Introduction
To discover new genes that play a crucial role in
common tumorigenesis regardless of tissue origin, we analyzed
commonly upregulated unknown genes in 242 normal and 300 tumor
samples originating from 11 different tissues using the Affymetrix
gene chip system. An Affymetrix fragment, later named cancer
upregulated gene (CUG) 2 was identified as a candidate gene that is
commonly upregulated in various tumor tissues such as ovary, liver,
lung and colon. This CUG2 was mapped to chromosome 6q22.32; it
spans ∼8.5 kb with a three-exon structure and encodes a 88-amino
acid poly-peptide (1). Further
study has revealed that CUG2 is a new component of centromere
required for a proper kinetochore function during cell division
(2). Of interest, CUG2 has been
shown to harbor an oncogenic effect in a transplanted model using
NIH3T3 cells expressing CUG2, in a manner similar to Ras (1). Although CUG2 overexpression leads to
activated Ras and MAPKs including p38 MAPK, which eventually
facilitates oncolytic reoviral replication (3), CUG2 contrastingly provides resistance
to oncolytic vesicular stomatitis virus infection through
activation of STAT1 as shown in our recent study (4).
Although STAT1 is well known as a master
transcription factor for IFN-related intracellular signaling,
leading to antiviral activity, several lines of evidence have shown
that STAT1 plays a role as an anti-oncogenic molecule in part by
upregulation of caspases (5,6),
cyclin-dependent kinase inhibitor 1A (7), the IFN-regulatory Factor 1 (IRF1)/p53
pathway (8), and downregulation of
the BCL2 family (9). In contrast,
emerging data reveal that in certain cellular contexts the
IFN/STAT1 pathway may facilitate tumor cell growth. Other studies
have reported that resistance to ionizing radiation and IFNs is
associated with constitutive overexpression of the IFN/STAT1
pathway in radio-resistant tumor cells (10). Recent studies have also
demonstrated that constitutive over-expression of STAT1 is
positively correlated with protection of tumor cells from genotoxic
stress such as treatment with doxorubicin (11), or cisplatin (12). Furthermore, since over-expression
of the IFN/STAT1 pathway is associated with poor prognosis in
different types of cancer, the IFN-related genes are suggested as
predictive markers for breast cancer patients resistant to the
adjuvant chemotherapy (13).
This study was initiated to explore further the
possible biological consequence of the activation of STAT1 mediated
by CUG2 during the development of cancer. We herein report that the
CUG2-induced activation of STAT1 promotes both enhanced cell
migration and resistance to an anticancer drug such as doxorubicin,
eventually contributing to the metastasis and progression of
cancer.
Materials and methods
Cell cultures
Colon26L5 cells, derived from mouse colon cancer and
modified so that they stably express either vector alone
(Colon26L5-Vec) or CUG2 (Colon26L5-CUG2), were cultured in DMEM
supplemented with 10% FBS, 1% penicillin and 1% streptomycin,
puromycin (Sigma-Aldrich, St. Louis, MO; 2 μg/ml) under 37°C
and 5% CO2. In addition, Colon26L5-CUG2 cells stably
suppressing STAT1 using shRNA (Colon26L5-CUG2-shSTAT1) and the
control cells (Colon26L5-CUG2-shCon) were cultured in the same
condition except with zeocin (Calbiochem, San Jose, CA, USA; 100
μg/ml).
Reagents and antibodies
For inhibition of protein kinases, wortmannin,
PD98059, SB203580 and Jak inhibitor I were purchased from
Calbiochem. For immunoblotting, anti-STAT1, STAT2, Jak1, Tyk2 and
their phospho-specific antibodies were acquired from Cell Signaling
Biotechnology (Danvers, MA, USA). In addition, anti-caspase-8
antibody was purchased from Cell Signaling Biotechnology.
Anti-β-actin antibody was obtained from Santa Cruz Biotechnology
(Santa Cruz, CA, USA) and anti-VSV glycoprotein (G) antibody was
obtained from Abcam (Cambridge, MA, USA).
Western blotting
Cells were harvested and lysed with lysis buffer
containing 1% NP-40 and protease inhibitors (Sigma-Aldrich). For
immunoblotting, proteins from whole cell lysates were resolved by
10 or 12% SDS-polyacrylamide gel electrophoresis (PAGE) and then
transferred to nitro cellulose membranes. Primary antibodies were
used at 1:1000 or 1:2000 dilutions, and secondary antibodies
conjugated with horse-radish peroxidase were used at 1:2000
dilutions in 5% nonfat dry milk. After the final washing, the
membranes were exposed for an enhanced chemiluminescence assay
using the LAS 4000 mini (Fuji, Tokyo, Japan).
Short interference RNA transfection
Cells were trypsinized and incubated overnight to
achieve 60–70% confluency before siRNA transfection. STAT1 siRNA
(commercially pre-made at Bioneer, Daejeon, Korea) or a negative
control siRNA (Bioneer) were mixed with Lipofectamine 2000
(Invitrogen, Carlsbad, CA, USA). The cells were incubated with the
transfection mixture for 6 h and then rinsed with DMEM containing
10% FBS. The cells were incubated for 48 h before harvest.
Cell migration assay
Migration assays were performed using 48-well Boyden
chambers (Neuroprobe, Gaitherburg, MD, USA) as described elsewhere
(14). The lower wells of the
chamber were filled with standard culture media. The chamber was
assembled using polycarbonate filters (Neuroprobe). Cells in
serum-free media (5×104 cells/well) were seeded in the
upper compartment of the chamber. After incubation for 24 h, cell
migration was quantified by counting the number of migrated cells
after staining with hematoxylin and eosin.
