Introduction
Colorectal cancer (CRC) is a result of uncontrolled
cell growth in the colon or rectum-parts of the large intestine, or
in the appendix. As of 2008 CRC is the second most common cause of
cancer in women and the third most common in men (1) with it being the fourth most common
cause of cancer death after lung, stomach and liver cancer (WHO,
2013). In Europe the 5-year survival for colorectal cancer is
<60%. In the developed world about a third of the people with
this malignancy die from it (2).
According to the National Cancer Registry of Korea,
age-standardized incidence rates increased from 27.0 to 50.2 per
100,000 for men and from 17.1 to 26.9 per 100,000 for women between
1999 and 2009. The overall incidence of CRC increased in both
gender annually from 1999 to 2009, while the incidence rates of the
most common cancers, such as stomach and liver cancer, decreased
during the same period (Korea National Cancer Information Center,
2013). According to one report, a total of 234,727 new cancer cases
and 73,313 cancer deaths are expected in Korea during 2012. Among
these cases, 32,215 are CRCs, followed by thyroid, stomach cancer,
and 8,195 cancer deaths are CRC-related (3). While >50% of CRC patients with
surgical resec tion is cured, 40–50% of these subjects eventually
experience recurrences and the possibility of re-operation is very
low (4). Because of high
prevalence and poor prognosis of CRC, there is a need to study
novel preventive treatment approaches for this malignancy.
Several flavonoids have been shown to interact with
purified topoisomerase I and II, suggesting that these compounds
may possess anticancer activities (5). It is well known that flavonoid
compounds exhibit strong antioxidant activity (6) and topoisomerase II inhibitory effects
(7) in the presence of OH
moieties. Patra and colleagues recently reported that MHY336, a
novel oxiranylchromenone derivative, showed potent topoisomerase II
inhibition through downregulation of topoisomerase IIα expression
and upregulation of p53 expression in p53 wild-type (p53-wt) LNCaP
prostate cancer cells (8). MHY336
is structurally similar to a flavonoid derivative but it has one
phenolic hydroxyl group substitution. They also demonstrated that
MHY336 markedly induced apoptotic cell death via
mitochondria-mediated intrinsic pathway in LNCaP cells. Moreover,
the cytotoxicity of MHY336 was more potent in p53-wt prostate
cancer cells than in p53 mutated (DU145) or deleted (PC3) cells.
Even though the role of p53 in the response to chemotherapeutic
agents is well established, the relationship between p53-driven
genes and drug sensitivity remains unclear. Here, we investigated
the role of p53 and its downstream effector p21, in determining
cytotoxicity of the MHY336 treatment in three isogenic variants of
human colon cancer HCT116 cells: p53+/+ (p53-wt),
p53−/− (p53-null) which has functional p21, and
p21−/− (p21-null) which has functional p53 (9).
Materials and methods
Chemicals
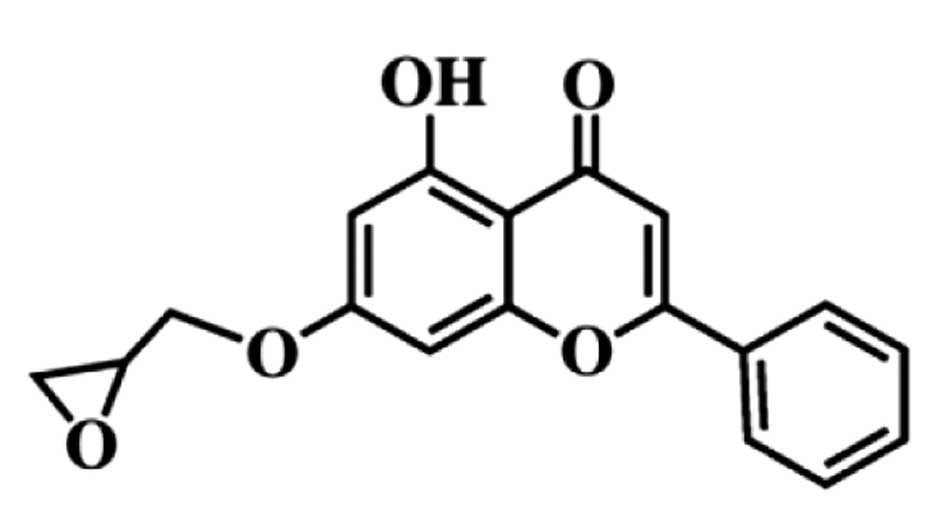
MHY336
[5-hydroxy-7-(oxiran-2-ylmethoxy)-2-phenyl-4H-chromen-4-one]
(Fig. 1) was kindly provided by
Professor Hyung Ryong Moon (Laboratory of Medicinal Chemistry,
College of Pharmacy, Pusan National University, Korea). MHY336 was
dissolved in sterile dimethyl sulfoxide (DMSO) to generate 10 mM
stock solution and stored at −20°C until use. Subsequent dilutions
were made in RPMI-1640 (Hyclone, Logan, UT, USA). The maximal
concentration of DMSO did not exceed 0.1% (v/v) in the treatment
range, where there was no influence on the cell growth. All other
chemicals with the highest purity available were from Sigma-Aldrich
Co. (St. Louis, MO, USA).
Cell culture
The human colorectal cancer cell lines HCT116
p53+/+ (p53-wt), p53−/− (p53-null) and
p21−/− (p21-null) were obtained from American Type
Culture Collection (Manassas, VA, USA) and kindly provided by
Professor Young-Chae Chang (Department of Pathology, Catholic
University of Daegu School of Medicine, Daegu, Korea) (9). These cells were cultured in RPMI-1640
(Hyclone) supplemented with 10% fetal bovine serum (FBS, Hyclone),
2 mM glutamine (Sigma-Aldrich), 100 U/ml penicillin (Hyclone), and
100 μg/ml streptomycin (Hyclone) at 37°C in a humidified 5%
CO2.
Cell viability assay
Cell viability was determined by MTT assay. For the
MTT assay, cells were seeded in a 24-well culture plate at a
density of 4×104 cells/well, cultured for 24 and 48 h in
the growth media and then treated with or without various reagents
for the indicated concentrations. The cells were incubated with 0.5
mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
(MTT, Sigma-Aldrich) at 37°C for 2 h. The formazan granules
generated by the live cells were dissolved in dimethyl sulfoxide
(DMSO), and the absorbance at 540 nm was monitored by using a
multi-well reader.
