Introduction
Thioredoxin (Trx) is a small redox protein (12 kDa)
with two redox-active cysteine residues (C32 and C35) in the active
site (1). There are two main
isoforms of Trx in mammalian cells; the cytoplasmic form, Trx-1 and
the mitochondrial form, Trx-2 (2).
These Trxs are reduced by thioredoxin reductase and NADPH following
the reduction of oxidative target proteins (3,4). Trx
affects cell growth and proliferation via regulating the redox
status in cells (5). Trx-1 is also
implicated in cell survival, tumor development and angiogenesis
(6,7). Numerous studies demonstrated that
overexpression of Trx is observed in many cancers such as gastric
and breast cancers (5,8). Therefore, Trx-1 inhibitors have been
considered as novel anticancer drugs. In particular, PX-12
(1-methylpropyl 2-imidazolyl disulfide) is an irreversible Trx-1
inhibitor, which has an anti-tumor effect (9). PX-12 decreased the activity of Trx-1
by thioalkylating the critical cysteine residue (Cys73) in this
protein or by increasing the dimerization of its oxidative form. In
addition, PX-12 downregulates the expression of vascular
endothelial growth factor via decreasing hypoxia-inducible
factor-1α, consequently inhibiting metastasis of cancer cells
(10,11). Recently, PX-12 has been clinically
tested in colorectal and pancreatic cancers (12,13).
Lung cancer is a major cause of cancer death in the
developed countries. Various novel therapeutic strategies are still
currently under consideration since the clinical use of cytotoxic
drugs is limited due to intrinsic or acquired resistant and
toxicity (14). An increase in
Trx-1 level is detected in lung cancer patients compared to the
control group (15). In addition,
it is reported that the high level of Trx-1 contributes to
chemo-resistance in lung cancer cells (16). However, little is known about the
cellular effect of PX-12 in lung cancer. Therefore, in the present
study we investigated the effects of PX-12 on cell growth and death
in human lung cancer A549 cells with respect to reactive oxygen
species (ROS) and glutathione (GSH) levels.
Materials and methods
Cell culture
Human lung adenocarcinoma A549 cells from the
American Type Culture Collection (ATCC, Manassas, VA, USA) were
maintained in a humidified incubator containing 5% CO2
at 37°C. A549 cells were cultured in RPMI-1640 (Sigma-Aldrich, St.
Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS;
Sigma-Aldrich) and 1% penicillin–streptomycin (Gibco BRL, Grand
Island, NY, USA). Cells were routinely grown in 100-mm plastic
tissue culture dishes (Nunc. Roskilde, Denmark) and harvested with
a solution of trypsin-EDTA (Gibco BRL) while in a logarithmic phase
of growth.
Reagents
PX-12 was purchased from Tocris Bioscience (Bristol,
UK) and was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich)
at 100 mM as a stock solution. The pan-caspase inhibitor
(Z-VAD-FMK; benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone),
caspase-3 inhibitor (Z-DEVD-FMK;
benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone), caspase-8
inhibitor (Z-IETD-FMK;
benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone) and caspase-9
inhibitor (Z-LEHDFMK;
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylke-tone) were
obtained from R&D Systems, Inc. (Minneapolis, MN, USA) and were
dissolved in DMSO at 10 mM to serve as stock solutions. NAC was
dissolved in the buffer [20 mM HEPES (pH 7.0)]. Based on the
previous studies (17,18), cells were pretreated with 15
μM caspase inhibitors or 2 mM NAC for 1 h prior to treatment
with PX-12. DMSO (0.03%) was used as a control vehicle and it did
not affect cell growth or death.
Growth inhibition assay
The effect of PX-12 on cell growth was determined by
measuring 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT; Sigma-Aldrich) absorbance in living cells as
described previously (19). In
brief, 1×104 cells per well were seeded in 96-well
microtiter plates (Nunc). Following exposure to the designated
doses of PX-12 for the indicated times, MTT solution [20 μl:
2 mg/ml in phosphate-buffered saline (PBS)] was added to each well
of the 96-well plates. The plates were additionally incubated for 3
h at 37°C. Medium was withdrawn from the plates by pipetting and
200 μl DMSO was added to each well to solubilize the
formazan crystals. The optical density was measured at 570 nm using
a microplate reader (Synergy™ 2, BioTek Instruments
Inc., Winooski, VT, USA).
Western blot analysis
The expression of proteins was evaluated using
western blot analysis, as previously described (20). In brief, 1×106 cells in
a 60-mm culture dish (Nunc) were incubated with the designated
doses of PX-12 for 72 h. The cells were then washed in PBS and
suspended in 5 Vol of lysis buffer (20 mM HEPES. pH 7.9, 20%
glycerol, 200 mM KCl, 0.5 mM EDTA, 0.5% NP40, 0.5 mM DTT, 1%
protease inhibitor cocktail). Supernatant protein concentrations
were determined using the Bradford method. Supernatant samples
containing 30 μg total protein were resolved by 15% SDS-PAGE
gels depending on the size of target proteins, transferred to
Immobilon-P PVDF membranes (Millipore, Billerica, MA, USA) by
electroblotting and then probed with anti-PARP, anti-c-PARP,
anti-Bcl-2, anti-Bax (Cell Signaling Technology Inc., Danvers, MA,
USA), anti-Trx-1 and anti-β-actin antibodies (Santa Cruz
Biotechnology, Santa Cruz, CA, USA). Membranes were incubated with
horseradish peroxidase-conjugated secondary antibodies. Blots were
developed using an ECL kit (Amersham, Arlington Heights, IL,
USA).
Measurement of Trx-1 activity
The Trx-1 activity was assessed using the
ProteoStat™ Thioredoxin-1 assay kit according to the
manufacturer’s instructions (EnZo Life Science, Plymouth Meeting,
PA, USA). In brief, 1×106 cells in 60-mm culture dish
(Nunc) were incubated with the indicated doses of PX-12 for 72 h.
The cells were then washed in PBS and suspended in 5 Vol of lysis
buffer (R&D Systems, Inc.). Protein concentrations were
determined using the Bradford method. Supernatant samples
containing 20 μg of total protein were used for
determination of Trx-1 activity, and added to each well in 96-well
microtiter plates (Nunc) with the insulin and DTT at 25°C for 30
min. The fluorescence intensity of each well was determined using a
fluorescence reader (Synergy 2).
