Introduction
Protein folding and degradation pathways are
strictly regulated in normal cells to avoid accumulation of
misfolded proteins and maintain protein homeostasis. When the
accumulation of misfolded proteins exceeds degradation, as often
occurs in damaged or aging cells or in cells exposed to chemical
agents that perturb protein folding, the unfolded protein response
(UPR) is elicited to re-establish protein homeostasis and prevent
apoptotic cell death (1). However,
the persistence of endoplasmic reticulum (ER) stress conditions can
switch on an apoptotic program, which finally results in cell
elimination (2,3). The development and the progression of
cancer is associated to the dysregulation of this process and the
persistent activation of the adaptive ER stress response. Indeed,
tumor cells are characterized by increased rates of protein
synthesis to fulfill the high metabolic demand of accelerated
proliferation and, thus, are chronically exposed to ER stress
conditions (4,5). In such a scenario, chronic ER stress
adaptive responses are involved in tumor progression, adaptation to
unfavorable environmental conditions and resistance to cytotoxic
agents and, therefore, are regarded as novel targets for the
development of new cancer therapeutics (6). Furthermore, recent studies suggest
that the ER stress response, besides being responsible for the
resistance to pharmacological agents that perturb protein
homeostasis (7), may favor
resistance to DNA damaging agents (8).
Our group has contributed to the demonstration that
the mitochondrial chaperone TRAP1 is involved in protection against
ER stress (9–11). Indeed, TRAP1 is an HSP90 homolog,
originally described as a protein responsible for the prevention of
mitochondrial apoptosis, due to its interaction with HSP90 and
cyclofilin D and its regulatory property on mitochondrial
transition pore (MTP) (12). As
TRAP1 is selectively upregulated in several human malignancies
(i.e., colorectal, breast and prostate carcinomas) (13–15),
its role in protecting against the cytotoxic activity of
antiblastic agents in human malignancies has been widely
demonstrated (13,16,17).
More recent evidence described the presence of TRAP1
on the outer side of the ER at the interface with mitochondria,
where this chaperone interacts with TBP7, an AAA-ATPase of the 19S
proteasomal subunit, and is responsible for the modulation of the
ER stress response and the quality control of specific
mitochondrial client proteins (9),
this resulting in protection against mitochondrial apoptosis
(18). Indeed, we recently
demonstrated that the ER-stress protecting activity of TRAP1 is
crucial for its antiapoptotic function and favors resistance
against taxanes in breast carcinoma (BC) cells (18). In this context, we observed that
ER-associated TRAP1 modulates the mitochondrial apoptotic pathway
by regulating the quality of specific client proteins and, among
others, 18 kDa sorcin, a mitochondrial protein involved in the
TRAP1 cytoprotective pathway (17), this likely representing a major
mechanism responsible for the survival response elicited by
ER-associated TRAP1 (9,18).
Based on these premises, we investigated the role of
ER-associated TRAP1 in favoring resistance to anthracyclins,
cytotoxic agents among the most effective anticancer drugs ever
developed (19,20). Indeed, it has been hypothesized
that the activation of the UPR in the ER is responsible for
resistance to genotoxic agents such as topoisomerase inhibitors and
platin derivatives (8,21). High expression of the ER chaperone
BiP/Grp78 is protective against apoptotic stimuli in cancer cells
(22), favors resistance to
anti-estrogen therapy and chemotherapeutics in several cancer cell
models (8,21,23)
and correlates with shorter overall survival in human prostate
(24) and lung cancer (22). The cytoprotective activity of
ER-associated TRAP1 against anthracyclins was investigated in BC
cells since our group recently demonstrated that TRAP1 and
BiP/Grp78 are co-upregulated in about 50% of human BCs (18) and considering that anthracyclins
are highly effective agents in these malignancies (25). Here, we report that TRAP1
regulation of ER stress response is critical in favoring resistance
to anthracyclins and provide evidence that this response involves
the modulation of PERK pathway.
Materials and methods
Cells, plasmids and chemicals
Human MCF7 BC cells were purchased from ATCC
(Manassas, VA, USA) and cultured in DMEM containing 10% (v/v) fetal
bovine serum in standard conditions. Cell line authentication was
performed by DNA profiling, according to ATCC product description.
Drug-resistant cells were selected as previously reported (18,26).
siRNAs of TRAP1 were purchased from Qiagen (Milan, Italy; cat. nos.
SI00115150 for TRAP1 and SI03650318 for negative control), diluted
to a final concentration of 20 nmol/l and transfected according to
the manufacturer’s protocol by using HiPerFect Transfection Reagent
(Qiagen). Constructs encoding for wild-type TRAP1, and
Δ1-59TRAP1-Myc and TBP7-Flag deletion mutants (9) were transiently transfected with
Polyfect Transfection reagent (Qiagen). Unless otherwise specified,
reagents were purchased from Sigma-Aldrich (Milan, Italy).
Immunoblot analysis and antibodies
Total cell lysates were obtained by homogenization
of cell pellets in cold lysis buffer (20 mM Tris, pH 7.5 containing
300 mM sucrose, 60 mM KCl, 15 mM NaCl, 5% (v/v) glycerol, 2 mM
EDTA, 1% (v/v) Triton X-100, 1 mM PMSF, 2 mg/ml aprotinin, 2 mg/ml
leupeptin and 0.2% (w/v) deoxycholate) for 1 min at 4°C and further
sonication for 30 sec at 4°C. Immunoblot analysis was performed as
previously reported (26,27). Briefly, equal amounts of protein
from cell lysates were separated by SDS-PAGE and transferred to a
nitrocellulose support (Bio-Rad, Hercules, CA, USA). Specific
proteins were detected by using the following antibodies: mouse
monoclonal anti-GAPDH (sc-47724, Santa Cruz Biotechnology, Segrate,
Italy), mouse monoclonal anti-TRAP1 (sc-13557, Santa Cruz
Biotechnology), rabbit polyclonal anti-caspase 12 (SPA-827;
StressGen, Milan, Italy), rabbit polyclonal anti-Grp94 (sc-11402,
Santa Cruz Biotechnology), rabbit polyclonal anti-phosphoPERK (Thr
981, sc-32577, Santa Cruz Biotechnology), rabbit monoclonal
anti-PERK (#3192, Cell Signaling Technology, Boston, MA, USA),
rabbit polyclonal anti-LC3B (#2775, Cell Signaling Technology),
mouse monoclonal anti-BiP/Grp78 (E-4, sc-166490, Santa Cruz
Biotechnology), mouse monoclonal anti-uniquitin (P4D1, sc-8017,
Santa Cruz Biotechnology), mouse monoclonal anti-Myc (9E10, sc-40,
Santa Cruz Biotechnology), and rabbit polyclonal anti-Flag (sc-807,
Santa Cruz Biotechnology) antibodies. Proteins were visualized with
an ECL detection system (Bio-Rad).