Wound healing assay
Cell migration was assessed using a scratch wound
assay (15). Briefly,
Colon26L5-CUG2-shCon or Colon26L5-CUG2-shSTAT cells were cultured
in six-well plates (5×105 cells/well). When the cells
were grown to 90% confluence, a single wound was made in the center
of the cell monolayer using a P-200 pipette tip. At 0 and 48 h of
incubation, the wound closure areas were visualized by
phase-contrast microscopy (Olympus, Tokyo, Japan) with a
magnification of ×100.
Reverse transcriptase-polymerase chain
reaction (RT-PCR)
Total RNA was extracted from cells using the RNeasy
mini kit (Qiagen, Valencia, CA, USA) in accordance with the
manufacturer’s instructions. Total RNA (3 μg) was converted
to cDNA using Superscript II reverse transcriptase (Invitrogen),
and PCR was performed using the specific primers (sense primer:
5′-GCGCTGTCGACCATAGTCTCCCAGAGG AAG-3′; anti-sense primer:
5′-CTAACCTCTGCTCTTCTTTAGAATTACCTTTGCTGC-3′). The cDNA products of
each reaction were diluted, and PCR was run at the optimized cycle
number. β-actin mRNA was used as an internal standard.
Statistical analysis
Data are presented as a means ± standard deviation.
Student’s t-test was used for statistical analysis, with p-value
<0.05 defined as significance.
Results
Akt-ERK signaling axis is involved in
CUG2-mediated STAT1 activation
As we earlier reported that CUG2, a novel oncogene,
induces activation of STAT1 in murine NIH3T3 fibroblast cells
(4), we next decided to explore
possible mechanisms by which the activation of STAT1 by CUG2 may be
involved in the development of cancer. To address this question, we
first established Colon26L5 cells, derived from a murine colon cell
line, stably expressing a vector only (Colon26L5-Vec), or human
CUG2 gene (Colon26L5-CUG2). Expression of human CUG2 transcripts
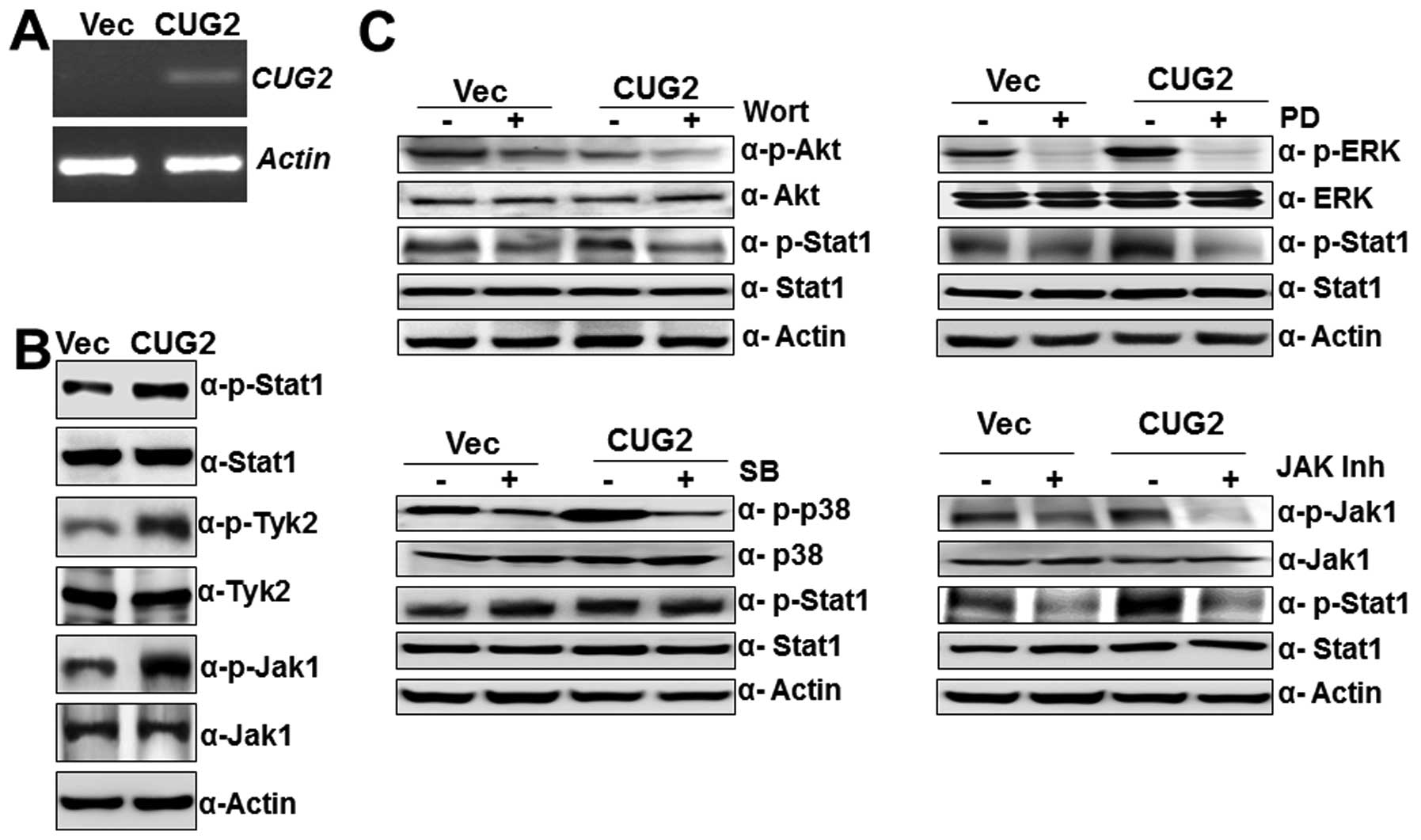
were confirmed by RT-PCR in Colon26L5-CUG2 cells (Fig. 1A). We then observed that
Colon26L5-CUG2 cells also exhibited higher levels of phosphorylated
STAT1 that were higher than in controls, as we had seen earlier in
NIH3T3 cells stably expressing CUG2 (NIHCUG2) cells. Furthermore,
phosphorylation of Jak1 and Tyk2, downstream molecules of type I
IFN signaling, was also seen when compared to Colon26L5-Vec cells
(Fig. 1B). The results indicate
that CUG2 expression activates STAT1 signaling in Colon26L5 cells
without ligand stimulation.