Annexin V staining
Annexin V-FITC is used to quantitatively determine
the percentage of cells within a population that are actively
undergoing apoptosis. The cells were treated under the appropriate
conditions for 24 h, subsequently harvested, trypsinized, washed
once in cold PBS, and suspended in 1X binding buffer
(Becton-Dickinson, San Jose, CA, USA Annexin V-FITC Apoptosis
Detection kit). The counted cells were stained in propidium iodide
(PI, Sigma-Aldrich) and Annexin V-FITC solution (Becton-Dickinson)
at room temperature for 15 min in the dark. The stained cells were
analyzed by flow cytometry within 1 h.
DNA fragmentation assay
Cells were lysed in a buffer, containing 5 mM
Tris-HCl (pH 7.5), 5 mM EDTA and 0.5% Triton X-100, for 30 min on
ice. Lysates were vortexed and cleared by centrifugation at 14,000
rpm for 20 min. Fragmented DNA in the supernatant was treated with
RNase, followed by proteinase K digestion,
phenol:chloroform:isoamyl alcohol mixture (25:24:1) extraction and
isopropanol precipitation. DNA was separated through a 1.5% agarose
gel, was stained with 0.1 μg/ml ethidium bromide, and was
visualized by UV source.
Cell cycle analysis
The DNA content was measured following the staining
of the cells with PI. The cells were treated under the appropriate
conditions for 24 h, subsequently trypsinized, washed once in cold
PBS, and then fixed in 70% ethanol at −20°C overnight. The fixed
cells were pelleted and stained in cold PI solution (50
μg/ml in PBS) at room temperature for 30 min in the dark.
The stained cells were analyzed by flow cytometry (FC500, Beckman
Coulter, Istanbul, Turkey).
Western blot analysis
The cells were treated with the appropriate
conditions, harvested, and washed with cold PBS. Total cells
lysates were lysed in lysis buffer [40 mM Tris (pH 8.0), 120 mM,
NaCl, 0.5% NP-40, 0.1 mM sodium orthovanadate, 2 μg/ml
aprotinin, 2 μg/ml leupeptin and 100 μg/ml PMSF].
Protein extracts were denatured by boiling at 100°C for 5 min in
sample buffer (0.5 M Tris-HCl, pH 6.8, 4% SDS, 20% glycerol, 0.1%
bromophenol blue, 10% β-mercaptoethanol). Equal amount of the total
proteins were subjected to 6–15% SDS-PAGE and transferred to PVDF.
The membranes were blocked with 5% non-fat dry milk in
Tris-buffered saline with Tween-20 buffer (TBS-T) (20 mM Tris, 100
mM NaCl, pH 7.5 and 0.1% Tween-20) for 1 h at room temperature.
Then, the membranes were incubated overnight at 4°C with primary
antibodies (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA).
The membranes were washed with TBS-T buffer and incubated for 1 h
with horseradish peroxidase-conjugated anti-rabbit or anti-mouse
immunoglobin (Santa Cruz). The membranes were washed with TBS-T
buffer. Antigen-antibody complexes were detected by the enhanced
chemiluminescence (ECL) detection system (GE Healthcare, Little
Chalfont, Bucks, UK).
Transient transfection
HCT116 p53-wt cells were seeded in 6-well culture
plate in antibiotic-free medium for 24 h and then transfected with
p53- or p21 siRNA using Lipofectamine 2000 (Invitrogen, Calsbad,
CA, USA) according to the manufacturer’s protocol. Control siRNA,
p53 siRNA and p21 siRNA were obtained from Bioneer (Sungnam,
Korea). The sequence of siRNA targeting p53 is: sense,
5′-CACUACAACU ACAUGUGUA-3′; antisense, 5′-UACACAUGUAGUUGUA GUG-3′
and p21 siRNA sequence is: sense, 5′-CUGUAC UGUUCUGUGUCUU-3′;
antisense, 5′-AAGACACAGA ACAGUACAG-3′. After 6-h transfection,
cells were cultured for 24 h in the growth media and then treated
with or without various reagents for the indicated
concentrations.
Statistical analysis
Results are expressed as the mean ± SE of three
separate experiments and analyzed by Student’s t-test. Means were
considered significantly different at p<0.05 or p<0.01.
Results
MHY336 has more potent cytotoxicity on
p53-wt cells than on p53-null and p21-null cells
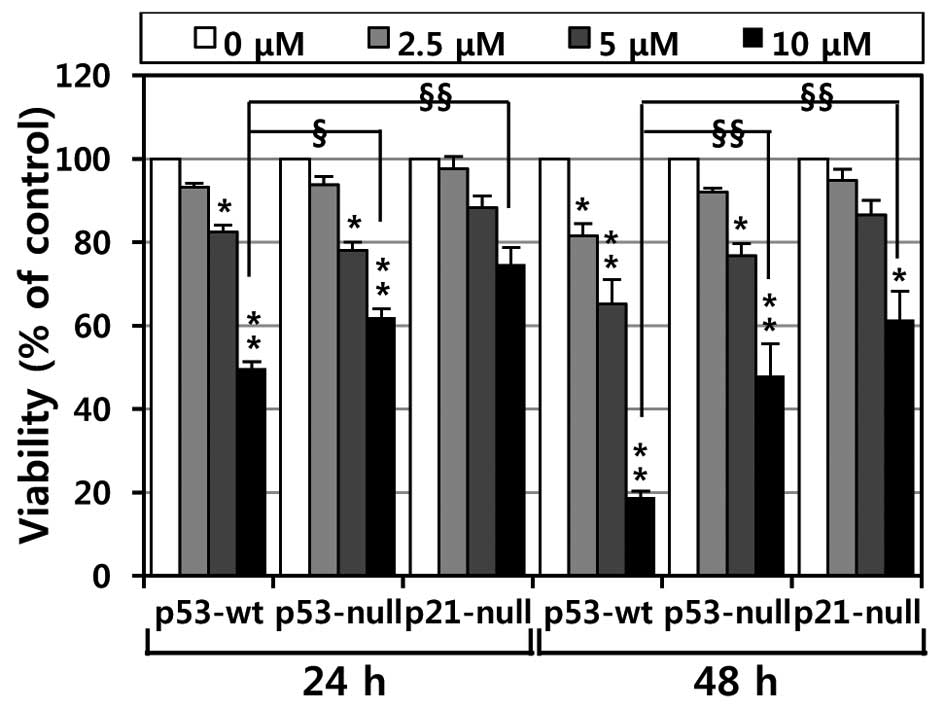
To investigate the effects of MHY336 on the
viability of p53-wt, p53-null and p21-null cells, the MTT assay was
performed. As shown in Fig. 2,
MHY336 showed concentration- or time-dependent cytotoxicity on
three isogenic variants. Moreover, we found a significant
difference in the IC50 among p53-wt, p53-null and
p21-null cells. The IC50 values of MHY336 on p53-wt
cells were ∼10 and 6 μM at 24 and 48 h, respectively.
