Introduction
Both clinical and epidemiological evidence show that
estrogens participate in the initiation and development of human
breast cancer (1,2). Understanding the role of both types
of estrogen receptor (ER) (ERα and ERβ), in the pathogenesis of
breast cancer is important, because effects of estrogen are
mediated through both of these ERs (3–6).
Although the function of ERα has been established and its remains
the most important marker of response to hormonal therapy in breast
cancer, the role of ERβ remains elusive with many conflicting
studies (7). The two ERs act in
distinct ways in several estrogen target cells and tissues
(8,9). There are two major conclusions to be
drawn from current research situation of ERs. First, ERα and ERβ
have different biological functions, which are indicated by their
distinct expression patterns and the different phenotypes reported
for the two ERs in knockout animals, respectively. Second, ERα and
ERβ have overlapping yet unique roles in estrogen signaling, as
judged from a number of gene expression profiling studies.
Mitofusin 2 (mfn2), also named as hyperplasia
suppressor gene for its antiproliferative effects, localizes to the
mitochondrial outer membrane and plays an essential role in
mitochondrial fusion, thus regulating mitochondrial morphology and
function. Chen et al(10)
recently demonstrated that mfn2 profoundly suppresses cell growth
and proliferation in multiple tumor cell lines and rat vascular
smooth muscle cells in vivo and in culture systems via
inhibition of the Ras-ERK MAPK signaling pathway. Also, there is
some evidence suggesting a protective effect of mfn2 in mammalian
cells (11–13).
There is a growing body of literature suggesting
that estrogen may modulate expression of some genes through a
non-classical pathway in which the ER interacts with other
transcription factors, a process referred to as transcription
factor cross-talk. In this pathway, the ER modulates the activities
of other transcription factors, such as activator protein (AP)-1,
by stabilizing their binding to DNA and/or recruiting coactivators
to the complex. DeNardo et al(14) identified sets of estrogen-induced
genes, including mfn2, whose promoters contain potential AP-1 sites
but no estrogen-responsive element (ERE) sequences, essential for a
classical model of estrogen action; these genes thus depend on AP-1
for their expression. Further characterization of the promoters
suggested that the ER regulated these genes through the
non-classical pathways mentioned above. However, the previous
study, unlike the study presented here, did not directly explore
the interaction between ERs and mfn2.
In the present study, we showed that ERβ inhibits
human breast cancer cell proliferation and migration by inducing
expression of mfn2. We report for the first time that ERβ acts
upstream of mfn2. Moreover, this observation indicated that mfn2
affects the proliferation and migration of human breast cancer
cells.
Materials and methods
Cell lines and groups
MCF-7, a human breast cancer cell line, was kindly
provided by Professor Mei-xiang Sang, Division of Scientific
Research, the Fourth Hospital of Hebei Medical University,
Shijiazhuang, China. Cells were cultured in growth medium
consisting of Dulbecco’s modified Eagle’s medium (DMEM) (Gibco-BRL,
USA) containing 4.5 g/l glucose, 2 mM L-glutamine, 5,000 IU/l
penicillin, 5 mg/l streptomycin, 125 U/l Fungizone, 2.2 g/l sodium
bicarbonate and 10% fetal bovine serum (FBS) pretreated by 5%
charcoal-dextran, in a 5% CO2 incubator. For experiments
carried out in serum-free conditions, cells were made quiescent by
culturing in serum-free medium for 24 h. DMEM with antibiotics and
glutamine, was supplemented with 0.5 g/l BSA (1). Cells were randomly divided into six
groups and cultured for 48 h with E2 (17β-estradiol, at doses of 0
mol/l group, 10−9 mol/l group, 10−8 mol/l
group, 10−7 mol/l group, 10−6 mol/l group and
10−5 mol/l) to determine the dose-dependent effects of
E2 on mfn2 and cell behavior. Cells of each group were cultured for
48 h in DMEM medium containing 10% FBS plus defferent dose of E2
without phenolsulfonphthalein (2).
To specially enhance mfn2 expression and explore its effect on
proliferation and migration of MCF-7 cells, cells were randomly
divided into four groups in gene transfection experiments as
follows: normal group (blank control), untransfected E2 group (E2),
control vector pEGFP-transfected E2 group (E2 plus control vector)
and pEGFP-mfn2-transfected E2 group (E2 plus mfn2 vector). Cells of
the three groups treated with E2 were cultured in DMEM with 10% FBS
plus 10−6 mol/l E2 for 48 h (3). To explore the effect of ERβ on mfn2
expression, cells were randomly divided into four groups as
follows: normal group (blank control), untransfected E2 group (E2),
control vector pEGFP-N1 E2 group (E2 plus control vector) and
pEGFP-N1-ESR2 E2 group (E2 plus ERβ vector). Cells of every group
were grown as described in group 2. Each experiment was repeated
six times.
Expression vectors and transient
transfection
The pEGFP-mfn2 and pEGFP-N1-ESR2 vectors and their
negative control vectors were purchased from Yingrun Biotechnology
Co. Ltd., (Changsha, China). pEGFP-mfn2 and pEGFP-N1-ESR2 plasmids
carry full-length mfn2 and ERβ genes, respectively. Transient
transfection of MCF-7 cells was carried out using Lipofectamine
2000 (Invitrogen Co., Carlsbad, CA, USA) according to the
manufacturer’s instructions. Briefly, MCF-7 cells were cultured in
6-well plates and the medium was changed the following day until
80% confluence was achieved. The cells were transfected with 4.0
μg vector DNA by 10 μl Lipofectamine 2000 in 2 ml
serum-free DMEM medium. At 6 h after transfection, the medium was
replaced by normal DMEM supplemented with 10% FBS, and cells were
cultured for 24 h. Cells were then cultured for 48 h in medium
containing 10% FBS and E2 to detect proliferation and migration of
MCF-7 cells (group 2) and mfn2 expression (group 3). The efficiency
of transfection was approximately 70% for all experimental
groups.
Western blot analysis
Protein extracted from MCF-7 cells was separated on
a 10% SDS-PAGE gel and then transferred onto PVDF membrane
(Millipore Corporation, Bedford, MA, USA). The membrane was blocked
for 1 h at 37°C with 5% BSA in Tris-buffered saline containing
0.05% Tween-20 (TBST). Next, the membrane was incubated at 4°C
overnight with primary antibodies for mfn2 (1:200, Santa Cruz
Biotechnology, Santa Cruz, CA, USA), ERβ (1:100, Santa Cruz
Biotechnology), and β-actin (1:1,000, Santa Cruz Biotechnology).
Subsequently, the membrane was rinsed three times with TBST
containing secondary antibody (1:5,000), treated with ECL solution
(Pierce, Rockford, IL, USA), and bands detected by exposing the
blots to X-ray film. For quantitative analysis (i.e., normalized
for β-actin), bands were evaluated with IPP 5.0 software.
Integrated optical density (IOD) of each band was measured, and
relative IOD calculated as the ratio of the target band IOD
compared to the IOD of the β-actin band.