We next explored which signaling pathways are
involved in CUG2-mediated STAT1 activation. To answer this
question, we treated Colon26L5-CUG2 cells with wortmannin (Akt
inhibitor, 7 μM), PD98059 (ERK inhibitor, 30 μM),
SB203580 (p38 MAPK inhibitor, 30 μM), or Jak1 inhibitor I
(80 μM). Inhibition of Jak1, an up-stream signaling molecule
of STAT1, reduced phosphorylation of STAT1 in Colon26L5-CUG2 cells
as expected (Fig. 1C). Moreover,
we found that treatment with wortmannin and PD98059 significantly
suppressed STAT1 phosphorylation whereas SB203580 treatment did not
significantly reduce phosphorylation of STAT1 in Colon26L5-CUG2
cells (Fig. 1C), indicating that a
cell proliferation signaling pathway is involved in CUG2-induced
STAT1 activation, but stress-related signaling may not be.
CUG2-mediated STAT1 promotes migration
and wound healing of Colon26L5 cells
To explore whether CUG2-mediated activation of STAT1
is directly related to tumor progress and metastasis, we first
examined cell migration in Colon26L5-CUG2 cells under suppression
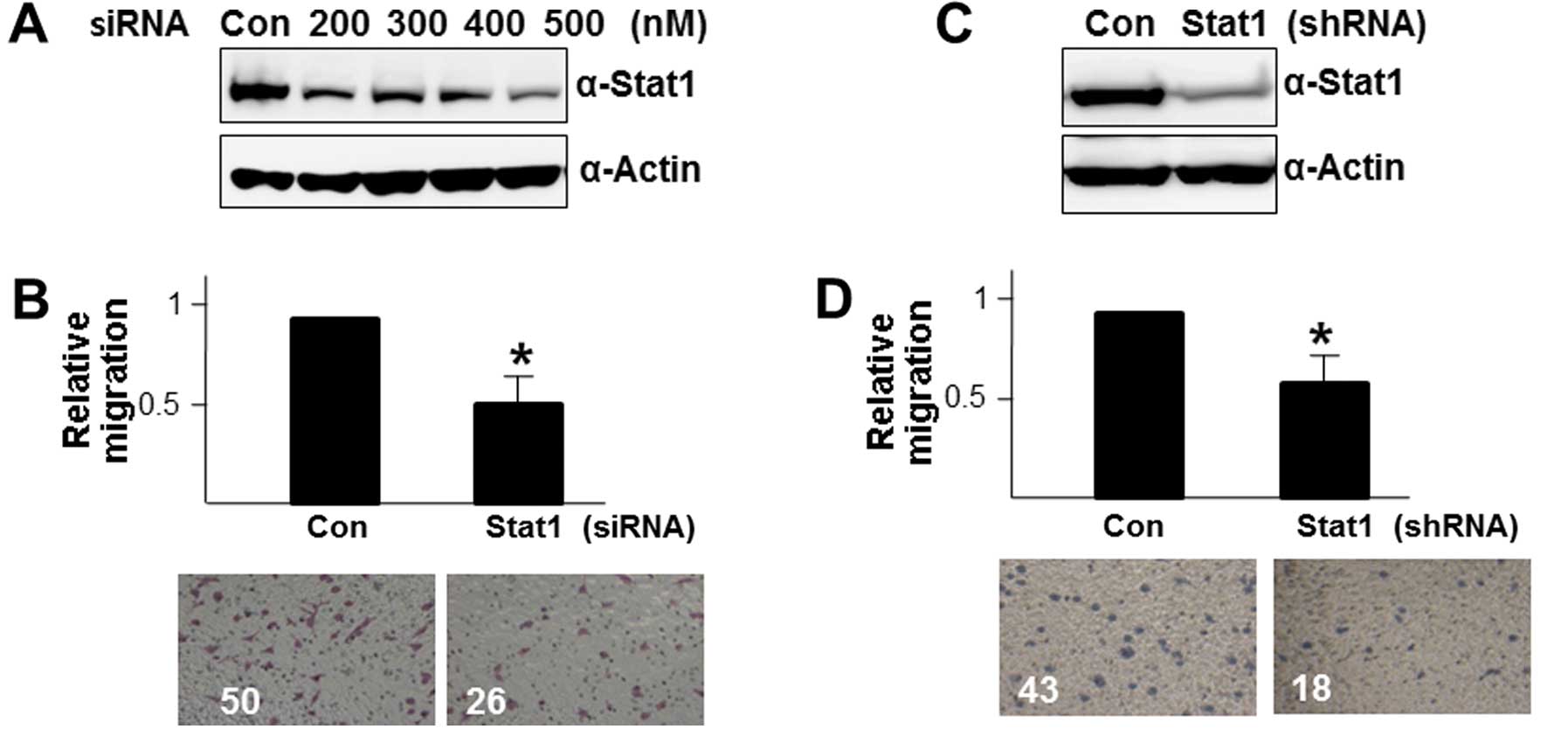
of STAT1. To address this issue, we transiently introduced STAT1
siRNA into Colon26L5-CUG2 cells and optimized the inhibitory
concentration of STAT1 siRNA at 500 nM (Fig. 2A). We then counted the cells
migrated from serum-free medium to serum-containing medium after
transfection of Colon26L5-CUG2 cells with STAT1 siRNA. We observed
that the number of migrating Colon26L5-CUG2 cells transfected with
STAT1 siRNA is significantly less than that of Colon26L5-CUG2 cells
transfected with control siRNA in the serum-containing well
(p<0.05; Fig. 2B), indicating
that STAT1 expression is required for the efficient migration of
colon cancer cells. To confirm this result, we introduced the
shSTAT1 vector into Colon26L5-CUG2 cells and established
Colon26L5-CUG2 cells stably suppressing STAT1
(Colon26L5-CUG2-shSTAT1; Fig. 2C).