However, the IC50 values of MHY336 were ∼15 and 10
μM at 24 and 48 h, respectively in p53-null cells and 20 and
15 μM at 24 and 48 h in p21-null cells, respectively. These
results indicate that p53-wt cells were more sensitive to MHY336
than p53-null and p21-null cells.
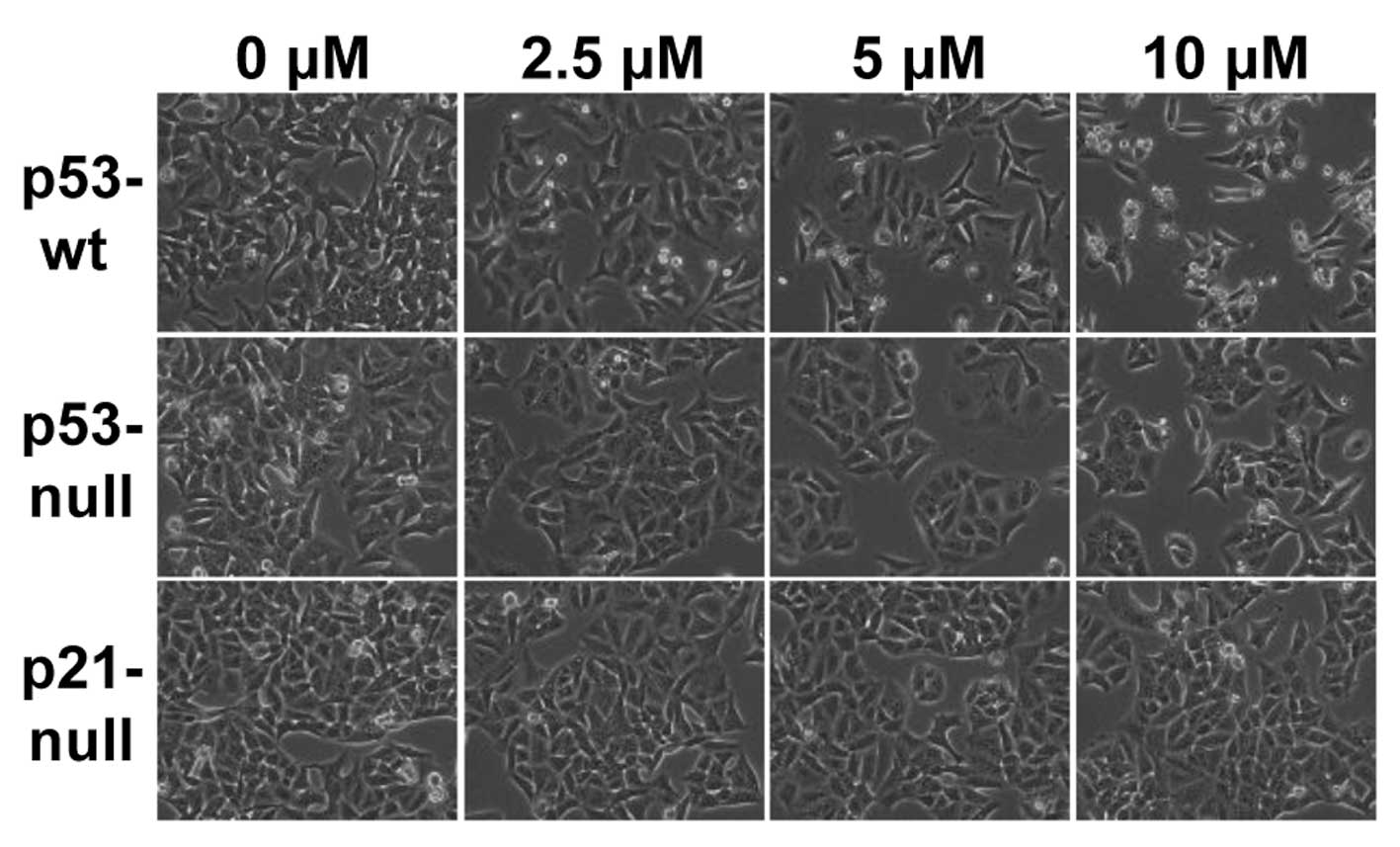
MHY336 induces morphological changes and
apoptosis p53/p21-dependently
To assess whether there are any morphological
changes in MHY336-treated cells, we examined the cells under a
phase-contrast light microscope after 24 h of incubation with or
without MHY336. As shown in Fig.
3, MHY336-untreated p53-wt, p53-null and p21-null cells spread
regularly in the culture plate and grew to near confluent. In
contrast, MHY336-treated p53-wt cells were shrunken and changed to
round form. Cell numbers were also decreased in a
concentration-dependent manner. MHY336-treated p53-null and
p21-null cells did not show any noteworthy morphological changes.