Semi-quantitative RT-PCR
Total RNA was extracted with TRIzol (Invitrogen Co.)
according to the manufacturer’s instructions. Total RNA (2
μg) was reverse transcribed using random primers and M-MLV
at 42°C for 1 h and then heated to 94°C for 5 min in a total
reaction volume of 20 μl. The PCR amplification began with a
5-min denaturation at 95°C, followed by 40 cycles of denaturation
at 95°C for 45 sec, annealing at 55°C for 45 sec and extension at
72°C for 60 sec. The final extension was set for 10 min at 72°C.
The products were electrophoresed on a 1.5% agarose gel, and the
levels of mfn2 mRNA were normalized with levels of GAPDH mRNA. All
PCR primers are shown in Table
I.
 | Table IPrimers and corresponding products
for mfn2 and GAPDH. |
Table I
Primers and corresponding products
for mfn2 and GAPDH.
| Gene | | Products (bp) |
|---|
| mfn2 | | |
| Sense |
5′-ATGCATCCCCACTTAAGCAC-3′ | 301 |
| Antisense |
5′-CCAGAGGGCAGAACTTTGTC-3′ |
| GAPDH | | |
| Sense |
5′-AACGGATTTGGTCGTATTG-3′ | 214 |
| Antisense |
5′-GCTCCTGGAAGATGGTGAT-3′ |
Immunofluorescence
MCF-7 cells were planted on cover slides in 6-well
plates. After fixing with 10% formalin at room temperature for 15
min, pretreating with 0.3% Triton X-100 for 20 min at 37°C and
blocking with goat serum for 30 min at 37°C, cells were incubated
with anti-mfn2 (1:200) overnight at 4°C. After washing with PBS for
three times, the slides was all incubated with FITC-conjugated
secondary antibody (1:200, Santa Cruz Biotechnology) for 2 h at
37°C. Then slides were viewed after being rinsed with PBS three
times.
Cell proliferation
Cell proliferation was measured using methyl
thiazolyl tetrazolium (MTT) shade selection experiments. Cells
(5×103 per well) were plated in triplicate in 96-well
plates and cultured for 24 h. Then,
3-2,5-dihydro-1-methyl-5h-tetrazole-5-thion sodium salt was added
for 4 h before absorbance was determined at 490 nm (SpectraMax,
Molecular Devices, Sunnyvale, CA, USA).
Measurement of cell migration
Cell migration was measured using a wound-healing
protocol developed and described in an earlier publication
(15).
Statistical analysis
The figure analysis was carried out by the software
of IPP. The quantitative data are presented as mean ± standard
deviation (SD). Statistical analyses were performed using one-way
analysis of variance (ANOVA) with Student-Newman-Keuls test.
Statistical differences were considered significant at a P-value of
<0.05.
Results
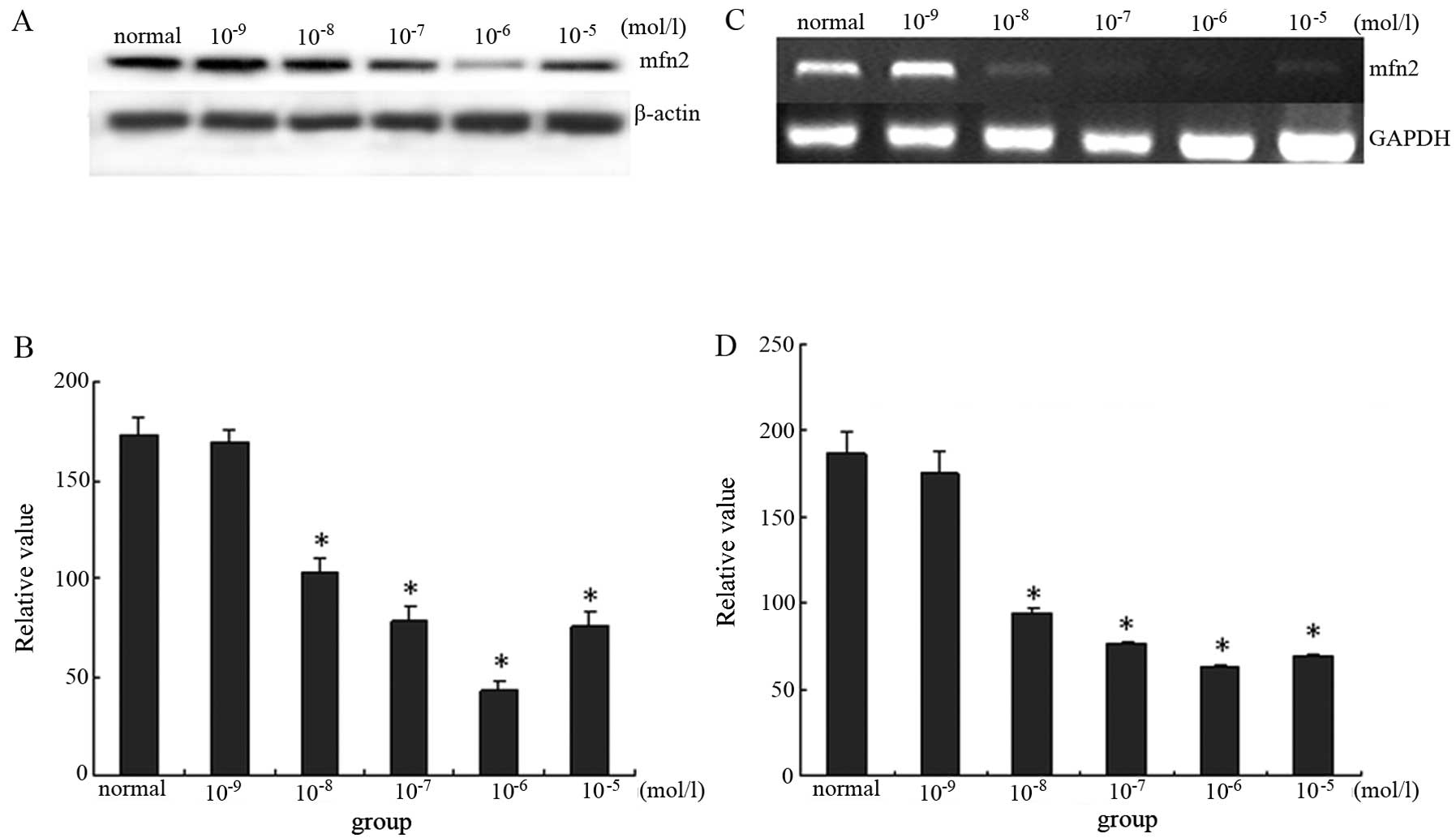
E2 downregulates expression of mfn2 in a
dose-dependent manner in MCF-7 cells
As described above, the ER might regulate mfn2
expression via a non-classical pathway. To observe the effect of
ERα on mfn2, MCF-7 cells, which primarily express ERα, were
cultured in medium containing E2. The effect of E2 on mfn2
expression through ERα was measured using immunoblotting for
protein levels and semi-quantitative RT-PCR for mRNA levels in
MCF-7 cells. E2 inhibited the expression of mfn2 in a
dose-dependent manner. Mfn2 was expressed at a higher level in
cells cultured with 10% FBS. When cells were pretreated with
10−9 mol/l, 10−8 mol/l, 10−7
mol/l, 10−6 mol/l and 10−5 mol/l group E2 for
48 h, protein expression of mfn2 decreased by 2.85, 40.00, 55.43,
74.29 and 57.14%, respectively. Thus, the lowest expression of mfn2
was seen in the 10−6 mol/l group. Similar changes were
seen when cells were analyzed by RT-PCR. These findings
demonstrated that E2 decreased mfn2 expression in a dose-dependent
manner at both the molecular and protein levels (Fig. 1).