We thereafter compared cell migration between
Colon26L5-CUG2-shSTAT1 and Colon26L5-CUG2-shCon cells as a control.
We observed the same pattern of cell migration as seen in
Colon26L5-CUG2 cells transiently transfected with STAT1 siRNA and
control siRNA (p<0.05; Fig.
2D). In parallel experiments, we also observed that suppression
of STAT1 expression with the shSTAT1 vector reduced migration of
NIH-CUG2 cells (data not shown).
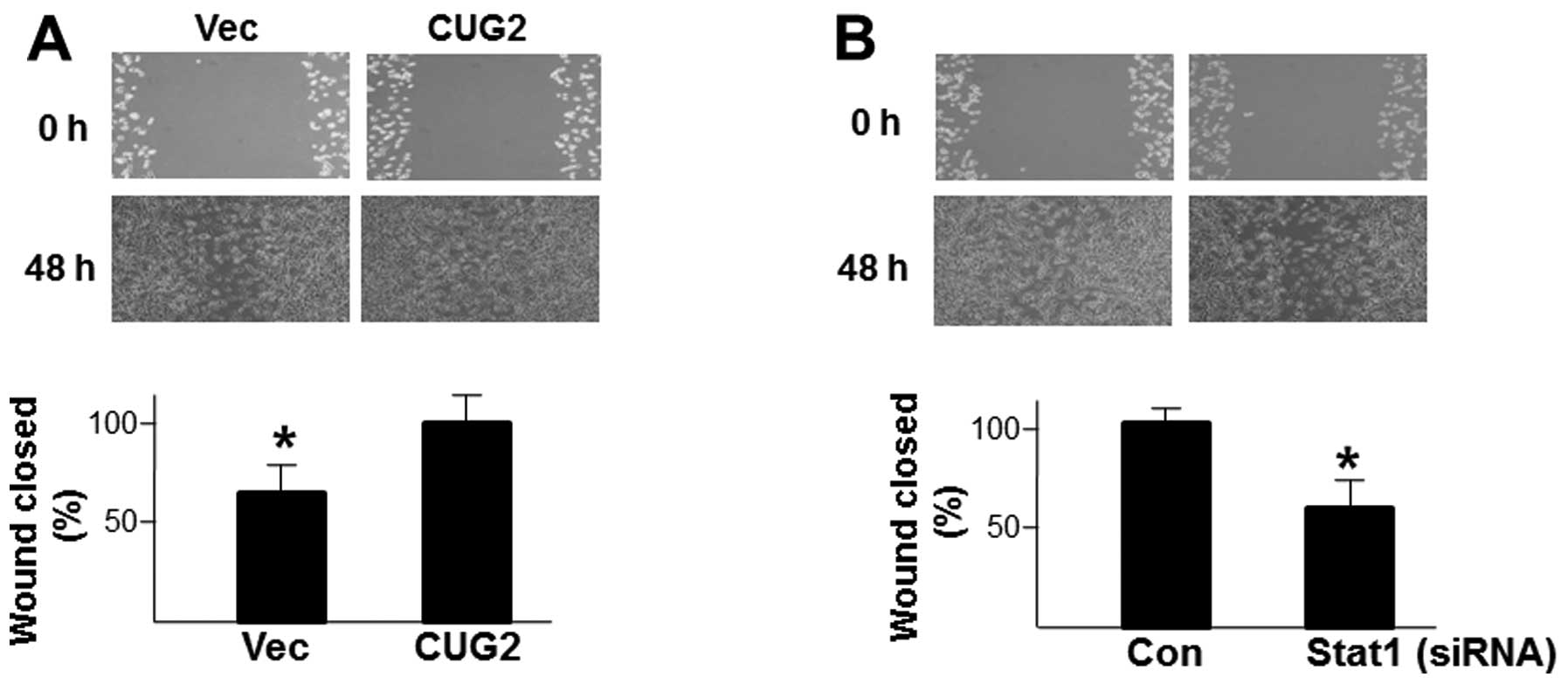
In addition, when we compared the capacity for cell
locomotion during wound healing between Colon26L5-Vec and
Colon26L5-CUG2 cells, we found that Colon26L5-CUG2 cells show
faster healing than Colon26L5-Vec cells (p<0.05; Fig. 3A). The result indicates that
upregulation of CUG2 elevates the ability of wound healing in colon
cells. To test whether STAT1 is a critical molecule in enhancing
the ability of wound healing, we examined wound healing in
Colon26L5 cells with suppression of STAT1 protein levels. We found
that Colon26L5-CUG2 cells transfected with control siRNA exhibited
faster healing from wounds than did Colon26L5-CUG2 cells
transfected with STAT1 siRNA (p<0.05; Fig. 3B). Based on these results, we
suggest that STAT1 is required for cell migration and wound healing
of colon cancer cells over-expressing CUG2.
