Introduction
Notch signaling is highly conserved throughout the
animal kingdom (1). It plays a
crucial role in embryonic development and is implicated in the
regulation of cellular proliferation, differentiation, apoptosis
and angiogenesis in adult organisms (2,3).
This pathway is essential for normal intestinal homeostasis by
controlling stem cell fate decisions (4). All Notch receptors and ligands as
well as several target genes (Hes1, 5, 6, 7 and Math1) are
expressed at various stages of development and differentiation in
mouse intestinal crypts, the niche of intestine stem/progenitor
cells (5). Inhibition of the Notch
signaling pathway via Hes1-depletion is accompanied by an increase
of the secretory cells at the expense of absorptive enterocytes
(6). Additionally, this pathway is
involved in cell cycle progression of crypt progenitor cells
(7). Because of its crucial role
in cell fate decision, it is not surprising that Notch signaling
has been implicated in colorectal cancer (CRC) development.
Overexpression of Notch receptors, ligands and downstream targets
is observed in most CRC tissues compared to normal colonic tissues
which is associated with activation of the pathway (8).
In mammals there are four Notch receptors (Notch 1,
2, 3, 4) and five Notch ligands (Jagged 1, 2, Delta-like 1, 3, 4),
all of which are transmembrane proteins. Two proteolytic cleavage
steps are required for activation of the receptor upon its
interaction with a ligand (9). The
first (S2) cleavage detaches the Notch extracellular domain and is
catalyzed by ADAM-family metalloproteases, ADAM10 or ADAM17/TACE
(10). The second (S3) cleavage is
executed by the γ-secretase complex, leading to the release of the
Notch intracellular domain (NICD) (11). The latter translocates to the
nucleus where it interacts with the transcriptional regulator RBPJ
and its co-activator Mastermind (Mam). This triggers transcription
of Notch target genes, such as those belonging to the Hes and Hey
families (12).
Several downstream effectors mediating the effects
of Notch on cellular proliferation and differentiation have been
described. One of them, p27Kip1, belongs to the CIP/KIP
family of cyclin-dependent kinase (CDK) inhibitors (CDKIs) which
also includes p21Cip1/WAF1 and p57Kip2. It
accumulates during quiescence (G0) and needs to be degraded before
a cell can enter the cell cycle. p27 is mainly regulated by
post-transcriptional mechanisms (13), including diverse phosphorylation
events and proteasomal degradation. To enable the cells to enter S
phase, nuclear p27 levels have to decrease which is realized by
SKP1-cullin-F-box protein (SCFSKP2) (14). The prerequisite for this
ubiquitination is a phosphorylation of p27 at threonine 187 (T187)
by the CDK2-cyclin E/A complex. Another ubiquitin ligase, called
Kip1 ubiquitination-promoting complex (KPC), accomplishes the
degradation of p27 in the cytoplasm during early G1 phase (15). This ubiquitination is also
phosphorylation-dependent since KIS and DYRK1 kinases can
phosphorylate nuclear p27 at serine 10 (S10), thereby allowing p27
binding to CRM1 (also known as exportin 1) via its nuclear export
signal (NES) (16). This leads to
the export of p27 to the cytoplasm where it becomes a target for
KPC-mediated proteasomal degradation. However, the precise
mechanisms governing the Notch-dependent regulation of p27 are far
from clear. Given the involvement of Notch in intestinal tissue
homeostasis, the Notch-p27 axis could play a pivotal role in the
development and progression of CRC.
CRC is on third and second place in terms of
incidence and mortality amongst all malignancies, and the
established therapies against advanced stages of the disease have
only limited efficacy. Because Notch signaling is essential for
stem cell fate decision, therapies targeting this pathway might
improve CRC treatment. Due to the crucial role of Notch signaling
in colon tissue, understanding the mechanisms of proliferation
activation and apoptosis inhibition by this pathway is of major
importance in order to develop novel promising antitumor therapies
for patients suffering from CRC. In this study, we characterized
the regulation of p27 by Notch1 and the effects of Notch1 signaling
on cell growth. Moreover, we investigated the potential beneficial
interference with Notch1 signaling for increasing the efficiency of
chemo- and radiotherapy for treatment of CRC.
Materials and methods
Materials
The following reagents were used: oxaliplatin
(Sigma, Deisenhofen, Germany, O9512), 5-fluorouracil (AppliChem,
Darmstadt, Germany, A7686,0005), cycloheximide (Calbiochem, San
Diego, CA, USA, 239763). The primary antibodies used in the
experiments were: anti-Notch1 (Santa Cruz, San Diego, CA, USA,
sc-6014), anti-p27 (Santa Cruz, sc-1641), anti-GAPDH (Santa Cruz,
sc-32233), anti-β-actin (Chemicon International, Temecula, CA, USA,
MAB1501), anti-KPC1 (Santa Cruz, sc-101122), anti-KPC2 (Abgent, San
Diego, CA, USA, AP5353b), anti-SKP2 (Cell Signaling, Danvers, MA,
USA, 4313), anti-PARP (BD Biosciences, San Jose, CA, USA, 556362),
anti-caspase-3 (Imgenex, San Diego, CA, USA, IMG-144A) and
anti-Ki67 (Dako, Glostrup, Denmark, M7240).
Cell culture
Human colorectal cancer cell lines were purchased
from ATCC (Rockville, MD, USA), expanded and frozen in aliquots
within four weeks. For the experiments described here, the cells
were thawed and cultured for no more than eight weeks. Cell lines
were authenticated by SNP profiling (17) and tested regularly for
contaminations by multiplex PCR performed in the core facility of
the DKFZ (18). All cell lines
except Caco2 were grown in RPMI medium (Invitrogen, Carlsbad, CA,
USA, 21875-034) containing 10% FCS (PAA, Piscataway, NJ, USA,
A15-151) and 1% penicillin/streptomycin (Invitrogen, 15140-122).
Caco2 cells were cultured in MEM medium (Invitrogen, 31095-029)
supplemented with 20% FCS, 1% penicillin/streptomycin, 1% MEM
non-essential amino acids (PAA, M11-003), 1 mM sodium pyruvate
(Sigma, S8636) and 1% GlutaMAX (Invitrogen, 35050-038). All cell
lines were cultured at 37°C in a 5% CO2 atmosphere.
Transfections
Knockdown of endogenous proteins was achieved by
transiently transfecting cell lines with short interfering RNA
(siRNA) oligonucleotides using Lipofectamine 2000 (Invitrogen,
11668-019). siRNA oligonucleotides for Notch1 were obtained from
Ambion [Austin, TX, USA, 144335 (si1), 108983 (si2)] and Dharmacon
[Lafayette, CO, USA, D-007771-08 (si3)]; p27 siRNA (J-003472-07),
KPC1 siRNA [D-007041-01 (si1), D-007041-03 (si2)], and SKP2 siRNA
[D-003324-07 (si1), D-003324-13 (si2)] were obtained from
Dharmacon. Non-targeting siRNA pool was used as a control
(Dharmacon, D-001810-10-20). The following siRNA concentrations
were used for transfection: Notch1 siRNAs, 25 nM; KPC1 and SKP2
siRNAs, 20 nM; p27 siRNA, 10 nM.
