Pyeongtaek, Korea). All animal studies were
approved, and followed the regulations of the Institutional Animal
Care and Use Committee in Konyang University.
Cell lines and reagents
Transgenic adenocarcinoma of the mouse prostate
(TRAMP) model-derived prostatic epithelial cell line (TRAMP-C2)
(23) and the metastatic human
prostate cancer cell line (PC3) were purchased from the American
Type Culture Collection (Manassas, VA, USA). TRAMP-C2 cells were
cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, San
Diego, CA, USA) with 10% fetal bovine serum, 1%
penicillin-streptomycin, 5% Nu-serum IV (Collaborative Biomedical
Products, Bedford, MA, USA), 5 μg/ml insulin (Sigma-Aldrich
Co., St. Louis, MO) and 10 nM dihydrotestosterone (Sigma-Aldrich
Co.) (24). PC3 cells were
maintained in RPMI-1640 medium supplemented with 10% fetal bovine
serum (Gibco) and 1% penicillin-streptomycin. Imiquimod was
purchased from InvivoGen (San Diego, CA, USA) and dissolved in
ultrapure water as a stock solution at a concentration of 5
mg/ml.
MTT assay
The effects of imiquimod (1–10 μg/ml) on
TRAMP-C2 cells growth were determined by MTT
(3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide,
Amresco®, Solon, OH, USA) assay. TRAMP-C2 cells were
plated into 48-well tissue culture plates at concentration of
5×104 cells/well. After 12 h, TRAMP-C2 cells were
treated with various concentration of imiquimod for 24, 48 and 72
h. The cell culture supernatant was removed and cells were
incubated with 5 mg/ml MTT solution for 4 h. At the end of
incubation, MTT solution was aspirated and then cells were mixed
with dimethyl sulfoxide (DMSO, Amresco). The dye absorption was
quantified using an automatic microplate spectrophotometer
(Berthold Technologies GmbH, Vienna, Austria) at 540 nm. For TLR7
inhibitor treatment, TRAMP-C2 cells were pretreated with various
concentration chloroquine for 2 h and then imiquimod (10
μg/ml) was added to the medium with chloroquine for 72 h.
Cell growth was determined by MTT assay as described above.
Flow cytometry
Imiquimod-treated TRAMP-C2 cells were stained with
PI (Sigma-Aldrich Co.) or PI/Annexin V-FITC (BD Bioscience,
Franklin Lakes, NJ) and analyzed by flow cytometry (BD LSR Flow
cytometer, San Jose, CA, USA).
Western blot analysis
TRAMP-C2 cells were plated into 60-mm culture dish
about 2×105 cells/well. After overnight incubation, the
culture medium was replaced with fresh media and cells were treated
with imiquimod (20 μg/ml). The cells were lysed 0, 12, 24
and 48 h after stimulation using lysis buffer with 1% Nonidet P-40,
complete protease inhibitor cocktail (Roche, Basel, Switzerland)
and 2 mM dithiothreitol. Cell lysates were incubated on ice for 30
min and centrifuged for 15 min at 13,000 rpm. The
protein-containing supernatant was harvested and the total protein
amount was quantified using Bradford assay kit (Bio-Rad, Hercules,
CA, USA). Cell lysates were added with sample buffer and loaded
onto a 10 or 12% SDS-PAGE gel. After electrophoresis, proteins were
transferred onto polyvinylidene fluoride (PVDF) membrane and
detected with the following antibodies: cleaved-PARP, caspase-3,
caspase-7, caspase-9, cyclinB1 and phospo-CDC2 (Abcam, Cambridge,
UK), p21 (Cell Signaling, Beverly, MA, USA), and anti-β-actin
(Santa Cruz Biotechnology, Santa Cruz, CA, USA) and goat
anti-rabbit IgG-HRP (Santa Cruz Biotechnology). The blots were
developed using ECL substrate (Thermo Scientific, Waltham, MA,
USA).
Cytokine production
The culture supernatants of imiquimod-treated
TRAMP-C2 cells with or without cloroquine pretreatment at the
concentration of 1, 10 and 50 nM were obtained and kept at −20°C
until cytokine measurement. The concentration of IL-6 in culture
supernatants was determined using a commercial DuoSet ELISA
Development kits (R&D Systems, Minneapolis, MN, USA) according
to the manufacturer’s instructions.
In vivo antitumor efficacy
C57BL/6 mice received subcutaneous (s.c.) single
injection of TRAMP-C2 cells (1×106 cells/mouse in 100
μl of injectable saline) into the shaven right flank and
tumor growth was monitored. On day 10 after tumor implantation,
imiquimod was injected intratumorally at 50 μg daily for 9
days. Tumors were injected in a different site for each treatment
day. Tumor length (L) and width (W) were measured and tumor weight
(WR) was calculated twice a week as follows: WR = 1/2 × L × W2.
Histological analysis
Tumor mass were removed, fixed in 10% formalin for
24 h, and processed in a standard alcohol-xylene series. The
tissues were then embedded in paraffin, and 3-μm sections
were prepared, each of which was stained with H&E
(Sigma-Aldrich Co.).
