Introduction
Breast cancer is one of the most common malignancies
in women. Approximately 15% of breast cancer cases belong to the
triple-negative breast cancer (TNBC) group, in which neither
estrogen/progesterone receptors nor HER2 expression can be detected
(1). Systemic treatment for
patients with triple-negative disease is currently limited to
chemotherapy, and survival of patients in this group is poor
compared to patients with other cancer subtypes. BRCA1 is known to
be involved in a number of DNA repair pathways, including DNA
double-strand break (DSB) repair through homologous recombination
(HR) (2,3), nucleotide excision repair (NER)
(4) and base excision repair (BER)
(5). Some TNBCs harbor defects in
DNA double-strand break repair by HR, such as BRCA1 dysfunction,
and are hypersensitive to the inhibition of poly (ADP-ribose)
polymerase (PARP) (6–9). However, BRCA-mutant tumors
represent only 2–3% of all breast cancers (10) and only 12.5% of TNBCs (1), which might restrict the therapeutic
utility of PARP inhibitor monotherapy. Cyclin-dependent kinase 1
(CDK1) is a master modulator of the initiation and transition
process through mammalian mitosis (11,12).
Previous studies have shown that CDK1 activity loss or its aberrant
expression are involved in the G2/M phase arrest in many tumor
types (13), and that CDK1
inhibition downregulates survival and induces apoptosis (14,15).
Besides, CDK1 phosphorylates BRCA1, and this is necessary for its
ability to efficiently form BRCA1 foci at DNA damage sites and
facilitate checkpoint activation (16). Therefore, CDK1 is considered as an
important therapeutic target (15). It is likely that the reduced CDK1
activity may also sensitize BRCA-proficient tumor cells to PARP
inhibition, facilitating the extension of the synthetic lethal
therapeutic option to a larger patient population. Here we show
that the cytotoxic effect of CDK1 and PARP inhibition, administered
in a sequential combination regimen, was superior over PARP
inhibition alone in the MDA-MB-231 BRCA-proficient breast cancer
cell line. Combined inhibition resulted in sustained DNA damage,
and this was paralleled by a dramatic G2/M cell cycle arrest and
the induction of cell death. CDK1 inhibition represents a plausible
strategy for expanding the utility of PARP inhibitors to
BRCA-proficient breast cancers.
Materials and methods
Cell lines and drugs
The MDA-MB-231, HCC1937, SK-BR-3, and MCF-7 human
breast cancer cell lines were obtained from the Cell Bank of the
Chinese Academy of Sciences (Shanghai, China). MDA-MB-231, SK-BR-3
and MCF-7 cells were grown in DMEM media (Invitrogen, Carlsbad, CA,
USA) with 10% fetal bovine serum (FBS) (HyClone, Logan, UT, USA),
and HCC1937 cells were cultured in RPMI-1640 (Invitrogen)
supplemented with 10% FBS. All cells were cultured in a 5%
CO2 incubator at 37°C.
AZD2281 (olaparib) was purchased from Selleck
Chemicals (Houston, TX, USA) and 100 mM stocks were prepared.
RO3306 was purchased from EMD Chemicals (Gibbstown, NJ, USA) and
diluted to a 10-mM stock solution. All stock solutions were
prepared using dimethyl sulfoxide (DMSO) as a solvent and stored at
−20°C.
Analysis of BRCA1 mutations
Genomic DNA was extracted from HCC1937 and
MDA-MB-231 using the TIANamp Genomic DNA Kit (Tiangen, Beijing,
China). The complete coding regions and the BRCA1
exon-intron boundaries were screened by a polymerase chain reaction
(PCR)-sequencing assay. The whole BRCA1 coding sequence was
amplified with 31 pairs of primers. Amplification of DNA fragments
was performed in a thermocycler (Gene Cycler™, Bio-Rad, Hercules,
CA, USA) in 25 μl reactions containing 30 ng of genomic DNA,
2.5 mM dNTP, 50 mM MgCl2, 10X PCR buffer, 0.5 μM
of each primer, and 1.25 units of AmpliTaq DNA polymerase (Promega,
Madison, WI, USA). The reactions were initially kept at 94°C for 3
min to activate the Taq DNA polymerase, followed by 30 sec
denaturation at 94°C, 30 sec annealing at a temperature suitable
for each primer pair, and 30 sec extension at 72°C. The PCR was run
for 35 cycles and a 10 min elongation step was performed at the
end. All fragments were sequenced using the BigDye Terminator Cycle
Sequencing Kit and an ABI 3730XL automated sequencer (Applied
Biosystems, Foster City, CA, USA). Each mutation was confirmed in
duplicate.
MTT assay and combination effect
Log phase cells (25,000) were seeded in 96-well
plates and incubated in a 37°C incubator with 5% CO2.
After 24 h, different concentrations of the PARP inhibitor AZD2281
in the case of the four cell lines, or of the CDK1 inhibitor RO3306
as a single agent in the case of MDA-MB-231, were administered to
determine the drug concentrations required to achieve a 50% growth
inhibition (GI50). For combination treatments, MDA-MB-231 cells
were treated with AZD2281 or with the sequential combination
regimen (RO3306 alone for 4 h followed by AZD2281 for an additional
72-h period) at the indicated doses. MTT (20 μl; 5 mg/ml
stock solution in saline) was added to each well and the cells were
incubated for 4 h. Supernatants were removed and formazan crystals
from viable cells were solubilized with 200 μl anhydrous
DMSO. The absorbance was detected with a 550 model microplate
reader at the 565 nm wavelength (17024, Bio-Rad).