Immunoblot analysis
Total cell lysates were prepared using lysis buffer
[120 mM NaCl, 50 mM Tris-HCl (pH 8.0), 5 mM EDTA, 0.5% Triton
X-100] containing phenylmethylsulfonylfluoride (1 mM), proteinase
inhibitors (Roche, Mannheim, Germany, 11697498001) and phosphatase
inhibitors (25 mM NaF, 200 μM NaVO3, 10 mM
NaPPi). For cytoplasmic and nuclear fractions cells were harvested
and processed with the Nuclear Extraction kit (Active Motif,
Rixensart, Belgium, 40010) according to the manufacturer’s
protocol. Protein concentration was measured using the Bradford
Assay (Bio-Rad, Munich, Germany, 500-0006). Soluble proteins (10–50
μg per lane) were separated on 10 or 12% SDS polyacrylamide
gels and blotted on nitro-cellulose membrane (Bio-Rad, 162-0115).
Membranes were incubated with primary and secondary antibody
(horseradish peroxidase-conjugated, Bio-Rad) which was visualized
using an enhanced chemiluminescence detection system (GE
Healthcare, Uppsala, Sweden, RPN2109).
Quantitative real-time PCR
Total RNA was isolated with the RNeasy Plus mini kit
(Qiagen, Germantown, MD, USA, 74134). A total of 1 μg RNA
was reverse-transcribed to cDNA using SuperScript II (Invitrogen,
18064-014) and Random Hexamers (Applied Biosystems, Carlsbad, CA,
USA, P14122). Quantitative real-time PCR was performed using
SYBR-Green® PCR Master mix (Applied Biosystems, 4309155)
and a 7300 Real-Time PCR system (Applied Biosystems). The following
primers were utilized: Notch1 5′-GGGCCCTGAATTTCA CTGT-3′ (forward),
5′-CGCAGAGGGTTGTATTGGTT-3′ (reverse); p27 5′-AAAAATCCGAGGTGCTTGG-3′
(forward), 5′-ACAGCCCGAAGTGAAAAGAA-3′ (reverse); KPC1 5′-GTC
CAAATGTTCTGGCAGGT-3′ (forward), 5′-TGAACCGCATC TTTTCCTCT-3′
(reverse); KPC2 5′-CATGTTGTAGGAGG GCAGGT-3′ (forward),
5′-CCCAAGATGGCTGATGTCTC-3′ (reverse); SKP2
5′-GAAGGGAGTCCCATGAAACA-3′ (forward), 5′-CCAGGAACTGCTCTCAAACC-3′
(reverse); 18S 5′-CATGGCCGTTCTTAGTTGGT-3′ (forward), 5′-ATGC
CAGAGTCTCGTTCGTT-3′ (reverse).
Flow cytometry
Flow cytometry analysis was performed on BD
Biosciences FACSCalibur flow cytometer using Cell Quest software.
For measuring cell death, cells were incubated with trypsin,
harvested, washed and stained with 50 μg/ml propidium iodide
(Sigma, P4170) in Nicoletti buffer containing 50 μg/ml RNase
(AppliChem, A3832,0050). For cell cycle distribution analysis,
cells were washed, incubated with trypsin, harvested, washed, fixed
in 75% ethanol for 1 h at 4°C, washed and stained with 50
μg/ml propidium iodide in PBS containing 50 μg/ml
RNase.
Immunohistochemistry
Cells were washed, incubated with trypsin,
harvested, washed and fixed in formaldehyde for 15 min at 37°C.
After washing, the cells were dissolved in 100% ethanol to which
30% FCS in PBS was added (in proportion 5:1). Paraffin-embedded
cells and tissue sections were dewaxed and rehydrated using xylene
and a series of graded alcohols, followed by heat-induced antigen
retrieval using a target retrieval solution (Dako, S2031) in a
pressure cooker for 15 min. The human tissue samples were provided
by the Tumor Tissue Bank of the NCT Heidelberg after approval by
the ethics committee of the University of Heidelberg. The
collection of tissue samples comprised 44 primary colorectal
adenocarcinomas with pT stadium pT3 and the following features: G2,
n=25; G3, n=19; pN0, n=25; pN1/2, n=19. Staining was done on an
automated staining system (Techmate 500, Dako) with
avidin-biotin-complex peroxidase technique using
aminoethylcarbazole for visualization and haematoxylin for
counterstaining. The sections were incubated with primary antibody
overnight at 4°C [anti-Notch1 (50 ng/ml), anti-p27 (5 μg/ml)
and anti-Ki67 (1:400)] and processed according to the
manufacturer’s instructions for the following kits: ChemMate
Detection kit (Dako, K5003), ChemMate Buffer kit (Dako, K5006),
Avidin/Biotin Blocking kit (Vector Laboratories, Burlingame, CA,
USA, SP-2001). For the immunohistochemical semi-quantitative
assessment of p27 expression, the staining intensity of
immunoreactive tumor cells was determined based on the following
scoring system: the intensity ranged from 0, negative; 1, low; 2,
medium to 3, high. For statistical analysis, negative and low p27
expression was regarded as ‘p27 low’ and medium and high p27
expression was regarded as ‘p27 high’. Proliferative activity of
the tumors was assessed by counting Ki67-positive cells in the
tumor tissue: ‘Ki67 low’ was attributed to tumors showing <50%
Ki67-positive tumor cells, and ‘Ki67 high’ tumors showing ≥50%
Ki67-positive tumor cells.
Clonogenicity assay
Cells were transfected with control, Notch1 and/or
p27 siRNA. After 24 h, 500 cells from each transfection were seeded
per well on 6-well plates. They were incubated for additional 6
days at 37°C and stained with crystal violet (Acros Organics, Geel,
Belgium, 229641000). Plates were scanned and the number of colonies
was counted using Image J Software (National Institute of Health,
Bethesda, MD, USA).
BrdU assay
Cells were transfected with control, Notch1 and/or
p27 siRNA. After 48 h, 5,000 cells were seeded per well on 96-well
plates. Following an additional 48 or 72 h incubation of the cells,
they were labeled with BrdU using Cell Proliferation Biotrak ELISA
System (GE Healthcare, RPN250) according to the manufacturer’s
instructions. The label was measured on Microplate Reader (Bio-Rad,
680) provided with Microplate Manager 5.2.1 software.
Pulse-chase experiment
For the pulse, the cells were washed with PBS and
incubated for 16 h in methionine/cysteine-free MEM, to which 500
μCi of [35S]-cysteine/methionine (Met-[35S]-label, Hartmann
Analytic, Braunschweig, Germany) was added per plate. After
labeling the cells were washed with PBS and chased with complete
MEM for the indicated times. The radiolabeled cells were lysed in
cold lysis buffer (30 mM Tris-HCl, 120 mM NaCl, 10% glycerol, 2 mM
EDTA, 2 mM KCl, 1% Triton X-100) containing protease inhibitors,
ultrasonified and centrifuged (14,000 × g, 15 min). Supernatants
were adjusted to the same protein content with lysis buffer and
pre-cleared with Sepharose® CL-4B beads (Sigma,
CL4B200). Samples were then incubated with 1.5 μg of p27
antibody overnight at 4°C. Immune complexes were precipitated with
rec-Protein G-Sepharose® 4B Conjugate beads (Invitrogen, 101241)
for 4 h at 4°C. Precipitates were washed five times with cold lysis
buffer. Proteins were released from the beads by incubation with
0.5 N NaOH for 1 h at 37°C. Finally protein samples were mixed with
Ultima Gold™ scintillation cocktail (Perkin-Elmer, Rodgau, Germany)
and counted for radioactivity in a liquid scintillation counter
with automatic quench correction (Perkin-Elmer). Non-specific
binding to the beads was determined by conducting the whole
immunoprecipitation procedure without p27 antibody. All
measurements were corrected for non-specific binding.