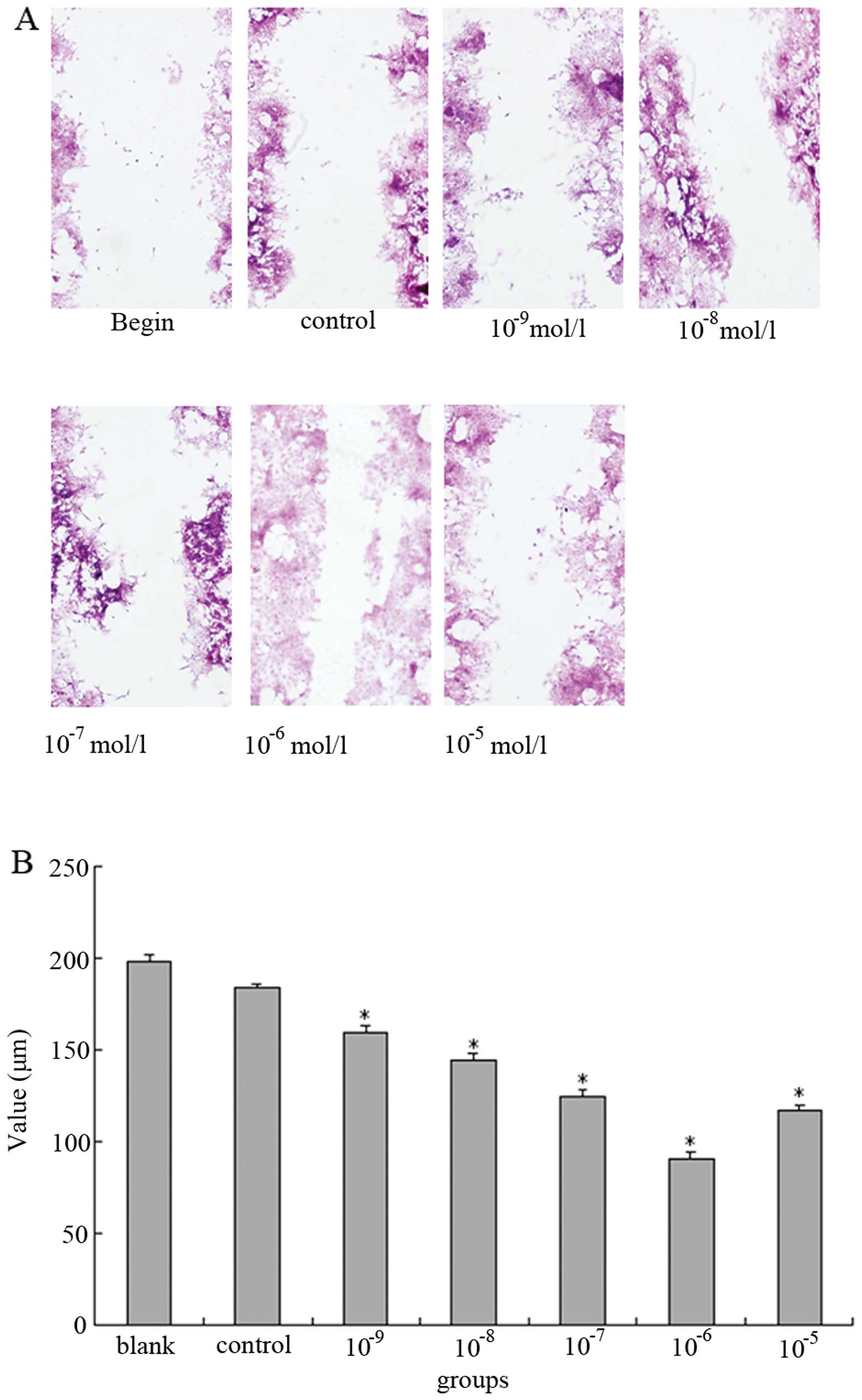
E2 enhances proliferation and migration
of MCF-7 cells
MCF-7 cells are the best-characterized ER-positive
cell line in terms of known genes regulated by estrogens that
promote proliferation. In order to confirm that E2 promotes
proliferation, E2-treated MCF-7 cells were examined using an MTT
assay. Absorbance of the MTT substrate at 490 nm for each dosage
group is showed in Table II.
Significant differences were seen among experimental groups and the
control group; the maximum effect of E2 on proliferation was seen
in the 10−6 mol/l group, where mfn2 was expressed at its
lowest level. These results demonstrated that E2 treatment resulted
in increased proliferation of MCF-7 cells, and that decreased mfn2
might play a positive role in this proliferation.
| Table IIE2 enhances proliferation of MCF-7
cells as quantified by MTT assay. |
Table II
E2 enhances proliferation of MCF-7
cells as quantified by MTT assay.
| Group (mol/l) | n | OD value (x ±
s) |
|---|
| Control | 6 | 0.45±0.18 |
|
10−9 | 6 | 0.54±0.10 |
|
10−8 | 6 |
0.62±0.16a |
|
10−7 | 6 |
0.71±0.15a |
|
10−6 | 6 |
0.97±0.06a |
|
10−5 | 6 |
0.89±0.11a |
To determine if E2 influenced cell motility, we
examined the ability of treated cells to migrate in a wound-healing
assay. In response to wounding the monolayer, the 10−6
mol/l group cells were able to almost completely heal the wound. In
contrast, the cells of other groups were unable to do so and
exhibited an obvious reduction in their rate of migration compared
to the 10−6 mol/l group. As compared with the
10−6 mol/l group cells, the reduction in the migration
rate of cells treated with E2 at 10−9 mol/l,
10−8 mol/l, 10−7 mol/l, 10−5 mol/l
and 0 mol/l were 64.3, 50.0, 31.4, 24.5 and 85.7%, respectively.
These results demonstrated that E2 also enhanced cell motility in a
dose-dependent manner (Fig.
2).