Cell cycle and sub-G1 cell analysis
Cell cycle and sub-G1 cell analysis were determined
by propidium iodide (PI, Ex/Em=488/617 nm; Sigma-Aldrich) staining
as described previously (20). In
brief, 1×106 cells in 60-mm culture dish (Nunc) were
incubated with the designated doses of PX-12 for 72 h. Total cells
including floating cells were then washed with PBS and fixed in 70%
(v/v) ethanol. Cells were washed again with PBS, then incubated
with PI (10 μg/ml) with simultaneous RNase treatment at 37°C
for 30 min. Cellular DNA content was measured using a FACStar flow
cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA) and analyzed
by using lysis II and cellfit software (Becton-Dickinson).
Annexin V-FITC/PI staining for cell death
detection
Apoptotic cell death was determined by staining
cells with Annexin V-fluorescein isothiocyanate (FITC, Invitrogen
Life Technologies, Camarillo, CA, USA; Ex/Em=488/519 nm) as
described previously (21). In
brief, 1×106 cells in 60-mm culture dish (Nunc) were
incubated with the designated doses of PX-12 for 72 h with or
without 15 μM each caspase inhibitor or 2 mM NAC. Cells were
washed twice with cold PBS and then resuspended in 500 μl
binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM NaCl, 2.5 mM
CaCl2) at a concentration of 1×106 cells/ml.
Annexin V-FITC (5 μl) and PI (1 μg/ml) were then
added and the cells were analyzed with a FACStar flow
cytometer.
Measurement of the mitochondrial membrane
potential (MMP; ΔΨm)
The MMP (ΔΨm) levels were measured by a
rhodamine 123 fluorescent dye (Sigma-Aldrich; Ex/Em=485/535 nm) as
described previously (21,22). In brief, 1×106 cells in
60-mm culture dish (Nunc) were incubated with the designated doses
of PX-12 for 72 h with or without 15 μM each caspase
inhibitor or 2 mM NAC. Cells were washed twice with PBS and
incubated with rhodamine 123 (0.1 μg/ml) at 37°C for 30 min.
Rhodamine 123 staining intensity was determined by a FACStar flow
cytometer. The cells that were rhodamine 123-negative were
indicated to have lost MMP (ΔΨm).
Transfection of cells with Bcl-2 and Bax
siRNAs
Gene silencing of Bax and Bcl-2 was performed using
a siRNA knockdown system. A non-specific control siRNA duplex
[5′-CCUACGCC ACCAAUUUCGU(dTdT)-3′], Bcl-2 siRNA duplex [5′-CAGA
AGUCUGGGAAUCGAU(dTdT)-3′] and Bax siRNA duplex
[5′-GCUGGACAUUGGACUUCCU(dTdT)-3′] were purchased from the Bioneer
Corp. (Daejeon, Korea). In brief, 2.5×105 cells in
6-well plates (Nunc) were incubated in RPMI-1640 supplemented with
10% FBS. Following 12 h, cells (∼30–40% confluence) in each well
were transfected with the control, Bax or Bcl-2 siRNA [80 pmol in
Opti-MEM (Gibco BRL)] using Lipofectamine 2000, according to the
manufacturer’s instructions (Invitrogen, Branford, CT, USA). One
day later, cells were treated with or without 20 μM PX-12
for additional 48 h. The transfected cells were collected and used
for western blot analysis, sub-G1 cells, Annexin V-FITC staining
and MMP (ΔΨm) level measurements.
Detection of intracellular ROS
levels
Intracellular ROS were detected by means of an
oxidation-sensitive fluorescent probe dye,
2′,7′-dichlorodihydrofluorescein diacetate (Ex/Em=495/529 nm;
Invitrogen Life Technologies) and dihydroethidium (DHE,
Ex/Em=518/605 nm; Invitrogen Life Technologies) as previously
described (21,23). DHE is highly selective for
O2•- among ROS. In brief, 1×106
cells in 60-mm culture dish (Nunc) were incubated with the
designated doses of PX-12 for indicated times with or without 15
μM each caspase inhibitor or 2 mM NAC. Cells were then
washed in PBS and incubated with 20 μM H2DCFDA or
DHE at 37°C for 30 min. H2DCFDA or DHE fluorescence was
assessed using a FACStar flow cytometer. ROS and
O2•- levels were expressed as mean
fluorescence intensity, which was calculated by CellQuest
software.
Detection of intracellular GSH
Cellular GSH levels were analyzed using a
5-chloromethylfluorescein diacetate dye (CMFDA, Ex/Em=522/595 nm;
Invitrogen Life Technologies) as previously described (23,24).
In brief, 1×106 cells in 60-mm culture dish (Nunc) were
incubated with the designated doses of PX-12 for 72 h with or
without 15 μM each caspase inhibitor or 2 mM NAC. Cells were
then washed with PBS and incubated with 5 μM CMFDA at 37°C
for 30 min. CMF fluorescence intensity was determined using a
FACStar flow cytometer. Negative CMF staining (GSH-depletion) of
cells is expressed as the percentage of (-) CMF cells.
Statistical analysis
Results represent the mean of at least three
independent experiments (mean ± SD). Data were analyzed using
Instat software (GraphPad Prism4, San Diego, CA, USA). The
Student’s t-test or one-way analysis of variance with post hoc
analysis using Tukey’s multiple comparison test was used for
parametric data. The statistical significance was defined as
p<0.05.
Results
Effects of PX-12 on the growth of A549
cells
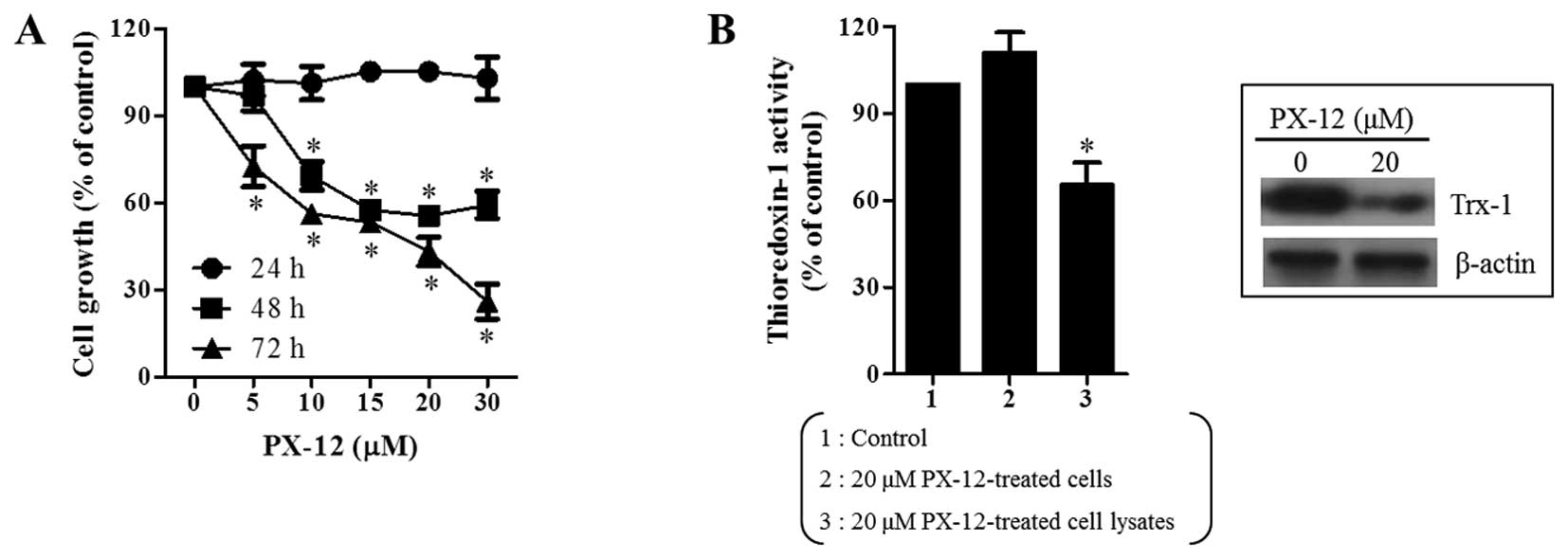
We first examined the effect of PX-12 on the growth
of A549 cells using MTT assays. After exposure to PX-12 for 24, 48
and 72 h, the growth of A549 cells dose-dependently decreased with
an IC50 of ∼20 μM at 48 and 72 h (Fig. 1A). In addition, it was observed
that PX-12 as a Trx-1 inhibitor decreased the activity of Trx-1 in
A549 cell lysate (Fig. 1B).