Results
Notch1 knockdown causes p27 stabilization
in CRC cells
Notch1 receptor overexpression and aberrant pathway
activation have previously been described in CRC (9). Additionally, it has been reported
that the effects of Notch1 on cell fate involve the cell cycle
regulator p27 (7,19). Therefore, we initially compared the
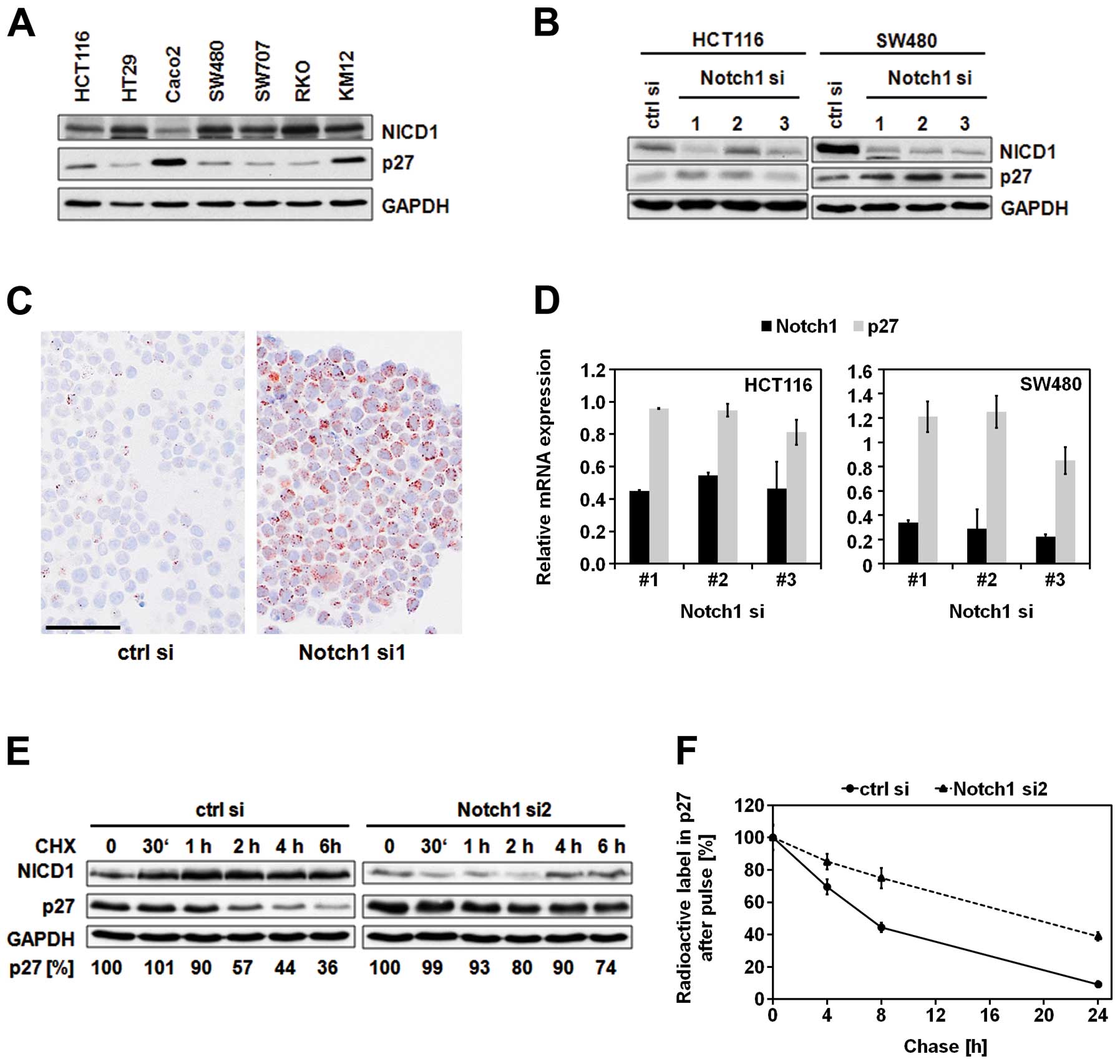
levels of activated Notch1 (NICD1) and p27 in different CRC cell
lines by immunoblot analysis (Fig.
1A). Amongst the seven analyzed cell lines, Caco2 cells showed
lowest NICD1 and highest p27 levels. Consistent with this
reciprocal correlation between p27 and Notch1 expression, knockdown
of Notch1 caused upregulation of p27 protein levels (Fig. 1B and C). Since the increased
protein levels of p27 were not caused by significant alterations in
p27 mRNA expression (Fig. 1D), we
further characterized the putative post-transcriptional regulation
of p27 by Notch1. Inhibition of protein translation using
cycloheximide indicated significant stabilization of p27 protein
over time when Notch1 was depleted (Fig. 1E). Consistently, pulse-chase
analysis revealed more than 2.5-fold prolongation of the p27
half-life (from 7 to 18 h) upon downregulation of Notch1 (Fig. 1F).