RNA extraction and semiquantitative and
Real-time RT-PCR analysis
Total RNA from cell pellets was extracted using the
TRIzol Reagent (Invitrogen, San Giuliano Milanese, Italy), For the
first strand synthesis of cDNA, 1 μg of RNA was used in a 20
μl reaction mixture utilizing a Transcriptor First Strand
cDNA Synthesis Kit (Roche, Mannheim, Germany). A total of 5
μl of cDNA sample were amplified using the LightCycler 480
SYBR-Green I Master (Roche) in an Light Cycler 480 (Roche). The
following primers were used: BiP/Grp78, forward
5′-GTGGAATGACCCGTCTGTC-3′, reverse 5′-CGTCTTTG GTTGCTTGGC-3′ (PCR
product 254 bp); GAPDH forward 5′-AGGCTGAGAACGGGAAGC-3′, reverse
5′-CCATGGTG GTGAAGACGC-3′ (PCR product 135 bp). Primers were
designed to be intron spanning. Reaction condition were as follows:
pre-incubation at 95°C for 5 min, followed by 45 cycles of 10 sec
at 95°C, 7 sec at 60°C, 10 sec at 72°C. GAPDH was chosen as an
internal control.
Apoptosis assay
Apoptosis was evaluated by cytofluorimetric analysis
of Annexin V and 7-amino-actinomycin-D (7-AAD)-positive cells using
the fluorescein isothiocyanate (FITC)-Annexin V/7-AAD kit (Beckman
Coulter, Milan, Italy). Stained cells were analyzed by using the
FACSCalibur™ (Becton-Dickinson). Ten thousand events were collected
per sample. Positive staining for Annexin V as well as double
staining for Annexin V and 7-AAD were interpreted as signs of,
respectively, early and late phases of apoptosis, as previously
reported (28).
Statistical analysis
The paired Student’s t-test was used to establish
the statistical significance between different levels of apoptosis
or gene expression in controls and treated cells or in transfected
cells and related scramble controls. Statistically significant
values (p<0.05) are reported in Fig. legends.
Results
The ER stress protecting activity of
TRAP1 is relevant for resistance of BC cells to anthracyclins
Since anthracyclins have been proposed to induce ER
stress conditions and Bip/Grp78 expression (8), we preliminarily confirmed that the
exposure of MCF7 cells to cytotoxic concentrations of both
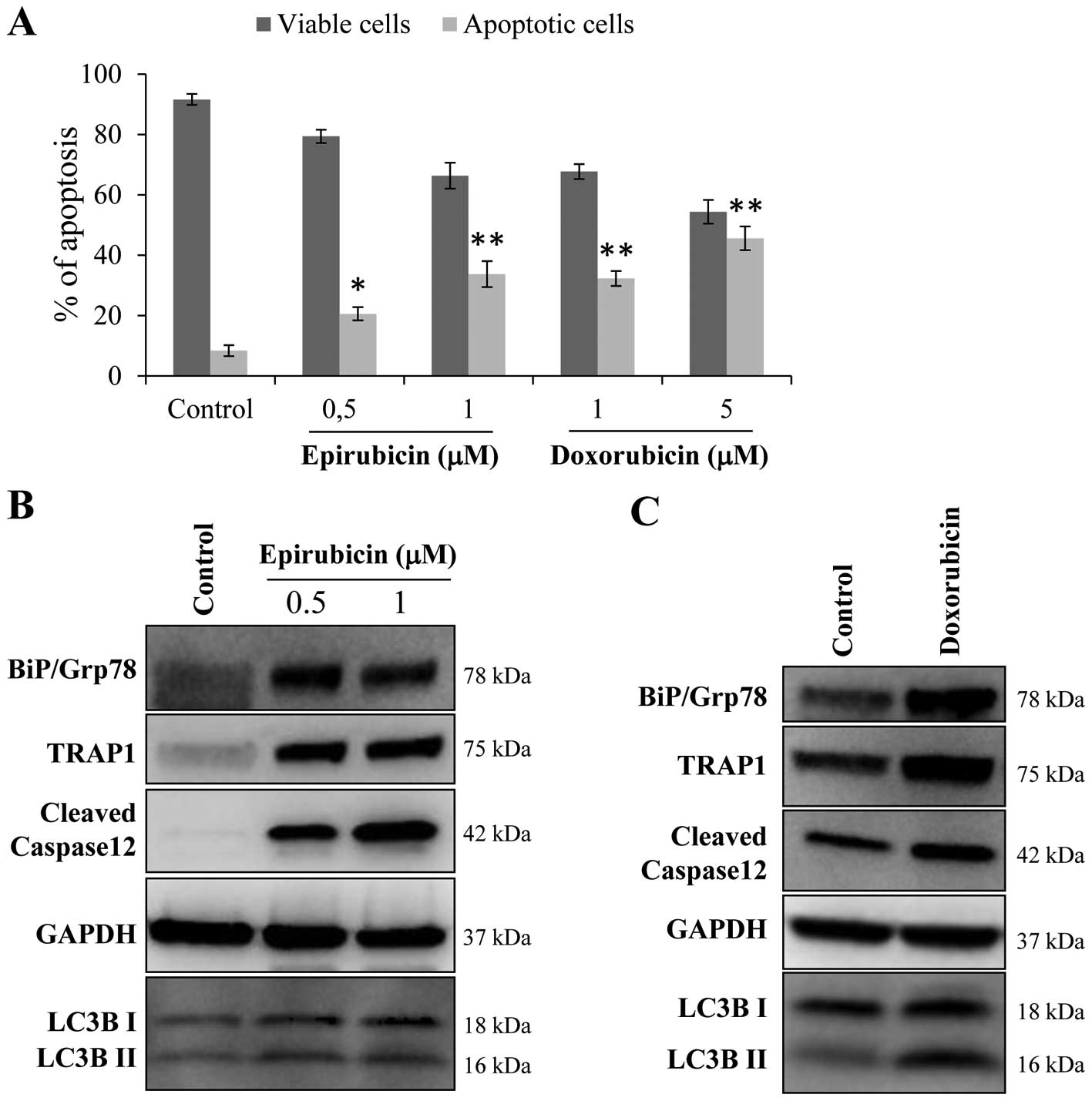
epirubicin and doxorubicin (Fig.