Moreover, PX-12 decreased the level of Trx-1 expression in A549
cells at 72 h (Fig. 1B).
Effects of PX-12 on cell cycle
distributions, cell death and MMP (ΔΨm) in A549
cells
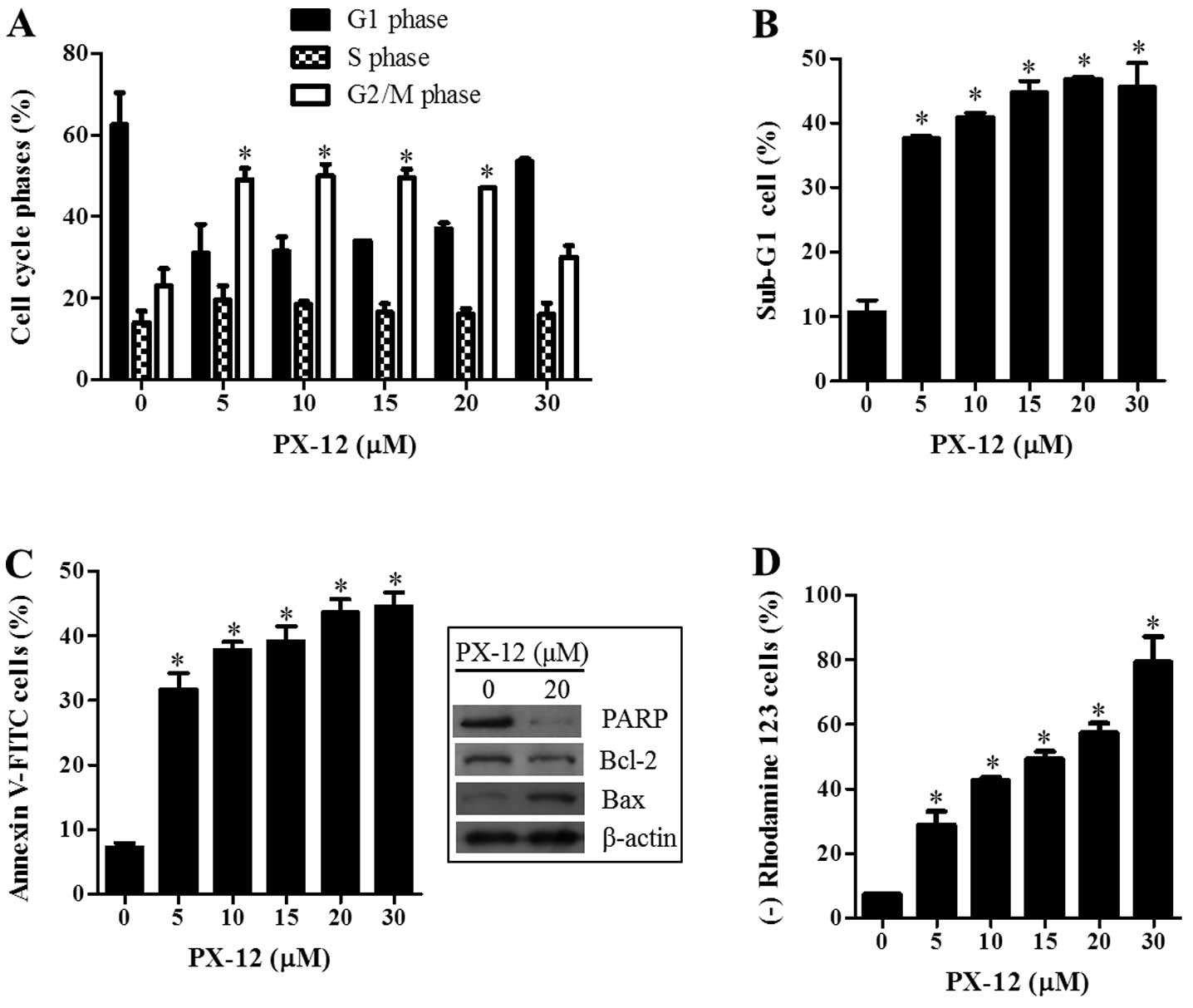
Because the growth inhibition of A549 cells caused
by PX-12 can be explained by an arrest during the cell cycle
progression, cell cycle distributions were examined at 72 h. As
shown in Fig. 2A, DNA flow
cytometric analysis indicated that 5-20 μM PX-12
significantly induced a G2/M phase arrest of the cell cycle in A549
cells. In addition, PX-12 increased the percentages of sub-G1 cells
in A549 cells in a dose-dependent manner at 72 h (Fig. 2B). This agent also increased the
numbers of Annexin V-FITC-positive cells in A549 cells (Fig. 2C). The intact of poly(ADP-ribose)
polymerase (PARP) and Bcl-2 levels were decreased by PX-12
(Fig. 2C). However, PX-12
increased Bax level in A549 cells at 72 h (Fig. 2C). Cell death is closely related to
the collapse of the MMP (ΔΨm) (25). As expected, the loss of MMP
(ΔΨm) was detected in PX-12 treated A549 cells (Fig. 2D).