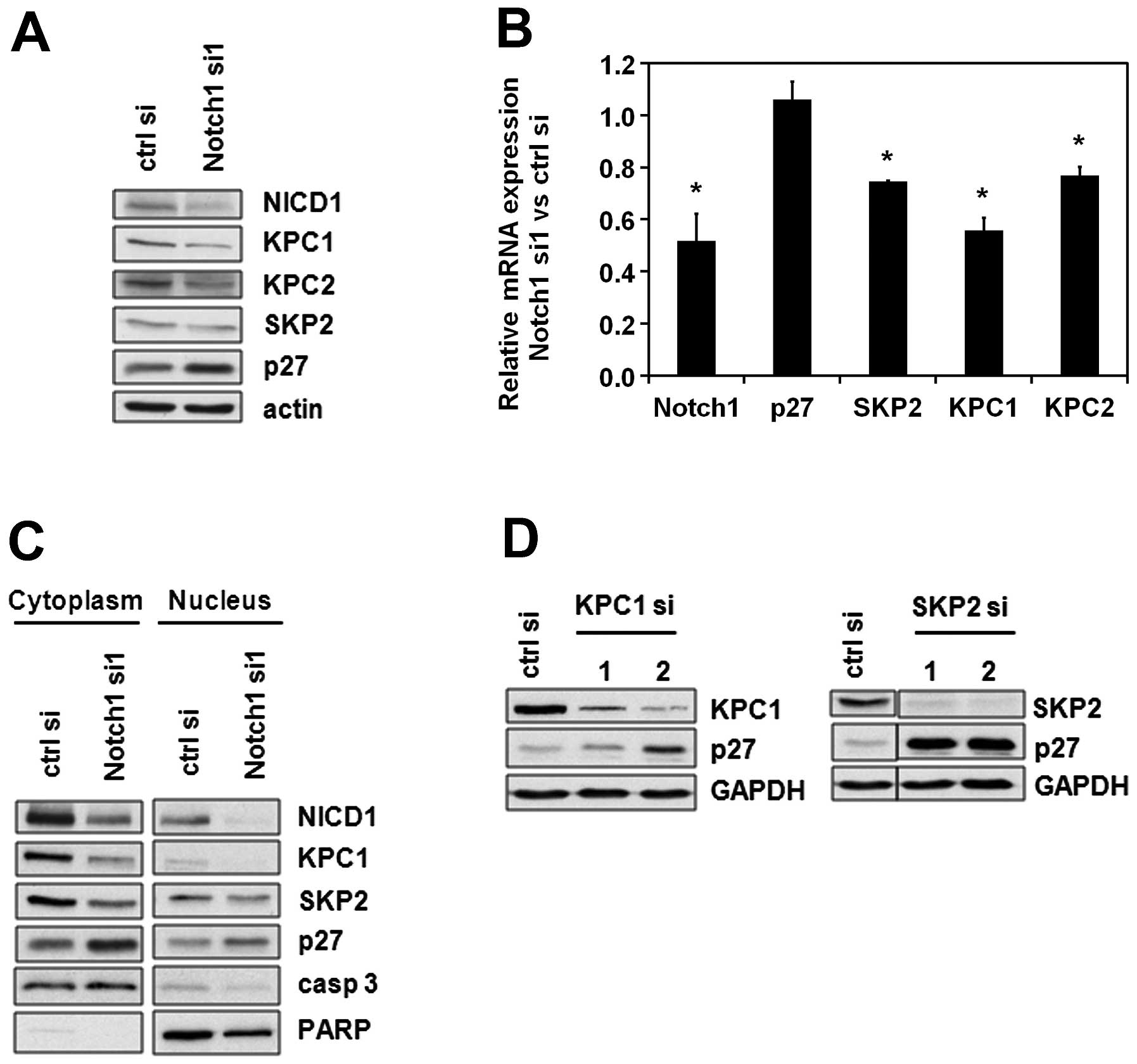
Notch1 regulates ubiquitin ligases
responsible for p27 degradation
The most prominent mode of regulating p27 levels
throughout the cell cycle is targeting p27 for proteasomal
degradation. This is accomplished via ubiquitination by the Kip1
ubiquitination-promoting complex (KPC) in the cytoplasm in early G1
phase (15) and by the
SKP1-CUL1-F-box protein (SCFSKP2) in the nucleus in late
G1 which allows the cell to proceed to S phase (20). Taking these key mechanisms of p27
regulation into consideration, we analyzed the effects of Notch1 on
the two subunits of the KPC-ubiquitin ligase, namely KPC1 and KPC2,
and on S-phase kinase-associated protein 2 (SKP2), the F-box
substrate-recognition subunit of SCFSKP2 complex. An
immunoblot analysis showed that KPC1, KPC2 and SKP2 were
downregulated after Notch1 depletion (Fig. 2A). This regulation occurs at least
partially on the transcriptional level, since SKP2, KPC1 and KPC2
mRNA levels were diminished upon Notch1 downregulation (Fig. 2B). A subcellular fractionation
revealed that in consequence of the regulation of both nuclear and
cytoplasmic p27-targeting ubiquitin ligases, the levels of p27
increase in both cellular fractions (Fig. 2C). Similar to Notch1 knockdown,
KPC1 and SKP2 knockdown resulted in an upregulation of p27
(Fig. 2D). Taken together, these
results suggest that the regulation of p27 by Notch1 is mediated by
KPC and/or SKP2.
p27 is a mediator of Notch1-dependent
growth arrest
Since p27 is one of the key inhibitors of the cell
cycle, we investigated the effects of its regulator, Notch1, on
cellular growth. Furthermore, we analyzed to which extent p27
contributes to these effects. Notch1 knockdown inhibited cell
proliferation as indicated by the decrease of BrdU-positive cells
which was maintained over time (Fig.
3A). This was accompanied by a G2/M phase arrest (Fig. 3B). Finally, colony formation was
inhibited by Notch1 downregulation (Fig. 3C). Importantly, all of these
Notch1-dependent effects on cellular growth and proliferation could
partially be compensated by knockdown of p27 (Fig. 3A–C). Further, to investigate the
impact of p27 expression on proliferation in human tumor tissue, 44
CRC patients’ samples were immunohistochemically analyzed. In
accordance with the data obtained from cell culture experiments,
samples expressing low levels of p27 were characterized by an
increased proliferation capacity and vice versa (Fig. 3D). Interestingly, an upregulation
of Notch1 and downregulation of p27 were observed at the
infiltration zones of the carcinomas (Fig. 3E).
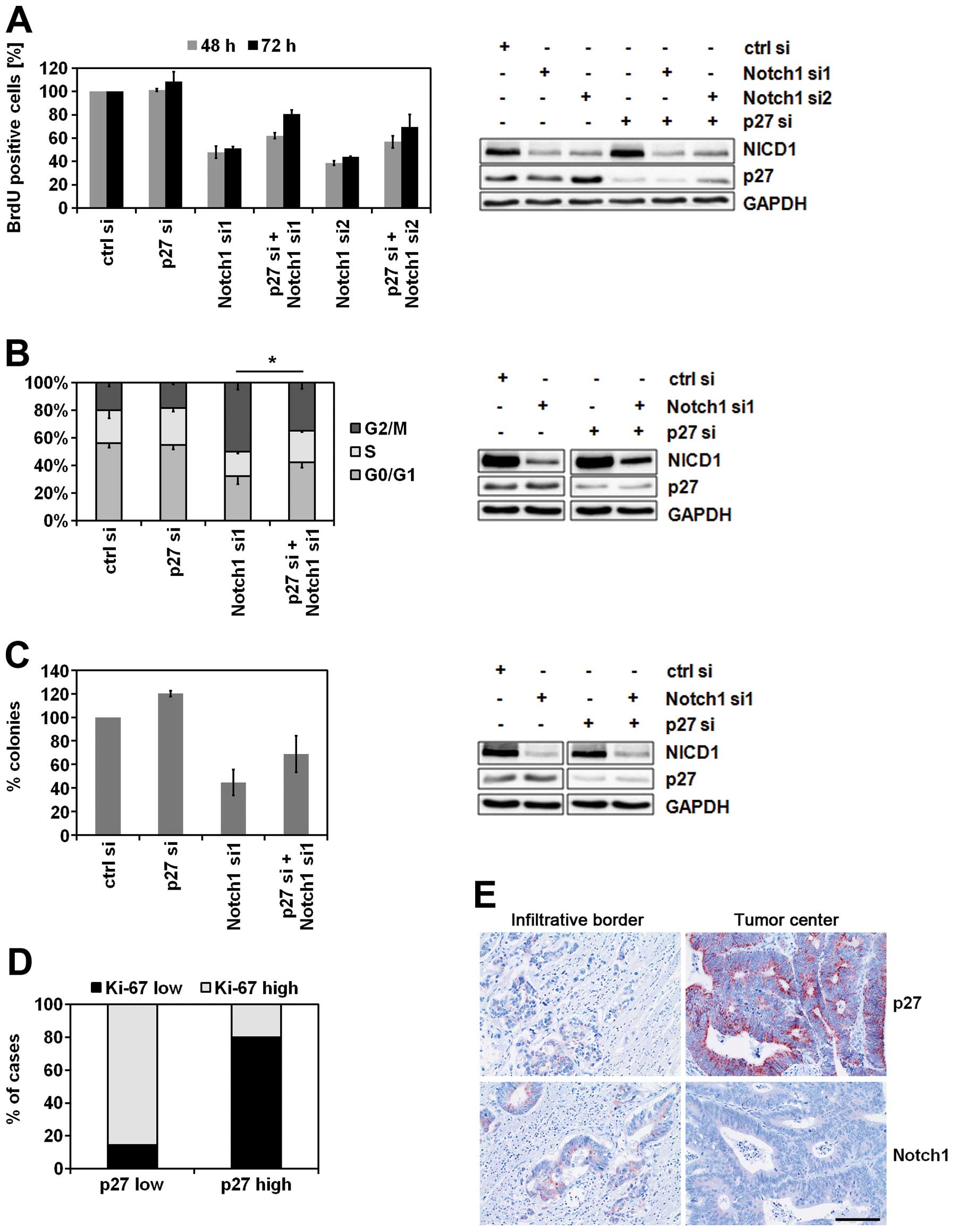 | Figure 3.p27 is a mediator of Notch1-dependent
growth arrest. (A) BrdU analysis of proliferating Caco2 cells.