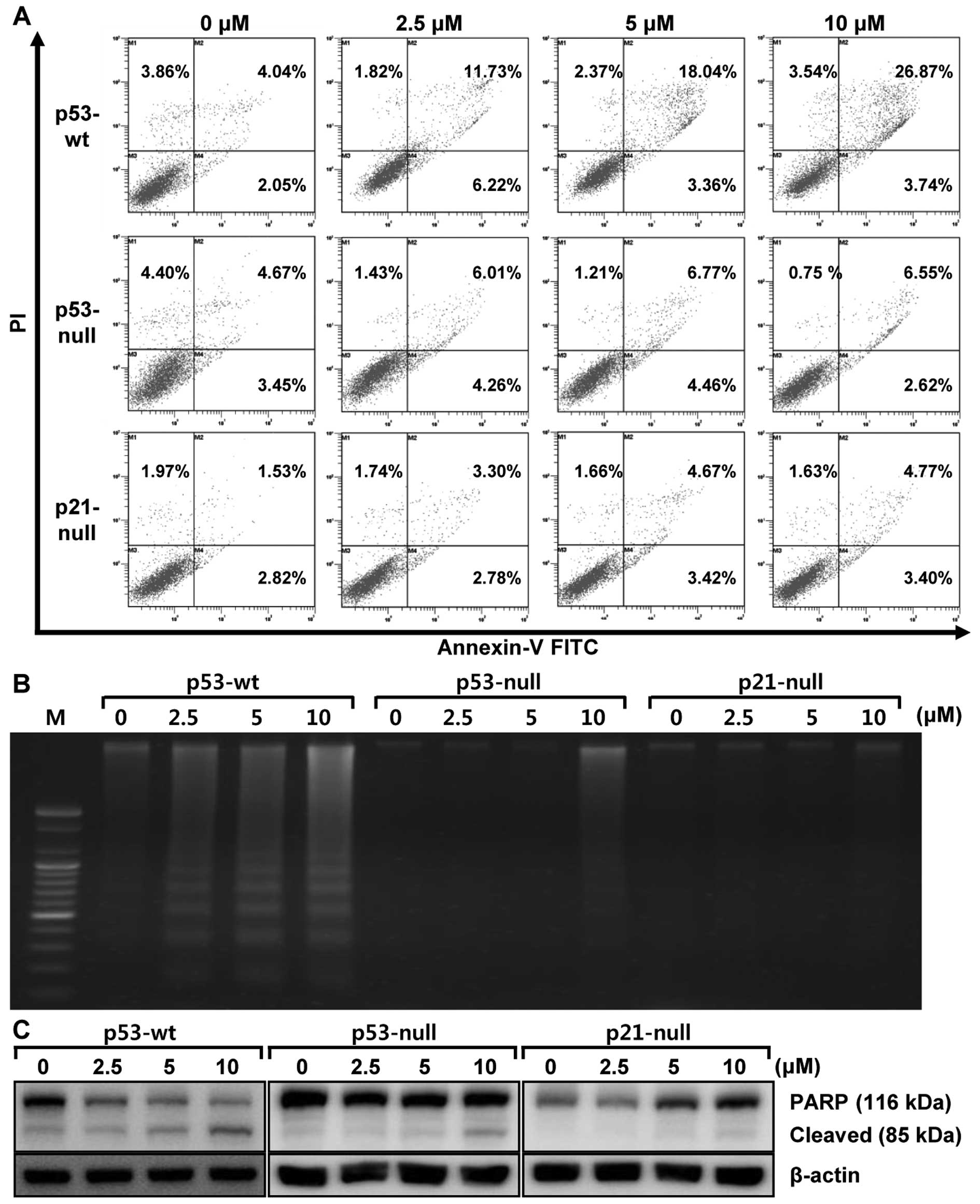
To confirm that these changes are attributed to apoptosis induced
by MHY336, flow cytometry analysis was performed in three isogenic
cells. As shown in Fig. 4A,
increase of early apoptosis (lower right quadrant) and late
apoptosis/necrosis (upper right quadrant) was clearly observed in a
concentration-dependent manner. The late apoptosis of MHY336 on
p53-wt cells increased from 4.04% (vehicle alone) to 26.87% (10
μM MHY336), whereas MHY336-treated p53-null and p21-null
cells did not show any variation. We also analyzed whether DNA
fragmentation, another hallmark of apoptosis, occurred in the
MHY336-treated isogenic cells. In p53-wt cells treated with MHY336
for 24 h, there was a typical ladder pattern of internucleosomal
fragmentation observed in a concentration-dependent manner after
agarose gel electrophoresis (Fig.
4B). On the other hand, DNA fragmentation was not identified in
either MHY336-treated p53-null or p21-null cells. Polypeptide
degradation, including poly(ADP-ribose) polymerase (PARP), was also
examined to see the possible involvement of apoptosis-associated
protease during the growth inhibition of the colon cancer cells.
PARP cleavage was evident by the appearance of the p85 PARP
cleavage fragment (Fig. 4C) and
clearly observed in p53-wt cells at the concentration of 5 and 10
μM of MHY336 treatments. MHY336-treated p53-null cells
showed lesser amount of the p85 PARP cleavage fragment in the 10
μM of MHY336 treatment, but not in p21-null cells.
Collectively, these results suggest that p53 and/or p21 are crucial
mediator(s) of MHY336-induced apoptosis.