The combination effect was evaluated by the
combination index (CI), which was calculated using the Calcusyn
software (Biosoft, Cambridge, UK). The definition of CI is as
follows: CI = (D)1/(Dx)1 +
(D)2/(Dx)2 +
(D)1(D)2/(Dx)1(Dx)2,
where (Dx)1 and (Dx)2 are the concentrations
of the individual drugs required to produce an X% effect alone, and
(D)1 and (D)2 are the concentrations of the
combination required to produce the same X% effect. The combination
effects were defined as follows: CI<1, synergistic effect; CI=1,
additive effect; and CI>1, antagonistic effect.
RNA interference
MDA-MB-231 cells were transiently transfected with
50 nM BRCA1 or CDK1 siRNA [5′-GCA GUG AAG AGA UAA AGA ATT-3′
(#1304, Shanghai GenePharma Co., Ltd, Shanghai, China) and 5′-GGG
GUU CCU AGU ACU GCA A dTdT-3′ (#2012, Ribo Bio Co., Ltd, Guangzhou,
China), respectively], or with a non-targeting control [5′-UUC UCC
GAA CGU GUC ACG UTT-3′ (#0420 Shanghai GenePharma Co., Ltd); and
5′-GUU CGC GUU ACG CGA GAU A dTdT-3′ (#7021, Ribo Bio Co., Ltd),
respectively], using the TurboFect siRNA transfection reagent
(Fermentas Life Sciences, Pittsburgh, PA, USA) in accordance to the
manufacturer’s instructions. Following a 6-h incubation period,
fresh AZD2281-containing growth medium was added and the MTT assay
was performed after an additional 72-h incubation period. For
western blot analysis, cells were harvested 48 h
post-transfection.
Cell cycle and apoptosis analysis
MDA-MB-231 cells (10×104 cells/ml) were
treated in 6-well plates with 50 μM AZD2281, 5 μM
RO3306, or the sequential combination regimen (5 μM RO3306
alone for 4 h followed by 50 μM AZD2281) for 48 h, as
described previously. For cell cycle analysis, cells were fixed in
70% ice-cold ethanol and stained with a propidium iodide (PI)
solution (25 μg/ml PI, 180 U/ml RNase and 0.1% Triton
X-100). For apoptosis analysis, staining was then performed using
the Annexin V-fluorescein isothio-cyanate apoptosis detection kit
(BestBio, Shanghai, China) according to the manufacturer’s
instruction. Both cell cycle distribution and apoptosis were
analyzed by flow cytometry (FC500; Beckman-Coulter, Brea, CA, USA)
and the results are displayed as histograms.
Cell shape assay
Cells were seeded directly in 6-well culture plates
at a density of 2×105 per well 24 h and treated with 50
μM AZD2281, 5 μM RO3306, or the sequential
combination regimen (5 μM RO3306 alone for 4 h followed by
50 μM AZD2281) for 48 h, as described previously. Cells were
then stained with DAPI and mounted to an inverted microscope
(Olympus, IX71).
Western blot analysis
Lysates were prepared from 4×105 cells by
dissolving pellets in 100 μl of lysis buffer (20 mM
Na2PO4 pH=7.4, 150 mM NaCl, 1% Triton X-100,
1% aprotinin, 1 mM phenylmethylsulfonyl fluoride, 10 mg/ml
leupeptin, 100 mM NaF and 2 mM Na3VO4).
Lysates were centrifuged at 14,000 rpm for 20 min. The supernatants
were collected, and the protein content was determined using the
Bio-Rad protein assay. Protein (30 μg) was loaded in each
well of a 6–15% SDS-PAGE gel. After resolving the proteins, they
were electrophoretically transferred to a nitrocellulose membrane
and incubated sequentially with primary antibody and horseradish
peroxidase-conjugated goat anti-mouse IgG (zs-2305, Zhongshan
Golden Bridge Biotechnology, Beijing, China) or goat anti-rabbit
IgG (zs-2301, Zhongshan Golden Bridge Biotechnology). After
washing, the bound antibody complex was detected using the LumiGLO
reagent (#7003, Cell Signaling Technology, Danvers, MA, USA) and
XAR film (Kodak, XBT-1) as described by the manufacturers. The
following primary antibodies were used: ER antibody (#1115-1,
Epitomics, Burlingame, CA, USA), PR antibody (#5132-1, Epitomics,),
HER2 antibody (#2165, Cell Signaling Technology), BRCA1 antibody
(sc-642, Santa Cruz Biotechnology, Santa Cruz, CA, USA),
phospho-BRCA1 antibody (#9009, Cell Signaling Technology), CDK1
antibody (#9116, Cell Signaling Technology), caspase-3 antibody
(#9662, Cell Signaling Technology), PARP antibody (sc-7150, Santa
Cruz Biotechnology), Bcl-2 antibody (#2870, Cell Signaling
Technology), Bax antibody (#2772, Cell Signaling Technology),
caspase-9 antibody (#9502, Cell Signaling Technology), caspase-8
antibody (#9746, Cell Signaling Technology), LC3 antibody
(NB100-2220, Novus Biologicals, Littleton, CO, USA), P62 antibody
(647702, BioLegend, San Diego, CA, USA), beclin 1 antibody (#3738,
Cell Signaling Technology), phospho-AKT (ser473) antibody (#4058,
Cell Signaling Technology), AKT antibody (#2967, Cell Signaling
Technology), phospho-mTOR (ser2448) antibody (#2971, Cell Signaling
Technology), mTOR antibody (#4517, Cell Signaling Technology), heat
shock protein 70 antibody (sc-24, Santa Cruz Biotechnology) and
glycer-aldehyde 3-phosphate dehydrogenase antibody (KC-5G4,
KangChen Bio-tech, Shanghai, China).