Effect of Bcl-2 and Bax siRNAs on cell
death and MMP (ΔΨm) in PX-12-treated A549 cells
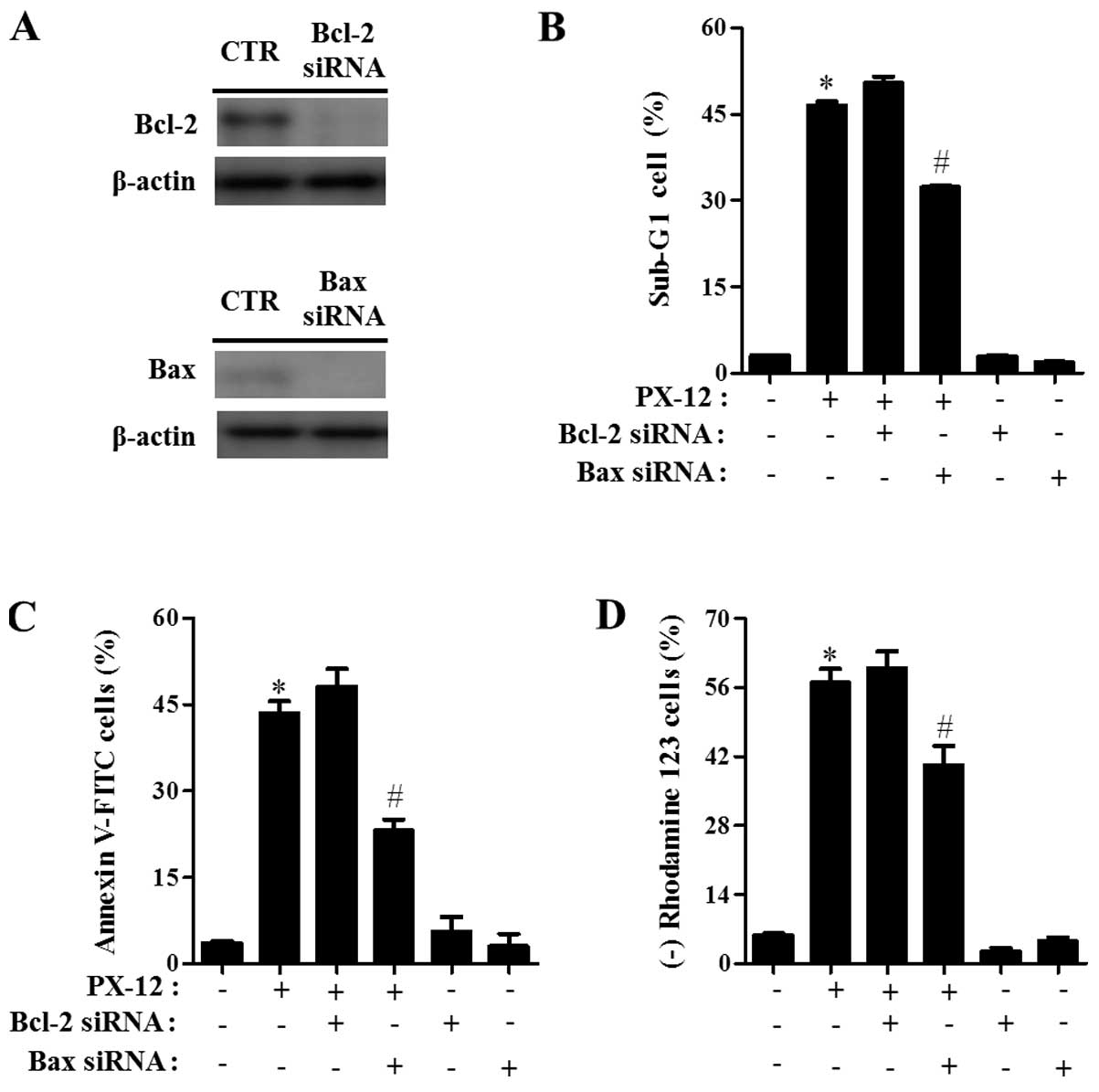
To investigate the effects of Bcl-2 and Bax on A549
cell death, A549 cells were transfected with either non-target
control siRNA, Bcl-2 or Bax siRNA. As shown in Fig. 3A, the expression of Bcl-2 and Bax
was clearly downregulated by the administration with each siRNA as
compared with cells transfected with control siRNA. While Bax siRNA
significantly prevented A549 cell death induced by PX-12, Bcl-2
siRNA slightly increased cell death in PX-12-treated A549 cells
(Fig. 3B and C). In addition, Bax
siRNA significantly attenuated the loss of MMP (ΔΨm) in
PX-12-treated A549 cells (Fig.
3D). However, Bcl-2 siRNA did not significantly affect the loss
of MMP (ΔΨm) caused by PX-12 (Fig. 3D).
Effects of PX-12 on the intracellular ROS
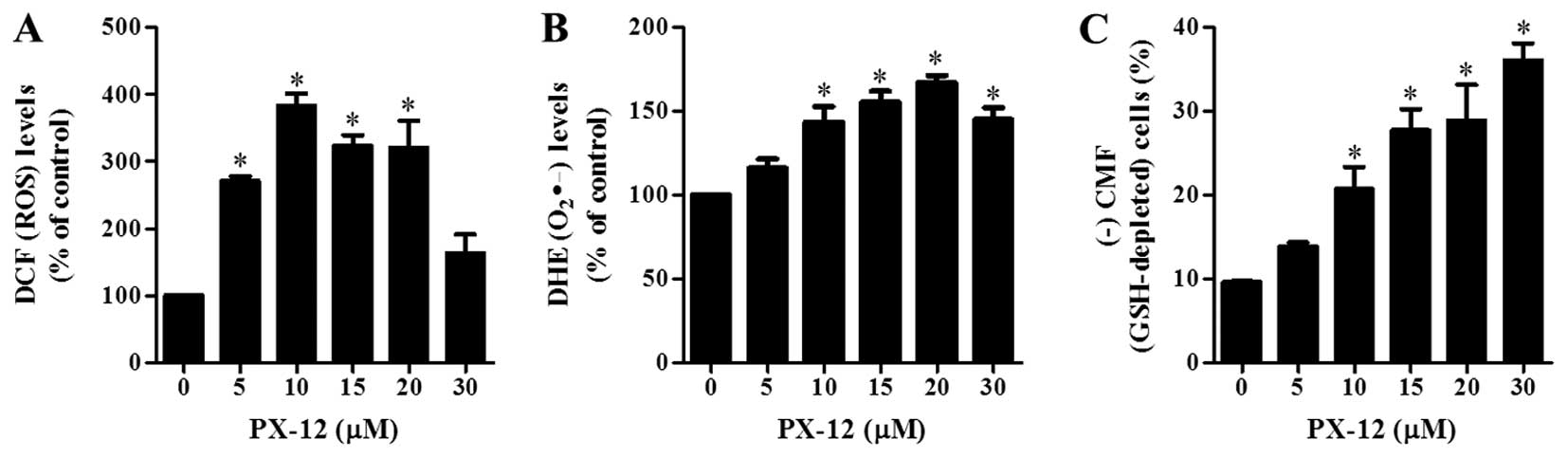
and GSH levels in A549 cells
The changes in intracellular ROS and GSH levels were
investigated in A549 cells treated with PX-12 at 72 h. As shown in
Fig. 4A, PX-12 increased the
intracellular ROS (DCF) levels in A549 cells at 72 h. Moreover, red
fluorescence derived from DHE reflecting the intracellular
O2•- levels significantly increased in
PX-12-treated A549 cells at 72 h (Fig.