1A) elicits an ER UPR characterized by high levels of BiP/Grp78
and the autophagy marker LC3, and the cleavage of caspase 12
(Fig. 1B). Since high expression
of BiP/Grp78 and adaptation to ER stress conditions have been
suggested to favor resistance to anthracyclins (8,21,29),
we questioned whether BC cells adapted to ER stress conditions may
be cross-resistant to doxorubicin and epirubicin. To this purpose
we used MCF7 cells chronically adapted to toxic concentrations of
the proteasome inhibitor, bortezomib, previously selected in our
laboratory (18) and characterized
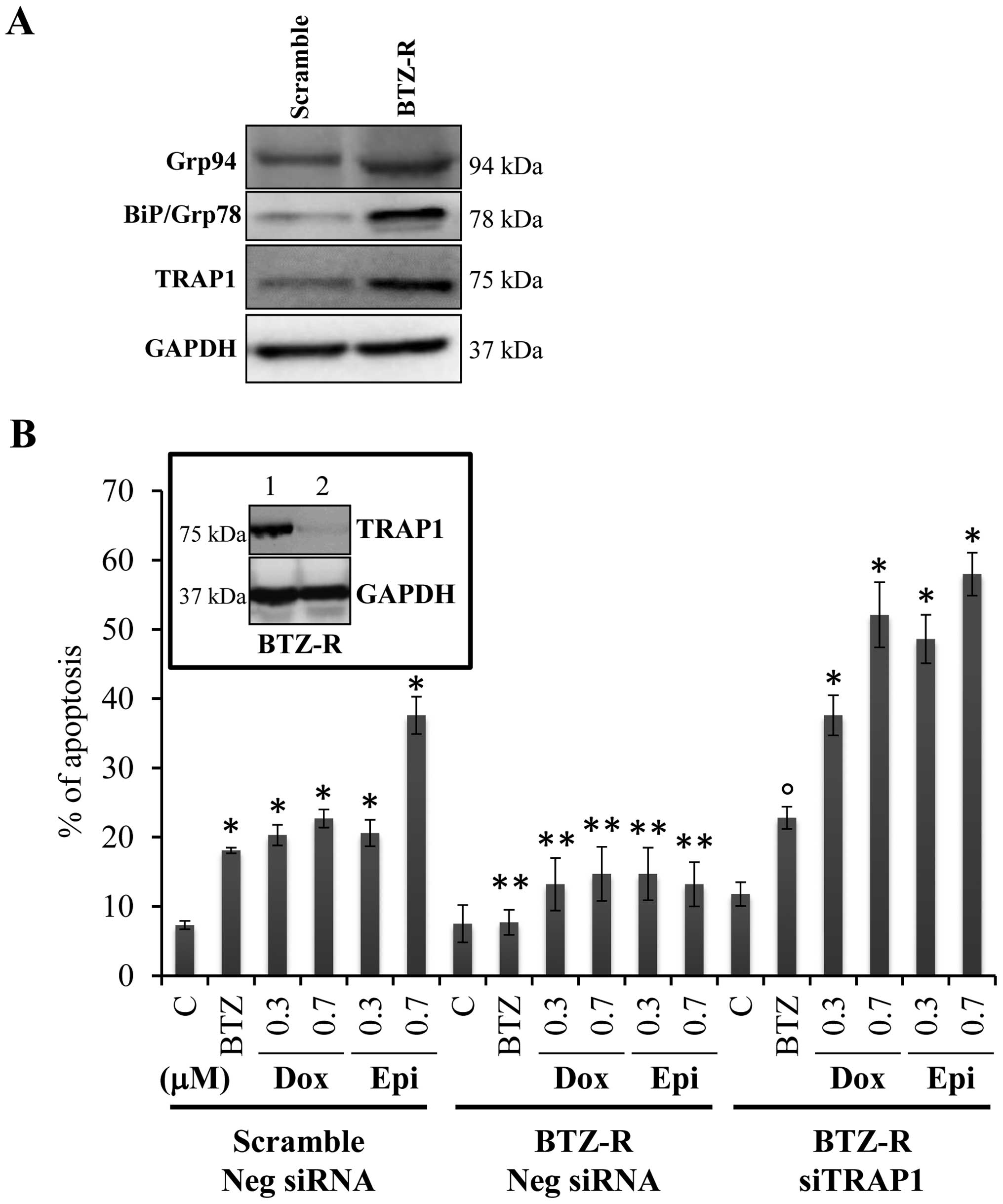
by increased BiP/Grp78 and Grp94 expression (Fig. 2A). Of note, cross-resistance was
observed upon exposure of bortezomib-resistant cells to cytotoxic
concentrations of doxorubicin and epirubicin (Fig. 2B). Since i) TRAP1 involvement in ER
stress protection was previously described (9); ii) the exposure of MCF7 cells to
anthracyclins resulted in the upregulation of TRAP1 (Fig. 1B); and iii) bortezomib-resistant
cells exhibited increased TRAP1 levels (Fig. 2A), we further investigated the role
of TRAP1 in favoring resistance to anthracyclins. To this aim,
TRAP1 was transiently interfered in bortezomib-resistant cells
(Fig. 2B, insert), this resulting
in re-establishment of sensitivity to epirubicin and doxorubicin
and bortezomib (Fig. 2B).
To characterize the involvement of TRAP1 in
resistance to anthracyclins we generated two further MCF7 cell
lines resistant to epirubicin and doxorubicin. Both
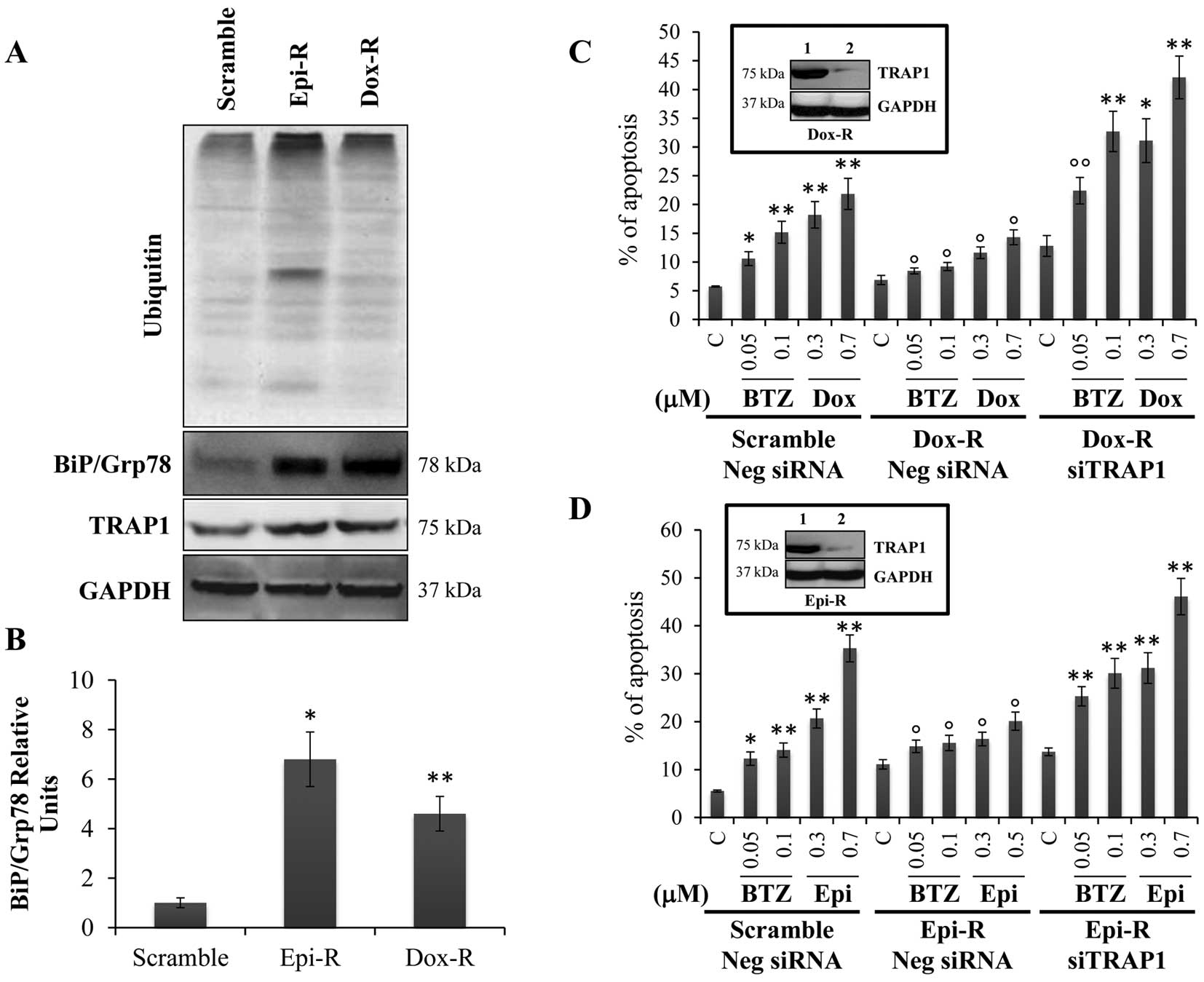
anthracyclin-resistant cell lines showed increase in total protein
ubiquitination and TRAP1 (Fig.