Confocal microscopy and indirect
immunofluorescence
MDA-MB-231 cells were grown on glass coverslips and
treated with 50 μM AZD2281, 5 μM RO3306, or 5
μM RO3306 alone for 4 h followed by 50 μM AZD2281 for
24 h. Cells were subsequently fixed in 4% paraformaldehyde and
stained overnight with primary antibodies against Rad51 (sc-8349,
Santa Cruz Biotechnology) or γ-H2AX (pSer139) (#NG1904671, Upstate
Biotechnology, Lake Placid, NY, USA). Afterwards, cells were washed
with phosphate-buffered saline and incubated for 1 h at room
temperature with either Alexa 488 (Invitrogen) or Alexa 555
(Invitrogen) secondary antibodies for Rad51 or γ-H2AX,
respectively. Cells were fixed with the ProLong gold antifade
reagent with 4′,6-diamidino-2-phenylindole (Invitrogen) and cured
at room temperature for 24 h before visualizing. The coverslips
were viewed with a laser-scanning confocal microscope (Olympus,
FV-1000).
Statistical analysis
All experiments were repeated three times. The
results of multiple experiments are given as the mean ± SE.
Statistical analysis was performed using the statistical software
package SPSS 17.0. p-values were calculated using a one way ANOVA
test and a Student’s t-test. p<0.05 was considered to indicate a
statistically significant difference.
Results
Cell line subtypes
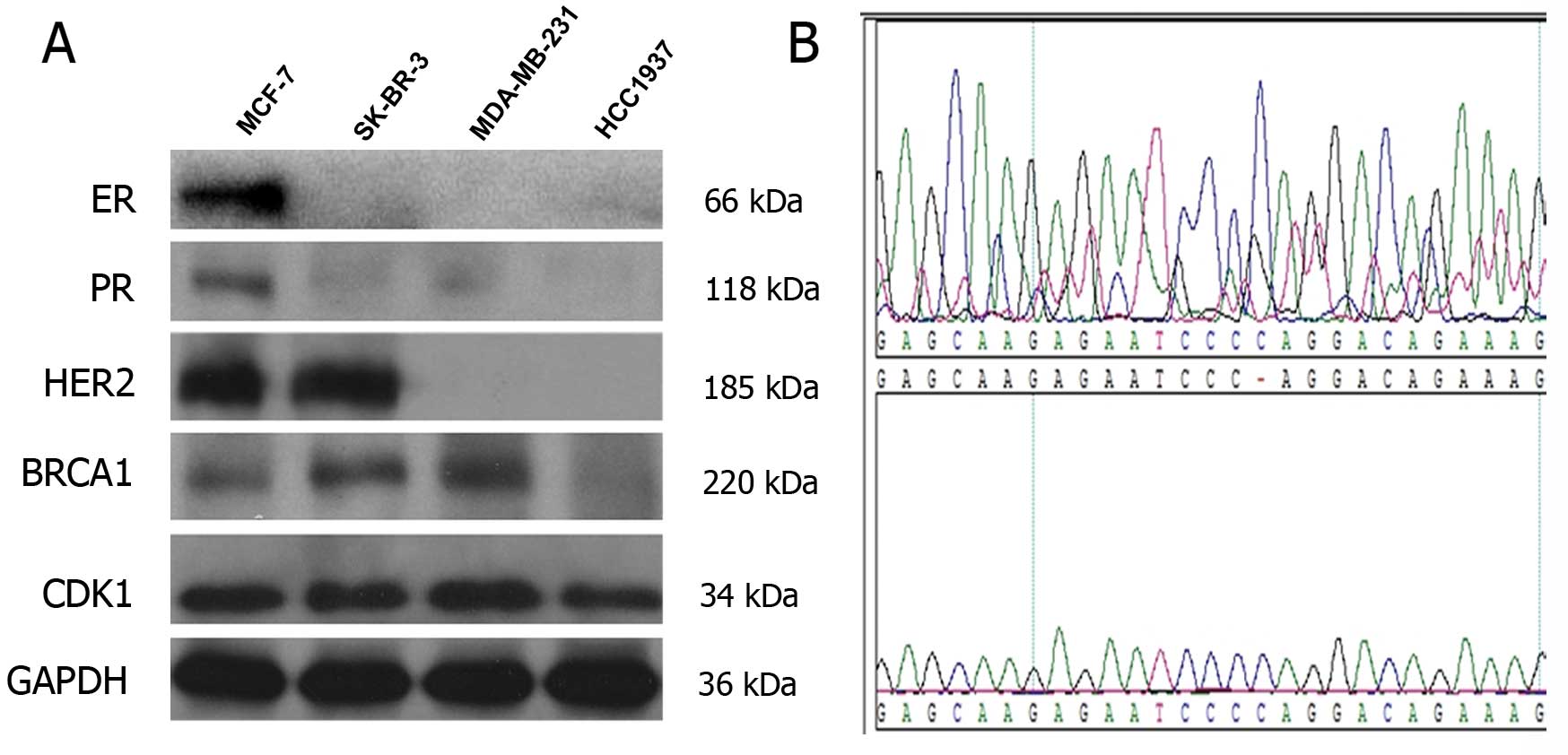
Initially, we used western blot assays to confirm
the characteristics of the four breast cancer cell lines. MCF-7 was
identified as the luminal subtype, SK-BR-3 as the HER2 subtype, and
MDA-MB-231 and HCC1937 as the triple negative subtypes. All cell
lines expressed CDK1 (Fig. 1A).
Besides, HCC1937 rather than MDA-MB-231 carried BRCA1
genetic mutations, harboring the 73–74insC exon (Fig. 1B).