Forty-eight hours after siRNA transfection, Caco2 cells were seeded
on 96-well plate and incubated for additional 48 or 72 h. Then, the
proliferating cells were labeled with BrdU and quantified
spectrophotometrically (n=2, mean ± SD) (left panel).
Representative immunoblot analysis of Caco2 lysates prepared in
parallel (right panel). Total soluble proteins (30 μg) were
separated per lane. (B) Cell cycle analysis of SW480 cells.
Seventy-two hours after siRNA-transfection the cells were stained
with PI and their cell cycle distribution was analyzed by flow
cytometry (n=3, mean ± SD, Student’s t-test, *P<0.05
for all three cell cycle phases) (left panel). Representative
immunoblot analysis of SW480 lysates prepared in parallel (right
panel). Total soluble proteins (30 μg) were separated per
lane. (C) Clonogenicity assay of HCT116 cells. Twenty-four hours
after transfection with the indicated siRNAs, 500 cells from each
transfection were seeded on a 6-well plate and incubated for
additional 6 days. Then the colonies were stained with crystal
violet and counted using Image J Software after scanning of the
plate (n=2, mean ± SD) (left panel). Representative immunoblot
analysis of HCT116 lysates prepared in parallel (right panel).
Total soluble proteins (30 μg) were separated per lane. (D)
Association between p27 expression and proliferative activity in
human CRC tissue samples. Tissue samples (n=44) were
immunohistochemically stained with anti-p27 antibody and anti-Ki67
antibody. The cut-off value for low/high Ki67 was 50% Ki67-positive
tumor cells (group ‘p27 low’, n=14; group ‘p27 high’, n=30). (E)
Immunohistochemical expression of Notch1 and p27 at the invasion
front of CRC. In contrast to central parts of the tumor, at the
infiltrative tumor border expression of p27 was typically low
whereas Notch1 was strongly expressed by carcinoma cells (bar, 50
μm). |
Notch1 knockdown synergistically promotes
cell death together with chemo- and radiotherapy
Various mechanisms for interference with Notch
signaling have been investigated in CRC therapy (reviewed in ref.
8) and γ-secretase inhibitors
(GSIs) have been tested in phase I and II clinical trials.
Therefore, we were interested in analyzing the potential
synergistic effects of Notch1 siRNA-mediated knockdown in
combination with chemo- and radiotherapy. Compared to GSIs, the
siRNA-mediated downregulation used in our study is specifically
limited to the interference with Notch1 signaling. As shown in
Fig. 4, siRNA-mediated suppression
of Notch1 significantly increased cell death induced by
5-fluorouracil (5-FU), oxaliplatin (OX) and ionizing radiation.
Notch1 knockdown itself induced slight to moderate increase of cell
death (control samples). In contrast, HCT116 cells showed only
slight additive effects of Notch1 knockdown and conventional
anticancer therapy (data not shown).
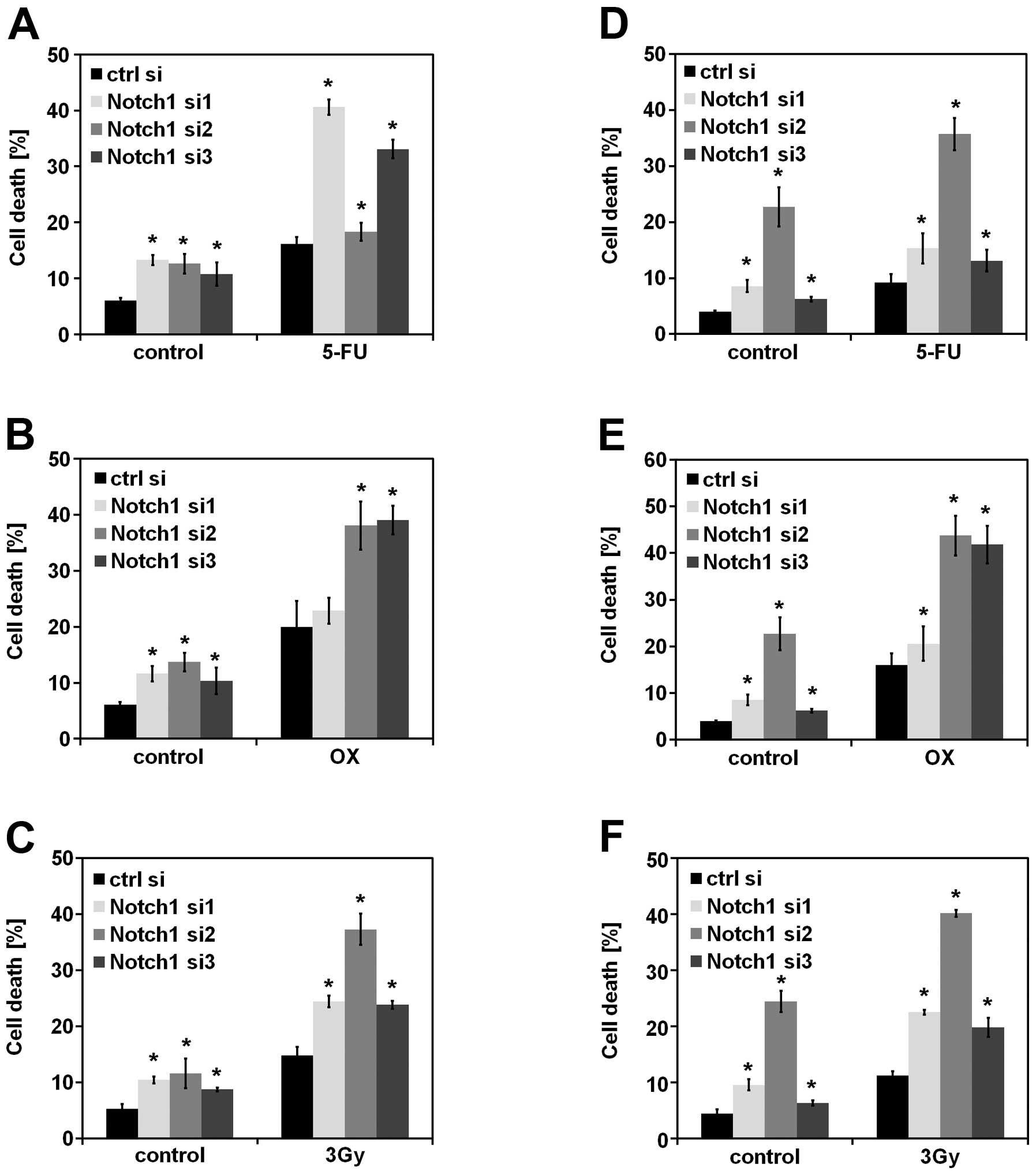 | Figure 4.Knockdown of Notch1 synergistically
increases cell death induced by chemo- and radiotherapy. (A, B and
C) SW480 and (D, E and F) SW707 cells were transfected with the
indicated siRNAs. Twenty-four hours later, the cells were treated
with a chemotherapeutic drug, 5-FU (200 μM) or oxaliplatin
(OX, 100 μM), or irradiated (3 Gy). After additional 48 h of
incubation, cell death was quantified by flow cytometry analysis
using PI staining (n=3, mean ± SD, Student’s t-test,
*P<0.01). P-values were calculated for each treatment
comparing the Notch1 siRNA-transfected cells to the control
siRNA-transfected cells. |
Discussion
Notch signaling plays a crucial role in cell
proliferation but the exact underlying mechanisms are far from
clear. Several studies have shown that in T-ALL and breast cancer
Notch1 regulates cell cycle and progression via its direct target
c-myc (21–24). Cyclin D1 was described to be a
direct target of Notch1 and Jag1-mediated Notch signaling in breast
cancer and RKE (rat kidney epithelial) cells (25–27),
and cyclin D3 was shown to be an essential mediator of Notch
tumorigenic role in T-ALL (28).
In addition to cyclin D1, CDK2 was also demonstrated to be
regulated by Notch in RKE cells (29). Accordingly, the CDKIs p21 and p57
are transcriptionally repressed by Notch signaling (7,30).
Further, Notch signaling was described to regulate p27
transcription in CRC initiating cells which ensured their
self-renewal (19).
In this study, we confirm that Notch1 suppresses the
expression of p27 in colorectal carcinoma cells. Various mechanisms
could account for this phenomenon. In fibroblasts and T-ALL cell
lines Notch1 signaling transcriptionally activates the expression
of ubiquitin ligase SKP2, thereby decreasing cellular p27 protein
