Introduction
Lung cancer is the leading cause of cancer-related
mortality worldwide, with a 5-year survival rate of only 20%.
Although patients diagnosed with early-stage disease have the
potential for cure with complete surgical resection, only 25% of
lung cancer cases are diagnosed at an early stage.
Small molecule inhibitors of epidermal growth factor
receptor tyrosine kinase (EGFR-TKI), such as gefitinib and
erlotinib (1,2), induce dramatic responses in certain
patients with non-small cell lung cancer (NSCLC) (3). EGFR mutation (short deletion in exon
19, L858R) positive patients have shown an impressive 60% response
rate, which exceeds the response rate for conventional chemotherapy
(4). However, the clinical success
of treatment with EGFR-TKIs is uniformly limited by the development
of acquired drug resistance. Two major mechanism of acquired
resistance have been identified as T790M mutation in EGFR gene and
gene amplification of MET oncogene (5,6).
Hence, new therapeutic strategies, such as the development of more
effective or alternative molecular-targeted agents against lung
cancer are eagerly awaited.
Recently, large-scale screening methods have been
applied to identify novel molecular targets. Mass spectrometry
targeting phosphorylated tyrosin on lung adenocarcinoma cells
harboring EGFR kinase domain mutation has been reported and
revealed that phosphorylation of several proteins, which have not
been identified in EGFR pathway, is strongly controlled by EGFR
protein (7). We performed similar
analysis and found that p190-A RhoGAP (p190) was one of common
proteins which include functional tyrosine controlled by EGFR
protein.
p190 is a 172-kDa protein that is encoded by the
GRLF1 gene located on 19q13.3. The direct action of p190 has
been reported to be a RhoGAP; converting Rho-GTP to Rho-GDP and
inactivating the Rho pathway (8).
Therefore, p190 has been thought to be a potent inhibitor of RhoA
(9). On the other hand, indirect
action of p190 has been reported to control the Ras pathway by
binding to p120RasGAP protein. p190 has a binding activity to
p120RasGAP and the activity is promoted by phosphorylation of
tyrosine (Y1105) of p190 (10). The activity of p120RasGAP is
reduced when p190 forms a complex with p120RasGAP (11). So far, p190 has been reported to
inhibit cell migration and invasion through negatively regulating
RhoA in some cell lines (12). On
the other hand, EGF-induced p190 phosphorylation has been reported
to play essential roles in Ras activation and cell proliferation
using different cell lines (13).
To date, only few reports are available on the function of p190 in
lung cancer. We investigated the roles of p190 in lung
adenocarcinoma cells and clarified whether p190 could be a
potential therapeutic target for lung cancer.
Materials and methods
Cell lines and culture
A549, H520 and H1975 were obtained from American
Type Culture Collection; ATCC (Manassas, VA, USA). PC-14 was
obtained from DS Pharma Niomedical Co., Ltd. (Suita, Osaka, Japan).
LK87, LCSC#1 and II-18 were obtained from Cell Resource Center for
Biomedical Research (Tohoku University, Sendai, Japan). All cells
were maintained in RPMI-1640 (Invitrogen, San Diego, CA, USA)
supplement with 10% FBS in a humidified 5% CO2 incubator at 37°C.
A549 and LK87 have wild-type (WT) EGFR and K-ras mutation (codon
12), PC-14 has EGFR mutation (exon 19 del), LCSC#1 and II-18 have
EGFR mutation (L858R) and H1975 has EGFR mutation (L858R and
T790M). H520, with squamous cell carcinoma histology, has both WT
EGFR. A549, H1975 and H520 have been reported to be
EGFR-TKI-resistant, whereas LCSC#1, PC-14 and II-18 to be
EGFR-TKI-sensitive. NHBE (normal human bronchial epithelial cell)
was obtained from Takara Bio Inc. (Otsu, Shiga, Japan) and was
grown in BEGM Bullet kit (Lonza Walkersville Inc., Walkersville,
MD, USA) according to the manufacturer’s instructions.
Western blot and immunoprecipitation
analyses
Protein samples were obtained from cells and
transferred onto an Immune-Blot PVDF Membrane (Bio-Rad, Hercules,
CA, USA). Proteins on the membranes were incubated with recommended
concentration of primary antibodies, horse-radish
peroxidase-labeled secondary antibodies and were visualized on
ImageQuant LAS 4000 mini (GE Healthcare, Buckinghamshire, UK) by
means of the ECL Western blot detection system.
For immunoprecipitation, the p190-A RasGAP antibody
was bound on to Protein A Dynabeads® (Invitrogen Dynal,
Oslo, Norway). The Protein A Dynabeads-p190-A RasGAP complex was
incubated with the cell lysates of lung cancer cells. Obtained
bead-protein complexes were eluted and analyzed by SDS-PAGE and
western blotting. Antibodies to EGFR, phospho-EGFR, p190,
phoshotyrosine, Src, phospho-Src family kinase, MEK, phospho-MEK,
Erk, phospho-ErK and β-actin were purchased from Cell Signaling
Technology, whereas antibodies to Ras, Rho and p120 were from
Abcam.
RNA interference (RNAi)
Cells were transfected with 5 nM siRNA directed to
p190-A RhoGAP (p190 siRNA; Silencer® Select Validated
siRNA, Applied Biosystems, Austin, TX, USA) or negative control
siRNA (NR siRNA; non-related siRNA, Applied Biosystems) using the
Lipofectamine RNAiMAX® (Invitrogen), according to the
manufacturer’s instructions. The target sequence of siRNA for p190
was GGCUGAUGUUGAUCUGCGATT.
cDNA synthesis and real-time PCR
cDNA synthesis using TaqMan® Gene
Expression Cells-to-Ct™ kits was performed according to the
manufacturer’s instructions. Semi-quantitative real-time RT-PCR was
performed by using the ABI PRISM 7000 HT sequence detection system
(Applied Biosystems). The Assays-on-Demand products purchased from
Applied Biosystems contained TaqMan minor groove binder probes
(6-FAM dye-labeled) combined with the primers for p190
(Hs00534180_m1). An Assay-on-Demand product for 18s-rRNA (4319413E)
was used as endogenous control.
Cell proliferation, migration and
invasion assays
Cell proliferation assay was performed using
CellTiter 96® AQueous One Solution Cell Proliferation
assay kit (Promega, Madison, WI, USA), according to the
manufacturer’s instructions. Transwell migration assay using
CytoSelect™ 24-well Cell Migration assay (Cell Biolabs, San Diego,
CA, USA) and Transwell invasion assay using CytoSelect™ 24-well
Cell Invasion assay (Cell Biolabs) were performed according to the
manufacturer’s instructions.
Cell cycle analysis
Cells were fixed with 70% ice cold-ethanol and
stored at −20°C. Then cells were incubated with 0.5% RNAse followed
by propidium iodide. Histograms were obtained by FACSCanto II (BD).