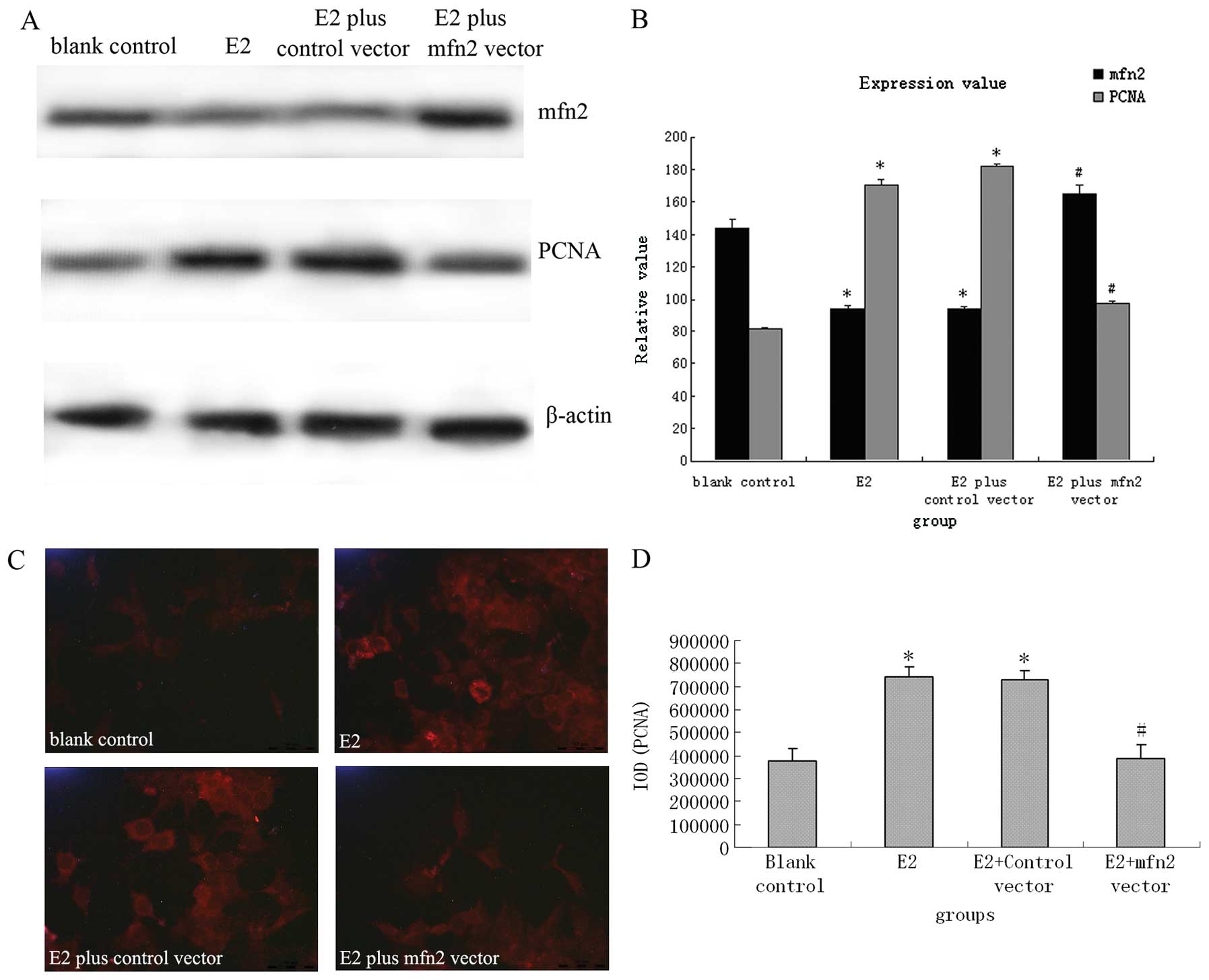
The mfn2 expression vector effectively
suppressed E2-induced upregulation of PCNA and migration in MCF-7
cells
A previous study demonstrated that mfn2 profoundly
suppresses cell growth and proliferation in multiple tumor cell
lines via inhibition of the Ras-ERK MAPK signaling pathway
(10). As described above, the
effect of E2 on MCF-7 cells might be partly dependent on inhibition
of mfn2. To investigate the involvement of mfn2 in E2-induced cell
proliferation and migration, MCF-7 cells were transfected with the
expression vector pEGFP-mfn2. As shown in Fig. 3A and B, normal cultured MCF-7 cells
(C) had standard expression levels of mfn2 and PCNA. However, both
untransfected MCF-7 cells stimulated with 10−6 mol/l E2
(E2), and control vector transfected cells stimulated with
10−6 mol/l E2 (E2+C) showed notably decreased mfn2
expression and enhanced PCNA expression. In comparison with MCF-7
cells transfected with control vector, mfn2 levels were increased
by 2.11-fold and PCNA levels were decreased by about 42.61% in
MCF-7 cells transfected with specific mfn2 expression vector
(E2+mfn2) (P<0.01). Consistent with the western blot analysis
results, immunofluorescence also revealed that the mfn2 vector
reversed E2-induced downregulation of PCNA protein (Fig. 3C and D).