CUG2-mediated STAT1 activation confers
Colon26L5 cells resistance to doxorubicin-induced apoptosis
To explore another possible biological consequence
of STAT1 activation induced by CUG2, we examined whether
upregulation of CUG2 provides resistance to an anticancer drug such
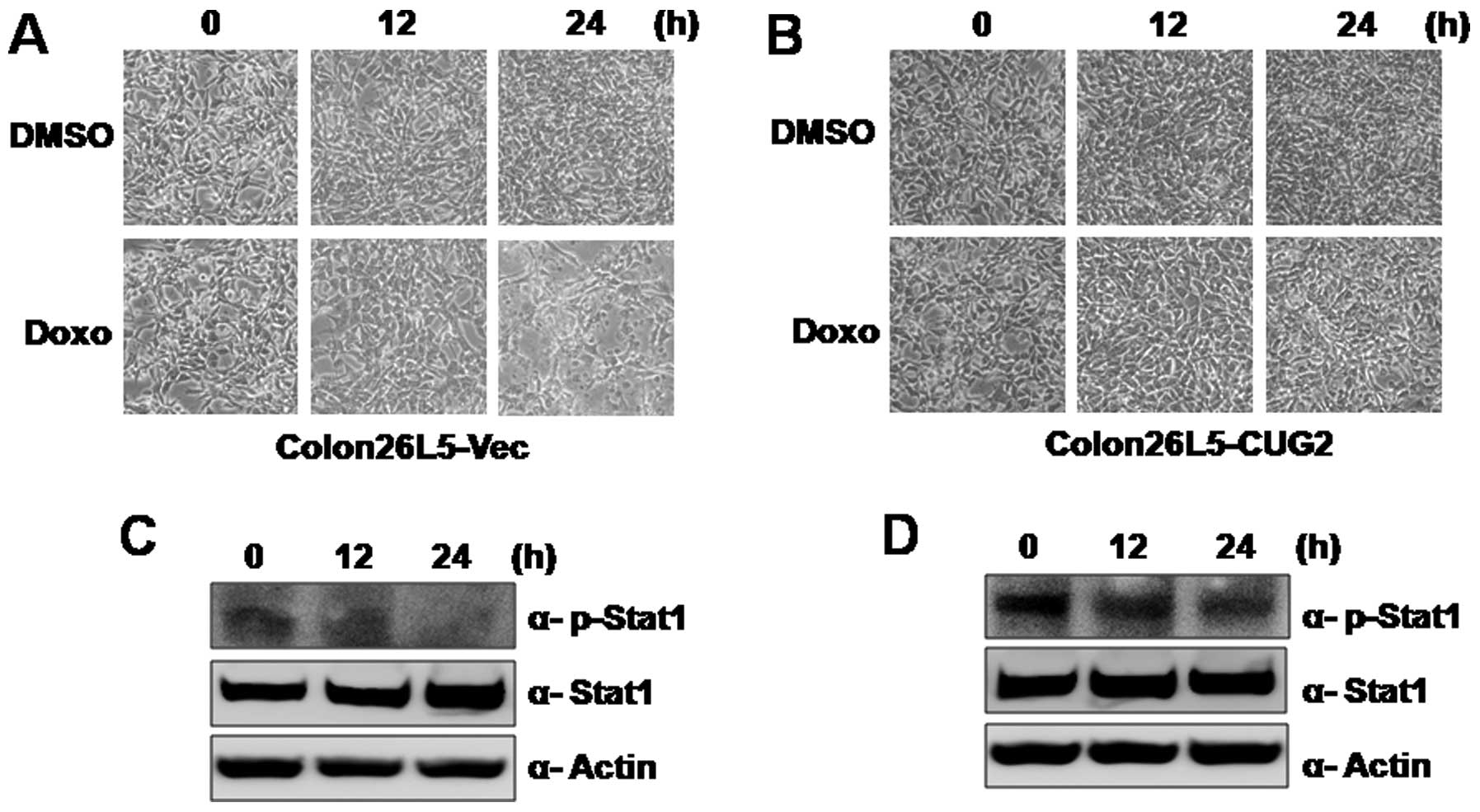
as doxorubicin. When Colon26L5-Vec and Colon26L5-CUG2 cells were
treated with doxorubicin, Colon26L5-Vec cells were highly
susceptible to cell death during doxorubicin treatment (Fig. 4A). However, Colon26L5-CUG2 cells
were resistant to doxorubicin (Fig.
4B). In addition, when we examined phosphorylation levels of
STAT1 in Colon26L5-Vec and Colon26L5-CUG2 cells, we found that
phosphorylation of STAT1 was decreased in Colon26L5-Vec cells while
the phosphorylated STAT1 level was maintained in Colon26L5-CUG2
cells during doxorubicin treatment for 24 h (Fig. 4C and D). This result suggests that
CUG2 confers resistance in Colon26L5 cells to doxorubicin through
STAT1 activation.