levels (20,31). In intestinal crypt progenitor cells
the transcriptional regulation of p27 by Notch1 signaling is
mediated by Hes1 (7). In our
study, the knockdown of Notch1 in the CRC cell lines did not cause
a decrease in Hes1 mRNA probably due to the compensatory signaling
by the other Notch receptors (data not shown). However, Notch1
knockdown induced a decrease in SKP2, KPC1 and KPC2 mRNA
expression. KPC1 is the catalytic subunit of the KPC E3
ubiquitin-protein ligase complex, while KPC2 is the non-catalytic
subunit of this complex. In contrast to SKP2, which is the
substrate-recognition subunit of the SCF E3 ubiquitin-protein
ligase complex, the KPC complex is localized in the cytoplasm. In
accordance with our data KPC has been shown to accomplish p27
degradation in different cell types such as fibroblasts, cells of
the nervous system and dendritic cells (15,32–36).
The KPC-mediated decrease of p27 in the cytoplasm is necessary for
the cells to proceed from G0 to G1 phase, while the SCF-mediated
downregulation of p27 in the nucleus is required for transition
from G1 to S phase of the cell cycle. Notch1 regulation of both E3
ubiquitin-protein ligase complexes accounts for the altered levels
of p27 in both the cytoplasmic and the nuclear cellular fractions.
While previous reports have shown that the SKP2 promoter contains a
NICD1-binding sequence, our study is the first one describing
Notch1-dependent transcriptional regulation of KPC1 and KPC2.
Further analyses revealed that the effects of Notch1
on cell cycle and cellular proliferation are at least partially
mediated by p27. Notch1 knockdown induced G2/M phase cell cycle
arrest, decreased proliferation and diminished colony formation.
However, the function of p27 might be supported by other cell cycle
inhibitors from the CDKI family such as p21 and p57. In fact, we
have observed a transcriptional regulation of p21 by Notch1
signaling leading to increase of p21 protein expression after
Notch1 knockdown (data not shown). Furthermore, transcriptional
regulation of p57 by the Notch target gene Hes1 has previously been
described in intestinal crypt progenitor cells (7). Hes1 was also shown to bind p57
promoter and repress p57 transcription, thereby avoiding senescence
in hepatocellular carcinoma (37).
Although the major role of p27 is to inhibit the
cell cycle, this is not its only function in the cell.
Interestingly, cytoplasmic localization of p27 has been associated
with enhanced cell motility because of reduced activity of the
small GTPase RhoA (38). Therefore
the subcellular localization is a crucial criterion whether p27
will play the role of a tumor suppressor or an oncogene. Highly
aggressive tumors are often characterized by reduced nuclear p27
which enhances proliferation and by mislocalized cytoplasmic p27
which drives invasion (39).
Several studies in CRC found that reduced p27 protein levels are
associated with poor outcome, including disease recurrence or death
(40–43). Others have reported that
cytoplasmic p27 correlated with high nuclear p27 staining and good
prognosis in CRC patients (44).
In addition to being used as prognostic marker, it has been
proposed that p27 has predictive potential for response to cancer
therapy. Increase of nuclear p27 levels or shift of p27 from the
cytoplasm to the nucleus might be a predictor for response to
inhibitors of p27 upstream regulators such as MEK/Src or PI3K/mTOR
respectively (45,46).
In addition to the inhibitory effects of Notch1
knockdown on proliferation and cell cycle regulation, our study
provides evidence, that high levels of Notch1 render CRC cells
resistant to cell death induced by cytotoxic drugs and ionizing
radiation. Notch signaling has previously been described to
regulate anti-apoptotic proteins such as Mcl-1, Bcl-2,
Bcl-xL and XIAP (47,48),
thereby inhibiting apoptosis. Additionally, Notch-dependent
inhibition of apoptosis can also take place through negative
regulation of p53 and PTEN (23,49).
Inhibition of Notch signaling has emerged as a promising future
therapeutic approach. GSIs are currently used in phase I and II
clinical trials for CRC patients (RO4929097, Roche, NCT01198535,
NCT01158274, NCT01131234; NCT01270438, NCT01116687) and other
malignancies such as breast cancer, pancreatic cancer, T-ALL, and
brain tumors (MK0752, Merck, NCT00645333, NCT01098344, NCT00100152,
NCT00572182). There is growing evidence that the combination of
Notch signaling inhibition and chemotherapy, radiotherapy or
inhibition of other pathways being crucial for tumor maintenance
will synergize together in order to kill CRC cells (50,51).