TUNEL assay
Sections were stained for apoptotic cells by a
modified terminal deoxynucleotidyl transferase-mediated dUTP-biotin
nick end-labeling (TUNEL) assay using the ApopTag Peroxidase In
Situ Apoptosis Detection Kit (Millipore, Billerica, MA, USA)
following the manufacturer’s instructions. Then sections were
counterstained with Methyl Green (Sigma-Aldrich Co.).
Statistical analysis
All assays were derived from at least three
independent experiments. Statistical comparisons among the
different values were perform using the Prism 5 GraphPad software
(San Diego, CA, USA). Data are presented as mean ± SD.
Results
Imiquimod inhibits the growth of TRAMP-C2
cells
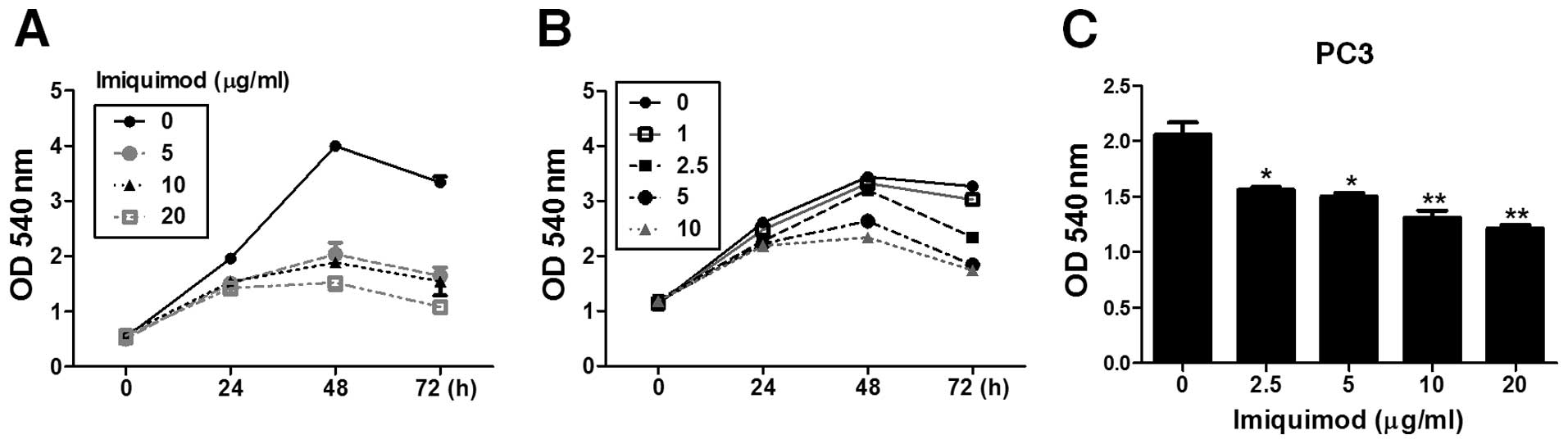
Inhibitory effect of imiquimod on the growth of
TRAMP-C2 cells was examined by MTT assay. The growth curve of
untreated cells increased rapidly by 48 h and slightly decreased at
72 h. Treatment at doses over 5 μg/ml of imiquimod delayed
the cell growth by 48 h (Fig. 1A).
To determine more definite dose-dependent effect of imiquimod on
the cell growth, the experiment was repeated with a narrow dose
range of imiquimod, 1 and 2.5 μg/ml of imiquimod did not
affect the cell growth by 48 h post-treatment (Fig. 1B). However, the cell growth was
more reduced by a treatment with 2.5 μg/ml of imiquimod at
72 h post-treatment, as compared with that in untreated cells
(Fig. 1B). The growth of PC3
cells, a human prostate cancer cell line, was inhibited by
imiquimod at 72 h post-treatment in a dose-dependent manner
(Fig. 1C).