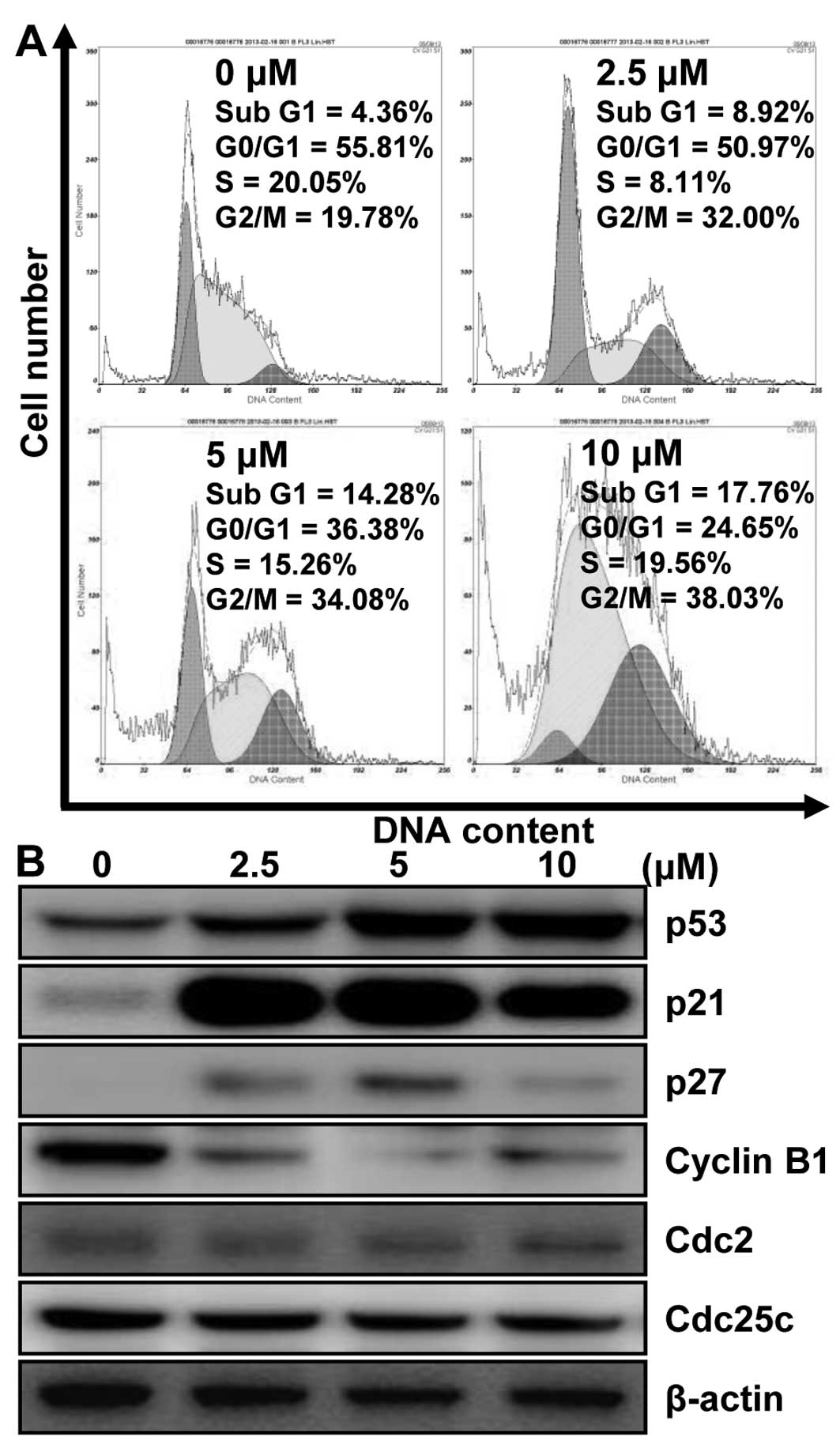
MHY336 modulates the cell cycle and its
regulatory proteins in p53-wt cells
To investigate whether MHY336 has an effect on the
cell cycle, we thus only used p53-wt cells. After 24-h incubation
with different concentrations of MHY336, p53-wt cells treated with
MHY336 were remarkably arrested in the G2/M phase. The proportions
of sub-G1 phase cells were increased as well (Fig. 5A). The population of the cells in
the G2/M phase was increased from 19.78% (vehicle alone) to 38.03%
(10 μM MHY336). The increase of cell population in G2/M
phase was accompanied by the decrease in G0/G1 cells. In addition,
after 24-h incubation with 10 μM MHY336, the fractions of
sub-G1 peak increased from 4.36% (vehicle alone) to 17.76% (10
μM MHY336). However, MHY336-treated p53-null and p21-null
cells did not show any changes (data not shown). These results
supported the MHY336 treatment for 24 h mainly induced the
inhibition of cell growth via G2/M phase arrest in the cell cycle.
To assess the effect of MHY336 on the intracellular protein
expression levels of G2/M phase in cell cycle, we performed western
blot analysis. As shown in Fig.
5B, the expression levels of cyclin B1, Cdc25c and Cdc2 were
decreased by MHY336 treatment in a concentration-dependent manner.
The induction of p21 is known to cause subsequent arrest in the
G1/G0 or G2/M phase of the cell cycle by binding of the
cyclin-cyclin dependent kinase (CDK) complex. The protein levels of
p21 and p27, CDK inhibitors, were increased in p53-wt cells by
MHY336 treatment concentration-dependently (Fig. 5B). In addition, protein levels of
p53 were also increased. Therefore, these results suggest that
MHY336 treatment induces G2/M phase arrest in cell cycle by
downregulating expressions of cyclin and CDKs, and by inducing p21
via p53-dependent pathway.
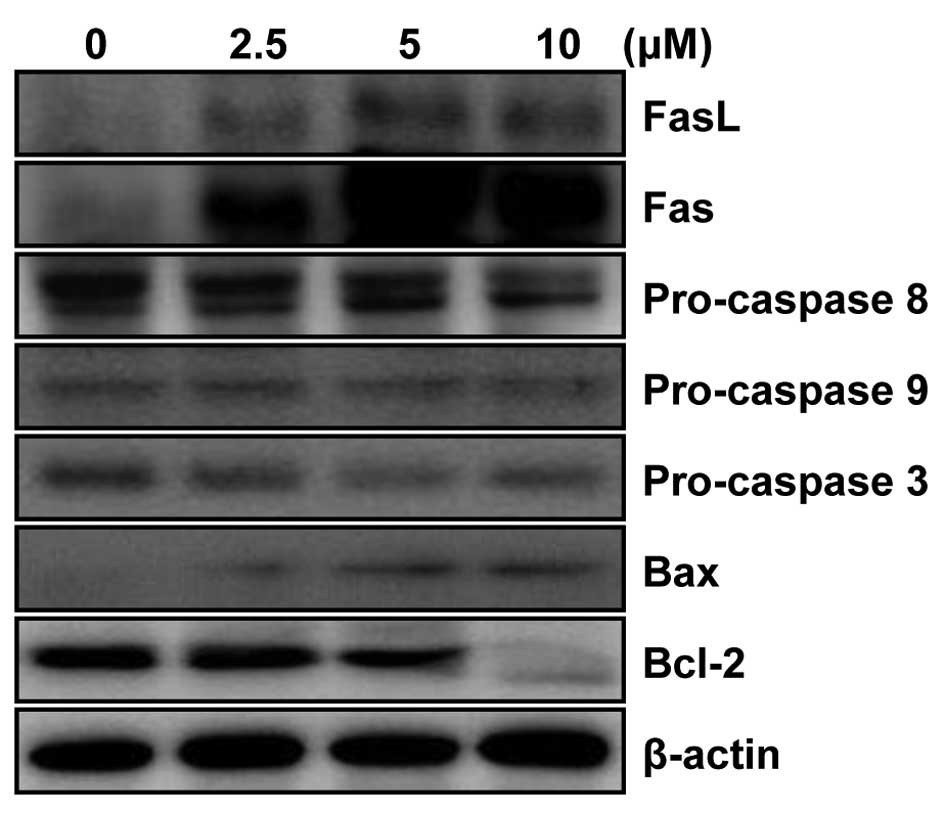
MHY336 modulates the expression levels of
apoptosis-related proteins in p53-wt cells
To determine whether the expression levels of
apoptosis-related proteins were modulated by MHY336, western blot
analysis was performed. As shown in Fig. 6, the levels of Fas as well as FasL
expression were significantly upregulated in a
concentration-dependent manner. In addition, significant activation
of pro-caspase-3 and -8 was observed, while there was little change
in procaspase 9. The expression level of Bax protein was markedly
upregulated, but Bcl-2 was downregulated in concentration-dependent
manner. These results, collectively, imply that MHY336 induces
apoptosis through both internal and external pathway in p53-wt
cells.