Doublets were excluded by scattergrams. Data were analyzed using
ModFit software.
Quantitative assay for GTP-bound Ras and
GTP-bound Rho
The levels of GTP-bound Ras and GTP-bound Rho were
detected with the Raf-1 Ras binding domain agarose (Cell Biolabs)
and GST-rohtekin pull-down assays (Cell Biolabs), respectively.
Real-time PCR on clinical samples
A total of 133 cDNA samples of pulmonary
adenocarcinomas (pathological stage IA–IIIB) and adjacent normal
lung tissue, were obtained along with clinicopathological data from
patients who had undergone surgery at the Miyagi Cancer Center or
Tohoku University Hospital. Quantitative RT-PCR was performed by an
ABI 7000 using TaqMan probes for p190. β-actin was used as an
internal control. Threshold cycles of prier probes were normalized
to β-actin and translated to relative values.
Immunohistochemical analysis
Expression of phospho-p190 protein in
paraffin-embedded clinical tissues was examined using the Linked
Streptavidin-Biotin method (Histofine SAB-PO, Nichirei Biosciences,
Japan). Rabbit anti-phosphop190 antibody (1:50 dilution; Abcam) was
used as a primary antibody. Two independent investigators evaluated
the immunohistochemical staining without knowledge of the
clinicopathological parameters. For semi-quantitative assessment of
the immunohistochemical results, the mean percentage of positive
tumor cells was determined in ≥10 random fields at x400
magnification in each section. It was graded as focal (<10%),
regional (11–50%), or diffuse (>50%). The intensity of the
phospho-p190 immunoreaction was graded weak, moderate or strong.
The mean percentage of positive tumor cells and the staining
intensity were then combined to produce a phospho-p190-IHC
result.
Survival and statistical analyses
Two-year disease-free survival (2-year DFS) was
calculated from the time of surgery to the patient’s last follow-up
or recurrence. DFS of patients was analyzed using Kaplan-Meier
survival curves and the log-rank test was used to examine the
statistical significance. All in vitro experiments were
performed in triplicate. Comparisons between 2 groups were carried
out using unpaired Student’s t-test; comparisons among 3 or more
groups were carried out using one-way ANOVA. Survival and
statistical analysis were carried out with the Prism 5 for Mac OS X
(GraphPad Software, USA). All P-values <0.05 were considered as
statistically significant.
Results
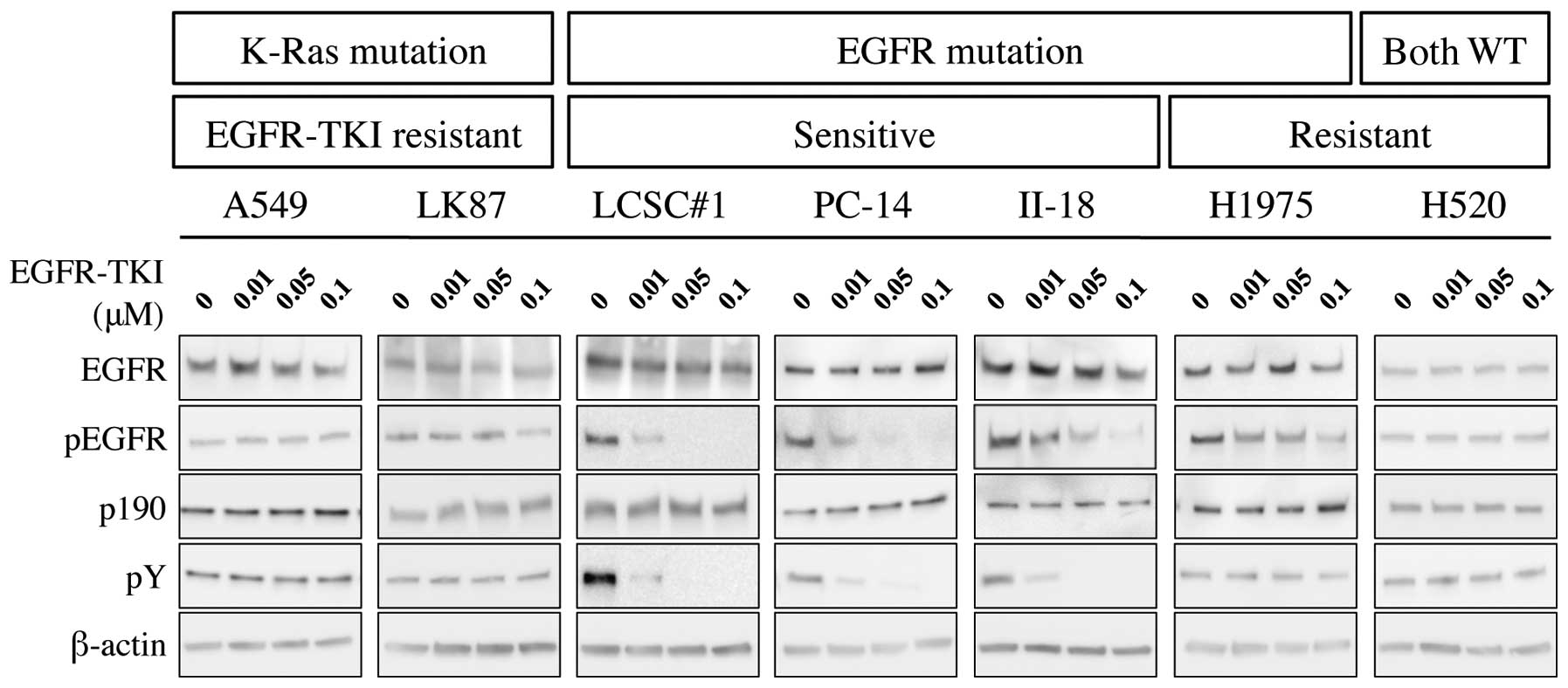
Western blotting showed that phosphorylation of EGFR
was inhibited by treatment with EGFR-TKI in EGFR-TKI-sensitive
cells; LCSC#1, PC-14 and II-18, but not in EGFR-TKI-resistant
cells; A549, LK87, H1975 and H520. Similarly, immunoprecipitation
analysis showed that phosphorylation of p190 was inhibited by
administration of EGFR-TKI (Fig.
1).