This is in fact the rationale of many ongoing clinical trials. For
example, the combination of cetuximab (a monoclonal antibody
blocking the EGFR) and the GSI RO4929097 is used in a phase I
clinical trial in patients with metastatic CRC (NCT01198535).
RO4929097 in combination with chemotherapy (oxaliplatin, leucovorin
calcium, 5-FU) and bevacizumab (a monoclonal antibody inhibiting
VEGF-A) is used in a phase II clinical trial in patients with the
same malignancy (NCT01270438). Since Notch signaling plays a
crucial role in CRC initiating cells (so-called colorectal cancer
stem cells), it further represents an attractive therapeutic target
especially with regard to the highly resistant cancer initiating
cells (52).
Unfortunately, GSIs target all Notch receptors as
well as the other substrates of the γ-secretase. Thus, significant
side-effects of GSIs, such as gastro-intestinal toxicity and
diarrhea, were observed in clinical trials (53) due to the inhibition of Notch
signaling causing the differentiation of the intestinal progenitor
cells into post-mitotic secretory goblet cells (54). Therefore, other methods for more
specific inhibition of individual players within the Notch
signaling pathway are explored. For instance, a monoclonal antibody
against the Delta-like ligand 4 (DLL4), called MEDI0639, is used in
a phase I clinical trial in advanced solid tumors (NCT01577745).
The expected antitumor effect of this antibody is due to inhibition
of tumor angiogenesis (3). Notch1
and Notch2 specific monoclonal antibodies (anti-NRR1 and anti-NRR2)
stabilizing the receptor negative regulatory region
(ADAM/S2-cleavage region) are holding promise for successful
specific inhibition of Notch signaling, thereby avoiding the severe
intestinal toxicity of the dual receptor inhibition (55).
In conclusion, our study provides initial evidence
that specific interference with Notch1 signaling could be
sufficient to sensitize CRC cells to 5-FU, oxaliplatin and ionizing
radiation, which are routinely used in the clinical practice.
Downregulation of Notch1 receptor leads to increased p27 levels and
analyzing the subcellular localization of this cell cycle inhibitor
might be important for predicting treatment success. Further
investigation on the mechanism of regulation of KPC1 and KPC2 by
Notch1 might also be an attractive topic. Collectively, we describe
possible mechanisms of regulation of CRC cell growth by Notch1
signaling and investigating these complex interactions is crucial
for using Notch1 inhibition for specifically targeting the death of
cancer cells with mild side-effects for the patients.
Acknowledgements
We thank Sarah Messnard and Heike
Conrad for their technical assistance. We further thank Stephan
Macher-Goeppinger for his expert advice on
immunohistochemistry.
References
|
1.
|
Bray SJ: Notch signalling: a simple
pathway becomes complex. Nat Rev Mol Cell Biol. 7:678–689. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Radtke F and Raj K: The role of Notch in
tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer.
3:756–767. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Benedito R, Roca C, Sorensen I, et al: The
notch ligands Dll4 and Jagged1 have opposing effects on
angiogenesis. Cell. 137:1124–1135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Fre S, Huyghe M, Mourikis P, Robine S,
Louvard D and Artavanis-Tsakonas S: Notch signals control the fate
of immature progenitor cells in the intestine. Nature. 435:964–968.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Schroder N and Gossler A: Expression of
Notch pathway components in fetal and adult mouse small intestine.
Gene Expr Patterns. 2:247–250. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Jensen J, Pedersen EE, Galante P, et al:
Control of endodermal endocrine development by Hes-1. Nat Genet.
24:36–44. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Riccio O, van Gijn ME, Bezdek AC, et al:
Loss of intestinal crypt progenitor cells owing to inactivation of
both Notch1 and Notch2 is accompanied by derepression of CDK
inhibitors p27Kip1 and p57Kip2. EMBO Rep. 9:377–383. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Qiao L and Wong BC: Role of Notch
signaling in colorectal cancer. Carcinogenesis. 30:1979–1986. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Zhang Y, Li B, Ji ZZ and Zheng PS: Notch1
regulates the growth of human colon cancers. Cancer. 116:5207–5218.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Zolkiewska A: ADAM proteases: ligand
processing and modulation of the Notch pathway. Cell Mol Life Sci.
65:2056–2068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Fortini ME: Gamma-secretase-mediated
proteolysis in cell-surface-receptor signalling. Nat Rev Mol Cell
Biol. 3:673–684. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Katoh M and Katoh M: Integrative genomic
analyses on HES/HEY family: Notch-independent HES1, HES3
transcription in undifferentiated ES cells, and Notch-dependent
HES1, HES5, HEY1, HEY2, HEYL transcription in fetal tissues, adult
tissues, or cancer. Int J Oncol. 31:461–466. 2007.PubMed/NCBI
|
|
13.
|
Chu IM, Hengst L and Slingerland JM: The
Cdk inhibitor p27 in human cancer: prognostic potential and
relevance to anti-cancer therapy. Nat Rev Cancer. 8:253–267. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Lu Z and Hunter T: Ubiquitylation and
proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2)
CDK inhibitors. Cell Cycle. 9:2342–2352. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Kamura T, Hara T, Matsumoto M, et al:
Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1)
at G1 phase. Nat Cell Biol. 6:1229–1235. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Ishida N, Hara T, Kamura T, Yoshida M,
Nakayama K and Nakayama KI: Phosphorylation of p27Kip1 on serine 10
is required for its binding to CRM1 and nuclear export. J Biol
Chem. 277:14355–14358. 2002. View Article : Google Scholar
|
|
17.
|
Castro F, Dirks WG, Fahnrich S,
Hotz-Wagenblatt A, Pawlita M and Schmitt M: High-throughput
SNP-based authentication of human cell lines. Int J Cancer.
132:308–314. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Schmitt M and Pawlita M: High-throughput
detection and multiplex identification of cell contaminations.
Nucleic Acids Res. 37:e1192009. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Sikandar SS, Pate KT, Anderson S, et al:
NOTCH signaling is required for formation and self-renewal of
tumor-initiating cells and for repression of secretory cell
differentiation in colon cancer. Cancer Res. 70:1469–1478. 2010.