4B). When intracellular GSH levels were measured in
PX-12-treated A549 cells using a CMFDA dye, PX-12 significantly
increased GSH-depleted cell number in A549 cells (Fig. 4C).
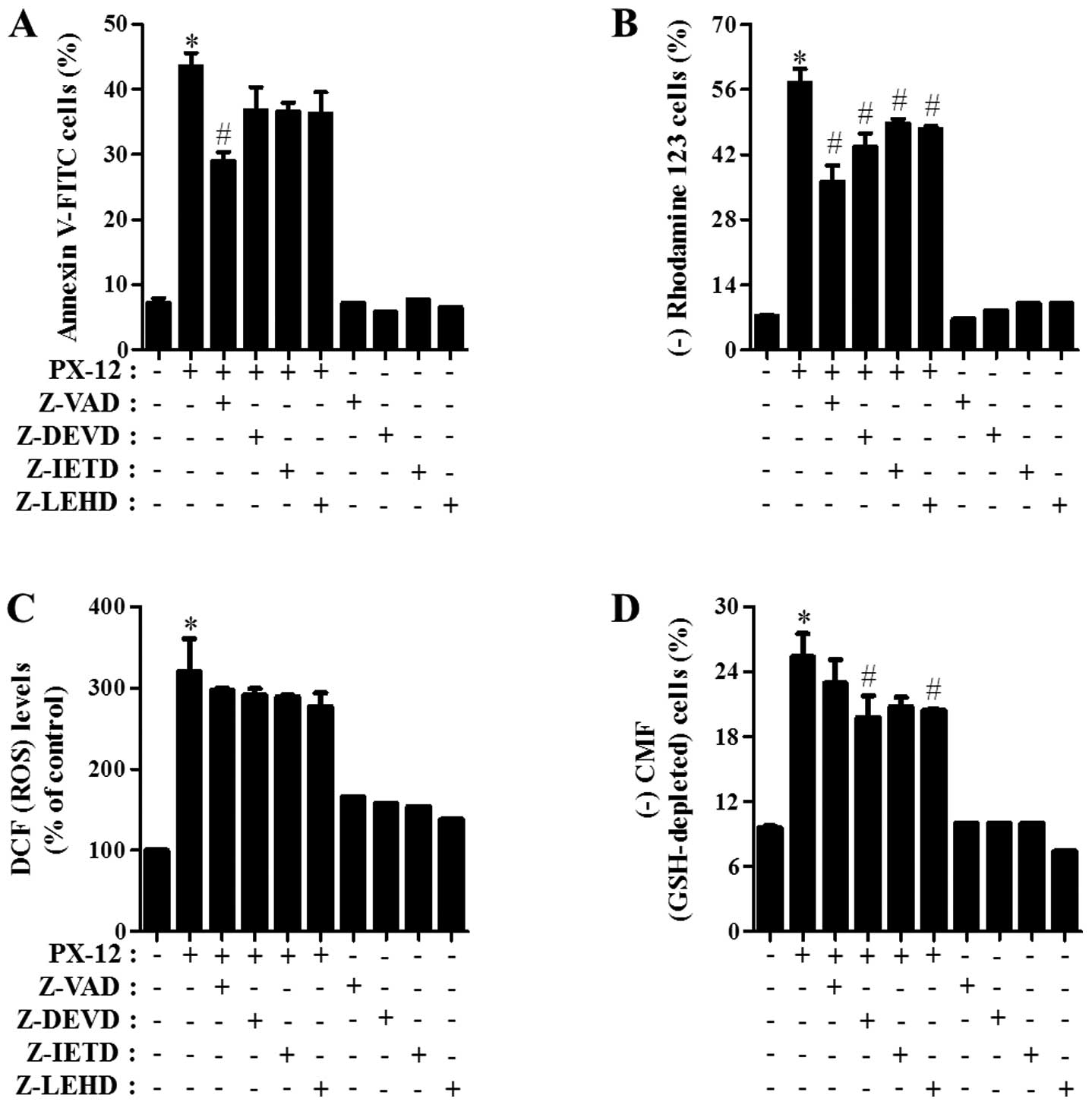
Effects of caspase inhibitors on cell
death, MMP (ΔΨm), ROS and GSH levels in PX-12-treated
A549 cells
Which caspase was involved in A549 cell death
induced by PX-12, was determined. For this experiment, we chose 20
μM PX-12 as a suitable dose to differentiate the levels of
cell death in the presence or absence of each caspase inhibitor.
Based on a previous study (17),
A549 cells were pretreated with 15 μM of each caspase
inhibitor for 1 h prior to treatment with PX-12. This dose did not
significantly affect cell death in the control A549 cells (Fig. 5A). Treatment with all the tested
caspase inhibitors (Z-VAD for pan-caspases, Z-DEVD for caspase-3,
Z-IETD for caspase-8 and Z-LEHD for caspase-9), especially Z-VAD
showed significant rescue of A549 cells from PX-12-induced
apoptosis at 72 h, as measured by the population of Annexin
V-FITC-positive cells (Fig. 5A).
Furthermore, all the caspase inhibitors significantly prevented the
loss of MMP (ΔΨm) caused by PX-12 (Fig. 5B).
Whether the intracellular ROS and GSH levels in
PX-12-treated A549 cells were changed by treatment with each
caspase inhibitor was investigated. As shown in Fig. 5C, all the caspase inhibitors
marginally reduced ROS levels in PX-12-treated A549 cells.
Moreover, these caspase inhibitors, especially Z-DEVD and Z-LEHD
prevented GSH depletion in these cells (Fig. 5D).