3A), as well as BiP/Grp78 upregulation at protein (Fig. 3A) and mRNA (Fig. 3B) levels. These
anthracyclin-resistant cell models, besides being resistant to the
respective selection agent, showed cross-resistance to the ER
stress agent bortezomib (Fig. 3C and
D). Consistently with previous results, the downregulation of
TRAP1 by siRNA in both anthracyclin-resistant cell lines (Fig. 3C and D, inserts) resulted in the
re-establishment of the sensitivity to doxorubicin and epirubicin,
as well as bortezomib (Fig. 3C and
D). These results suggest that adaptation to ER stress and
anthracyclins share common mechanisms and that TRAP1 is part of
this process.
ER-associated TRAP1 is responsible for
cytoprotection against anthracyclins
Previous evidence suggests that TRAP1 anti-apoptotic
function relies on a dual mechanism: a direct regulation of
cyclophillin D folding and MTP opening within mitochondria
(12) and a quality control on
specific mitochondrial anti-apoptotic client proteins in the ER
(18). Thus, we questioned whether
this cytoprotective activity of ER-associated TRAP1 is relevant in
favoring resistance to anthracyclins. To this purpose we used BC
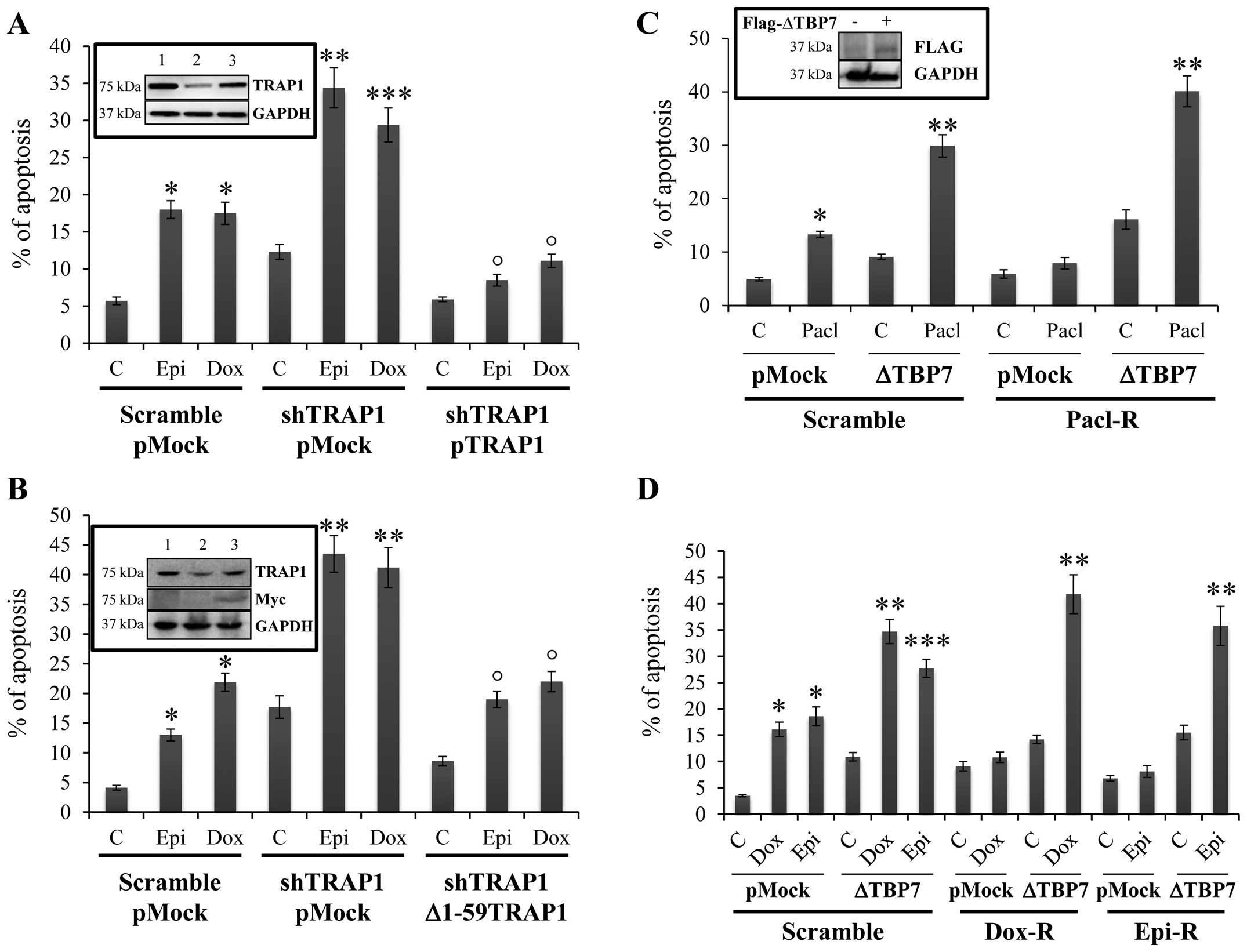
MCF7 cells with low TRAP1 levels (shTRAP1 MCF7 cells, Fig. 4A, insert), transiently transfected
with TRAP1 expression vector (Fig.