Sensitivity to PARP inhibition
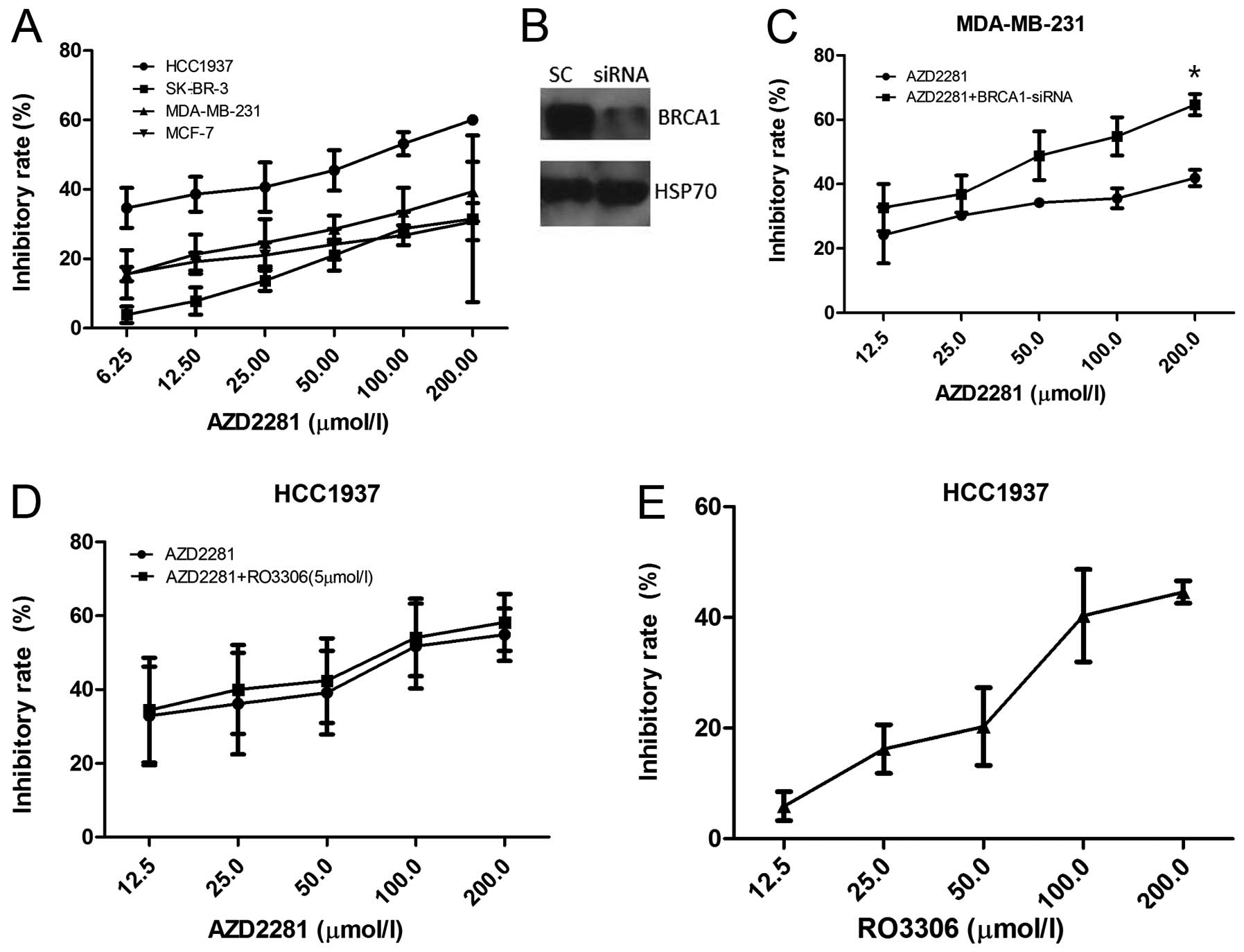
First, we examined the viability of breast cancer
cell lines by the MTT assay in the presence of the PARP inhibitor
AZD2281. The BRCA1 mutated cell line HCC1937 was the most
sensitive, with GI50 of 68.19 μM at 72 h, followed by
MDA-MB-231, MCF-7 and SK-BR-3 (Fig.
2A). After BRCA1 siRNA transfection, the effects of AZD2281 in
MDA-MB-231 were dramatically enhanced (p=0.047, Fig. 2B and C). Furthermore, HCC1937 cells
were exposed either to AZD2281 or RO3306 alone or to 5 μM
RO3306 for 4 h, followed by exposure to AZD2281 for 72 h, and the
inhibition rates were nearly the same between AZD2281 and the
combination group (p=0.547, Fig. 2D
and E), confirming that AZD2281-caused cell growth inhibition
depends on the BRCA1 function.