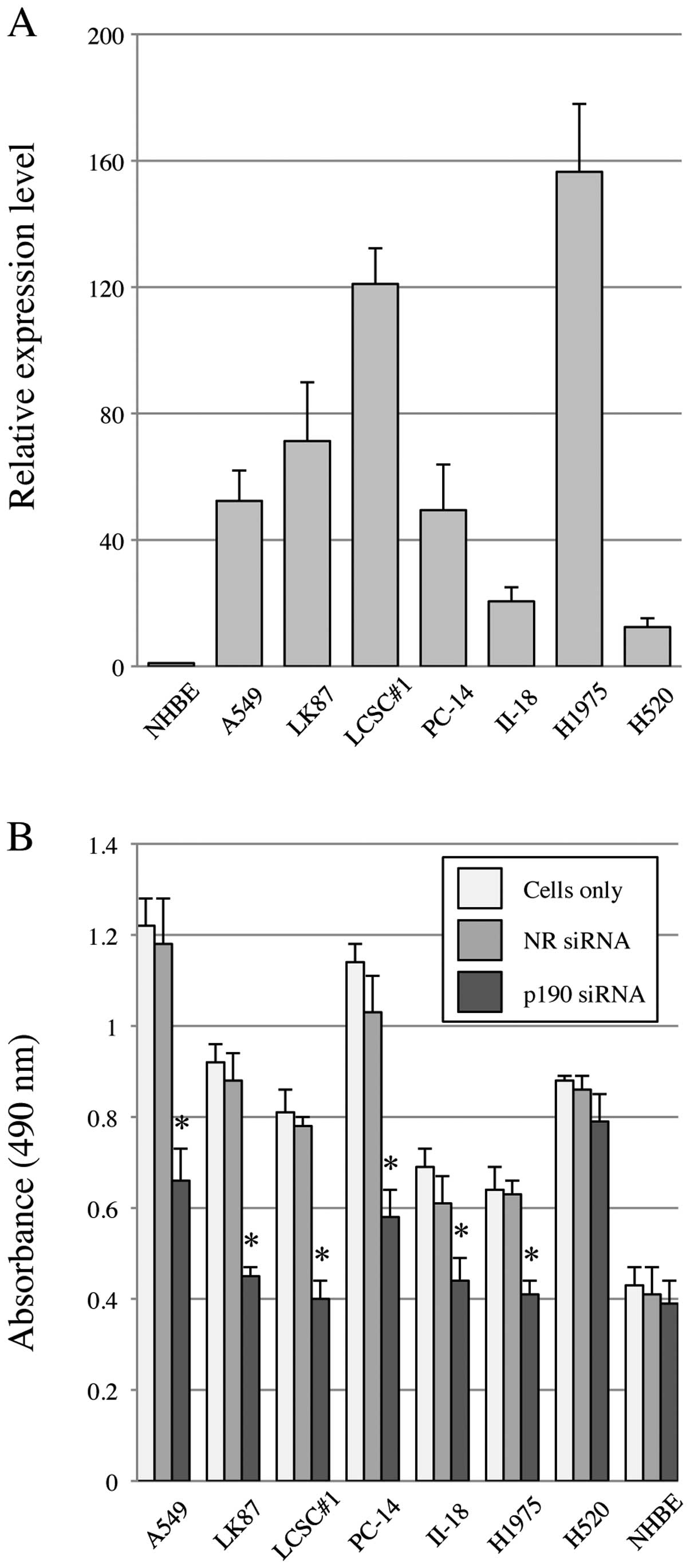
Since no chemical inhibitor for p190-tyrosine
phosphorylation is currently available, we performed p190 knockdown
using siRNA (p190 RNAi). Before knockdown, we confirmed that all
lung cancer cell lines in this study show high levels of p190 mRNA
expression by RT-PCR, ∼20–160-fold higher than that of NHBE
(Fig. 2A). To test the effect on
cell proliferation after p190 RNAi, MTS assay was performed. The
results showed that cellular proliferation of all lung
adenocarcinoma cell lines in this study was significantly
suppressed by p190 RNAi, while such suppression was not observed in
H520 and NHBE (Fig. 2B).
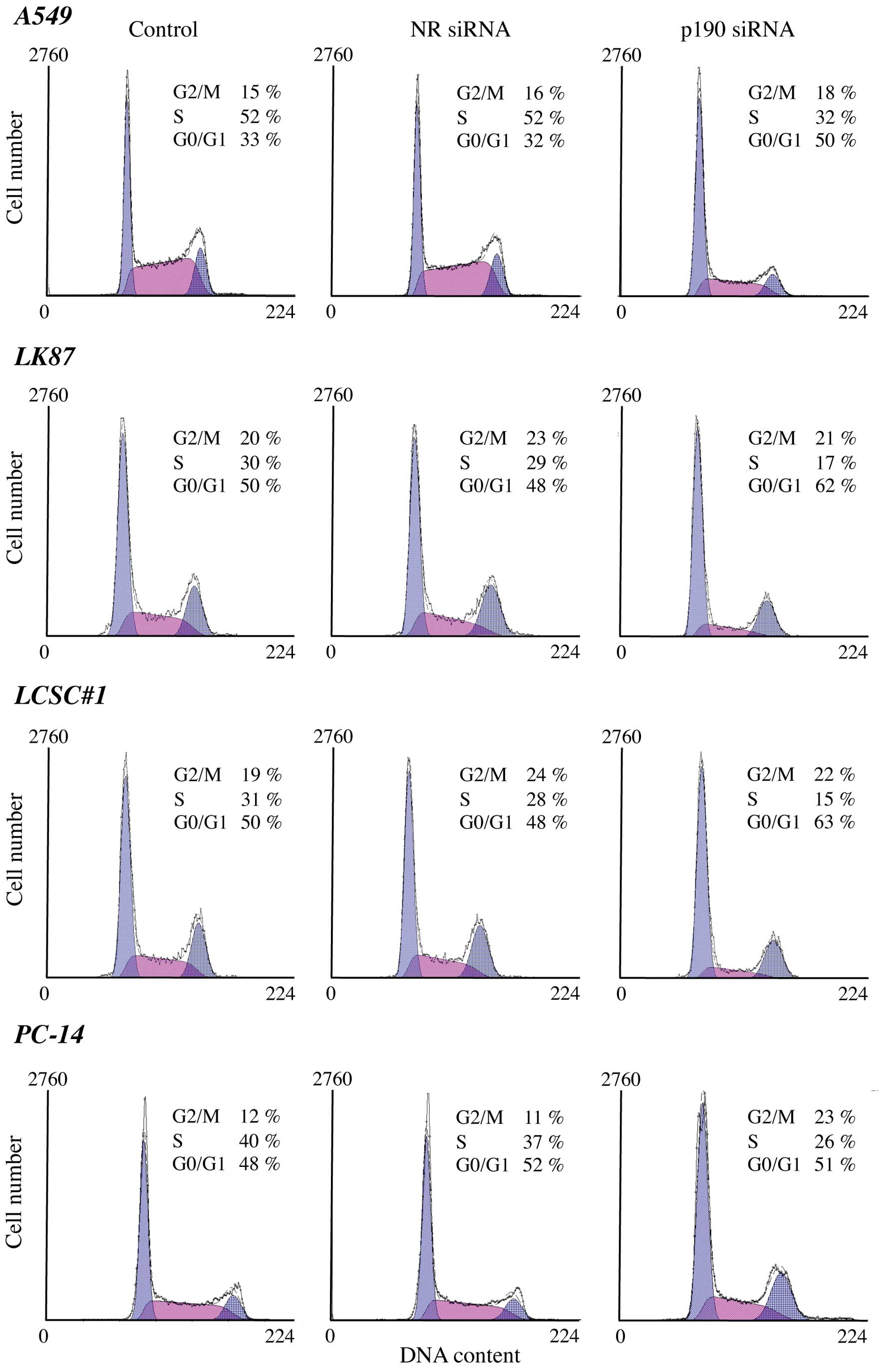
To understand the mechanism of the growth
suppression we further performed cell cycle analyses using lung
cancer cells after p190 RNAi. A decrease in the S population was
observed after p190 RNAi in all lung adenocarcinoma cell lines but
not in the squamous cell carcinoma H520 (Fig. 3). These results indicate that p190
RNAi led the lung adenocarcinoma cells to cell cycle arrest.