4A, insert) or the Δ1-59TRAP1 deletion mutant (Fig. 4B, insert), which lacks the
mitochondrial targeting sequence and, thus is unable to enter into
mitochondria, while still able to bind TBP7 and protect from ER
stress (9,18). We questioned whether this TRAP1
mutant was able to protect from apoptosis induced by the ER stress
agent, bortezomib and/or anthracyclins. Interestingly, we observed
a significant cytoprotective activity toward the proteasome
inhibitor, as well as doxorubicin and epirubicin due to
re-expression of both wild-type TRAP1 (Fig. 4A) and the mitochondrial
import-defective TRAP1 mutant (Fig.
4B). In order to further support the cytoprotective function of
ER-associated TRAP1/TBP7 pathway we used a TBP7 deletion mutant,
which was previously generated by our group, lacking TRAP1 binding
site and, characterized for a dominant negative function over the
endogenous TBP7 protein function (9). The TBP7 deletion mutant was
transfected in scramble MCF7 (Fig.
4C, insert) and in MCF7 cells adapted to docetaxel (Fig. 4C), a cytotoxic which acts by
inducing ER stress (30) or in
MCF7 cells adapted to anthracyclins (Fig. 4D). Consistently with previous
results, the ΔTBP7 mutant increased apoptotic cell death in
scramble cells upon paclitaxel (Fig.
4C) and doxorubicin and epirubicin (Fig. 4D) treatment, as well as
re-established drug sensitivity in docetaxel- (Fig. 4C) and epirubicin- and
doxorubicin-resistant (Fig. 4D)
cell models. According to our previous observations, these results
are the consequence of the disabling of this cytoprotective pathway
due to the disruption of TRAP1/TBP7 interaction upon transfection
with the TBP7 deletion mutant (9).
The modulation of PERK phosphorylation by
TRAP1 correlates with resistance to anthracyclins
A major cytoprotective signal elicited by ER stress
conditions is the phosphorylation of PERK which is responsible for
the ATF4-dependent preferential translation of genes involved in
cell metabolism and cytoprotective functions (31) and, among other, Bip/Grp78 (32). Since PERK upregulation in tumor
cells has been recently proposed to induce the resistance to
doxorubicin (33), we questioned
whether the cytoprotective function of TRAP1 toward anthracyclins
may involve the modulation of PERK pathway. In preliminary
experiments, we observed the inhibition of PERK phosphorylation in
MCF7 cells upon impairment of TRAP1/TBP7 pathway by the
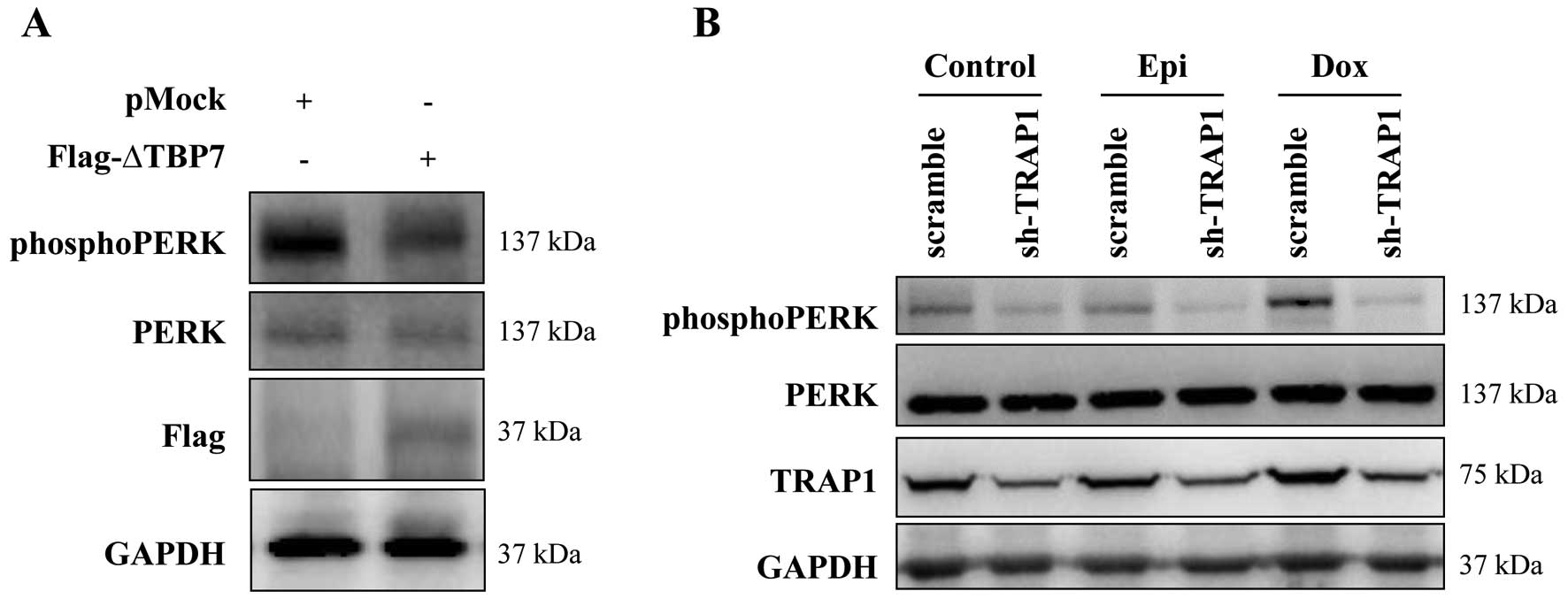
transfection of the ΔTBP7 deletion mutant (Fig. 5A). Since a reduced phosphorylation
of PERK was also observed in TRAP1 KD MCF7 cells compared to the
scramble counterpart (Fig. 5B),
these cell lines were exposed to a short-term treatment with
doxorubicin and epirubicin. Of note, shTRAP1 cells failed to
sustain PERK phosphorylation in response to anthracyclins (Fig. 5B), suggesting that the low
expression of TRAP1 is likely responsible for the inability to
elicit a cytoprotective response upon DNA damaging agents.