Sensitivity to CDK1 inhibition
In order to examine whether AZD2281 combined with
RO3306 was superior over PARP inhibition alone in the
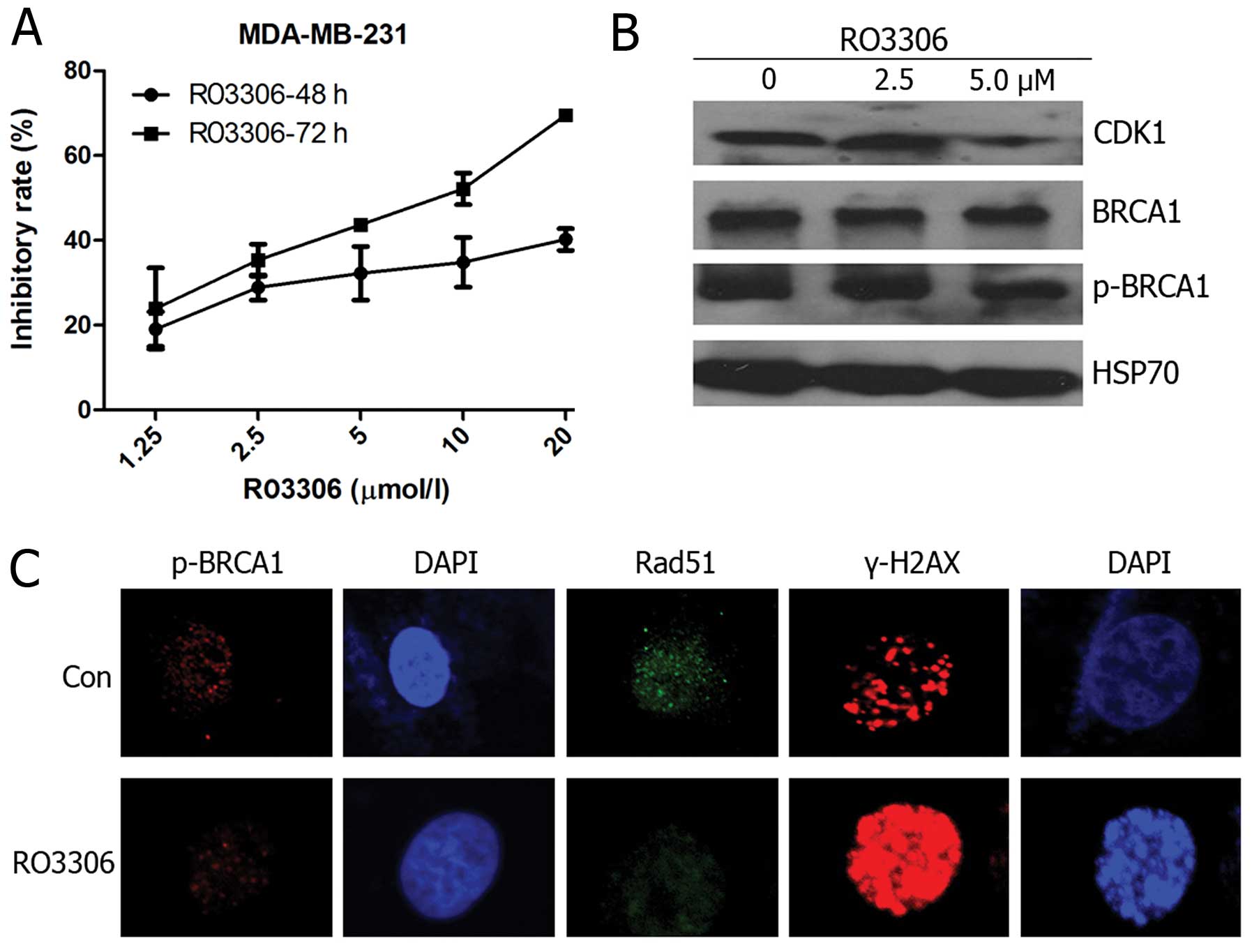
BRCA-proficient breast cancer cell line MDA-MB-231, we first
determined the effect of RO3306 on reduction in cell counts and the
GI50 value of 55.72 μM at 48 h and 7.11 μM at 72 h
(Fig. 3A). Western blot analysis
to determine the BRCA1 phosphorylation status at Ser1524 was
performed following the 48-h treatment regimen. Upon RO3306
treatment, Ser1524 phosphorylation levels decreased significantly
in a dose-dependent manner (Fig.
3B), confirming that the observed effect of RO3306 was
associated with inhibition of the targeted BRCA1 phosphorylation
rather than total BRCA1. Rad51 foci represent a crucial component
of the HR repair machinery (17)
and γ-H2AX is an early response marker for DSB DNA damage signaling
(18). We assayed p-BRCA1, Rad51,
and γ-H2AX foci formation by immunofluorescence. After a 24-h
treatment with 5 μM RO3306, decreased p-BRCA1 and Rad51 foci
levels and an increase in γ-H2AX were found (Fig. 3C).