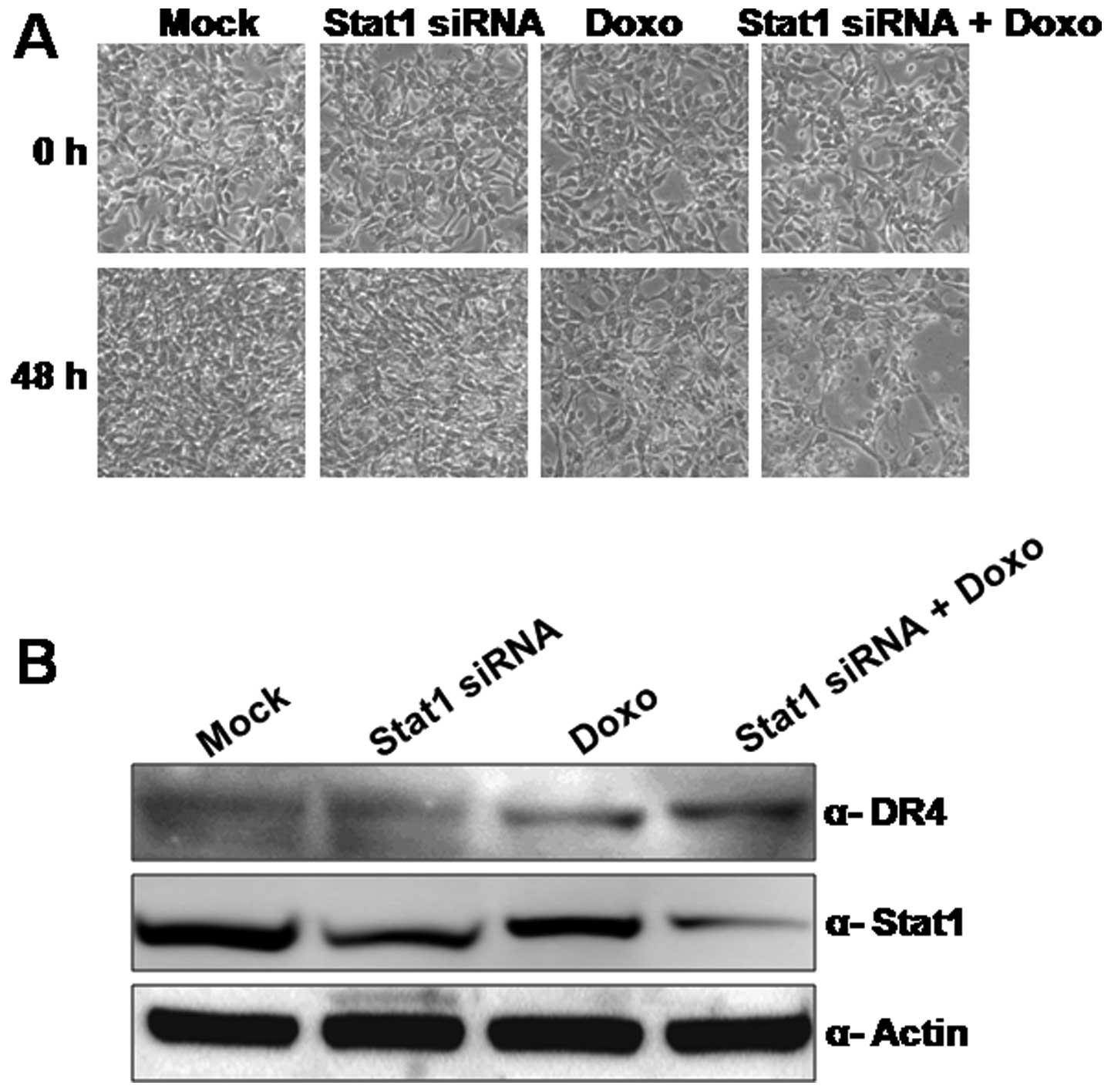
To confirm whether CUG2-mediated activation of STAT1
affects survival of colon cancer cells under doxorubicin treatment,
we treated Colon26L5-CUG2 cells with doxorubicin alone, STAT1 siRNA
alone or doxorubicin plus STAT1 siRNA. Colon26L5-CUG2 cells
survived in up to 500 μg/ml doxorubicin for 48 h (Fig. 5A). However, the combined treatment
sensitized doxorubicin-induced cell death. We observed cell death
beginning from 24 h post-treatment of doxorubicin under suppression
of STAT1 expression (data not shown) and clearly found extensive
cell death of Colon26L5-CUG2 cells at 48 h. This cell death
resulted from apoptosis induced by doxorubicin because increase of
death receptor (DR) 4 expression was found in the co-treatment
(Fig. 5B). Furthermore, we found
that NIH-CUG2-shSTAT1 cells were more susceptible to doxorubicin
than the NIH-CUG2-shCon cells (data not shown). These results
indicate that STAT1 confers resistance to doxorubicin-induced
apoptosis in Colon26L5-CUG2 cells. The result suggests that STAT
may be an important anticancer therapeutic target in cancer cells
overexpressing CUG2.
Discussion
Many studies have reported that STAT1 plays a role
as tumor suppressor. For example, one study has shown that STAT1 is
an important component of ErB2/Neu signaling that determines the
transforming capacity of the receptor tyrosine kinase (16). STAT1 knockout mice were more
susceptible to chemical carcinogenesis than their wild-type
counterparts (17). Double
knockout STAT1−/−, p53−/− mice exhibited a
higher incidence of tumor formation and a broader spectrum of
tumors than p53−/− knockout mice (17). Furthermore, STAT1 activation
inhibited cell proliferation and enhanced the induction of
apoptosis in tumor cells treated with IFN-γ or TNF-α via
upregulation of p21, a cyclin-dependent kinase inhibitor (7,17).