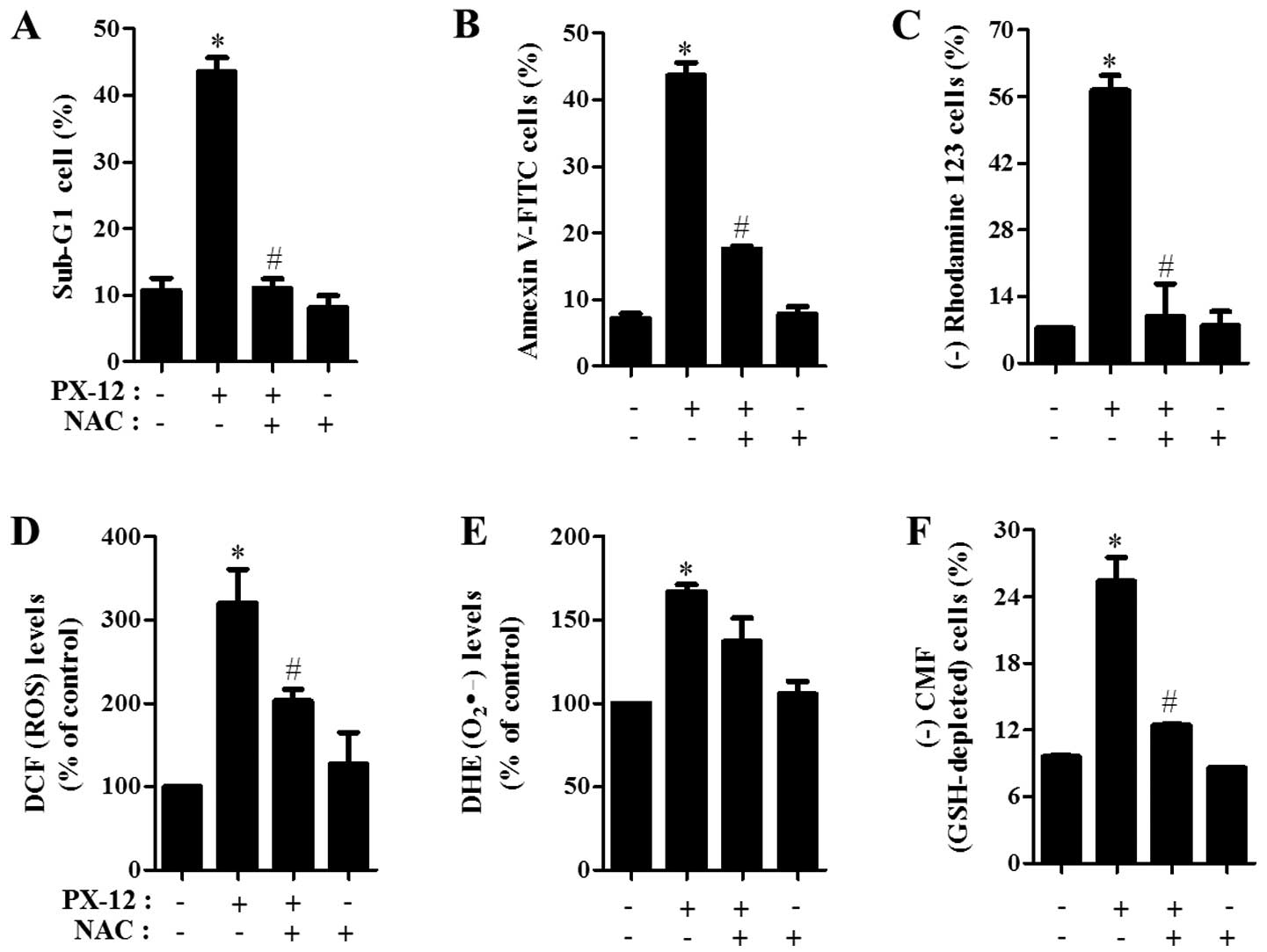
Effects of NAC on cell death and MMP
(ΔΨm), ROS and GSH levels in PX-12-treated A549
cells
Next, the effects of NAC on cell death and MMP
(ΔΨm) in 20 μM PX-12-treated A549 cells were
assessed at 72 h. As shown in Fig. 6A
and B, NAC significantly decreased the percentages of sub-G1
cells and Annexin V-FITC-positive cells in PX-12-treated A549
cells. With respect to MMP (ΔΨm), NAC significantly
attenuated the loss of MMP (ΔΨm) caused by PX-12
(Fig. 6C).
Furthermore, it was determined whether the
intracellular ROS and GSH levels in PX-12-treated A549 cells were
changed by treatment with NAC. NAC markedly decreased ROS levels
including O2•- in PX-12-treated A549 cells at
72 h (Fig. 6D and E). In relation
to GSH levels, NAC markedly prevented GSH depletion caused by PX-12
in A549 cells (Fig. 6F).
Discussion
We investigated the effects of PX-12 in A549 lung
cancer cells on cell growth and death in relation to ROS and GSH
level. It was observed that PX-12 decreased the activity of Trx-1
in A549 cell lysate, indicating that PX-12 directly inhibited the
activity of Trx-1 in this lysate. In addition, the level of Trx-1
expression was downregulated in PX12-treated A549 cells. Since the
tight binding of PX-12 to Trx-1 leads to compositional changes in
Trx-1 protein, it is possible that Trx-1 can be degraded via a
ubiquitination system. However, the activity of Trx-1 was not
reduced in PX-12-treated A549 cells at 72 h. These results suggest
that the activity of Trx-1 does not closely correspond with its
level and the different effects of PX-12 on the activity of Trx-1
in cell lysate and cells are probably derived from the different
functional bioavailability of this drug.
PX-12 inhibited the growth of A549 cells time- and
dose-dependently. PX-12 significantly induced a G2/M phase arrest
of the cell cycle in A549 cells. Similarly, it has been found that
PX-12 induces a G2/M phase arrest in cancer cells such as breast
cancer, B-cell lymphoma (1,26)
and Calu-6 lung cancer cells (unpublished data). Therefore, the
G2/M phase arrest in PX-12-treated cancer cells was an underlying
mechanism to suppress cell growth and prolifeation. PX-12 also
induced apoptosis in A549 cells and this drug strongly triggered
the loss of MMP (ΔΨm). These results supported that
apoptosis is closely related to the collapse of MMP
(ΔΨm) (27).