The downregulation of TRAP1 and the
simultaneous induction of ER stress by pharmacological agents
result in additive apoptotic effects
Since our data suggest that TRAP1 protects from ER
stress and that this function is relevant to elicit an
anti-apoptotic response upon ER stress conditions, we questioned
whether the inhibition of TRAP1 may be clinically relevant in
combination with pharmacological ER stress inducers (i.e.,
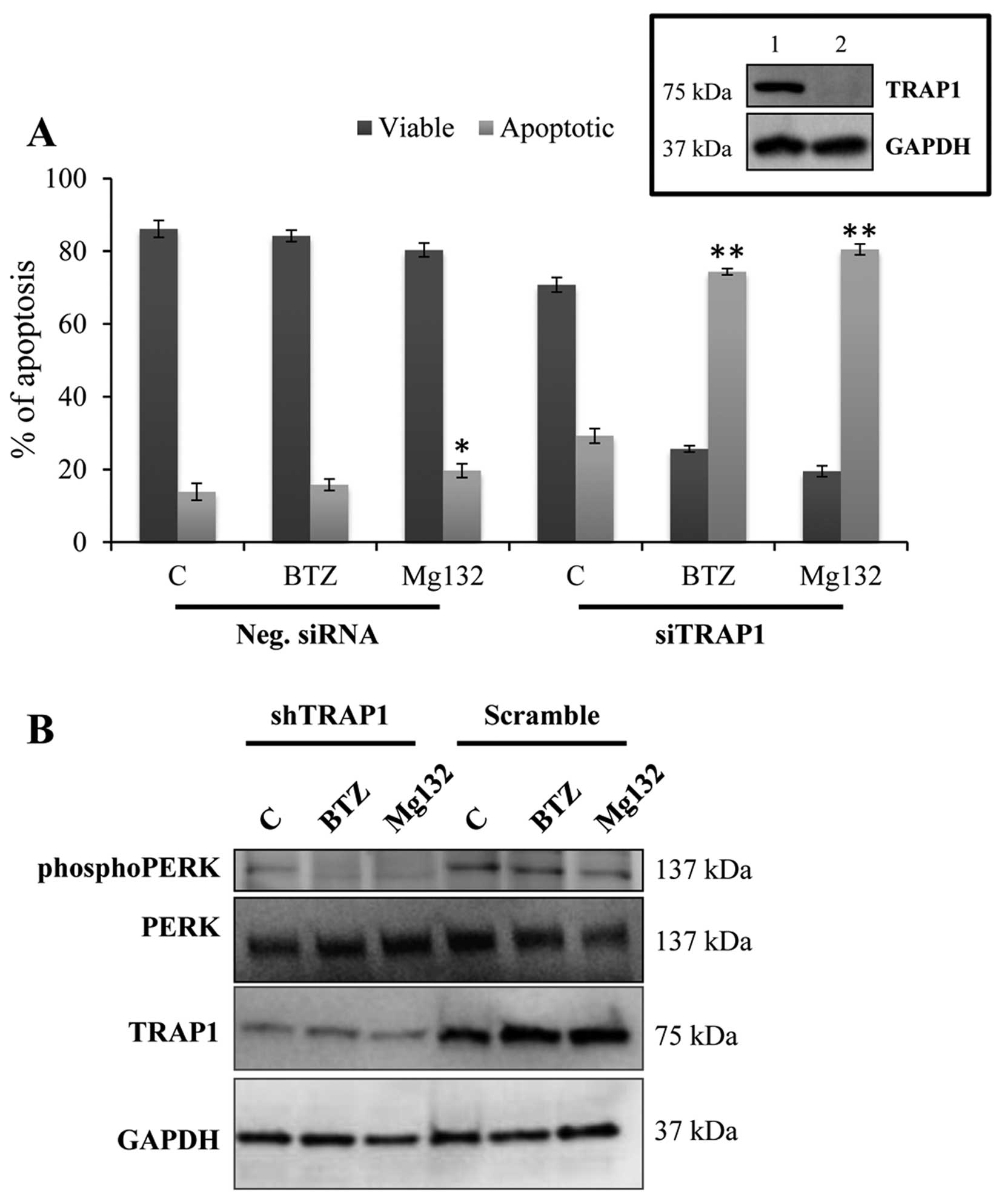
bortezomib and Mg132). The inhibition of TRAP1 was achieved by
molecular strategies (i.e., transient downregulation by siRNA),
this resulting by itself in reduced cell viability in MCF7 cells
(Fig. 6A). Furthermore, the
simultaneous inhibition of the proteasome activity with
subcytotoxic concentrations of bortezomib and Mg132 resulted in a
significant increase of apoptotic cell death (Fig. 6A). In agreement with our previous
results, a lower activation of PERK phosphorylation in
TRAP1-interefered cells compared to scramble cells is observed with
the inability to sustain PERK phoshorylation in response to
proteasome inhibitors (Fig. 6B).
These results suggest that strategies that combine TRAP1 inhibition
with agents that induce ER stress may achieve clinically-relevant
synergistic/additive activities.
Discussion
Cancer cells are exposed to chronic ER stress
conditions and are, thus, characterized by the persistent
activation of the ER UPR, with constitutive upregulation of several
molecular chaperones and other cytoprotective genes, as well as
activation of stress-related pathways. While excessive or prolonged
UPR results in apoptosis (2,3),
mild chronic ER stress conditions are beneficial for cancer cells,
favoring the adaptation to unfavorable environments and being
responsible for anti-apoptotic responses which ultimately lead to
drug resistance (7). In such a
context, recent evidence suggests that the ER UPR is responsible
for inducing resistance to genotoxic agents and, among others,
topoisomerase inhibitors (8).
Indeed, the upregulation of the ER chaperone BiP/Grp78 protects
against the cytotoxic activity of doxorubicin and cisplatin in
human melanoma and gastric cancer cells (8,21).
Furthermore, genotoxic agents, besides their well known DNA
damaging activity, favor the activation/upregulation of
stress-related pathways that are responsible for cytoprotective
responses (32). However, the
mechanistic link between the ER stress response and prevention of
apoptosis induced by DNA damaging agents is still poorly
understood.
A putative role of TRAP1 in the ER stress control
was initially suggested by Takemoto et al (10) who demonstrated that mitochondrial
TRAP1 is associated with the activation of the UPR (10). Immediately after, this novel TRAP1
function was documented at molecular level showing that the
selective targeting of HSP90 chaperones in mitochondria of human
tumor cells triggers an organelle UPR and compensatory autophagy
and that disabling this general adaptive pathway could potentially
be used in the treatment of human tumors (11). In parallel studies, our group
demonstrated that TRAP1, originally described as a mitochondrial
protein, is also localized at the interface between ER and
mitochondria where it is involved in crosstalk between these
organelles, in this subcellular location TRAP1 interacts with TBP7,
and plays a role in protection from ER stress, through a
co-traslational regulation of protein ubiquitination and a quality
control on specific mitochondrial client proteins (9,34).
Furthermore, this ER stress protecting activity by TRAP1 is crucial
for its anti-apoptotic function: indeed, the quality control on
specific mitochondrial proteins and, among others, 18 kDa sorcin
represents an additional mechanism used by this HSP90 chaperone to
prevent apoptosis through the mitochondrial pathway (18). Based on the evidence that TRAP1
regulates the ER stress response and represents a key player in
coupling organelle proteostasis and cell survival, through the
regulation of the mitochondrial apoptotic pathway (9–11,18),
we explored its role in favoring resistance to anthracyclins in BC
cells.