View Article : Google Scholar
|
|
20.
|
Sarmento LM, Huang H, Limon A, et al:
Notch1 modulates timing of G1-S progression by inducing SKP2
transcription and p27 Kip1 degradation. J Exp Med. 202:157–168.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Sharma VM, Calvo JA, Draheim KM, et al:
Notch1 contributes to mouse T-cell leukemia by directly inducing
the expression of c-myc. Mol Cell Biol. 26:8022–8031. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Weng AP, Millholland JM, Yashiro-Ohtani Y,
et al: c-Myc is an important direct target of Notch1 in T-cell
acute lymphoblastic leukemia/lymphoma. Genes Dev. 20:2096–2109.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Palomero T, Sulis ML, Cortina M, et al:
Mutational loss of PTEN induces resistance to NOTCH1 inhibition in
T-cell leukemia. Nat Med. 13:1203–1210. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Klinakis A, Szabolcs M, Politi K, Kiaris
H, Artavanis-Tsakonas S and Efstratiadis A: Myc is a Notch1
transcriptional target and a requisite for Notch1-induced mammary
tumorigenesis in mice. Proc Natl Acad Sci USA. 103:9262–9267. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Ling H, Sylvestre JR and Jolicoeur P:
Notch1-induced mammary tumor development is cyclin D1-dependent and
correlates with expansion of pre-malignant multipotent duct-limited
progenitors. Oncogene. 29:4543–4554. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Cohen B, Shimizu M, Izrailit J, et al:
Cyclin D1 is a direct target of JAG1-mediated Notch signaling in
breast cancer. Breast Cancer Res Treat. 123:113–124. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Stahl M, Ge C, Shi S, Pestell RG and
Stanley P: Notch1-induced transformation of RKE-1 cells requires
up-regulation of cyclin D1. Cancer Res. 66:7562–7570. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Sicinska E, Aifantis I, Le Cam L, et al:
Requirement for cyclin D3 in lymphocyte development and T cell
leukemias. Cancer Cell. 4:451–461. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Ronchini C and Capobianco AJ: Induction of
cyclin D1 transcription and CDK2 activity by Notch(ic): implication
for cell cycle disruption in transformation by Notch(ic). Mol Cell
Biol. 21:5925–5934. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Gotte M, Greve B, Kelsch R, et al: The
adult stem cell marker Musashi-1 modulates endometrial carcinoma
cell cycle progression and apoptosis via Notch-1 and p21WAF1/CIP1.
Int J Cancer. 129:2042–2049. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Dohda T, Maljukova A, Liu L, et al: Notch
signaling induces SKP2 expression and promotes reduction of p27Kip1
in T-cell acute lymphoblastic leukemia cell lines. Exp Cell Res.
313:3141–3152. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Hara T, Kamura T, Kotoshiba S, et al: Role
of the UBL-UBA protein KPC2 in degradation of p27 at G1 phase of
the cell cycle. Mol Cell Biol. 25:9292–9303. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Lu Y, Adegoke OA, Nepveu A, et al: USP19
deubiquitinating enzyme supports cell proliferation by stabilizing
KPC1, a ubiquitin ligase for p27Kip1. Mol Cell Biol. 29:547–558.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Zhao J, Zhang S, Wu X, et al: KPC1
expression and essential role after acute spinal cord injury in
adult rat. Neurochem Res. 36:549–558. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Lu C, Huang X, Zhang X, et al: miR-221 and
miR-155 regulate human dendritic cell development, apoptosis, and
IL-12 production through targeting of p27kip1, KPC1, and SOCS-1.
Blood. 117:4293–4303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Susaki E, Nakayama K and Nakayama KI:
Cyclin D2 translocates p27 out of the nucleus and promotes its
degradation at the G0-G1 transition. Mol Cell Biol. 27:4626–4640.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Giovannini C, Gramantieri L, Minguzzi M,
et al: CDKN1C/P57 is regulated by the Notch target gene Hes1 and
induces senescence in human hepatocellular carcinoma. Am J Pathol.
181:413–422. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
See WL, Heinberg AR, Holland EC and Resh
MD: p27 deficiency is associated with migration defects in
PDGF-expressing gliomas in vivo. Cell Cycle. 9:1562–1567. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Wander SA, Zhao D and Slingerland JM: p27:
a barometer of signaling deregulation and potential predictor of
response to targeted therapies. Clin Cancer Res. 17:12–18. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Noguchi T, Kikuchi R, Ono K, Takeno S,
Moriyama H and Uchida Y: Prognostic significance of p27/kip1 and
apoptosis in patients with colorectal carcinoma. Oncol Rep.
10:827–831. 2003.PubMed/NCBI
|
|
41.
|
Manne U, Jhala NC, Jones J, et al:
Prognostic significance of p27(kip-1) expression in colorectal
adenocarcinomas is associated with tumor stage. Clin Cancer Res.
10:1743–1752. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Wu JT, Kakar S, Nelson RL, et al:
Prognostic significance of DCC and p27Kip1 in colorectal cancer.
Appl Immunohistochem Mol Morphol. 13:45–54. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Rosati G, Chiacchio R, Reggiardo G, De
Sanctis D and Manzione L: Thymidylate synthase expression, p53,
bcl-2, Ki-67 and p27 in colorectal cancer: relationships with tumor
recurrence and survival. Tumour Biol. 25:258–263. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Watson NF, Durrant LG, Scholefield JH, et
al: Cytoplasmic expression of p27(kip1) is associated with a
favourable prognosis in colorectal cancer patients. World J
Gastroenterol. 12:6299–6304. 2006.PubMed/NCBI
|
|
45.
|
Gysin S, Lee SH, Dean NM and McMahon M:
Pharmacologic inhibition of RAF→MEK→ERK signaling elicits
pancreatic cancer cell cycle arrest through induced expression of
p27Kip1. Cancer Res. 65:4870–4880. 2005.
|
|
46.
|
Hong F, Larrea MD, Doughty C, Kwiatkowski
DJ, Squillace R and Slingerland JM: mTOR-raptor binds and activates
SGK1 to regulate p27 phosphorylation. Mol Cell. 30:701–711. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Fassl A, Tagscherer KE, Richter J, et al:
Notch1 signaling promotes survival of glioblastoma cells via
EGFR-mediated induction of anti-apoptotic Mcl-1. Oncogene.
31:4698–4708. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Liu WH, Hsiao HW, Tsou WI and Lai MZ:
Notch inhibits apoptosis by direct interference with XIAP
ubiquitination and degradation. EMBO J. 26:1660–1669. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Beverly LJ, Felsher DW and Capobianco AJ:
Suppression of p53 by Notch in lymphomagenesis: implications for
initiation and regression. Cancer Res. 65:7159–7168. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Meng RD, Shelton CC, Li YM, et al:
gamma-Secretase inhibitors abrogate oxaliplatin-induced activation
of the Notch-1 signaling pathway in colon cancer cells resulting in
enhanced chemosensitivity. Cancer Res. 69:573–582. 2009. View Article : Google Scholar
|
|
51.
|
Akiyoshi T, Nakamura M, Yanai K, et al:
Gamma-secretase inhibitors enhance taxane-induced mitotic arrest
and apoptosis in colon cancer cells. Gastroenterology. 134:131–144.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Pannuti A, Foreman K, Rizzo P, et al:
Targeting Notch to target cancer stem cells. Clin Cancer Res.
16:3141–3152. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Real PJ, Tosello V, Palomero T, et al:
Gamma-secretase inhibitors reverse glucocorticoid resistance in T
cell acute lymphoblastic leukemia. Nat Med. 15:50–58. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Van Es JH, van Gijn ME, Riccio O, et al:
Notch/gamma-secretase inhibition turns proliferative cells in
intestinal crypts and adenomas into goblet cells. Nature.
435:959–963. 2005.PubMed/NCBI
|
|
55.
|
Wu Y, Cain-Hom C, Choy L, et al:
Therapeutic antibody targeting of individual Notch receptors.
Nature. 464:1052–1057. 2010. View Article : Google Scholar : PubMed/NCBI
|