Reduced CDK1 activity sensitizes cells to
PARP inhibition
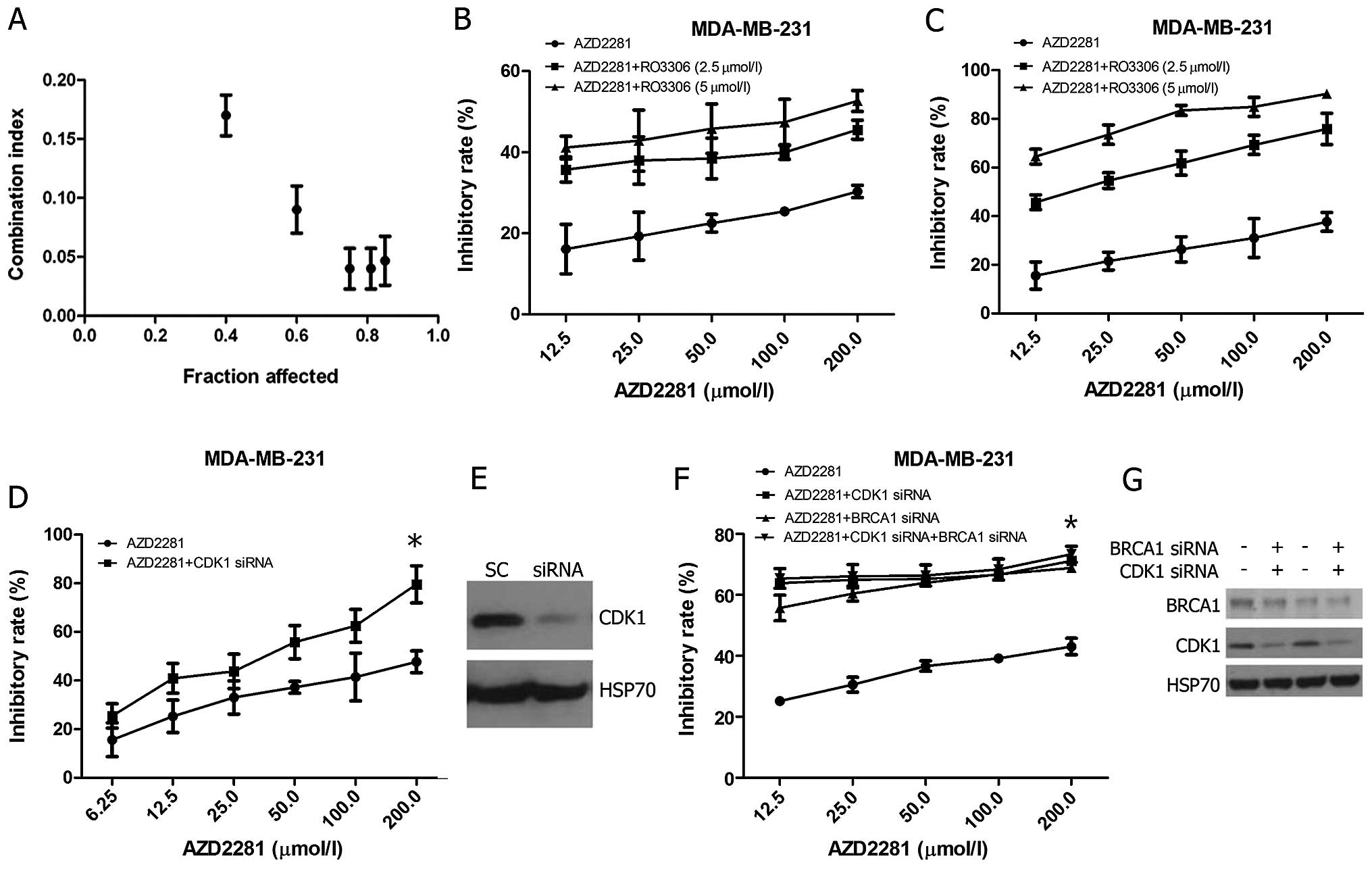
We investigated whether CDK1 inhibition could
potentiate the growth inhibitory effects of PARP inhibition. First,
the CI values of AZD2281: RO3306 = 40:1, 20:1, 10:1, 5:1, 2.5:1,
2:1, 1:1, 1:2, 1:4 and 1:8 were detected. The results showed that
the 10:1 ratio exerted the strongest synergistic effect, with the
combination index of 0.077 (Fig.
4A). Subsequently, we studied whether significant growth
inhibition could be observed upon combining sub-optimal RO3306
doses (doses ≤GI50). When used as a single agent over 72 h, 2.5 and
5 μM RO3306 concentrations reduced MDA-MB-231 cell growth by
approximately 35 and 43%, respectively (Fig. 3A); hence, these doses were
selected. Results showed that the combination reduced GI50 of
AZD2281 in a time- and dose-dependent manner, combined with 5
μM RO3306, the GI50 significantly reduced to 5.25 μM
for 72 h (Fig. 4B and C). CDK1
silencing (Fig. 4E) and sequential
treatment with AZD2281 resulted in a significant reduction of cell
growth as compared to AZD2281 alone (p=0.046, Fig. 4D), which confirmed the specificity
of the CDK1 inhibitors. In addition, if reduced CDK1 activity
sensitized cells to PARP inhibition mainly through BRCA1
abrogation, CDK1 depletion should not sensitize BRCAl-deficient
cells further. In the absence of CDK1 siRNA, BRCA1 depletion
sensitized MDA-MB-231 cells to AZD2281 to a similar degree as
depletion of CDK1. No further reduction in the inhibition of
viability was observed after AZD2281 treatment in cells that were
depleted for both BRCA1 and CDK1 (Fig.
4F and G).