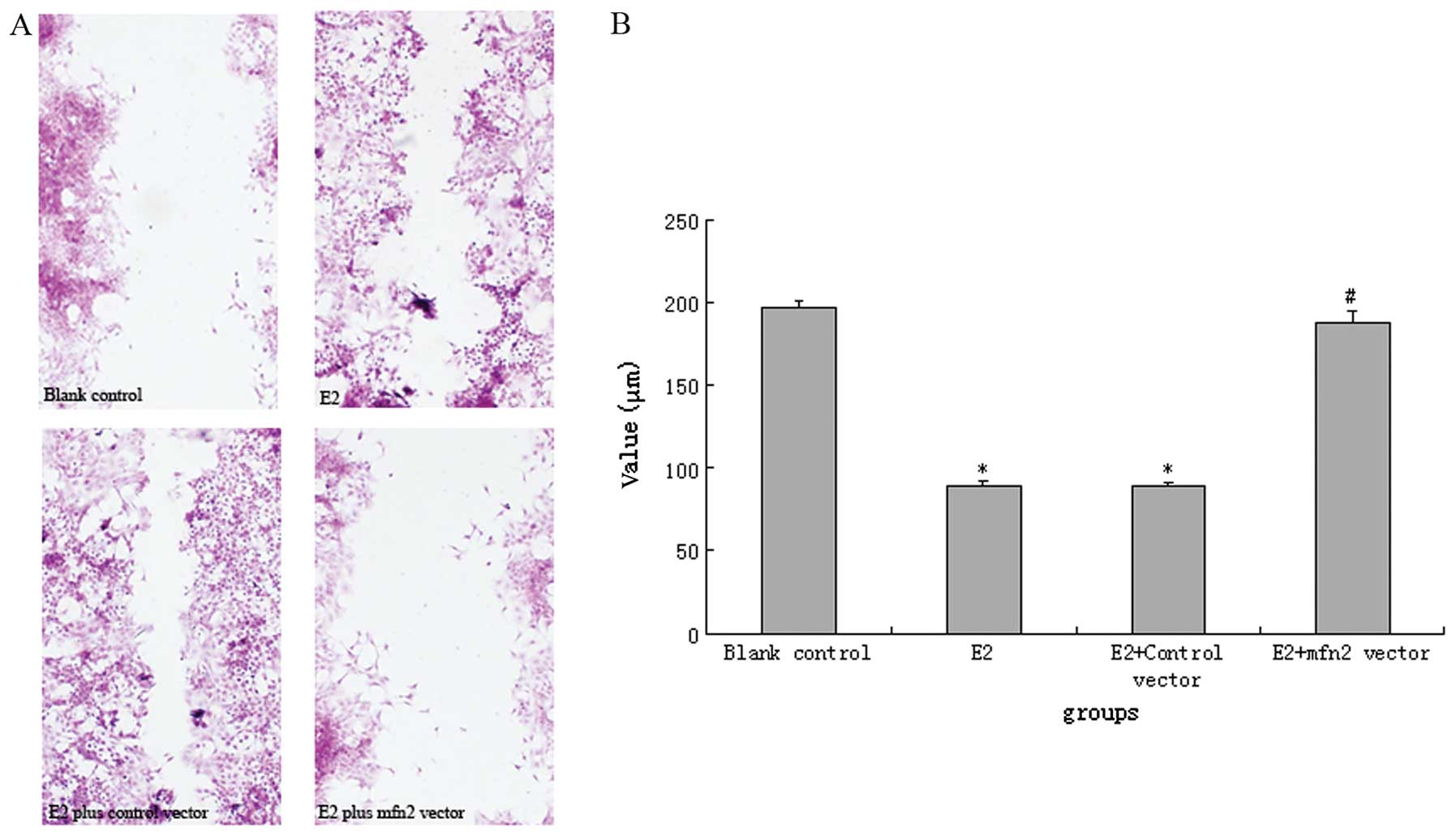
Cells transfected with the mfn2 vector showed
moderate resistance to E2 stimulation. In comparison with
E2-stimulated untransfected or control vector-transfected cells,
mfn2 expression vector-transfected cells demonstrated decreased
cell migration (Fig. 4). The
wound-healing assay indicated that MCF-7 cells and control
vector-transfected cells stimulated by E2 almost completely healed
the wound, as compared with unstimulated cells. However, this
alteration was reversed by transfection with the mfn2 expression
vector.
ERβ ameliorates E2-induced mfn2
downregulation in MCF-7 cells
As stated previously, estrogen’s effects are
mediated through two ERs, ERα and ERβ (3–6). We
hypothesized that ERβ might also act upstream of mfn2, because mfn2
is identically regulated by E2. To test our hypothesis, MCF-7 cells
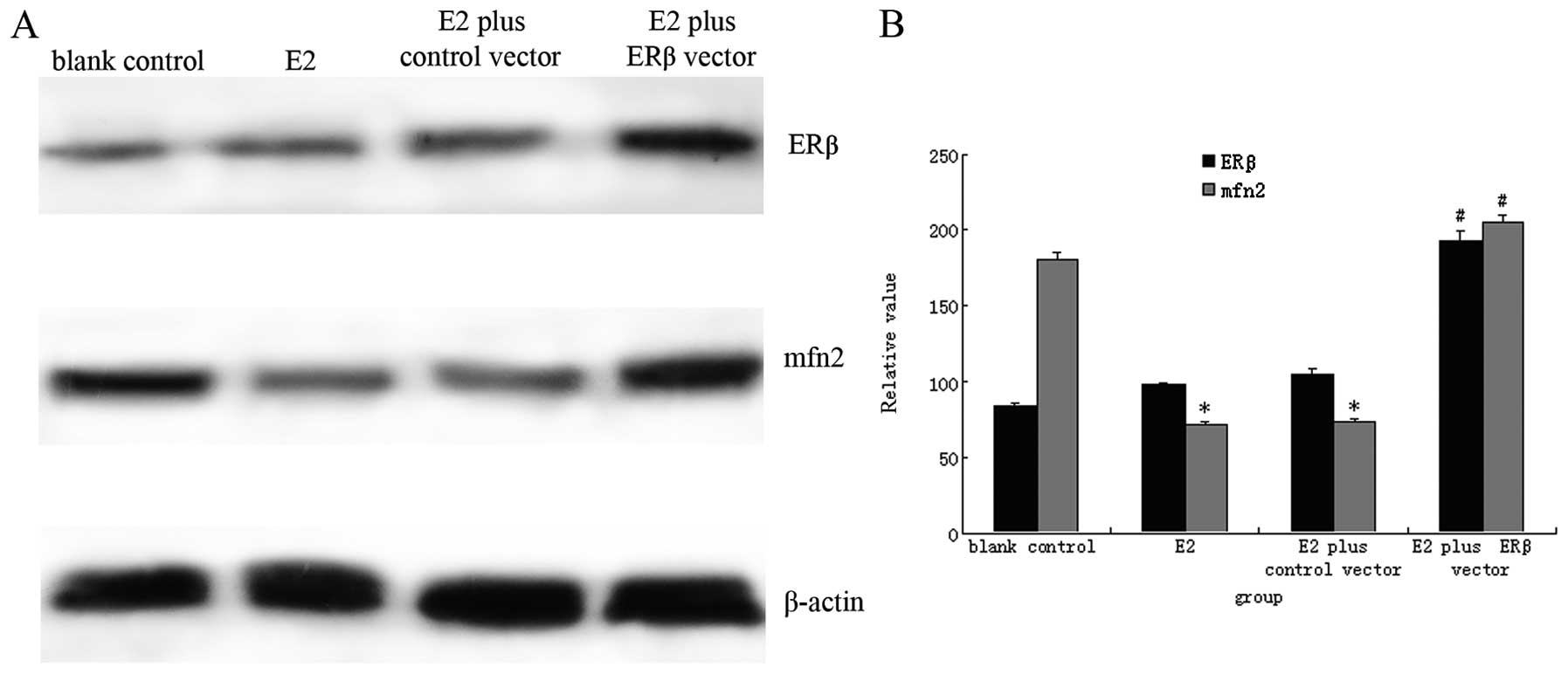
were transfected with an ERβ expression vector. As seen in Fig. 5, MCF-7 cells transfected with the
ERβ vector showed high ERβ protein expression after stimulation
with E2 for 24 h. However, no changes in ERβ protein levels were
found in MCF-7 cells transfected with blank control vector or in
untransfected MCF-7 cells. In comparison with MCF-7 cells
transfected with blank vector, ERβ protein was increased by about
2.25-fold in MCF-7 cells transfected with the ERβ vector
(P<0.01). Cells transfected with the ERβ vector showed
antagonistic effects on E2 stimulation; mfn2 protein was
upregulated in these cells as compared with blank
vector-transfected MCF-7 cells and untransfected cells treated with
E2. These results indicated that MCF-7 cells transfected with the
ERβ vector showed moderate resistance to E2 stimulation and
subsequent decreased downregulation of mfn2 protein (Fig. 5).