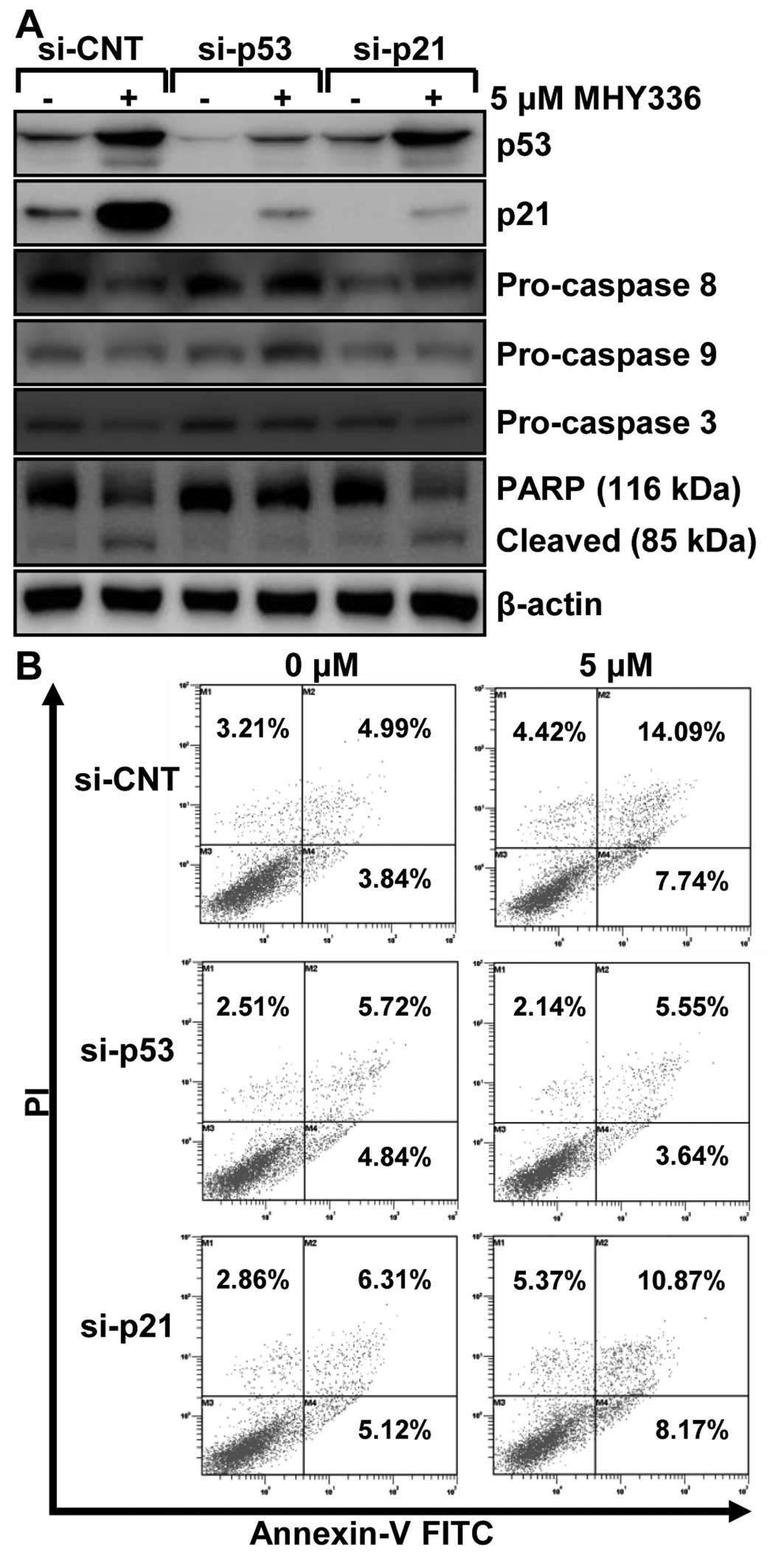
p53 and p21 are required for
MHY336-induced apoptosis in p53-wt cells
To examine the role of p53 and p21 in MHY336-induced
apoptosis, we performed knockdown of the p53 or p21 using siRNA in
p53-wt cells. As shown in Fig. 7A,
p53 siRNA completely inhibited the expression levels of p53 and
p21, whereas p21 siRNA suppressed only p21 in p53-wt cells. These
results showed that p53 silencing led to abrogation of 5 μM
MHY336-induced activation of pro-caspase-3 and cleavage of PARP. On
the other hand, p21-silencing partially resulted in repression of
activation of pro-caspase-3 and 8, and subsequently PARP cleavage.
Furthermore, after cells were double stained with Annexin V and PI
for apoptosis and necrosis, the results revealed that the p53
knockdown p53-wt cells significantly blocked apoptosis of MHY336
and the p21 knockdown of p53-wt cells incompletely inhibited
apoptosis of MHY336 treatment (Fig.
7B). Based on these data, we suggest that p53 and p21 are
important regulators of MHY336-induced apoptosis.