Imiquimod leads to G2/M cell
cycle arrest in TRAMP-C2 cells
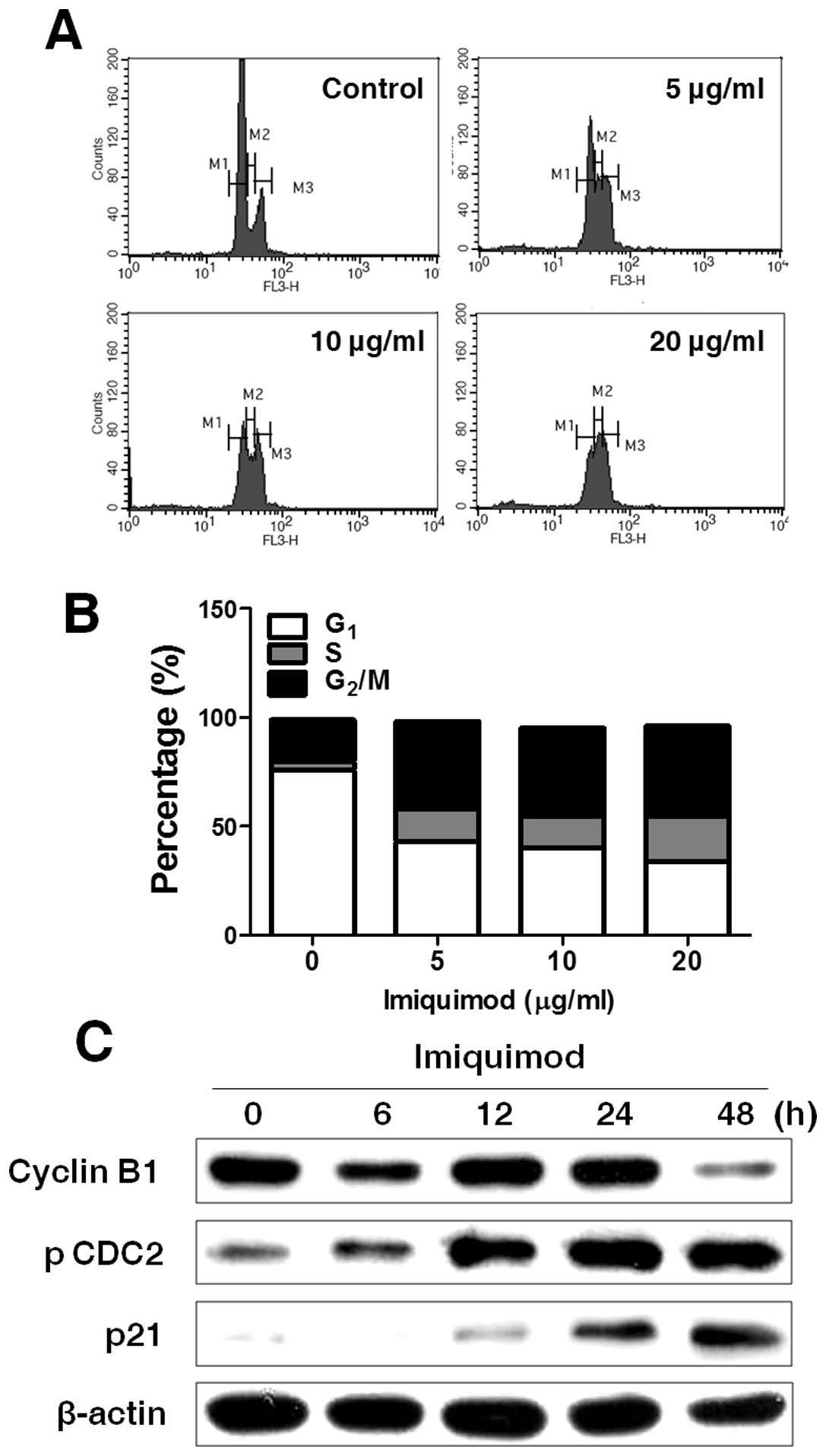
We next investigated the effect of imiquimod on the
cell cycle arrest in TRAMP-C2 cells. The cells were treated with
various doses of imiquimod for 48 h and DNA contents for cell cycle
arrest were determined by FACS analysis. The cell percentage in
G1 phase was reduced in imiquimod-treated TRAMP-C2 cells
(Fig. 2A and B). In contrast,
imiquimod increased the cell percentage in G2/M phase
dose-dependently, as compared with untreated cells (Fig. 2A and B). In addition, western blot
analysis was performed to determine the changes of expression or
activation of G2/M cell cycle-specific markers. Results
showed that the expression of cyclin B1, which promotes nuclear
accumulation and initiation of mitosis (25), was decreased in TRAMP-C2 cells by
imiquimod at 48 h post-treatment (Fig.
2C). Moreover, imiquimod enhanced the phosphorylation of CDC2,
which is a master regulatory kinase on the control of the
G2/M transition (26),
from 6 h after treatment (Fig.
2C). The expression of cell cycle regulator p21 was also
increased by imiquimod from 12 h after treatment (Fig. 2C). These findings suggest that
imiquimod may suppress the growth of prostate cancer cells via
G2/M cell cycle arrest.
Imiquimod induces mitochondria-dependent
apoptosis in prostate cancer cells
Previous studies showed that imiquimod can induce
direct apoptosis in various cancer cells (13,17,18,20).