The present study demonstrated that i) the ER stress
response is responsible for inducing resistance to epirubicin and
doxorubicin; ii) ER-associated TRAP1 plays a key role in mediating
this adaptive response to anthracyclins; and iii) this process is
likely mediated by TRAP1 regulation of PERK pathway. In support of
these conclusions, our data showed a cross-resistance between
bortezomib and anthracyclins in both ER stress- and
anthracyclin-adapted BC cell models and a regulatory function by
TRAP1 in this process. Of note, TRAP1 was upregulated upon
short-term exposure to anthracyclins and in chronically-adapted
drug-resistant cell lines and its silencing favored the
re-establishment of drug sensitivity.
A major observation is the evidence that this
cytoprotective response toward anthracyclins is driven by
ER-associated TRAP1 and its ability to prevent ER stress. The
evidence obtained by transfecting the TRAP1 mutant defective for
mitochondrial import in TRAP1 KD cells and the TBP7 dominant
negative deletion mutant in drug-resistant cell lines supports the
notion that the ER stress protective function of TRAP1 is critical
in favoring resistance to anthracyclins. Furthermore, our data
suggest that this cytoprotective activity of TRAP1 likely relies on
the modulation of PERK pathway. Indeed, PERK is an important stress
sensor of the UPR, responsible for mediating phosphorylation of the
α subunit of the eukaryotic translation initiation factor eIF2
(31), this leading in turn to
global inhibition of protein synthesis and parallel ATF4-dependent
preferential translation of genes involved in cell metabolism and
cytoprotective functions (31). In
this scenario, previous studies suggested that PERK represents a
nodal point between ER stress and cell response to
chemotherapeutics, being responsible for resistance to doxorubicin,
through the interaction with the ER luminal protein ERp29 (33). Our study suggests that TRAP1
modulates the activity of PERK pathway and that this regulation is
likely crucial for protection from cytotoxics. In such a scenario,
high TRAP1 levels in tumor cells may represent a prerequisite for
enabling an efficient cytoprotective ER stress response and this
may represent a major mechanism responsible for drug resistance.
Specifically in this report, this function is linked, for the first
time, to our knowledge, to protection toward anthracyclins, agents
that primarily act by inhibiting topisomerase and inducing DNA
damage. Interestingly, previous studies by our group have shown
TRAP1 involvent in the protection against taxanes (18), agents that disregulate protein
homeostasis (35,36). In this perspective, our studies
highlight the relevance of the ER stress protective activity of
TRAP1, as complementary/additive respect to its well known
anti-apoptotic mitochondrial function (36), in favoring resistance to apoptosis
through the regulation of protein quality and the modulation of
mitochondrial apoptotic pathway. Thus, TRAP1 targeting in the ER
may be regarded as a strategy to disable the crosstalk between ER
and mitochondria and, eventually, revert resistance to
cytotoxics.
Novel HSP90/TRAP1 inhibitors are under preclinical
evaluation, as valuable anticancer agents to be used as single
agents or in combination with other anticancer drugs in the
treatment of human malignancies (37). We observed that the simultaneous
targeting of the ER cytoprotective pathway by the downregulation of
TRAP1, which is a much more specific approach compared to chemical
ER stress inducers, and the parallel inhibition of the proteasome
activity results in a dramatic increase in apoptotic cell death.
This additive/synergistic activity between TRAP1 silencing and
stress agents represents the proof of principle that the disabling
of cyto-protective responses upon TRAP1 suppression, and, among
others, PERK phosphorylation, may enhance the cytotoxic activity of
agents that target the ER response (i.e., ER stress agents,
taxanes, anthracyclins). Thus, TRAP1 inhibition may represent a
novel strategy to selectively block relevant cyto-protective
pathways used by cancer cells to escape apoptosis and acquire a
drug resistant phenotypes. These observation may be clinically
relevant in human BCs that are characterized by the co-upregulation
of TRAP1 and BiP/Grp78 in about 50% of cases and likely use these
adaptive responses to acquire resistance to apoptosis (18). Furthermore, specific BC subtypes,
i.e. HER2-positive or triple negative breast cancers, may represent
ideal human tumor models to evaluate anti-HSP90 chaperone therapy,
based on the evidence that HER2 in a well known HSP90 client
protein (38,39), whereas triple negative BCs are
aggressive drug-resistant malignancies which lack specific
molecular targets for treatment (40). In such a perspective, our data
provide the preclinical rationale for evaluating TRAP1/HSP90
targeting as a strategy to revert resistance to apoptosis in
selected human malignancies.
Abbreviations:
|
TRAP1
|
TNF receptor-associated protein 1;
|
|
HSP
|
heat shock proteins;
|
|
KD
|
knockdown;
|
|
PERK
|
PRKR-like endoplasmic reticulum
kinase;
|
|
UPR
|
unfolded protein response;
|
|
ER
|
endoplasmic reticulum;
|
|
shRNA
|
short-hairpin RNA;
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
Acknowledgements
This study was supported by grants
from the Associazione Italiana per la Ricerca sul Cancro (AIRC,
IG13128) to M.L. and F.E., the Italian Ministry of Health
(GR-2010-2310057 E66I10000220001) to F.M. and POR Campania FSE
2007-2013, project crème to F.E.
References
|
1.
|
Walter P and Ron D: The unfolded protein
response: from stress pathway to homeostatic regulation. Science.
334:1081–1086. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Schröder M and Kaufman RJ: The mammalian
unfolded protein response. Annu Rev Biochem. 74:739–789. 2005.
|
|
3.
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Ellgaard L and Helenius A: Quality control
in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 4:181–191.
2003. View
Article : Google Scholar
|
|
5.
|
Ron D: Translational control in the
endoplasmic reticulum stress response. J Clin Invest.
110:1383–1388. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Tassone P, Tagliaferri P, Fulciniti MT, Di
Martino MT and Venuta S: Novel therapeutic approaches based on the
targeting of microenvironment-derived survival pathways in human
cancer: experimental models and translational issues. Curr Pharm
Des. 13:487–496. 2007. View Article : Google Scholar
|
|
7.
|
Wilson TR, Johnston PG and Longley DB:
Anti-apoptotic mechanisms of drug resistance in cancer. Curr Cancer
Drug Targets. 9:307–319. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Reddy RK, Mao C, Baumeister P, Austin RC,
Kaufman RJ and Lee AS: Endoplasmic reticulum chaperone protein
GRP78 protects cells from apoptosis induced by topoisomerase
inhibitors: role of ATP binding site in suppression of caspase-7
activation. J Biol Chem. 278:20915–20924. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Amoroso MR, Matassa DS, Laudiero G, et al:
TRAP1 and the proteasome regulatory particle TBP7/Rpt3 interact in
the endoplasmic reticulum and control cellular ubiquitination of
specific mitochondrial proteins. Cell Death Differ. 19:592–604.