An ERβ expression vector effectively
suppressed E2-induced enhancement of proliferation and migration in
MCF-7 cells
The results above showed that mfn2 negatively
regulated E2-induced proliferation and migration of MCF-7 cells and
ERβ acted as an upstream signal of mfn2; therefore, we hypothesized
that ERβ could also inhibit proliferation and migration of MCF-7
cells. To investigate this hypothesis, MCF-7 cells were transfected
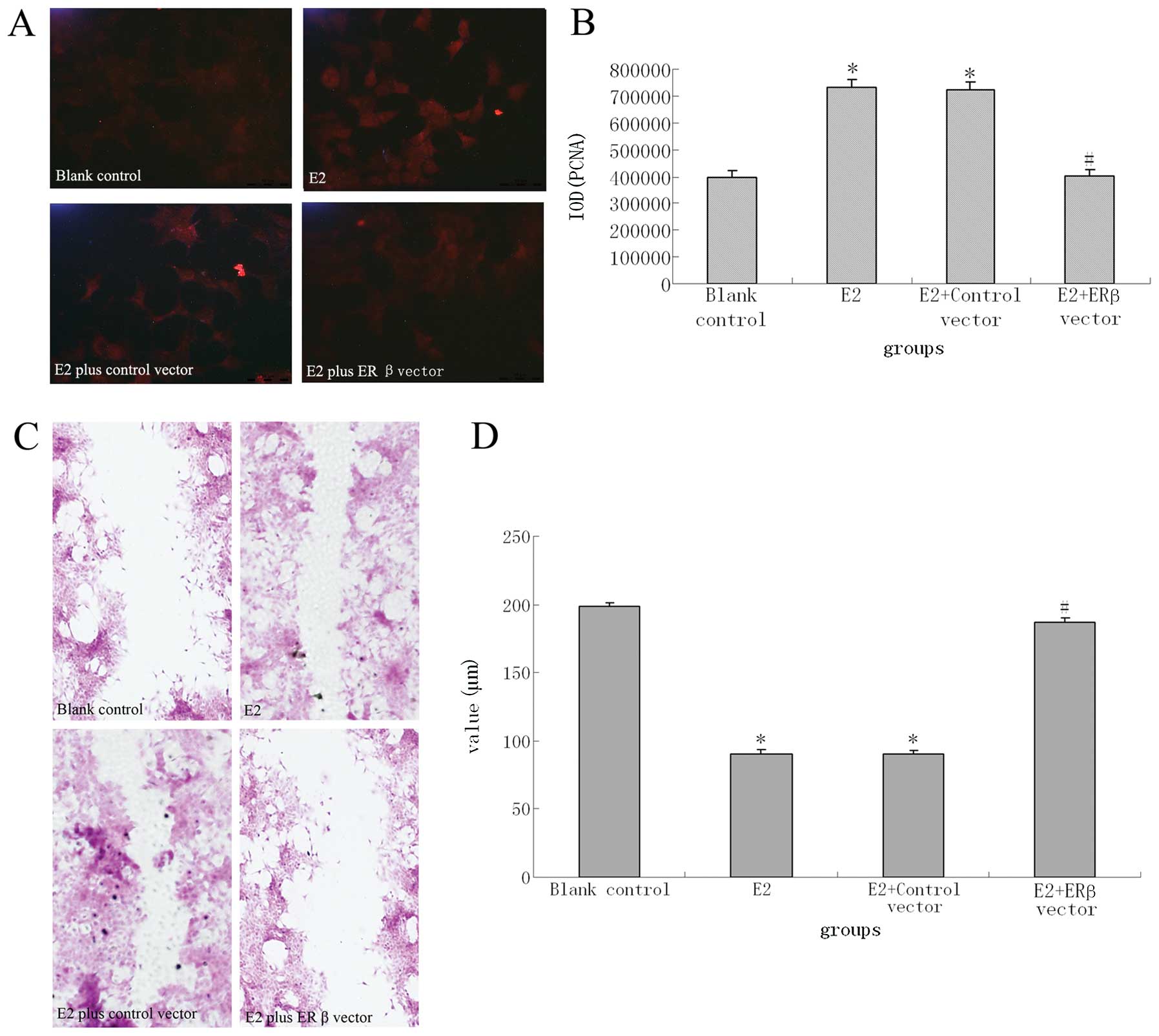
with the ERβ expression vector (pEGFP-N1-ESR2). An MTT assay was
used to examine the proliferation of MCF-7 cells. As shown in
Table III, there were significant
differences of absorbance of MTT substrate at 490 nm between
experimental groups and the control group. MCF-7 cells transfected
with the ERβ expression vector showed moderate resistance to E2
stimulation and did not exhibit the enhanced proliferation
demonstrated by blank vector-transfected and untransfected MCF-7
cells cultured with medium containing E2. The same results can also
be seen in the immunofluorescence detection of PCNA expression
(Fig. 6A and B).
| Table IIIERβ inhibits the E2-induced
proliferation of MCF-7 cells as quantified by MTT assay. |
Table III
ERβ inhibits the E2-induced
proliferation of MCF-7 cells as quantified by MTT assay.
| Group | n | OD value (x ±
s) |
|---|
| Blank control | 6 | 0.44±0.03 |
| E2 | 6 | 0.92±0.06a |
| E2 plus control
vector | 6 | 0.91±0.02a |
| E2 plus ERβ
vector | 6 | 0.50±0.04b |
To determine if ERβ influenced cell motility, we
examined the ability of transfected cells to migrate in a
wound-healing assay. As showed in Fig.
6C and D, there was decreased cell migration of ERβ expression
vector-transfected cells as compared with E2-stimulated
untransfected cells and control vector-transfected cells. In fact,
the wound-healing assay indicated that MCF-7 cells stimulated by E2
and cells transfected with control vector expressed a stronger
ability to heal the wound as compared with normal group cells
untreated with E2. However, this ability was reversed by
transfection with the ERβ vector.
Discussion
It is acknowledged that ERα and ERβ have distinct
roles in breast cancer cells. Although the majority considered that
ERα promotes proliferation and migration in breast cancer cells and
the function of ERα had been clearly elucidated, the exact roles of
ERβ in the pathogenesis of breast cancer are unclear. In fact, the
function of ERβ in the pathogenesis and development of breast
cancer is contradictory. Some studies indicate that ERβ may
function as a tumor suppressor and that the loss of ERβ may promote
breast carcinogenesis (16–18);
however, there are other studies suggesting that ERβ may promote
cell proliferation and breast tumor formation (19,20).