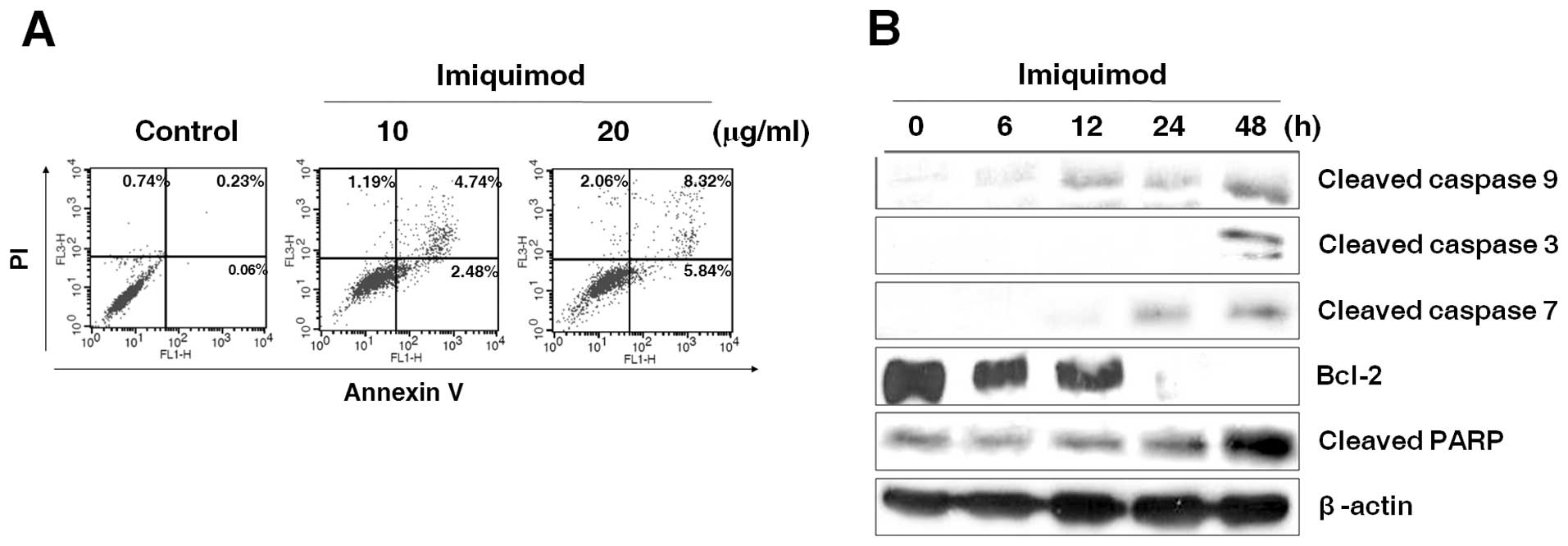
To investigate whether imiquimod induces apoptosis in prostate
cancer cells, TRAMP-C2 cells were treated with 10 or 20
μg/ml of imiquimod for 48 h, stained with PI and Annexin V,
and analyzed by flow cytometry. The percentage of Annexin
V-positive cells was increased by the treatment of imiquimod
dose-dependently (Fig. 3A). In
addition, we examined the change of molecules associating with
mitochondrial-dependent apoptosis pathway by western blot analysis.
The anti-apoptotic molecule bcl-2 expression in TRAMP-C2 cells was
reduced by imiquimod in a time-dependent manner (Fig. 3B). Imiquimod also led to cleavage
of caspase-9, caspase-3 and caspase-7 by 48 h (Fig. 3B). Moreover, cleaved form of PARP
was also increased by imiquimod in TRAMP-C2 cells (Fig. 3B). These findings indicate that
imiquimod may lead to apoptosis in prostate cancer cells via the
intrinsic pathway.
A TLR7 inhibitor chloroquine does not
restore the growth inhibition by imiquimod in prostate cancer
cells
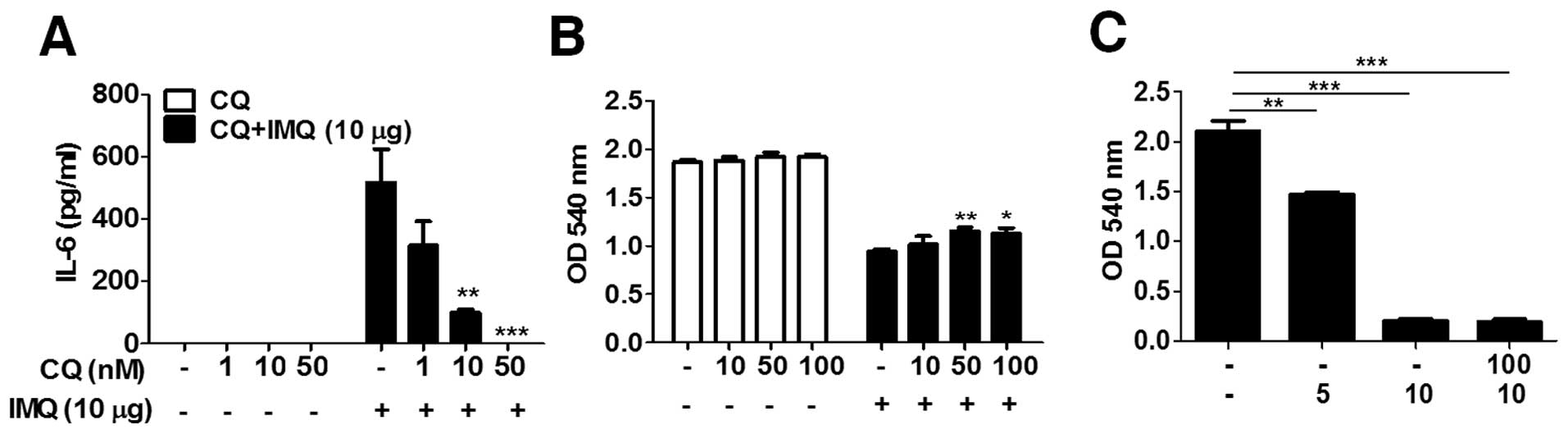
Although imiquimod has been described as a TLR7
agonist, it is still unclear whether TLR7 is required for its
antitumor effect. To clarify this, we performed an inhibitor assay
using chloroquine, which is an anti-malarial drug to block
activation of endosomal TLRs such as TLR3, 7 and 9 by inhibiting
endosomal acidification (27). Our
preliminary experiment showed that imiquimod enhanced the gene
expression of TLR7 and induced IL-6 production in TRAMP-C2 cells
(data not shown). As shown in Fig.