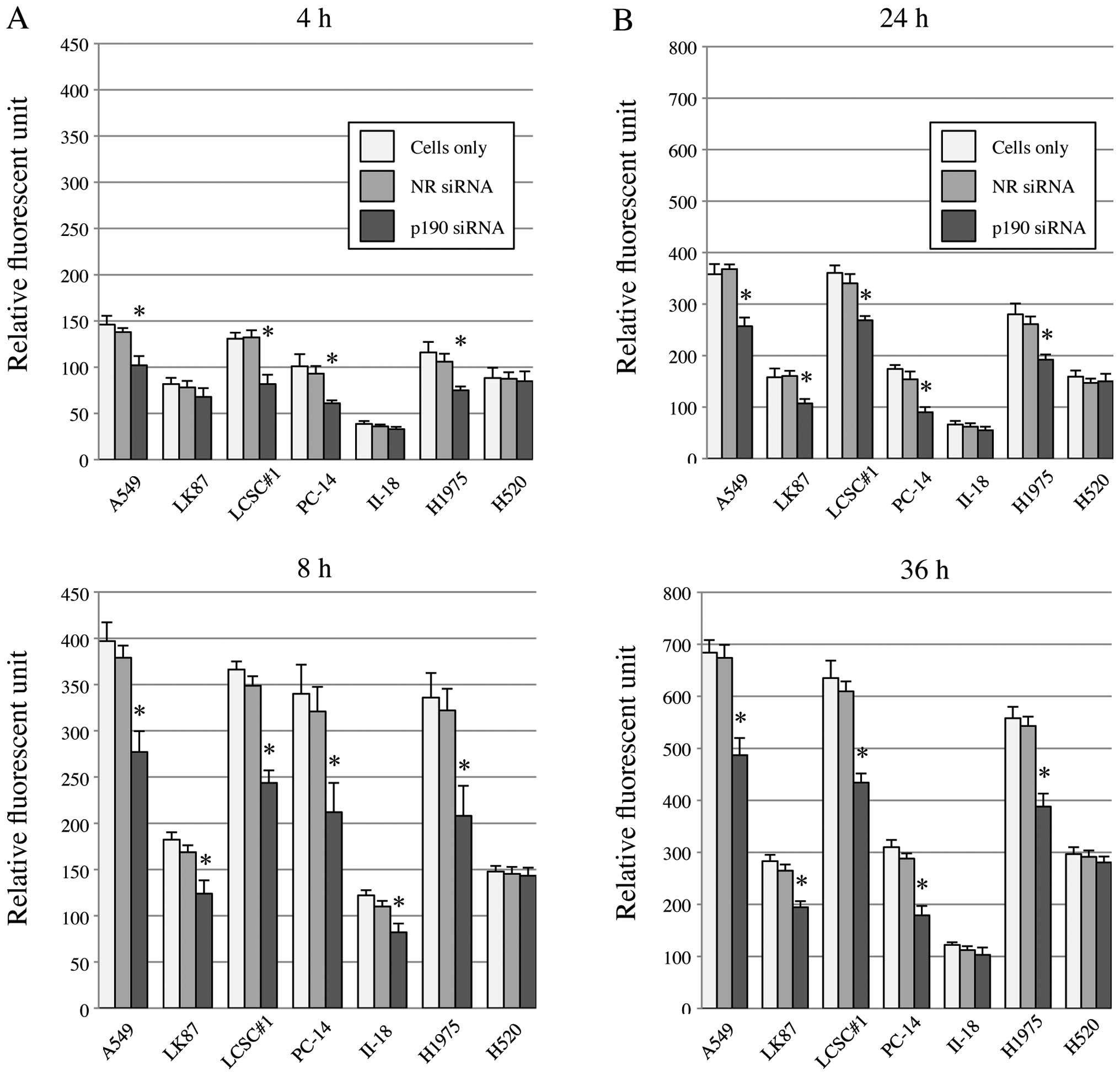
p190 is a RhoGAP, and reportedly is
involved in control of cell invasion and migration
To clarify this function in lung cancer cell lines,
we knocked down p190 and tested invasion and migration of lung
cancer cells with high levels of p190 mRNA. By p190 RNAi, both cell
invasion and migration were significantly reduced in A549, LK87,
LCSC#1, PC-14 and H1975 (Fig. 4).
In II-18 cell and H520, the difference was not detected probably
due to original nature of low activity of cell
invasion/migration.