Discussion
This study was conducted to investigate the role of
p53 and its downstream target p21, in the response of HCT116
colorectal cancer cells to MHY336 treatment. To achieve this goal,
we used three isogenic variants of the HCT116 colon cancer cells:
p53-wt, p53-null and p21-null. Treatment of p53-wt cells with
MHY336 resulted in the maximum apoptotic response whereas p53-null
cells were more resistant, and p21-null cells were the most
resistant to apoptosis. These results clearly indicate that p53 and
subsequently p21 has a pivotal role in sensitization of colon
cancer cells to apoptosis in response to cytotoxic agents, such as
MHY336. Our data suggest that the increased cytotoxicity in p53-wt
cells to MHY336 may be due to the presence of the transcription
factor and tumor suppressor p53.
Although the role of p53 as an important mediator of
apoptosis is well established under some conditions, the downstream
effectors of the p53 pathway in apoptosis are not well known.
Several p53-inducible genes which are involved in apoptosis have
been identified, such as mdm-2 (10), GADD45 (11), BAX (12), and p21 (13,14).
Previous evidence suggests that, in addition to cell cycle arrest,
p21 may have a role in apoptosis in both p53-dependent and
p53-independent pathways (15,16).
p53-induced apoptosis results from overlapping downstream pathways
that both suppress mitogenic and survival signaling and promote
proapoptotic signaling (17).
Therefore, p53 can upregulate the proapoptotic Bcl-2 family member
Bax (12) and possibly
transcriptionally repress the anti-apoptotic protein Bcl-2
(18). Therefore, p53-mediated
upregulation of Bax and probably concomitant downregulation of
Bcl-2 are pivotal in shifting the ratio of Bax/Bcl-2 in the cell
and ultimately favoring apoptosis.
Normally, p53 plays a major role as both a cell
cycle regulator and an inducer of apoptosis in response to a
variety of cellular stresses (8,19).
As expected, p53-wt cells exhibited a typical G2/M phase arrest via
p21, a CDK inhibitor, upregulation and induced apoptosis
consequently via both extrinsic and intrinsic pathways as described
previously (8). On the other hand,
the p53-null cells displayed a loss of the growth-inhibitory and
apoptotic effects of MHY336, indicating that the p53 gene plays a
crucial role in mediating the effects of MHY336. We also found an
absence of p21 in p53-null cells, suggesting the lack of a
functional p53 protein could lead to loss of p21 protein levels in
response to MHY336 exposure. This was also manifested in a
significant reduction of apoptosis in p53-null cells on MHY336
treatment. This study thus suggests that p21 plays an important
role in exerting the apoptotic activity of MHY336 in a
p53-dependent manner.
p21, a cyclin-dependent kinase inhibitor, is
commonly associated with the G1 checkpoint and G2/M phase, its
association with inhibiting the expression of the Cdc2/cyclin B1
complex has been also demonstrated (20,21).
p21 transcription can be regulated through p53-dependent (22) and p53-independent pathways
(23). In the present study, p21
was increased by MHY336 treatment in p53-wt cells, along with
significant increase in the protein levels of p53. Therefore, the
upregulation of p21, and the downregulation of cyclin B1, Cdc2, and
Cdc25c may be one of the molecular mechanisms by which MHY336
inhibited p53-wt cell growth and induced cell cycle arrest.
p53 stimulates a wide network of signals that act
through two major apoptotic pathways. The extrinsic, death receptor
pathway triggers the activation of a caspase cascade, and the
intrinsic, mitochondrial pathway shifts the balance in the Bcl-2
family towards the pro-apoptotic members, promoting the formation
of the apoptosome, and consequently caspase-mediated apoptosis
(24). Wild-type p53 can bind to
the specific p53 binding site in the topoisomerase II promoter.
During cellular stress or after DNA double-strand breaks, the
upregulation of wild-type p53 activates the transcriptional
downregulation of topoisomerase II (8,25–27).
Therefore, we speculate that MHY336 treatment in p53-wt cells
induce DNA damage, which would result in upregulation of p53 and
trigger the p53-dependent apoptotic cell death via inhibition of
topoisomerase II activity in HCT116 colon cancer cells and LnCaP
prostate cancer cells as previously reported (8).
Apoptosis, a controlled and energy-dependent
process, is the best-described form of programmed cell death. There
are two major apoptotic pathways, which are the extrinsic pathway
and the intrinsic pathway (28).
The extrinsic pathway is initiated by the binding of transmembrane
death receptors [Fas, tumor necrosis factor receptor 1 (TNFR1),
TNF-related apoptosis-inducing ligand (TRAIL) receptors] with
cognate extracellular ligands (29). Ligand receptors recruit adaptor
proteins [TNFR-associated death domain (TRADD) and Fas-associated
death domain (FADD)], which interact with and trigger the
activation of caspase-8. Activated caspase-8 thereby stimulates the
effector caspase-3, -6 and -7 which ultimately execute apoptosis
(30). The intrinsic pathway is
domi nated by the Bcl-2 family of proteins which govern the release
of cytochrome c from the mitochondria (31,32).
Cytochrome c stimulates the apoptosome formation (Apaf-1,
dATP,cyto chrome c and caspase-9) followed by activation of
caspase-9, which in turn causes the activation of the ‘executioner’
caspases (-3, -6 and -7) (31,33,34).
In conclusion, we have clearly shown, using three
isogenic variants, that MHY336 induces growth arrest and apoptosis
primarily via a p53-dependent pathway that necessarily involves the
function of p21. Taken together, these results suggest that the
novel compound MHY336 may be useful in the chemo prevention and/or
treatment of colon cancer.
Acknowledgements
This study was supported by the
National Research Foundation of Korea (NRF) grant funded by the
Korea government (MSIP) (No. 2009-0083538). We thank Aging Tissue
Bank for providing research information.
References
|
1.
|
Merika E, Saif MW, Katz A, Syrigos K and
Morse M: Review. Colon cancer vaccines: an update. In Vivo.