Compromising CDK1 and PARP activities
induces cell cycle arrest and apoptosis
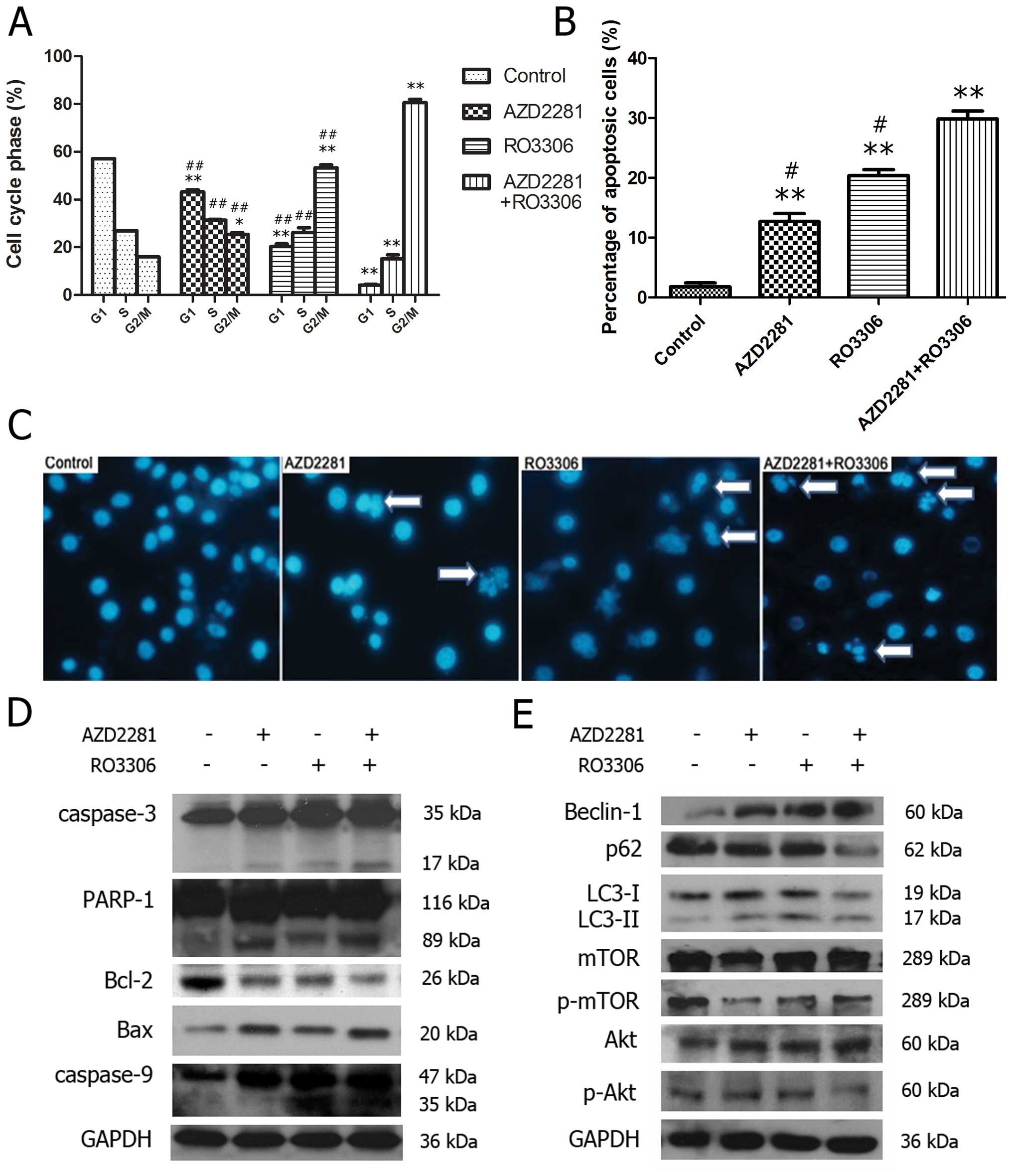
As shown in Fig.
5A, compared to the control treatment (57.10±3.55%), 50
μM AZD2281, 5 μM RO3306, and 5 μM RO3306
combined with 50 μM AZD2281 reduced the percentage of cells
in the G1 phase to 43.20±1.56%, 20.30±1.95% and 4.13±0.68%,
respectively (p<0.001). The percentage of G2/M cells was
15.93±3.68% in the control group and increased to 25.43±1.71,
53.27±2.21, and 80.63 ±2.25%, respectively (p<0.001).
Furthermore, p-values derived from t-tests comparing
AZD2281-treated cells versus cells treated with RO3306 followed by
AZD2281 showed a significant percentage of the cells exhibited cell
cycle alterations (p<0.001). These results showed that both
AZD2281 and RO3306 induced G2/M phase arrest, and the combination
exerted a stronger one. To identify whether CDK1 and PARP
inhibition induced apoptosis, treated cells were stained with
Annexin V-FITC/PI and the population of apoptotic cells was
analyzed by flow cytometry. As seen in Fig. 5B, exposure to 5 μM RO3306
followed by 50 μM AZD2281 significantly increased the
proportion of apoptotic cells. In the control group, 1.80±1.08% of
the cells were positive for Annexin V-FITC staining, while AZD2281,
RO3306 and RO3306 followed by AZD2281 resulted in 12.70±2.26,
20.40±1.71 and 29.83±2.34%, individually (p<0.001). Furthermore,
combination treatments resulted in a significant increase in
apoptosis compared to AZD2281 or RO3306 alone (p<0.05). DAPI
staining revealed that apoptotic bodies were most typically
observed in the combination group (Fig. 5C). To test whether the sequential
combination therapy involves increased caspase activation relative
to a single agent, we analyzed both the cleavage of the PARP and
caspase-3. Western blot analysis demonstrated that although
caspase-3 and PARP were modestly cleaved after AZD2281 or RO3306
treatment alone, the levels of cleaved fragments were prominent
when both drugs were applied (Fig.
5D). We further tested the expression levels of the
proapoptotic protein Bax and the antiapoptotic proteins Bcl-2,
results showed that the combination caused a more dramatic Bcl-2
downregulation and Bax upregulation than either drug used alone.
These data indicated that both AZD2281 and RO3306 induced apoptosis
and that the combination enhanced this effect. Additionally,
caspase-9 rather than caspase-8 (data not shown) was cleaved to
produce a 37 kDa fragment, indicating that the apoptosis occurred
due to a mitochondrial-dependent caspase pathway.
Compromising CDK1 and PARP activities
induces autophagy
The growth inhibition rate was about 50% for the
combined treatment at 48 h (Fig.
4B), while the apoptosis rate was 29.83+2.34% (Fig. 5B). Therefore, it was interesting to
test whether other types of cell death were involved. We found that
although cells treated with individual drugs showed a very small
accumulation of LC3II and beclin 1, cells treated with RO3306
followed by AZD2281 had a strong band indicating an increase in
both proteins, and a concomitant P62 reduction (Fig. 5E), showing that not only AZD2281,
but also RO3306 induces autophagy and that the combination
exacerbates this effect. The exposure to RO3306, followed by
AZD2281, inhibited AKT and mTOR phosphorylation, indicating that
autophagy was mainly induced by the PI3K/AKT/mTOR pathway
inhibition.