2012. View Article : Google Scholar
|
|
10.
|
Takemoto K, Miyata S, Takamura H, Katayama
T and Tohyama M: Mitochondrial TRAP1 regulates the unfolded protein
response in the endoplasmic reticulum. Neurochem Int. 58:880–887.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Siegelin MD, Dohi T, Raskett CM, et al:
Exploiting the mitochondrial unfolded protein response for cancer
therapy in mice and human cells. J Clin Invest. 121:1349–1360.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kang BH, Plescia J, Dohi T, Rosa J, Doxsey
SJ and Altieri DC: Regulation of tumor cell mitochondrial
homeostasis by an organelle-specific Hsp90 chaperone network. Cell.
131:257–270. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Costantino E, Maddalena F, Calise S, et
al: TRAP1, a novel mitochondrial chaperone responsible for
multi-drug resistance and protection from apoptotis in human
colorectal carcinoma cells. Cancer Lett. 279:39–46. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Leav I, Plescia J, Goel HL, et al:
Cytoprotective mitochondrial chaperone TRAP-1 as a novel molecular
target in localized and metastatic prostate cancer. Am J Pathol.
176:393–340. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Fang W, Li X, Jiang Q, et al:
Transcriptional patterns, biomarkers and pathways characterizing
nasopharyngeal carcinoma of Southern China. J Transl Med. 6:322008.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Landriscina M, Maddalena F, Laudiero G and
Esposito F: Adaptation to oxidative stress, chemoresistance, and
cell survival. Antioxid Redox Signal. 11:2701–2716. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Landriscina M, Laudiero G, Maddalena F, et
al: Mitochondrial chaperone Trap1 and the calcium binding protein
Sorcin interact and protect cells against apoptosis induced by
anti-blastic agents. Cancer Res. 70:6577–6586. 2010. View Article : Google Scholar
|
|
18.
|
Maddalena F, Sisinni L, Lettini G, et al:
Resistance to paclitxel in breast carcinoma cells requires a
quality control of mitochondrial antiapoptotic proteins by TRAP1.
Mol Oncol. 7:895–906. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Hande KR: Clinical applications of
anticancer drugs targeted to topoisomerase II. Biochim Biophys
Acta. 1400:173–184. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Minotti G, Menna P, Salvatorelli E, Cairo
G and Gianni L: Anthracyclines: molecular advances and
pharmacologic developments in antitumor activity and
cardiotoxicity. Pharmacol Rev. 56:185–229. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Jiang CC, Mao ZG, Avery-Kiejda KA, Wade M,
Hersey P and Zhang XD: Glucose-regulated protein 78 antagonizes
cisplatin and adriamycin in human melanoma cells. Carcinogenesis.
30:197–204. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Chae YC, Caino MC, Lisanti S, et al:
Control of tumor bioenergetics and survival stress signaling by
mitochondrial HSP90s. Cancer Cell. 22:331–344. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Scriven P, Coulson S, Haines R,
Balasubramanian S, Cross S and Wyld L: Activation and clinical
significance of the unfolded protein response in breast cancer. Br
J Cancer. 101:1692–1698. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Tan SS, Ahmad I, Bennett HL, et al: GRP78
up-regulation is associated with androgen receptor status,
Hsp70-Hsp90 client proteins and castrate-resistant prostate cancer.
J Pathol. 223:81–87. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Khasraw M, Bell R and Dang C: Epirubicin:
is it like doxorubicin in breast cancer? A clinical review Breast.
21:142–149. 2012.PubMed/NCBI
|
|
26.
|
Barone C, Landriscina M, Quirino M, et al:
Schedule-dependent activity of 5-fluorouracil and irinotecan
combination in the treatment of human colorectal cancer: in vitro
evidence and a phase I dose-escalating clinical trial. Br J Cancer.
96:21–28. 2007. View Article : Google Scholar
|
|
27.
|
Landriscina M, Fabiano A, Altamura S, et
al: Reverse transcriptase inhibitors down-regulate cell
proliferation in vitro and in vivo and restore thyrotropin
signaling and iodine uptake in human thyroid anaplastic carcinoma.
J Clin Endocrinol Metab. 90:5663–5671. 2005. View Article : Google Scholar
|
|
28.
|
Maddalena F, Laudiero G, Piscazzi A, et
al: Sorcin induces a drug-resistant phenotype in human colorectal
cancer by modulating Ca(2+) homeostasis. Cancer Res. 71:7659–7669.
2011.PubMed/NCBI
|
|
29.
|
Feng R, Zhai WL, Yang HY, Jin H and Zhang
QX: Induction of ER stress protects gastric cancer cells against
apoptosis induced by cisplatin and doxorubicin through activation
of p38 MAPK. Biochem Biophys Res Commun. 406:299–304. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Mhaidat NM, Thorne R, Zhang XD and Hersey
P: Involvement of endoplasmic reticulum stress in Docetaxel-induced
JNK-dependent apoptosis of human melanoma. Apoptosis. 13:1505–1512.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Harding HP, Zhang Y and Ron D: Protein
translation and folding are coupled by an
endoplasmic-reticulum-resident kinase. Nature. 397:271–274. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Harding HP, Novoa I, Zhang Y, et al:
Regulated translation initiation controls stress-induced gene
expression in mammalian cells. Mol Cell. 6:1099–1108. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Farmaki E, Mkrtchian S, Papazian I,
Papavassiliou AG and Kiaris H: ERp29 regulates response to
doxorubicin by a PERK-mediated mechanism. Biochim Biophys Acta.
1813:1165–1171. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Matassa DS, Amoroso MR, Crudele V, et al:
Translational control in the stress adaptive response of cancer
cells: a novel role for the heat shock protein TRAP1. Cell Death
Dis. 4:e851 View Article : Google Scholar : 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Wang J, Yin Y, Hua H, et al: Blockade of
GRP78 sensitizes breast cancer cells to microtubules-interfering
agents that induce the unfolded protein response. J Cell Mol Med.
13:3888–3897. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Kang BH: TRAP1 regulation of mitochondrial
life or death decision in cancer cells and mitochondria-targeted
TRAP1 inhibitors. BMB Rep. 45:1–6. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Neckers L, Kern A and Tsutsumi S: Hsp90
inhibitors disrupt mitochondrial homeostasis in cancer cells. Chem
Biol. 14:1204–1206. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Xu W, Mimnaugh E, Rosser MF, et al:
Sensitivity of mature Erbb2 to geldanamycin is conferred by its
kinase domain and is mediated by the chaperone protein Hsp90. J
Biol Chem. 276:3702–3708. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Basso AD, Solit DB, Munster PN and Rosen
N: Ansamycin antibiotics inhibit Akt activation and cyclin D
expression in breast cancer cells that overexpress HER2. Oncogene.
21:1159–1166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Brouckaert O, Wildiers H, Floris G and
Neven P: Update on triple-negative breast cancer: prognosis and
management strategies. Int J Womens Health. 4:511–520.
2012.PubMed/NCBI
|