Regardless of these contradictions, the majority of studies focus
on the classical model of estrogen signaling through ERs, ERα and
ERβ, in which ERs act at ERE-containing promoters. In the classical
model, ligand-activated ER binds specifically to DNA at EREs
through its DNA binding domain and brings coactivators and
corepressors to the transcription site via its activator function
(AF)-1 and AF-2 domains. However, an increasing number of studies
show that estrogen also modulates gene expression by a second
mechanism in which the ER interacts with other transcription
factors through a process referred to as transcription factor
cross-talk. In this case, the ER modulates the activities of other
transcription factors such as activator protein (AP)-1, or SP-1 by
stabilizing their binding to DNA and/or recruiting coactivators to
the complex (21,23). DeNardo et al(14) reported a model of estrogen-ER
activation of AP-1 through interaction with existing coactivator
complexes that in turn stabilize the entire complex and/or induce
this complex into a higher state of activity. They also identified
6 estrogen-induced/AP-1 dependent genes, including mfn2, which
might fit this model. However, their conclusions were only
speculative, as they did not provide detailed data or investigated
the interaction of ERs and mfn2 in vitro. In this study, we
investigated the role of ERβ in estradiol-induced proliferation and
migration of human breast cancer cells and studied whether mfn2
participated in this behavior.
First, we explored whether E2 (17β-estradiol)
affected proliferation and migration of MCF-7 cells, a human breast
cancer cell line primarily expressing ERα and thus mimicking the
majority of ER-positive breast tumors. Similar to some previous
studies that revealed that E2 affected biological behavior of human
breast cells (24–26), our results showed that both the
proliferation and migration abilities of MCF-7 were significantly
increased when cultured with increasing doses of E2. Furthermore,
regulation was in a dose-dependent manner, with the maximum effect
seen in the 10−6 mol/l group. These data suggest that E2
and ERα are positive regulators of MCF-7 cells.
Whether mfn2 was involved in the initiation and
progression of human breast cancer has not been previously
reported. To investigate the role of mfn2, we observed the
expression of mfn2 in MCF-7 cells cultured within defferent doses
of E2. Interestingly, we found that E2 could decrease mfn2
expression in a dose-dependent manner, and that the changes in mfn2
levels were correlated with the proliferation and migration of
MCF-7 cells. These results indicated that mfn2 might negatively
regulate estradiol-induced proliferation and migration of MCF-7
cells. Furthermore, we found that introduction of mfn2 blocked the
response of MCF-7 cells to E2. Thus, mfn2 plays an important
regulatory role in E2-induced proliferation and migration of MCF-7
cells. Considering the reports that mfn2 is one of the
estrogen-induced/AP-1 dependent genes (14), the above results suggested that
mfn2 might negatively regulate E2-induced MCF-7 cell proliferation
and migration by a non-classical pathway. Mfn2, a
proliferation-inhibiting gene, targets to the outer membrane of
mitochondria. The mfn2 gene was found to play roles in the
inhibition of cellular proliferation and the promotion of apoptosis
(10) and exhibits antitumor
activity in a wide range of cancer cell lines (27–29),
suggesting that mfn2 may be important in the development of human
cancers. Again, the present study also provided a potential target
for prevention or treatment for breast cancer patients with ERα
positive expression.
Approximately 70% of breast tumors express ERβ, and
most tumors coexpress both ERα and ERβ (30,31).
However, whether ERβ is involved in E2-induced downregulation of
mfn2 is still unknown. Clearly, additional studies are needed to
clarify the role of ERβ in breast cancer. In the present study, we
introduced ERβ into MCF-7 cells and investigated the effects of ERβ
on proliferation and migration of MCF-7 cells as well as its
effects on mfn2 expression. Our studies demonstrate that ERβ
changes the phenotype of MCF-7 cells in response to E2. In
ERα-expressing MCF-7 cells, E2 causes proliferation and migration,
as well as suppression of mfn2. In contrast, when ERβ is expressed
along with ERα, MCF-7 cells are directed to antitumor pathways and
high levels of mfn2 even in the presence of estrogens. These
results suggest that ERβ can alter the response of MCF-7 to
estrogens and demonstrate that ERβ may function as a tumor
suppressor through the mfn2 pathway. Many cell-based studies
suggest that ERβ acts as a negative modulator of ERα action. When
ERα and ERβ are co-transfected into ER negative (ER-) cells, ERβ
inhibits ERα transcriptional activity and decreases the sensitivity
of the cells to E2 (6,32). ERβ also lowers both ERα mRNA and
protein levels in MCF-7 cells, thus indirectly influencing function
of ERα (33,34). ERβ overexpression in MCF-7 breast
cancer cells can not only inhibit ERα regulation of a subset of
genes involved in DNA replication, cell-cycle regulation, and
proliferation (35,36), but also inhibit cell proliferation
in response to E2 (34,36–38),
in part by increasing expression of antiproliferative genes
(p21Cip1 and p27Kip1). Our results were quite
similar to these reports, and minor deference lie in downstream
factors. There may be diverse mechanisms for the effect of ERβ on
the response of ERα to E2. Recently, some studies revealed in
series that the responses of breast cancer cell lines to
17β-estradiol are dependent on the ERα/ERβ ratio (39,40).
Most importantly, ERβ might regulate mfn2 expression directly in a
non-classical pathway similar to ERα. Therefore, further studies
are needed to determine the exact mechanisms of interaction between
ERs, ERα and ERβ, and mfn2, especially to delineate the mechanism
of action through experiments such as in-depth promoter analysis
and CHIP.
References
|
1
|
MacGregor JI and Jordan VC: Basic guide to
the mechanisms of antiestrogen action. Pharmacol Rev. 50:151–196.
1998.PubMed/NCBI
|
|
2
|
Sommer S and Fuqua SA: Estrogen receptor
and breast cancer. Semin Cancer Biol. 11:339–352. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Green S, Walter P, Greene G, et al:
Cloning of the human oestrogen receptor cDNA. J Steroid Biochem.
24:77–83. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kuiper GG, Enmark E, Pelto-Huikko M, et
al: Cloning of a novel receptor expressed in rat prostate and
ovary. Proc Natl Acad Sci USA. 93:5925–5930. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Katzenellenbogen BS, Montano MM, Ediger
TR, et al: Estrogen receptors: selective ligands, partners, and
distinctive pharmacology. Recent Prog Horm Res. 55:163–193.
2000.PubMed/NCBI
|
|
6
|
Nilsson S, Makela S, Treuter E, et al:
Mechanisms of estrogen action. Physiol Rev. 81:1535–1565. 2001.
|
|
7
|
Speirs V, Carder PJ, Lane S, et al:
Oestrogen receptor β: what it means for patients with breast
cancer. Lancet Oncol. 5:174–181. 2004.
|
|
8
|
Dahlman-Wright K, Cavailles V, Fuqua SA,
et al: International Union of Pharmacology. LXIV Estrogen
receptors. Pharmacol Rev. 58:773–781. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Harris HA: Estrogen receptor-β: recent
lessons from in vivo studies. Mol Endocrinol. 21:1–13. 2007.
|
|
10
|
Chen KH, Guo X, Ma D, et al: Dysregulation
of HSG triggers vascular proliferative disorders. Nat Cell Biol.