4A, chloroquine inhibited imiquimod-induced production of IL-6
in the cells in a dose-dependent manner. Although statistically
significant, the restorative effect of chloroquine on
imiquimod-induced inhibition of cell growth was minor (Fig. 4B). In an experiment with longer
incubation time (9 days), chloroquine did not restore the growth
inhibition by imiquimod in TRAMP-C2 cells (Fig. 4C).
Imiquimod inhibits in vivo growth of
prostate cancer in mice
In vivo antitumor efficacy of imiquimod was
evaluated in a mouse model s.c. implanted with TRAMP-C2 cells. On
day 10 after tumor implantation, mice were daily treated with PBS
or imiquimod (50 μg) by intratumoral injection for 9 days.
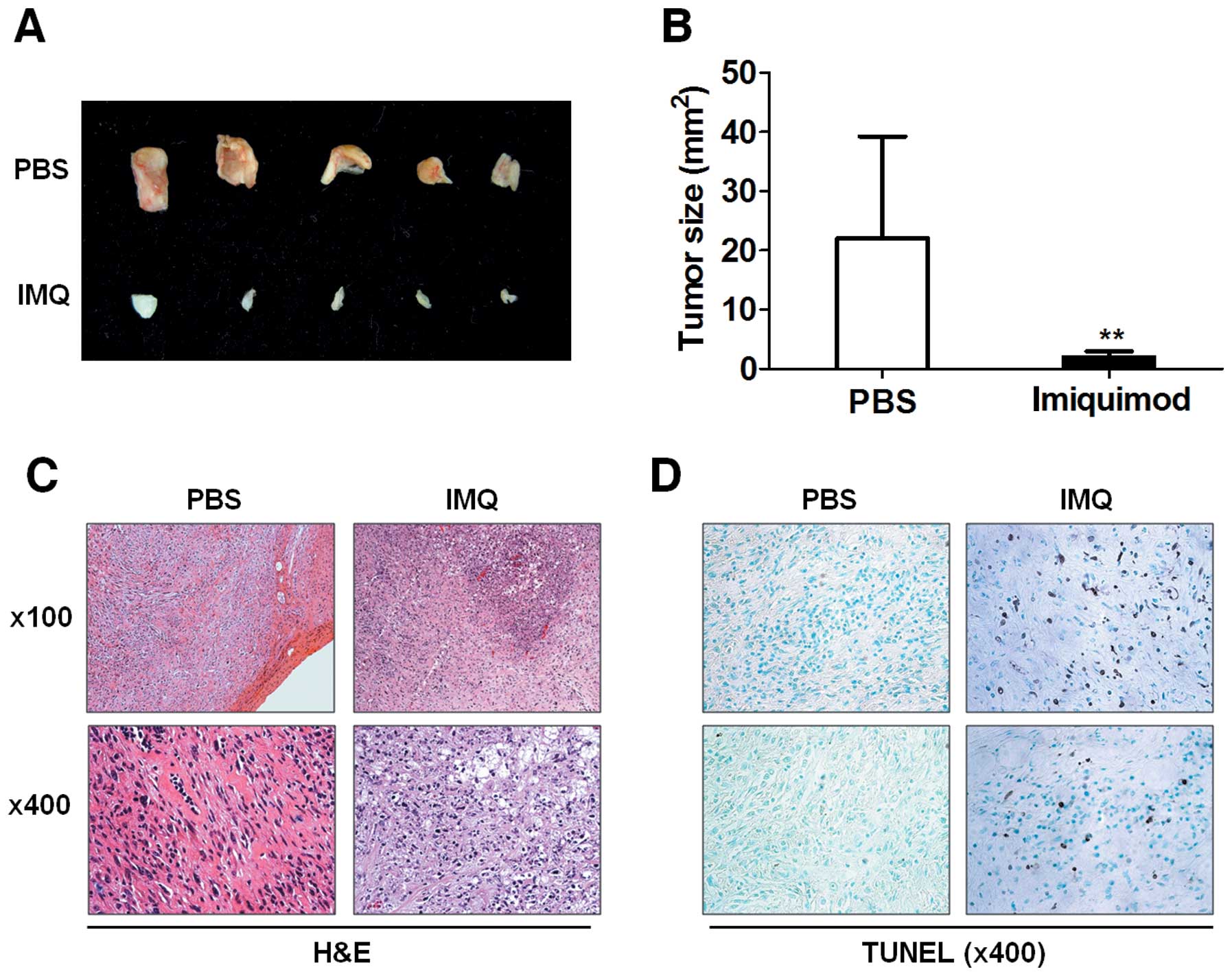
Tumor size was significantly reduced in imiquimod-treated mice, as
compared with PBS-treated group (Fig.