In Ras-transformed cells, activation of STAT1 also inhibited
Ras-MAPK kinase signaling and reduced expression of RhoA, Cdc42,
and Rac1 GTPases (18). In
addition, STAT1 phosphorylation at tyrosine 701 activating p27Kip1,
a cyclin-dependent kinase inhibitor, has also been reported
(19).
However, herein we add complexity to the earlier
findings by reporting that CUG2 contributes to the tumor
progression of colon cancer cells by the enhancement of cell
migration and cell survival during DNA damage, apparently via
activation of STAT1. Thus, we suggest that STAT1 can also play an
unexpected role as a proto-oncogene, as we found that suppression
of STAT1 with siRNA or shRNA vector reduced cell migration and
enhanced doxorubicin-induced apoptosis in colon cells
overexpressing CUG2. Our proposal is supported by other studies
showing that constitutive activation of STAT1 accelerates
metastasis of melanoma cancer cells and confers resistance to drug-
or ionization-induced cell death of melanoma (20). Another doxorubicin-resistant
myeloma cell line also exhibited a higher abundance of
phosphorylated STAT1 (11).
STAT1 signaling was reported to be involved in
resistance to the platinum drug AMD473 in ovarian cancer (12). Further studies showed that
knockdown of STAT1 led to significant suppression of tumor growth
and enhancement of sensitivity to irradiation and drug treatment
compared to untreated tumor cells (11,21).
These changes were accompanied by alternative patterns of gene
expression such as glycolysis/gluconeogenesis, the citric acid
cycle, and oxidative phosphorylation. In particular, STAT1
expression had the most significant influence on the
glycolysis/gluconeogenesis pathway, suggesting that STAT1-dependent
expression of the energy metabolic pathway is associated with tumor
growth and resistance to stress such as irradiation (22). We will therefore investigate
metabolic pathways such as glycolysis/gluconeogeneis, the citric
acid cycle, and oxidative phosphorylation in both
Colon26L5-CUG2-shSTAT1 and Colon26L5-CUG2-shCon cells using
microarrays in our next study.
We observed that CUG2 expression constitutively
activates STAT1 in NIH3T3 and Colon25L5 cells and that the Akt-ERK
signaling pathway is involved in this event but p38MAPK is not.
However, we do not know the detailed mechanism by which CUG2
stimulates STAT1 through Akt-ERK signaling. Of note, a recent study
has shown that CUG2 interacts with nucleophosmin (NPM1/B23) protein
(23). NPM1/B23 protein has
multiple functions including cell growth, proliferation, resistance
to stress and cancer development (24,25).
Based on these findings, we speculate that CUG2, a component of
kinetochores, plays a role in the development of cancer at least in
part through its interaction with NPM1/B23 protein. Cancerous cells
mediated by the CUG2-NPM1/B23 signaling axis produce growth factors
and cytokines including EGF and IL-6, which can then deliver
signals to the secretory or neighboring cells in an autocrine or a
paracrine manner. Moreover, it is known that not only IFN signaling
but also growth factor signaling (such as EGF) can activate STAT1
(26). Taken together, we propose
that the interaction of CUG2 with NPM1/B23 protein can activate EGF
release for cell proliferation, leading thereby to STAT1
activation. To answer more precisely how CUG2 overexpression might
activate STAT1 through its interaction with NPM1/B23 protein will
require further studies.
Since we demonstrated enhancement of cell migration
as consequence of STAT1 activation by CUG2, we examined genes
related to cell migration using microarray assays. We found that
transcripts of CCL5, CCL9 and CXCL10 increased 12-, 6.8-, and
4.5-fold, respectively, in Colon26L5-CUG2 cells compared to those
in Colon26L5-Vec cells (data not shown). Many lines of evidence
have supported that CCL5, CCL9 and CXCL10 are involved in
metastasis of various cancer cells (27–29),
and have shown that IFN signaling induces upregulation of these
chemokines (30–32). We therefore assume that upregulated
chemokines through the STAT1-IFN axis enhance migration of colon
cancer cells. Based on our results, we suggest that CUG2 enhances
metastasis and drug resistance through STAT1 activation, which
eventually contributes to tumor progression.
Acknowledgements
This study was supported by a grant
from the National R&D Program for Cancer Control, Ministry of
Health and Welfare, Republic of Korea (1120140) and the World Class
University Program (R31-2008-000-20004-0) through National Research
Foundation funded by the Korean government.
References
|
1.
|
Lee S, Gang J, Jeon SB, et al: Molecular
cloning and functional analysis of a novel oncogene,
cancer-upregulated gene 2 (CUG2). Biochem Biophys Res Commun.
360:633–639. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Kim H, Lee M, Lee S, et al:
Cancer-upregulated gene 2 (CUG2), a new component of centromere
complex, is required for kinetochore function. Mol Cells.
27:697–701. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Park EH, Park EH, Cho IR, et al: CUG2, a
novel oncogene confers reoviral replication through Ras and p38
signaling pathway. Cancer Gene Ther. 17:307–314. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Malilas W, Koh SS, Srisuttee R, et al:
Cancer upregulated gene 2, a novel oncogene, confers resistance to
oncolytic vesicular stomatitis virus through STAT1-OASL2 signaling.
Cancer Gene Ther. 20:125–132. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Chin YE, Kitagawa M, Kuida K, Flavell RA
and Fu XY: Activation of the STAT signaling pathway can cause
expression of caspase 1 and apoptosis. Mol Cell Biol. 17:5328–5337.
1997.PubMed/NCBI
|
|
6.
|
Kumar A, Commane M, Flickinger TW, Horvath
CM and Stark GR: Defective TNF-alpha-induced apoptosis in
STAT1-null cells due to low constitutive levels of caspases.