A high ratio of Bax to Bcl-2 has been known to be a
main trigger in the collapse of MMP (ΔΨm) and apoptosis
in cells (28). Likewise, the
levels of Bcl-2 and Bax were downregulated and upregulated in
PX-12-treated A549 cells, respectively. Moreover, the
administration of Bax siRNA prevented A549 cell death caused by
PX-12 whereas that of Bcl-2 siRNA did not strongly affect apoptosis
and MMP (ΔΨm) in PX-12 treated A549 cells. Therefore,
PX-12 seemed to induce apoptosis in A549 cells depending on the
upregulation of Bax protein. When determined which caspases were
involved in apoptosis in PX-12-treated A549 cells, all the tested
caspase inhibitors, especially Z-VAD prevented PX-12-induced A549
cell death. These data demonstrated that mitochondrial intrinsic
pathway as well as death receptor extrinsic pathway together are
necessary for the complete induction of apoptosis in PX-12-treated
A549 cells. In addition, all the caspase inhibitors markedly
attenuated the loss of MMP (ΔΨm) caused by PX-12. These
results suggest that the loss of MMP (ΔΨm) induced by
PX-12 is a crucial step to fully induce apoptosis in A549
cells.
PX-12 as an inhibitor of Trx-1 can affect the status
of redox in cells. It is reported that PX-12 induces oxidative
stress (29). Likewise, the
intracellular ROS levels including O2•-
significantly increased in PX-12-treated A549 cells at 72 h. All
caspase inhibitors showing anti-apoptotic effects attenuated ROS
levels in PX-12-treated A549 cells. Furthermore, NAC markedly
prevented apoptotic cell death and the loss of MMP (ΔΨm)
in PX-12-treated A549 cells, accompanied by decreasing ROS levels
including O2•- in these cells. Taken
together, these results suggest that PX-12-induced cell death is
mediated by oxidative stress. GSH is an important intracellular
antioxidant that protects cells from damage caused by free
radicals, peroxides and toxins. It is able to clear away
O2•- and provide electrons for glutathione
peroxidase to reduce H2O2 to H2O.
Apoptotic effects are inversely comparative to GSH content
(30,31). Similarly, PX-12 increased the
percentages of GSH-depleted cells in A549 cells at 72 h. NAC and
caspase inhibitors markedly prevented the depletion of GSH in
PX-12-treated A549 cells. However, BSO, which is an inhibitor of
GSH synthesis, did not affect apoptotic cell death in A549 cells
(data not shown). Therefore, the loss of GSH content seemed to be
necessary but not sufficient to induce apoptosis in PX-12-treated
A549 cells.
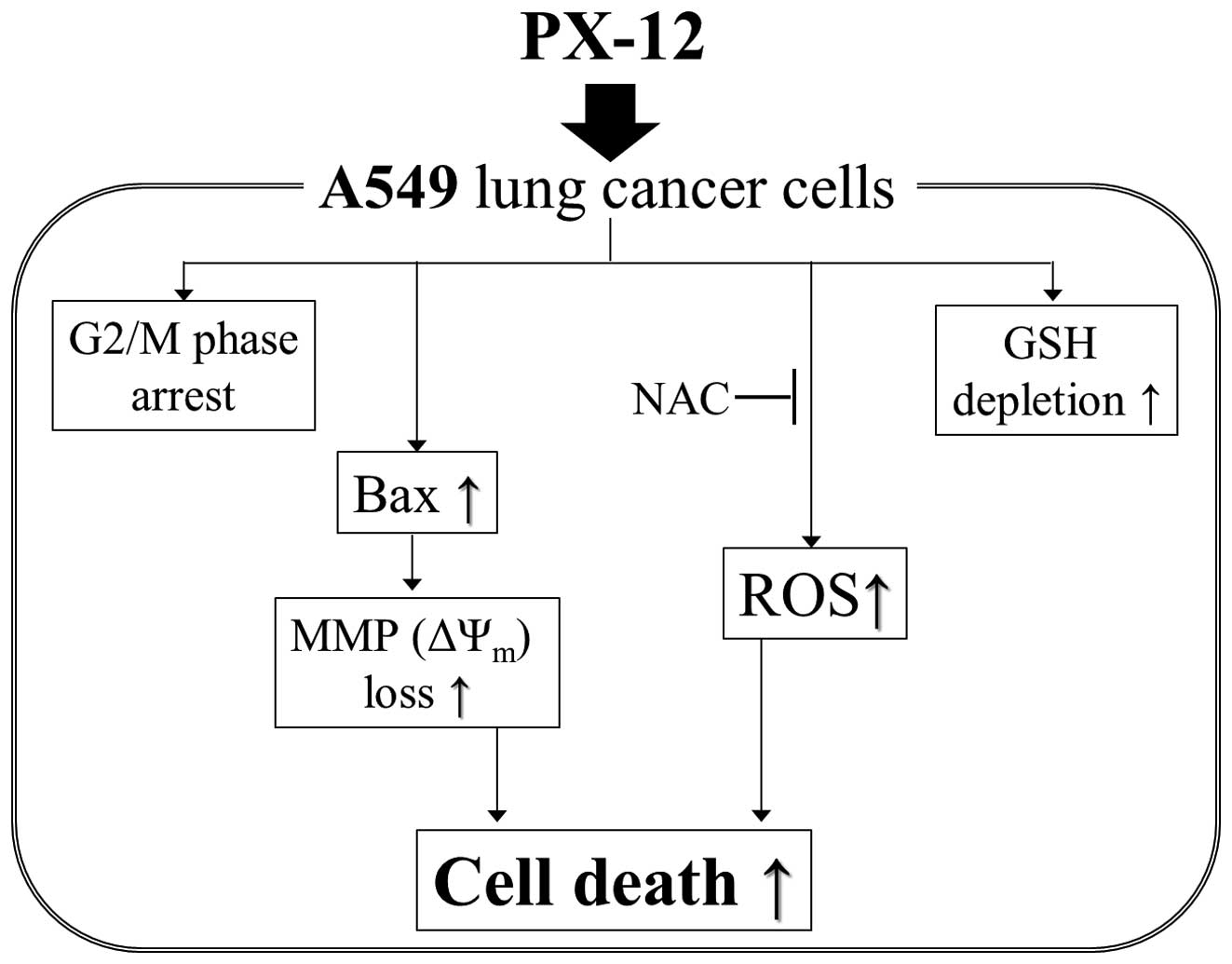
In conclusion, depicted in Fig. 7, it is the first report that PX-12
inhibited the growth of A549 lung cancer cells via G2/M phase
arrest, and Bax-mediated and ROS-dependent apoptosis.
Acknowledgements
This study was supported by the
National Research Foundation of Korea (NRF) grant funded by the
Korea government (MSIP) (No. 2008-0062279) and supported by the
Basic Science Research Program through the National Research
Foundation of Korea (NRF) funded by the Ministry of Education
(2013006279).