5A and B). Histologically, live tumor cells were compactly
grown in PBS-treated mice (Fig.
5C). In contrast, in imiquimod-treated mice, necrotic area was
broadly observed and cellular components were mostly dead cells
(Fig. 5C). TUNEL staining revealed
that apoptotic cells were increased by intratumoral injection of
imiquimod in TRAMP-C2 cell-implanted mice (Fig. 5D).
Discussion
Although imiquimod is known as a TLR7 agonist, it
can also induce cellular signaling TLR7-independently. Imiquimod
can induce transcriptional activation of proinflammatory factors
through adenosine receptor signaling (28). In addition, it activates p38, ERK
and JNK MAPKs and induces apoptosis in primary keratinocytes
independently of TLR7 and MyD88 (29). Schön et al suggest that
imiquimod triggers inflammatory response via TLR7/8 or adenosine
receptor signaling, whereas it induces direct apoptosis
independently (28). In this
study, an endosomal TLR inhibitor chloroquine inhibited
imiquimod-induced production of IL-6 in TRAMP-C2 cells, whereas it
did not affect cell growth inhibition by imiquimod. These results
suggest that imiquimod may primarily induce growth inhibition in
prostate cancer cells through a TLR7-independent mechanism.
Regulation of cell cycle and apoptosis can be
important targets for cancer chemotherapy (30). The anti-proliferative effects of
imidazoquinolines family are mediated by cell cycle arrest and/or
apoptosis in various cancer cells (31,32).
However, the pattern of cell cycle arrest caused by
imidazoquinolines is different between the studies. An
imidazoquinoline derivative NVP-BEZ235 inhibits the growth of T
cell acute lymphoblastic leukemia in the
G0/G1 phase of the cell cycle (33). In addition, imiquimod and
resiquimod treatment of cancer cells decreased cell proliferation
through G1/S phase arrest of the cell cycle through
opioid growth factor receptor (OGFr) pathway (32). In contrast, treatment of EAPB0203,
a member of the imidazo[1,2-a]quinoxalines, inhibited cell growth
through G2/M cell cycle arrest and apoptosis in
HTLV-I-transformed and HTLV-I-negative malignant T cells and fresh
ATL cells (31). In the present
study, flow cytometry and western blot analysis revealed that
imiquimod induces cell cycle arrest at G2/M phase in
TRAMP-C2 cells. It remains to be clarified whether different
pattern of imidazoquinolines-induced cell cycle arrest is due to
difference of cancer cell types or structural difference of
imidazoquinolines.
It is well known that imiquimod induces direct
apoptosis in various cancer cells (12,13,16,34).
The mechanism of imiquimod-induced apoptosis in tumor cells seems
to be mitochondria-dependent. In melanoma cells, the pro-apoptotic
activity of imiquimod was independent of cell surface death
receptors including CD95, TNF receptors or TRAIL (TNF-related
apoptosis-inducing ligand) receptors (12,13).
Rather, it was depended on Bcl-2 degradation because melanoma cells
overexpressing Bcl-2 were relatively resistant against
imiquimod-induced apoptosis as compared with their sham-transfected
control cells (13). Apoptosis of
tumor cells was abrogated by inhibition of caspase activation
(13). Moreover, blocking the
functions of membrane-bound death receptors did not affect the
pro-apoptotic activity of imiquimod (12). Imiquimod resulted in release of
mitochondrial cytochrome c into the cytosol, which is a
process that eventually leads to activation of caspase-9 and
caspase-3 by proteolytic cleavage (35). In this study, imiquimod led to
Bcl-2 degradation and cleavage of caspase-9, which are critical for
intrinsic apoptosis. Therefore, as in other types of cancer,
imiquimod seems to induce apoptosis in prostate cancer cells via
mitochondria-dependent pathway.
In this study, intratumoral injection of imiquimod
effectively inhibited in vivo tumor growth in mice s.c.
implanted with TRAMP-C2 cells. Although the number of apoptotic
cells was increased in imiquimod-treated mice, in vivo
antitumor action mechanism of imiquimod seems to be more complex.
Several studies have demonstrated that cytokines such as type I IFN
or cytotoxic T cells could participate in tumor destruction in
vivo induced by imiquimod. Imiquimod can induce a profound
tumor-directed cellular immune response (17,36).