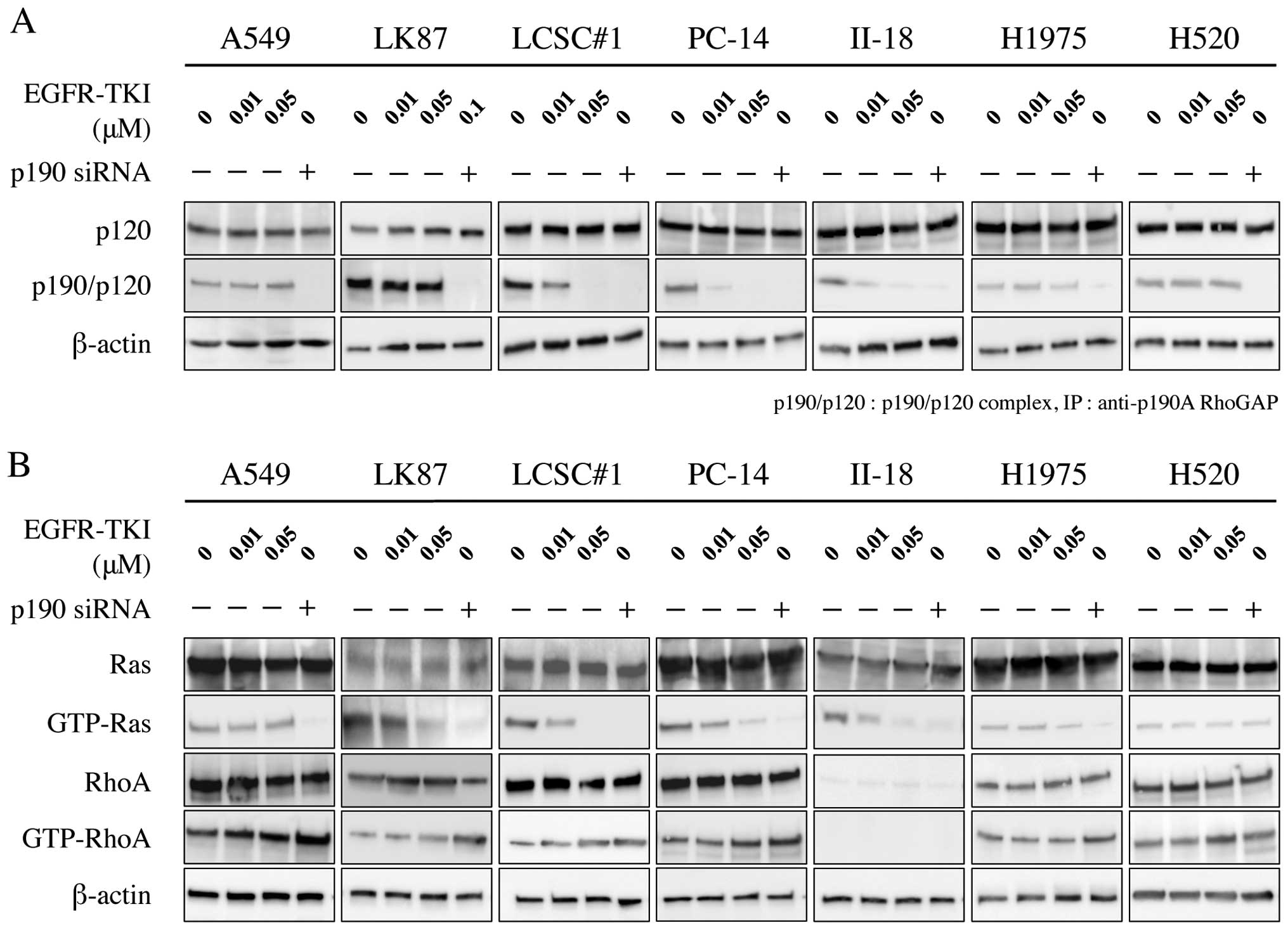
Phosphorylation of p190 at Y1105 is known
to promote formation of a p190/p120 complex. Formation of the
complex has been reported to downregulate the RasGAP activity of
p120, leading to an elevation of Ras activity. These complexes
upregulate the RhoGAP activity of p190, leading to a repression of
Rho activity. We investigated the effect of both EGFR-TKI and p190
RNAi on the p190/p120 complex formation, Ras activation and Rho
inactivation in lung cancer cell lines. Immunoprecipitation
analysis showed that p190/p120 complex formation was inhibited in
EGFR-TKI-sensitive cell lines by EGFR-TKI treatment in a
dose-dependent manner, but not in EGFR-TKI-resistant cell lines. On
the other hand, formation of the complex was inhibited by p190 RNAi
in all lung cancer cell lines (Fig.
5A). Ras inactivation was parallel to disruption of p190/120
complex by EGFR-TKI or p190 RNAi in all adenocarcinoma cell lines,
while Rho activation was inverse to disruption of p190/120 complex
in the adenocarcinoma cell lines except II-18 (Fig. 5B). In II-18 cells, RhoA activation
could not be evaluated due to low internal protein level of
RhoA.
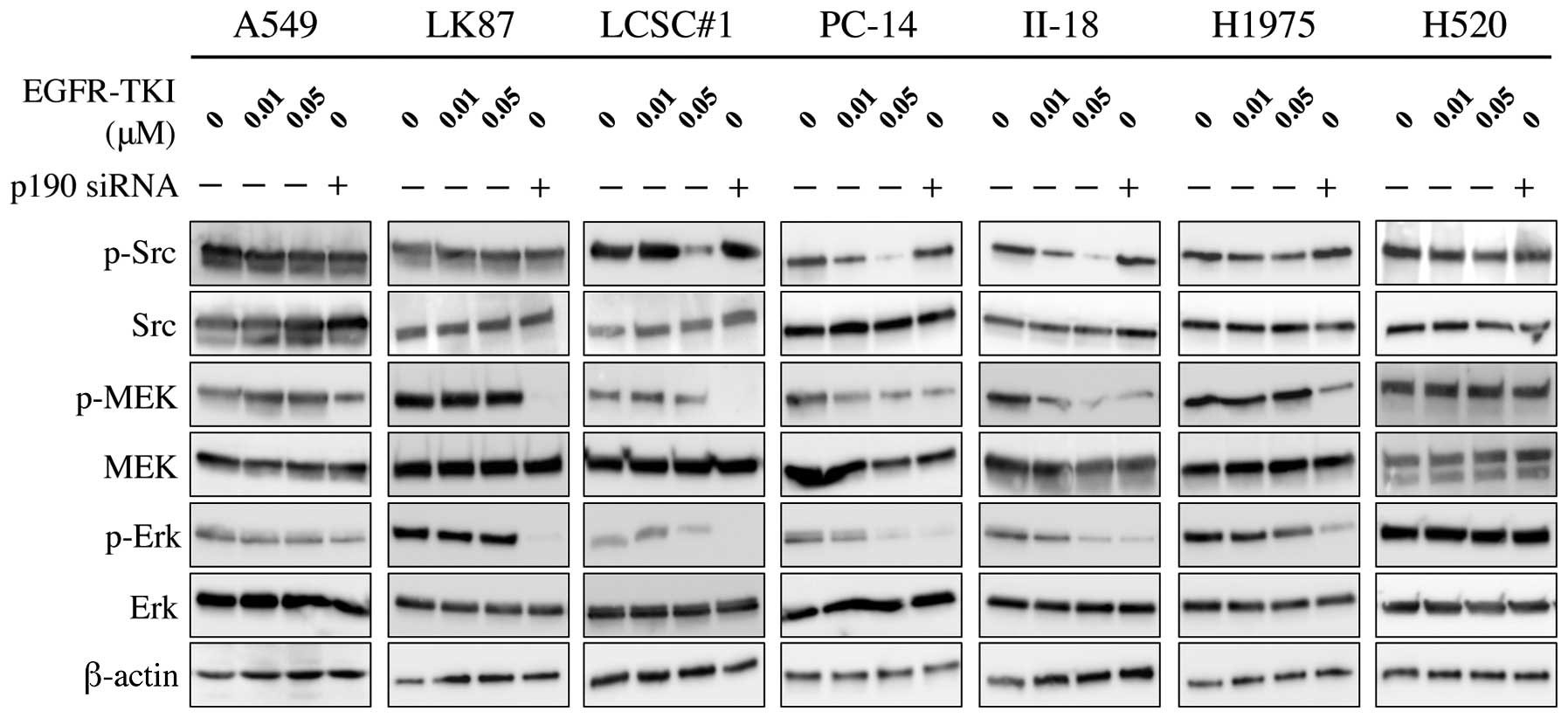
Phosphorylation of MEK and Erk, downstream molecules
of Ras, were inhibited by p190 RNAi in all adenocarcinoma cell
lines, although the effect is relatively small in A549. In
contrast, phosphorylation of Src, a potential upstream molecule of
p190, was inhibited by EGFR-TKI in EGFR-TKI-sensitive cells but not
in EGFR-TKI-resistant cells, while no effect of p190 RNAi was
observed (Fig. 6).