Science. 278:1630–1632. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Chin YE, Kitagawa M, Su WC, You ZH,
Iwamoto Y and Fu XY: Cell growth arrest and induction of
cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1.
Science. 272:719–722. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Townsend PA, Scarabelli TM, Davidson SM,
Knight RA, Latchman DS and Stephanou A: STAT-1 interacts with p53
to enhance DNA damage-induced apoptosis. J Biol Chem.
279:5811–5820. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Stephanou A, Brar BK, Knight RA and
Latchman DS: Opposing actions of STAT-1 and STAT-3 on the Bcl-2 and
Bcl-x promoters. Cell Death Differ. 7:329–330. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Khodarev NN, Beckett M, Labay E, Darga T,
Roizman B and Weichselbaum RR: STAT1 is overexpressed in tumors
selected for radioresistance and confers protection from radiation
in transduced sensitive cells. Proc Natl Acad Sci USA.
101:1714–1719. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Fryknas M, Dhar S, Oberg F, et al: STAT1
signaling is associated with acquired crossresistance to
doxorubicin and radiation in myeloma cell lines. Int J Cancer.
120:189–195. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Roberts D, Schick J, Conway S, et al:
Identification of genes associated with platinum drug sensitivity
and resistance in human ovarian cancer cells. Br J Cancer.
92:1149–1158. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Weichselbaum RR, Ishwaran H, Yoon T, et
al: An interferon-related gene signature for DNA damage resistance
is a predictive marker for chemotherapy and radiation for breast
cancer. Proc Natl Acad Sci USA. 105:18490–18495. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Park HD, Lee Y, Oh YK, et al: Pancreatic
adenocarcinoma up regulated factor promotes metastasis by
regulating TLR/CXCR4 activation. Oncogene. 30:201–211. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Liang CC, Park AY and Guan JL: In vitro
scratch assay: a convenient and inexpensive method for analysis of
cell migration in vitro. Nat Protoc. 2:329–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Raven JF, Williams V, Wang S, et al: Stat1
is a suppressor of ErbB2/Neu-mediated cellular transformation and
mouse mammary gland tumor formation. Cell Cycle. 10:794–804. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Kaplan DH, Shankaran V, Dighe AS, et al:
Demonstration of an interferon gamma-dependent tumor surveillance
system in immunocompetent mice. Proc Natl Acad Sci USA.
95:7556–7561. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Wang S and Koromilas AE: Stat1 is an
inhibitor of Ras-MAPK signaling and Rho small GTPase expression
with implications in the transcriptional signature of Ras
transformed cells. Cell Cycle. 8:2070–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Wang S, Raven JF, Durbin JE and Koromilas
AE: Stat1 phosphorylation determines Ras oncogenicity by regulating
p27 kip1. PLoS One. 3:e34762008. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Khodarev NN, Roach P, Pitroda SP, et al:
STAT1 pathway mediates amplification of metastatic potential and
resistance to therapy. PLoS One. 4:e58212009. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Rickardson L, Fryknas M, Dhar S, et al:
Identification of molecular mechanisms for cellular drug resistance
by combining drug activity and gene expression profiles. Br J
Cancer. 93:483–492. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Pitroda SP, Wakim BT, Sood RF, et al:
STAT1-dependent expression of energy metabolic pathways links
tumour growth and radioresistance to the Warburg effect. BMC Med.
7:682009. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Chun Y, Park B, Koh W, et al: New
centromeric component CENP-W is an RNA-associated nuclear matrix
protein that interacts with nucleophosmin/B23 protein. J Biol Chem.
286:42758–42769. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Okuwaki M: The structure and functions of
NPM1/Nucleophsmin/B23, a multifunctional nucleolar acidic protein.
J Biochem. 143:441–448. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Grisendi S, Mecucci C, Falini B and
Pandolfi PP: Nucleophosmin and cancer. Nat Rev Cancer. 6:493–505.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Khodarev NN, Roizman B and Weichselbaum
RR: Molecular pathways: interferon/stat1 pathway: role in the tumor
resistance to genotoxic stress and aggressive growth. Clin Cancer
Res. 18:3015–3021. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Velasco-Velazquez M, Jiao X, De La Fuente
M, et al: CCR5 antagonist blocks metastasis of basal breast cancer
cells. Cancer Res. 72:3839–3850. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Kitamura T, Fujishita T, Loetscher P, et
al: Inactivation of chemokine (C-C motif) receptor 1 (CCR1)
suppresses colon cancer liver metastasis by blocking accumulation
of immature myeloid cells in a mouse model. Proc Natl Acad Sci USA.
107:13063–13068. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Lee JH, Kim HN, Kim KO, et al: CXCL10
promotes osteolytic bone metastasis by enhancing cancer outgrowth
and osteoclasto-genesis. Cancer Res. 72:3175–3186. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Nakano M, Fujii T, Hashimoto M, et al:
Type I interferon induces CX3CL1 (fractalkine) and CCL5 (RANTES)
production in human pulmonary vascular endothelial cells. Clin Exp
Immunol. 170:94–100. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Nardi V, Naveiras O, Azam M and Daley GQ:
ICSBP-mediated immune protection against BCR-ABL-induced leukemia
requires the CCL6 and CCL9 chemokines. Blood. 113:3813–3820. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Ohmori Y and Hamilton TA: Cooperative
interaction between interferon (IFN) stimulus response element and
kappaB sequence motifs controls IFN gamma- and
lipopolysaccharide-stimulated transcription from the murine IP-10
promoter. J Biol Chem. 268:6677–6688. 1993.
|