Sullivan et al revealed that antitumorigenic effect of
imiquimod is mediated by upregulation of local IFN-α levels
(16). Moreover, a recent in
vivo study showed that imiquimod strongly enhances
antigen-specific activation of antitumoral CD8+ T cells
(37). Collectively, it is likely
that in vivo antitumor effect of imiquimod is achieved by
direct apoptosis and enhancement of immune responses.
To our knowledge, this study is the first attempt to
demonstrate that a direct treatment of imiquimod can significantly
inhibit the growth of prostate cancer. Our current study revealed
that imiquimod suppressed the proliferation of both mouse
(TRAMP-C2) and human (PC3) prostate cancer cells in a
TLR7-independent manner. Treatment of imiquimod resulted in
G2/M phase cell cycle arrest and intrinsic apoptosis.
Finally, we showed that imiquimod effectively inhibited tumor
growth in mice s.c. implanted with TRAMP-C2 cells. These results
suggest that imiquimod can be an effective therapeutic against
locally generated prostate cancer.
Acknowledgements
This study was supported by a program
(grant no. 2010-0002626) for Basic Research in Science and
Engineering and by the World Class Institute (WCI) program (grant
no. 2009-002) of the National Research Foundation of Korea (NRF)
funded by the Ministry of Education, Science and Technology of
Korea (MEST).
References
|
1
|
Jemal A, Murray T, Ward E, et al: Cancer
statistics, 2005. CA Cancer J Clin. 55:10–30. 2005. View Article : Google Scholar
|
|
2
|
De Marzo AM, Platz EA, Sutcliffe S, et al:
Inflammation in prostate carcinogenesis. Nat Rev Cancer. 7:256–269.
2007.
|
|
3
|
Pienta KJ and Smith DC: Advances in
prostate cancer chemotherapy: a new era begins. CA Cancer J Clin.
55:300–318. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maluf FC, Smaletz O and Herchenhorn D:
Castration-resistant prostate cancer: systemic therapy in 2012.
Clinics. 67:389–394. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Feldman BJ and Feldman D: The development
of androgen-independent prostate cancer. Nat Rev Cancer. 1:34–45.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rosenberg JE, Weinberg VK, Kelly WK, et
al: Activity of second-line chemotherapy in docetaxel-refractory
hormone-refractory prostate cancer patients. Cancer. 110:556–563.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miller R, Gerster J, Owens M, Slade H and
Tomai M: Review article imiquimod applied topically: a novel immune
response modifier and new class of drug. Int J Immunopharmacol.
21:1–14. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Von Krogh G, Lacey C, Gross G, Barrasso R
and Schneider A: European course on HPV associated pathology:
guidelines for primary care physicians for the diagnosis and
management of anogenital warts. Sex Transm Infect. 76:162–168.
2000.PubMed/NCBI
|
|
9
|
Sidky YA, Borden EC, Weeks CE, Reiter MJ,
Hatcher JF and Bryan GT: Inhibition of murine tumor growth by an
interferon-inducing imidazoquinolinamine. Cancer Res. 52:3528–3533.
1992.PubMed/NCBI
|
|
10
|
Hengge UR, Roth S and Tannapfel A: Topical
imiquimod to treat recurrent breast cancer. Breast Cancer Res
Treat. 94:93–94. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Beutner KR, Geisse JK, Helman D, Fox TL,
Ginkeld A and Owens ML: Therapeutic response of basal cell
carcinoma to the immune response modifier imiquimod 5% cream. J Am
Acad Dermatol. 41:1002–1007. 1999.
|
|
12
|
Schön M, Bong AB, Drewniok C, et al:
Tumor-selective induction of apoptosis and the small-molecule
immune response modifier imiquimod. J Natl Cancer Inst.
95:1138–1149. 2003.PubMed/NCBI
|
|
13
|
Schön MP, Wienrich BG, Drewniok C, et al:
Death receptor-independent apoptosis in malignant melanoma induced
by the small-molecule immune response modifier imiquimod. J Invest
Dermatol. 122:1266–1276. 2004.PubMed/NCBI
|
|
14
|
Berman B, Sullivan T, De Araujo T and
Nadji M: Expression of Fas-receptor on basal cell carcinomas after
treatment with imiquimod 5% cream or vehicle. Br J Dermatol.