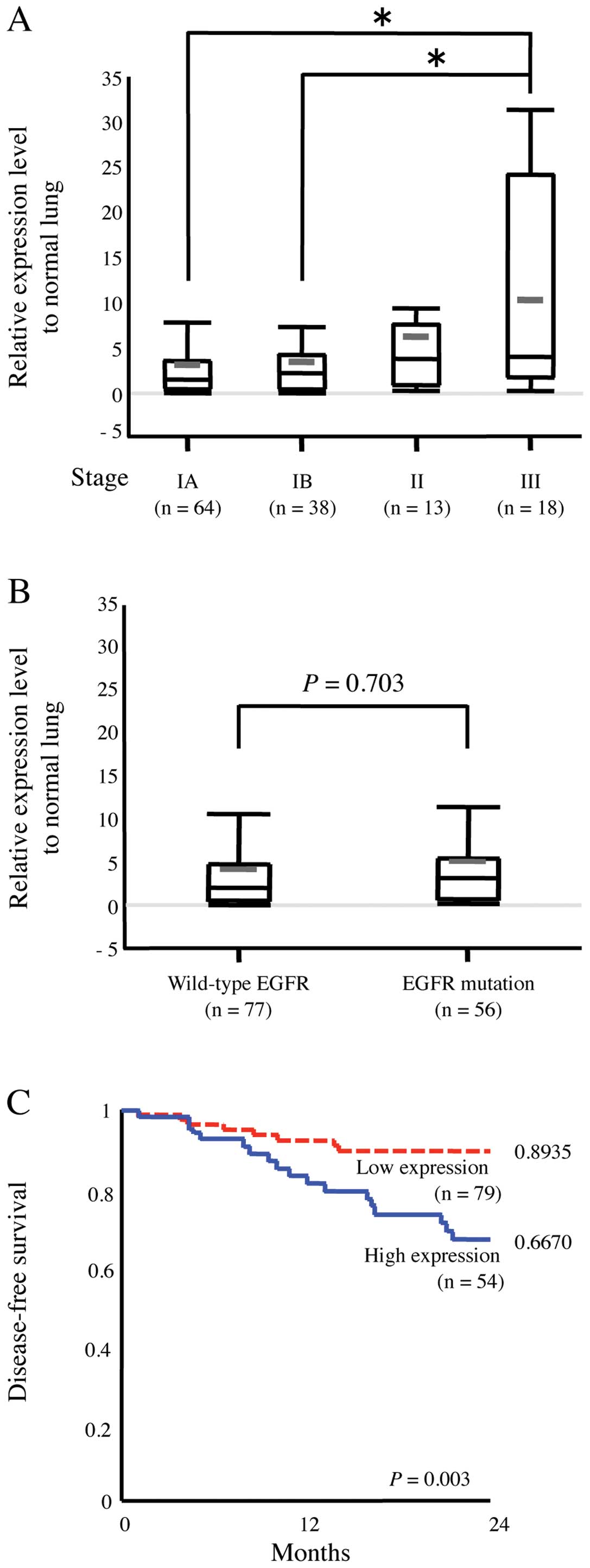
We also studied the expression level of p190 in
clinical samples. Relative expression level of p190 mRNA in tumor
to paired normal lung tissue was tested by RT-PCR in 133 human lung
adenocarcinoma tissue samples (Table
I). High level of p190 mRNA was detected in majority of samples
of lung adenocarcinoma. Pathological stage III patients (n=18) had
significantly higher mRNA level of p190 compared with stage IA or
IB patients (P<0.05) (Fig. 7A).
Whereas EGFR mutation existed in 56 samples, there was no
association between p190 mRNA level and EGFR mutation of the tumors
(P=0.703) (Fig. 7B). Furthermore,
we analyzed a cohort of 133 patients with outcome data. By ROC
curve analysis, we determined a threshold of p190 mRNA expression
level as three times the level of corresponding normal tissue of
the patients. High expression of p190 is associated with poor
disease-free survival (P=0.003) (Fig.
7C).
 | Table I.Characteristics of the 133 lung
adenocarcinoma patients whose surgical specimens were studied by
RT-PCR. |
Table I.
Characteristics of the 133 lung
adenocarcinoma patients whose surgical specimens were studied by
RT-PCR.
| Characteristics | No. of patients
(%) |
|---|
| Age at surgery | |
| <70 | 67 (50) |
| ≥70 | 66 (50) |
| Gender | |
| Male | 64 (48) |
| Female | 69 (52) |
| Pathological
stage | |
| IA | 64 (48) |
| IB | 38 (29) |
| II | 13 (10) |
| III | 18 (13) |
| EGFR mutation | |
| Positive | 56 (43) |
| L858R | 22 (17) |
| Exon19
deletion | 30 (23) |
| Other
mutations | 4 (3) |
| Negative | 77 (57) |
| p190 relative
expression level | |
| <3 | 79 (59) |
| ≥3 | 54 (41) |
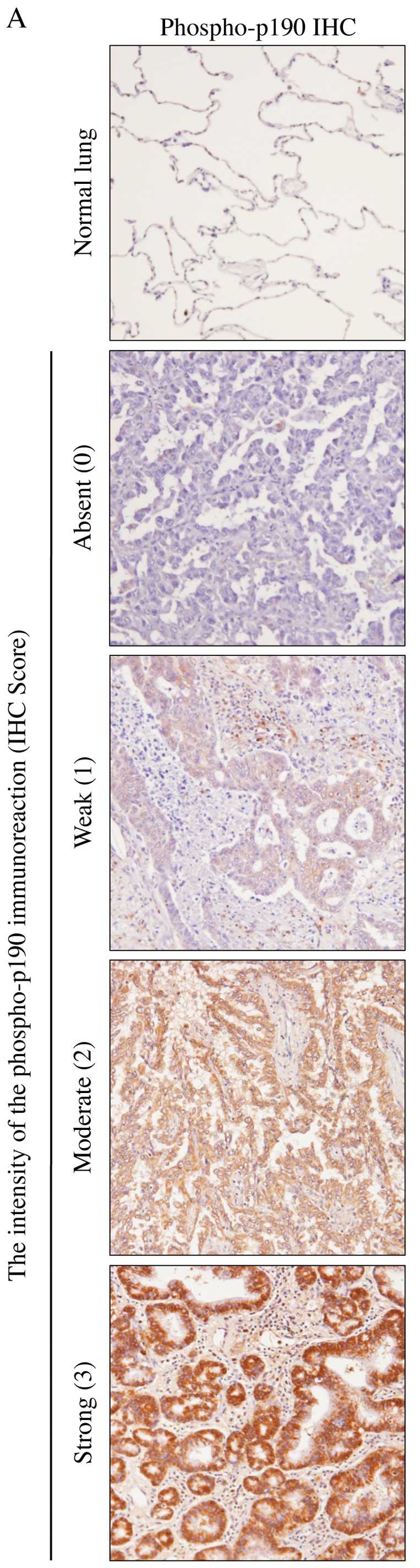
Phosphorylated-p190 (phospho-p190) was evaluated
immunohistochemically in 52 adenocarcinoma specimens.