References
|
1.
|
Li C, Thompson MA, Tamayo AT, et al:
Over-expression of Thioredoxin-1 mediates growth, survival, and
chemoresistance and is a druggable target in diffuse large B-cell
lymphoma. Oncotarget. 3:314–326. 2012.PubMed/NCBI
|
|
2.
|
Yang J, Li C, Ding L, Guo Q, You Q and Jin
S: Gambogic acid deactivates cytosolic and mitochondrial
thioredoxins by covalent binding to the functional domain. J Nat
Prod. 75:1108–1116. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Chae JS, Gil Hwang S, Lim DS and Choi EJ:
Thioredoxin-1 functions as a molecular switch regulating the
oxidative stress-induced activation of MST1. Free Radic Biol Med.
53:2335–2343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Ungerstedt J, Du Y, Zhang H, Nair D and
Holmgren A: In vivo redox state of human thioredoxin and redox
shift by the histone deacetylase inhibitor suberoylanilide
hydroxamic acid (SAHA). Free Radic Biol Med. 53:2002–2007. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Lim JY, Yoon SO, Hong SW, Kim JW, Choi SH
and Cho JY: Thioredoxin and thioredoxin-interacting protein as
prognostic markers for gastric cancer recurrence. World J
Gastroenterol. 18:5581–5588. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Pramanik KC and Srivastava SK: Apoptosis
signal-regulating kinase 1-thioredoxin complex dissociation by
capsaicin causes pancreatic tumor growth suppression by inducing
apoptosis. Antioxid Redox Signal. 17:1417–1432. 2012. View Article : Google Scholar
|
|
7.
|
Dunn LL, Buckle AM, Cooke JP and Ng MK:
The emerging role of the thioredoxin system in angiogenesis.
Arterioscler Thromb Vasc Biol. 30:2089–2098. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Cha MK, Suh KH and Kim IH: Overexpression
of peroxiredoxin I and thioredoxin 1 in human breast carcinoma. J
Exp Clin Cancer Res. 28:932009. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Wondrak GT: Redox-directed cancer
therapeutics: molecular mechanisms and opportunities. Antioxid
Redox Signal. 11:3013–3069. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Welsh SJ, Williams RR, Birmingham A,
Newman DJ, Kirkpatrick DL and Powis G: The thioredoxin redox
inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin
inhibit hypoxia-induced factor 1alpha and vascular endothelial
growth factor formation. Mol Cancer Ther. 2:235–243. 2003.
|
|
11.
|
Mukherjee A and Martin SG: The thioredoxin
system: a key target in tumour and endothelial cells. Br J Radiol.
81(Spec1): S57–S68. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Baker AF, Adab KN, Raghunand N, et al: A
phase IB trial of 24-hour intravenous PX-12, a thioredoxin-1
inhibitor, in patients with advanced gastrointestinal cancers.
Invest New Drugs. 31:631–641. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Ramanathan RK, Kirkpatrick DL, Belani CP,
et al: A Phase I pharmacokinetic and pharmacodynamic study of
PX-12, a novel inhibitor of thioredoxin-1, in patients with
advanced solid tumors. Clin Cancer Res. 13:2109–2114. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Petty RD, Nicolson MC, Kerr KM,
Collie-Duguid E and Murray GI: Gene expression profiling in
non-small cell lung cancer: from molecular mechanisms to clinical
application. Clin Cancer Res. 10:3237–3248. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Fernandes AP, Capitanio A, Selenius M,
Brodin O, Rundlof AK and Bjornstedt M: Expression profiles of
thioredoxin family proteins in human lung cancer tissue:
correlation with proliferation and differentiation. Histopathology.
55:313–320. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Wangpaichitr M, Sullivan EJ,
Theodoropoulos G, et al: The relationship of thioredoxin-1 and
cisplatin resistance: its impact on ROS and oxidative metabolism in
lung cancer cells. Mol Cancer Ther. 11:604–615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Han YH, Kim SZ, Kim SH and Park WH:
Pyrogallol inhibits the growth of lung cancer Calu-6 cells via
caspase-dependent apoptosis. Chem Biol Interact. 177:107–114. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Han YH and Park WH: The effects of
N-acetyl cysteine, buthionine sulfoximine, diethyldithiocarbamate
or 3-amino-1,2,4-triazole on antimycin A-treated Calu-6 lung cells
in relation to cell growth, reactive oxygen species and
glutathione. Oncol Rep. 22:385–391. 2009.
|
|
19.
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: Effects of carbonyl cyanide p-(trifluoromethoxy)
phenylhydrazone on the growth inhibition in human pulmonary
adenocarcinoma Calu-6 cells. Toxicology. 265:101–107. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
You BR and Park WH: Zebularine inhibits
the growth of HeLa cervical cancer cells via cell cycle arrest and
caspase-dependent apoptosis. Mol Biol Rep. 39:9723–9731. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Han YH, Moon HJ, You BR and Park WH: The
effect of MG132, a proteasome inhibitor on HeLa cells in relation
to cell growth, reactive oxygen species and GSH. Oncol Rep.
22:215–221. 2009.PubMed/NCBI
|
|
22.
|
Han YH, Kim SH, Kim SZ and Park WH:
Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) as an
O2(*-) generator induces apoptosis via the
depletion of intracellular GSH contents in Calu-6 cells. Lung
Cancer. 63:201–209. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Han YH and Park WH: Propyl gallate
inhibits the growth of HeLa cells via regulating intracellular GSH
level. Food Chem Toxicol. 47:2531–2538. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
You BR and Park WH: Gallic acid-induced
lung cancer cell death is related to glutathione depletion as well
as reactive oxygen species increase. Toxicol In Vitro.
24:1356–1362. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Griffiths EJ: Mitochondria - potential
role in cell life and death. Cardiovasc Res. 46:24–27. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Vogt A, Tamura K, Watson S and Lazo JS:
Antitumor imidazolyl disulfide IV-2 causes irreversible G(2)/M cell
cycle arrest without hyperphosphorylation of cyclin-dependent
kinase Cdk1. J Pharmacol Exp Ther. 294:1070–1075. 2000.PubMed/NCBI
|
|
27.
|
Yang J, Liu X, Bhalla K, et al: Prevention
of apoptosis by Bcl-2: release of cytochrome c from mitochondria
blocked. Science. 275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Lee YJ, Kim JH, Chen J and Song JJ:
Enhancement of metabolic oxidative stress-induced cytotoxicity by
the thioredoxin inhibitor 1-methylpropyl 2-imidazolyl disulfide is
mediated through the ASK1-SEK1-JNK1 pathway. Mol Pharmacol.
62:1409–1417. 2002. View Article : Google Scholar
|
|
30.
|
Estrela JM, Ortega A and Obrador E:
Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci.
43:143–181. 2006. View Article : Google Scholar
|
|
31.
|
You BR and Park WH: Arsenic trioxide
induces human pulmonary fibroblast cell death via increasing ROS
levels and GSH depletion. Oncol Rep. 28:749–757. 2012.PubMed/NCBI
|