24:607–628. 2010.PubMed/NCBI
|
|
2.
|
Cunningham D, Atkin W, Lenz HJ, et al:
Colorectal cancer. Lancet. 375:1030–1047. 2010. View Article : Google Scholar
|
|
3.
|
Jung KW, Won YJ, Kong HJ, Oh CM, Seo HG
and Lee JS: Prediction of cancer incidence and mortality in Korea,
2013. Cancer Res Treat. 45:15–21. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Luu C, Arrington AK, Schoellhammer HF,
Singh G and Kim J: Targeted therapies in colorectal cancer:
surgical considerations. J Gastrointest Oncol. 4:328–336.
2013.PubMed/NCBI
|
|
5.
|
Lopez-Lazaro M, Willmore E and Austin CA:
The dietary flavonoids myricetin and fisetin act as dual inhibitors
of DNA topoisomerases I and II in cells. Mutat Res. 696:41–47.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Cao G, Sofic E and Prior RL: Antioxidant
and prooxidant behavior of flavonoids: structure-activity
relationships. Free Radic Biol Med. 22:749–760. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Constantinou A, Mehta R, Runyan C, Rao K,
Vaughan A and Moon R: Flavonoids as DNA topoisomerase antagonists
and poisons: structure-activity relationships. J Nat Prod.
58:217–225. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Patra N, De U, Kang JA, et al: A novel
epoxypropoxy flavonoid derivative and topoisomerase II inhibitor,
MHY336, induces apoptosis in prostate cancer cells. Eur J
Pharmacol. 658:98–107. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Jeong JH, Kang SS, Park KK, Chang HW,
Magae J and Chang YC: p53-independent induction of G1 arrest and
p21WAF1/CIP1 expression by ascofuranone, an isoprenoid
antibiotic, through downregulation of c-Myc. Mol Cancer Ther.
9:2102–2113. 2010.PubMed/NCBI
|
|
10.
|
Oliner JD, Kinzler KW, Meltzer PS, George
DL and Vogelstein B: Amplification of a gene encoding a
p53-associated protein in human sarcomas. Nature. 358:80–83. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Kastan MB, Zhan Q, el-Deiry WS, et al: A
mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is
defective in ataxia-telangiectasia. Cell. 71:587–597. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Miyashita T and Reed JC: Tumor suppressor
p53 is a direct transcriptional activator of the human bax gene.
Cell. 80:293–299. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Harper JW, Adami GR, Wei N, Keyomarsi K
and Elledge SJ: The p21 Cdk-interacting protein Cip1 is a potent
inhibitor of G1 cyclin-dependent kinases. Cell. 75:805–816. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Xiong Y, Hannon GJ, Zhang H, Casso D,
Kobayashi R and Beach D: p21 is a universal inhibitor of cyclin
kinases. Nature. 366:701–704. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Waldman T, Kinzler KW and Vogelstein B:
p21 is necessary for the p53-mediated G1 arrest in human cancer
cells. Cancer Res. 55:5187–5190. 1995.PubMed/NCBI
|
|
16.
|
Agrawal S, Agarwal ML, Chatterjee-Kishore
M, Stark GR and Chisolm GM: Stat1-dependent, p53-independent
expression of p21 (waf1) modulates oxysterol-induced apoptosis. Mol
Cell Biol. 22:1981–1992. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Aneja R, Ghaleb AM, Zhou J, Yang VW and
Joshi HC: p53 and p21 determine the sensitivity of
noscapine-induced apoptosis in colon cancer cells. Cancer Res.
67:3862–3870. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Haldar S, Negrini M, Monne M, Sabbioni S
and Croce CM: Downregulation of bcl-2 by p53 in breast cancer
cells. Cancer Res. 54:2095–2097. 1994.PubMed/NCBI
|
|
19.
|
Wang XW and Harris CC: p53
tumor-suppressor gene: clues to molecular carcinogenesis. J Cell
Physiol. 173:247–255. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Niculescu AB III, Chen X, Smeets M, Hengst
L, Prives C and Reed SI: Effects of p21 (Cip1/Waf1) at both the
G1/S and the G2/M cell cycle transitions: pRb is a critical
determinant in blocking DNA replication and in preventing
endoreduplication. Mol Cell Biol. 18:629–643. 1998.PubMed/NCBI
|
|
21.
|
Baus F, Gire V, Fisher D, Piette J and
Dulic V: Permanent cell cycle exit in G2 phase after DNA damage in
normal human fibroblasts. EMBO J. 22:3992–4002. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
el-Deiry WS, Tokino T, Velculescu VE, et
al: WAF1, a potential mediator of p53 tumor suppression. Cell.
75:817–825. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Gartel AL and Tyner AL: Transcriptional
regulation of the p21(WAF1/CIP1) gene. Exp Cell Res. 246:280–289.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Haupt S, Berger M, Goldberg Z and Haupt Y:
Apoptosis - the p53 network. J Cell Sci. 116:4077–4085. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Sandri MI, Isaacs RJ, Ongkeko WM, et al:
p53 regulates the minimal promoter of the human topoisomerase
IIalpha gene. Nucleic Acids Res. 24:4464–4470. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Wang Q, Zambetti GP and Suttle DP:
Inhibition of DNA topoisomerase II alpha gene expression by the p53
tumor suppressor. Mol Cell Biol. 17:389–397. 1997.PubMed/NCBI
|
|
27.
|
Valkov NI and Sullivan DM: Tumor p53
status and response to topoisomerase II inhibitors. Drug Resist
Updat. 6:27–39. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Yang SY, Sales KM, Fuller B, Seifalian AM
and Winslet MC: Apoptosis and colorectal cancer: implications for
therapy. Trends Mol Med. 15:225–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Peter ME: The flip side of FLIP. Biochem
J. 382:e1–e3. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Ashkenazi A: Targeting the extrinsic
apoptosis pathway in cancer. Cytokine Growth Factor Rev.
19:325–331. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Cory S and Adams JM: The Bcl2 family:
regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Kuwana T, Mackey MR, Perkins G, et al:
Bid, Bax, and lipids cooperate to form supramolecular openings in
the outer mitochondrial membrane. Cell. 111:331–342. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Oliver L and Vallette FM: The role of
caspases in cell death and differentiation. Drug Resist Updat.
8:163–170. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Iannolo G, Conticello C, Memeo L and De
Maria R: Apoptosis in normal and cancer stem cells. Crit Rev Oncol
Hematol. 66:42–51. 2008. View Article : Google Scholar : PubMed/NCBI
|