Immunohistochemical staining showed that 92% (48 of 52) of
pulmonary adenocarcinoma samples were positive for phospho-p190
(Table II). The staining intensity
was also stronger in tumor cells than that in normal cells
(Fig. 8A). No statistically
significant difference was found between IHC score and clinical
outcome of lung adenocarcinoma patients (Table II). However, there were
statistically significant association between histological features
and phospho-p190 staining using Fisher’s exact-test (P=0.013)
(Fig. 8B).
| Table II.Immunohistochemical staining of
phospho-p190 protein in the 52 lung adenocarcinoma specimens. |
Table II.
Immunohistochemical staining of
phospho-p190 protein in the 52 lung adenocarcinoma specimens.
| Characteristics | Total no. | No. IHC score
| P-value |
|---|
| 0 or 1 | 2 or 3 |
|---|
| Age at surgery | | | | 0.092 |
| <70 | 23 | 15 | 8 | |
| ≥70 | 29 | 11 | 18 | |
| Gender | | | | 0.10 |
| Male | 25 | 9 | 16 | |
| Female | 27 | 16 | 11 | |
| Pathological
stage | | | | 1 |
| IA, IB | 41 | 21 | 20 | |
| II–IV | 11 | 5 | 6 | |
| EGFR mutation | | | | 0.78 |
| Positive | 28 | 15 | 13 | |
| Negative | 24 | 11 | 13 | |
| p190 relative mRNA
level | | | | <0.005 |
| <3 | 30 | 21 | 9 | |
| ≥3 | 22 | 5 | 17 | |
Discussion
In this study, we found three novel findings. First,
we clearly demonstrated that phosphorylation of p190 is involved in
the EGFR signal pathway. Previous mass spectrometry screening
suggested that tyrosine (Y1105) phosphorylation of p190
is affected by EGFR-TKI. Here, we discovered that one point
detected by previous mass spectrometry analysis is actually on the
line of EGFR signaling pathway by detailed western blot analysis.
We highlighted unique functional characteristics of p190RhoGAP, a
sort of switch of Ras and Rho pathway and how EGFR-TKI affects this
switch in lung adenocarcinoma cells. As briefly described in the
introduction, p190 is one of the GTPase activating proteins (GAPs)
and converts RhoA, small GTPases, from a GTP-bound active form into
a GDP-bound inactive form (14).
In addition, previous studies have reported that activated p190
positively regulates the Ras pathway, by forming complex with
p120RasGAP which converts Ras-GTP to Ras-GDP (8,10).
We demonstrated that p190 played a crucial role in regulation of
Ras and Rho in lung adenocarcinoma cells. Furthermore, we
demonstrated that, by p190 RNAi, the same molecular behavior, such
as Ras inactivation and Rho activation to EGFR-TKI was observed not
only in EGFR-TKI-sensitive cells but also in EGFR-TKI-resistant
cells.
Second, we showed that cell proliferation as well as
cell migration/invasion is significantly impaired by p190 RNAi in
lung adenocarcinoma cell lines. Our results of cell cycle analysis
revealed that suppression of proliferation in lung adenocarcinoma
cells was caused by cell cycle arrest, not by apoptosis.
Interestingly, arrest patterns, such as G1/S or G2/M was different
in cells. At present, it is not possible to explain clearly the
reason of the difference, underlying different molecular background
between cells is possible reason. As previously reported, apoptosis
is induced in lung adenocarcinoma cells harboring EGFR kinase
mutation by treatment of EGFR-TKI (15). The p190ARhoGAP-Ras pathway is not
responsible or is not enough for induction of apoptosis in
EGFR-TKI-sensitive cells. Nevertheless, it is noteworthy that
significant suppression of cell proliferation is observed by p190
RNAi in EGFR-TKI-resistant cells; even in A549 and LK87 harboring
KRAS mutation which generates constitutively-active Ras protein
(16).
By western blot analysis, we showed that RhoA
protein was activated by p190 RNAi, while Ras protein is
inactivated, thus discordant molecular signaling, both promoting
and inhibiting cell proliferation, were generated simultaneously.
Results of MTS assay indicates that Ras signaling rather than Rho
signaling is predominant on cell proliferation in lung
adenocarcinoma cells. KRAS mutation has been reported to exist in
∼20% of lung cancer patients and to be mutually exclusive with EGFR
mutations (17). Although the
mutations in EGFR correlate with sensitivity of the tumors to
EGFRTKIs, K-ras mutations are associated with primary resistance to
EGFR-TKIs (18). This result
indicates that p190 is a promising molecular target for lung
adenocarcinoma regardless of the mutation type of lung cancer.
Invasion and metastasis are the leading causes of
treatment failure and the most significant predictor of poor
clinical outcome in patients with lung cancer (19). Increased motility is the first step
in invasion and metastasis. RhoA expression is associated with poor
prognosis especially in breast cancer (20), hepatic carcinoma (21) and pancreatic cancer (22). Our study showed that GTP-binding
active form RhoA existed in the six lung adenocarcinoma cell lines,
except II-18 by p190 RNAi. On the basis of this result, we expected
that ability of invasion and migration would be increased in lung
adenocarcinoma cell lines by p190 RNAi. Whereas, our study
disclosed that in vitro invasion/migration has been
significantly inhibited in lung adenocarcinoma cells, except II-18.
One possibility is that, in lung adenocarcinoma cells, cross-talk
between Rho family of GTPases, such as RhoA, Rac1 and Cdc42 exists
and control activation of cell invasion and migration (23,24).
Alternatively, cell invasion and migration might be affected by
inactivation of Ras followed by cell cycle arrest (25).
Third, overexpression of p190 mRNA was detected from
all lung adenocarcinoma cell lines. In addition, high expression of
p190 mRNA significantly correlated to advanced pathological stage
and was also associated with poor disease-free survival.