6:872–883. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Neuspiel M, Zunino R, Gangaraju S, et al:
Activated mitofusin 2 signals mitochondrial fusion, interferes with
Bax activation, and reduces susceptibility to radical induced
depolarization. J Biol Chem. 280:25060–25070. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Santel A, Frank S, Gaume B, et al:
Mitofusin-1 protein is a generally expressed mediator of
mitochondrial fusion in mammalian cells. J Cell Sci. 116:2763–2774.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sugioka R, Shimizu S and Tsujimoto Y:
Fzo1, a protein involved in mitochondrial fusion, inhibits
apoptosis. J Biol Chem. 279:52726–52734. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
DeNardo DG, Kim HT, Hilsenbeck S, et al:
Global gene expression analysis of estrogen receptor transcription
factor cross talk in breast cancer: identification of
estrogen-induced/activator protein-1-dependent genes. Molecular
Endocrinology. 19:362–378. 2005. View Article : Google Scholar
|
|
15
|
Ma L, Liu YP, Geng CZ, et al: Low-dose
epirubicin inhibits ezrin-mediated metastatic behavior of breast
cancer cells. Tumori. 97:400–405. 2011.PubMed/NCBI
|
|
16
|
Ström A, Hartman J, Foster JS, et al:
Estrogen receptor beta inhibits 17beta-estradiol-stimulated
proliferation of the breast cancer cell line T47D. Proc Natl Acad
Sci USA. 101:1566–1571. 2004.PubMed/NCBI
|
|
17
|
Secreto FJ, Monroe DG, Dutta S, et al:
Estrogen receptor alpha/beta isoforms, but not betacx, modulate
unique patterns of gene expression and cell proliferation in Hs578T
cells. J Cell Biochem. 101:1125–1147. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen L, Qiu J, Yang C, et al:
Identification of a novel estrogen receptor beta1 binding partner,
inhibitor of differentiation-1, and role of ERbeta1 in human breast
cancer cells. Cancer Lett. 278:210–219. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jensen EV, Cheng G, Palmieri C, et al:
Estrogen receptors and proliferation markers in primary and
recurrent breast cancer. Proc Natl Acad Sci USA. 4:42001.PubMed/NCBI
|
|
20
|
Speirs V, Malone C, Walton DS, et al:
Increased expression of estrogen receptor β mRNA in
tamoxifen-resistant breast cancer patients. Cancer Res.
59:5421–5424. 1999.
|
|
21
|
Kushner PJ, Agard DA, Greene GL, et al:
Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol.
74:311–317. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang W, Dong L, Saville B, et al:
Transcriptional activation of E2F1 gene expression by 17β-estradiol
in MCF-7 cells is regulated by NF-Y-Sp1/estrogen receptor
interactions. Mol Endocrinol. 13:1373–1387. 1999.
|
|
23
|
Safe S: Transcriptional activation of
genes by 17β-estradiol through estrogen receptor-Sp1 interactions.
Vitam Horm. 62:231–252. 2001.
|
|
24
|
Ru Lee W, Chen CC, Liu S, et al: 17
beta-estradiol (E2) induces cdc25A gene expression in breast cancer
cells by genomic and non-genomic pathways. J Cell Biochem.
99:209–220. 2006.PubMed/NCBI
|
|
25
|
Yoshioka H, Hiromori Y, Aoki A, et al:
Possible aryl hydrocarbon receptor-independent pathway of 2, 3, 7,
8-tetrachlorodibenzo-p-dioxin-induced antiproliferative response in
human breast cancer cells. Toxicol Lett. 211:257–265. 2012.
View Article : Google Scholar
|
|
26
|
Singh KP, Treas J, Tyagi T, et al: DNA
demethylation by 5-aza-2-deoxycytidine treatment abrogates 17
beta-estradiol-induced cell growth and restores expression of DNA
repair genes in human breast cancer cells. Cancer Lett. 316:62–69.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang W, Zhou D, Wei J, et al: Hepatitis B
virus X protein inhibits p53-mediated upregulation of mitofusin-2
in hepatocellular carcinoma cells. Biochem Biophys Res Commun.
421:355–360. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jin B, Fu G, Pan H, et al: Anti-tumour
efficacy of mitofusin-2 in urinary bladder carcinoma. Med Oncol.
28(Suppl 1): S373–S380. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rehman J, Zhang HJ, Toth PT, et al:
Inhibition of mitochondrial fission prevents cell cycle progression
in lung cancer. FASEB J. 26:2175–2186. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dotzlaw H, Leygue E, Watson PH, et al:
Expression of estrogen receptor-β in human breast tumors. J Clin
Endocrinol Metab. 82:2371–2374. 1997.
|
|
31
|
Fuqua SA, Schiff R, Parra I, et al:
Estrogen receptor β protein in human breast cancer: correlation
with clinical tumor parameters. Cancer Res. 63:2434–2439. 2003.
|
|
32
|
Pettersson K, Delaunay F and Gustafsson
JA: Estrogen receptor β acts as a dominant regulator of estrogen
signaling. Oncogene. 19:4970–4978. 2000.
|
|
33
|
Matthews J, Wihlen B, Tujague M, et al:
Estrogen receptor (ER) β modulates ER α-mediated transcriptional
activation by altering the recruitment of c-Fos and c-Jun to
estrogen-responsive promoters. Mol Endocrinol. 20:534–543.
2006.
|
|
34
|
Chang EC, Frasor J, Komm B, et al: Impact
of estrogen receptor β on gene networks regulated by estrogen
receptor α in breast cancer cells. Endocrinology. 147:4831–4842.
2006.
|
|
35
|
Lin CY, Strom A, Li Kong S, et al:
Inhibitory effects of estrogen receptor beta on specific
hormone-responsive gene expression and association with disease
outcome in primary breast cancer. Breast Cancer Res. 9:R252007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Williams C, Edvardsson K, Lewandowski SA,
et al: A genome-wide study of repressive effects of estrogen
receptor beta on estrogen receptor alpha signaling in breast cancer
cells. Oncogene. 27:1019–1032. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Paruthiyil S, Parmar H, Kerekatte V, et
al: Estrogen receptor β inhibits human breast cancer cell
proliferation and tumor formation by causing a G2 cell cycle
arrest. Cancer Res. 64:423–428. 2004.
|
|
38
|
Behrens D, Gill JH and Fichtner I: Loss of
tumourigenicity of stably ER β-transfected MCF-7 breast cancer
cells. Mol Cell Endocrinol. 274:19–29. 2007.PubMed/NCBI
|
|
39
|
Nadal-Serrano M, Sastre-Serra J, Pons DG,
et al: The ERalpha/ERbeta ratio determines oxidative stress in
breast cancer cell lines in response to 17beta-estradiol. J Cell
Biochem. 113:3178–3185. 2012. View Article : Google Scholar
|
|
40
|
Sastre-Serra J, Nadal-Serrano M, Pons DG,
et al: The effects of 17β-estradiol on mitochondrial biogenesis and
function in breast cancer cell lines are dependent on the ERα/ERβ
ratio. Cell Physiol Biochem. 29:261–268. 2012.
|