149:59–61. 2003.
|
|
15
|
Smith KJ, Germain M and Skelton H:
Squamous cell carcinoma in situ (Bowen’s disease) in renal
transplant patients treated with 5% imiquimod and 5% 5-fluorouracil
therapy. Dermatol Surg. 27:561–564. 2001.
|
|
16
|
Sullivan TP, Dearaujo T, Vincek V and
Berman B: Evaluation of superficial basal cell carcinomas after
treatment with imiquimod 5% cream or vehicle for apoptosis and
lymphocyte phenotyping. Dermatol Surg. 29:1181–1186. 2003.
|
|
17
|
Smith EB, Schwartz M, Kawamoto H, et al:
Antitumor effects of imidazoquinolines in urothelial cell carcinoma
of the bladder. J Urol. 177:2347–2351. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schwartz MJ, Liu H, Hwang DH, Kawamoto H
and Scherr DS: Antitumor effects of an imidazoquinoline in renal
cell carcinoma. Urology. 73:1156–1162. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hayashi T, Crain B, Corr M, et al:
Intravesical Toll-like receptor 7 agonist R-837: optimization of
its formulation in an orthotopic mouse model of bladder cancer. Int
J Urol. 17:483–490. 2010. View Article : Google Scholar
|
|
20
|
Asakura M and Miura H: Imiquimod 5% cream
for the treatment of nasal lesion of metastatic renal cell
carcinoma. Dermatol Ther. 24:375–377. 2011.
|
|
21
|
Liu H, Schwartz MJ, Hwang DH and Scherr
DS: Tumour growth inhibition by an imidazoquinoline is associated
with c-Myc down-regulation in urothelial cell carcinoma. BJU Int.
101:894–901. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kauffman EC, Liu H, Schwartz MJ and Scherr
DS: Toll-like receptor 7 agonist therapy with imidazoquinoline
enhances cancer cell death and increases lymphocytic infiltration
and proinflammatory cytokine production in established tumors of a
renal cell carcinoma mouse model. J Oncol. 2012:1032982012.
View Article : Google Scholar
|
|
23
|
Foster BA, Gingrich JR, Kwon ED, Madias C
and Greenberg NM: Characterization of prostatic epithelial cell
lines derived from transgenic adenocarcinoma of the mouse prostate
(TRAMP) model. Cancer Res. 57:33251997.PubMed/NCBI
|
|
24
|
Touny LH and Banerjee PP: Identification
of both Myt 1 and Wee 1 as necessary mediators of the p21
independent inactivation of the cdc 2/cyclin B1 complex and growth
inhibition of TRAMP cancer cells by genistein. Prostate.
66:1542–1555. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Toyoshima F, Moriguchi T, Wada A, Fukuda M
and Nishida E: Nuclear export of cyclin B1 and its possible role in
the DNA damage-induced G2 checkpoint. EMBO J. 17:2728–2735. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
O’Connor PM: Mammalian G1 and G2 phase
checkpoints. Cancer Surv. 29:151–182. 1997.
|
|
27
|
Sainathan SK, Bishnupuri KS, Aden K, et
al: Toll-like receptor-7 ligand imiquimod induces type I interferon
and antimicrobial peptides to ameliorate dextran sodium
sulfate-induced acute colitis. Inflamm Bowel Dis. 18:955–967. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schon MP, Schon M and Klotz KN: The small
antitumoral immune response modifier imiquimod interacts with
adenosine receptor signaling in a TLR7-and TLR8-independent
fashion. J Invest Dermatol. 126:1338–1347. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Drobits B, Holcmann M, Amberg N, et al:
Imiquimod clears tumors in mice independent of adaptive immunity by
converting pDCs into tumor-killing effector cells. J Clin Invest.
122:575–585. 2012. View
Article : Google Scholar
|
|
30
|
Hartwell LH and Kastan MB: Cell cycle
control and cancer. Science. 266:1821–1828. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Moarbess G, El-Hajj H, Kfoury Y, et al:
EAPB0203, a member of the imidazoquinoxaline family, inhibits
growth and induces caspase-dependent apoptosis in T-cell lymphomas
and HTLV-I-associated adult T-cell leukemia/lymphoma. Blood.
111:3770–3777. 2008. View Article : Google Scholar
|
|
32
|
Zagon IS, Donahue RN, Rogosnitzky M and
Mclaughlin PJ: Imiquimod upregulates the opioid growth factor
receptor to inhibit cell proliferation independent of immune
function. Exp Biol Med (Maywood). 233:968–979. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chiarini F, Grimaldi C, Ricci F, et al:
Activity of the novel dual phosphatidylinositol 3-kinase/mammalian
target of rapamycin inhibitor NVP-BEZ235 against T-cell acute
lymphoblastic leukemia. Cancer Res. 70:8097–8107. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Meyer T, Nindl I, Schmook T, Ulrich C,
Sterry W and Stockfleth E: Induction of apoptosis by Toll-like
receptor-7 agonist in tissue cultures. Br J Dermatol. 149:9–13.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cryns V and Yuan J: Proteases to die for.
Genes and development. 12:1551–1570. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Imbertson LM, Beaurline JM, Couture AM, et
al: Cytokine induction in hairless mouse and rat skin after topical
application of the immune response modifiers imiquimod and S-28463.
J Invest Dermatol. 110:734–739. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rechtsteiner G, Warger T, Osterloh P,
Schild H and Radsak MP: Cutting edge: priming of CTL by
transcutaneous peptide immunization with imiquimod. J Immunol.
174:2476–2480. 2005. View Article : Google Scholar : PubMed/NCBI
|