Unexpectedly, EGFR mutation did not correlate with either mRNA
level of p190 or immunostaining results of phospho-p190. This might
suggest that p190 receives also other signals than the EGFR pathway
in vivo. By immunohistochemical study, almost all (92%)
pulmonary adenocarcinoma specimens were positive for phospho-p190
protein. Overexpression of p190 mRNA was observed in the patients
with early stage lung cancer, and phospho-p190 staining was
observed in almost all patients of lung cancer suggesting that
overexpression of p190 may be involved in early carcinogenesis of
lung adenocarcinoma. Interestingly, lepidic type histology, which
is generally recognized to show less invasive nature, showed
negative correlation to expression of phospho-p190 protein.
In conclusion, phosphorylation of p190 is regulated
by EGFR in lung adenocarcinoma cells and p190 involves cell
proliferation and migration/invasion through controlling the Ras
signaling pathway. Studies on clinical specimens suggest that p190
is associated with aggressive nature of the tumors. p190 shows
promise as a novel molecular target for treatment of lung
adenocarcinoma including tumors with acquired resistance to
EGFR-TKI or with K-ras mutation.
Acknowledgements
The authors thank Drs Nobuhiro Tanuma,
Hiroteru Miyashita, Takayuki Nakagawa, Hisashi Ohishi, Sumiko
Maeda, Masafumi Noda, Tetsu Sado and Yasushi Hoshikawa for their
technical assistance and advice.
References
|
1.
|
Baselga J and Averbuch SD: ZD1839
(‘Iressa’) as an anticancer agent. Drugs. 60:33–40. 2000.
|
|
2.
|
Shepherd FA, Rodrigues Pereira J, Ciuleanu
T, et al: Erlotinib in previously treated non-small-cell lung
cancer. N Engl J Med. 2:123–132. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Kris MG, Natale RB, Herbst RS, et al:
Efficacy of gefitinib, an inhibitor of the epidermal growth factor
receptor tyrosine kinase, in symptomatic patients with non-small
cell lung cancer: a randomized trial. JAMA. 16:2149–2158. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Inoue A, Suzuki T, Fukuhara T, et al:
Prospective phase II study of gefitinib for chemotherapy-naive
patients with advanced non-small-cell lung cancer with epidermal
growth factor receptor gene mutations. J Clin Oncol. 21:3340–3346.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Yun CH, Mengwasser KE, Toms AV, et al: The
T790M mutation in EGFR kinase causes drug resistance by increasing
the affinity for ATP. Proc Natl Acad Sci USA. 6:2070–2075. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 5827:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Guha U, Chaerkady R, Marimuthu A, et al:
Comparisons of tyrosine phosphorylated proteins in cells expressing
lung cancer-specific alleles of EGFR and KRAS. Proc Natl Acad Sci
USA. 37:14112–14117. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Arthur WT and Burridge K: RhoA
inactivation by p190RhoGAP regulates cell spreading and migration
by promoting membrane protrusion and polarity. Mol Biol Cell.
9:2711–2720. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Tikoo A, Czekay S, Viars C, et al: p190-A,
a human tumor suppressor gene, maps to the chromosomal region
19q13.3 that is reportedly deleted in some gliomas. Gene. 1:23–31.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Roof RW, Haskell MD, Dukes BD, Sherman N,
Kinter M and Parsons SJ: Phosphotyrosine (p-Tyr)-dependent and
-independent mechanisms of p190 RhoGAP-p120 RasGAP interaction: Tyr
1105 of p190, a substrate for c-Src, is the sole p-Tyr mediator of
complex formation. Mol Cell Biol. 12:7052–7063. 1998.PubMed/NCBI
|
|
11.
|
Moran MF, Polakis P, McCormick F, Pawson T
and Ellis C: Protein-tyrosine kinases regulate the phosphorylation,
protein interactions, subcellular distribution and activity of
p21ras GTPase-activating protein. Mol Cell Biol. 4:1804–1812.
1991.
|
|
12.
|
Kusama T, Mukai M, Endo H, et al:
Inactivation of Rho GTPases by p190 RhoGAP reduces human pancreatic
cancer cell invasion and metastasis. Cancer Sci. 9:848–853. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Shen CH, Chen HY, Lin MS, et al: Breast
tumor kinase phosphorylates p190RhoGAP to regulate rho and ras and
promote breast carcinoma growth, migration and invasion. Cancer
Res. 19:7779–7787. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Moon SY and Zheng Y: Rho GTPase-activating
proteins in cell regulation. Trends Cell Biol. 1:13–22. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Costa DB, Halmos B, Kumar A, et al: BIM
mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung
cancers with oncogenic EGFR mutations. PLoS Med. 10:1669–1679.
2007.PubMed/NCBI
|
|
16.
|
Mitchell CE, Belinsky SA and Lechner JF:
Detection and quantitation of mutant K-ras codon 12 restriction
fragments by capillary electrophoresis. Anal Biochem. 1:148–153.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Forbes S, Clements J, Dawson E, et al:
Cosmic 2005. Br J Cancer. 2:318–322. 2006. View Article : Google Scholar
|
|
18.
|
Massarelli E, Varella-Garcia M, Tang X, et
al: KRAS mutation is an important predictor of resistance to
therapy with epidermal growth factor receptor tyrosine kinase
inhibitors in non-small-cell lung cancer. Clin Cancer Res.
10:2890–2896. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Detterbeck FC, Boffa DJ and Tanoue LT: The
new lung cancer staging system. Chest. 1:260–271. 2009. View Article : Google Scholar
|
|
20.
|
Ma L, Liu YP, Geng CZ, Wang XL, Wang YJ
and Zhang XH: Over expression of RhoA is associated with
progression in invasive breast duct carcinoma. Breast J. 1:105–107.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Xiaorong L, Wei W, Liyuan Q and Kaiyan Y:
Underexpression of deleted in liver cancer 2 (DLC2) is associated
with overexpression of RhoA and poor prognosis in hepatocellular
carcinoma. BMC Cancer. 8:2052008. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Dittert DD, Kielisch C, Alldinger I, et
al: Prognostic significance of immunohistochemical RhoA expression
on survival in pancreatic ductal adenocarcinoma: a high-throughput
analysis. Hum Pathol. 7:1002–1010. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Takenawa T and Miki H: WASP and WAVE
family proteins: key molecules for rapid rearrangement of cortical
actin filaments and cell movement. J Cell Sci. 10:1801–1809.
2001.PubMed/NCBI
|
|
24.
|
Tsubakimoto K, Matsumoto K, Abe H, et al:
Small GTPase RhoD suppresses cell migration and cytokinesis.
Oncogene. 15:2431–2440. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Downward J: Targeting RAS signalling
pathways in cancer therapy. Nat Rev Cancer. 1:11–22. 2003.
View Article : Google Scholar : PubMed/NCBI
|