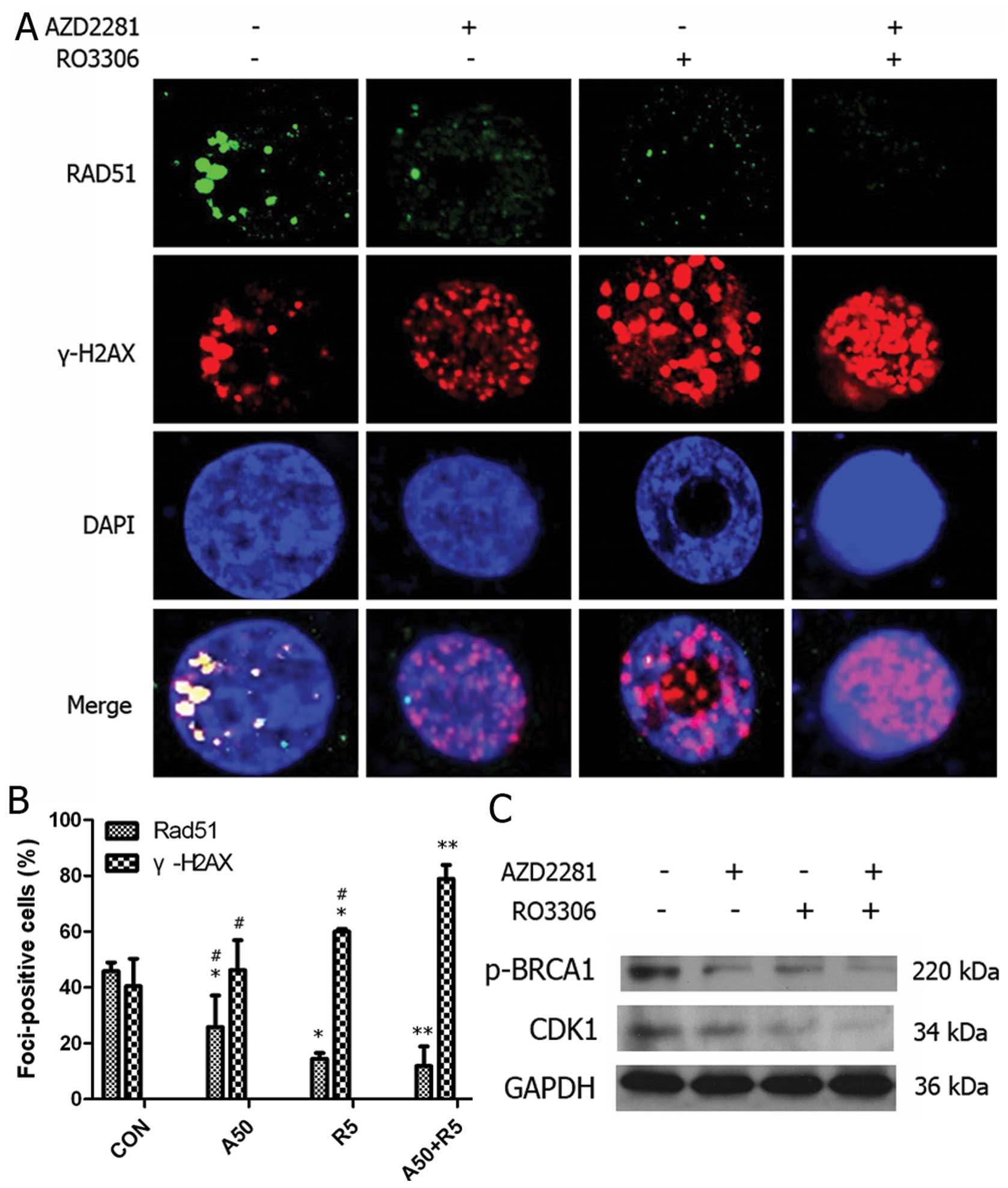
Compromising the CDK1 and PARP activities
causes BRCA1 dysfunction and DNA damage
We investigated the effect of AZD2281 and RO3306 on
DNA damage by staining for Rad51 and γ-H2AX to test whether
CDK1-inhibited cells would be sensitive to PARP inhibition
similarly to BRCA1-deficient cells. As shown in Fig. 6A and B, BRCA1-proficient MDA-MB-231
cells were unable to form Rad51 foci in response to RO3306 followed
by AZD2281 treatment, as compared to cells treated with AZD2281
alone (p=0.040) rather than RO3306 (p=0.663), confirming that the
homologous recombination defect resulted in unrepaired
recombinogenic lesions and cell death. No significant differences
existed in the number of γ-H2AX foci between control cells and
cells treated with AZD2281, suggesting that AZD2281 alone did not
induce DSB. In contrast, the number of γ-H2AX foci increased
dramatically in cells undergoing the sequential combination
treatment as compared to AZD2281 (p=0.001), a finding that was
supported by Paull et al (19), who reported that γ-H2AX foci formed
normally in response to DSBs in mammalian cells. We further
confirmed the mechanism by showing that BRCA1, p-BRCA1 and CDK1
expression decreased upon RO3306 followed by AZD2281 treatment
(Fig. 6C).
Discussion
The study of the molecular subclasses of breast
cancer suggests that treatments should be targeted more selectively
to improve outcomes. Currently, a major challenge is to identify
such targets and more effective therapeutic regimens for TNBCs.
BRCA1 is one of the highly penetrant breast cancer
susceptibility genes, and the BRCA1 protein fulfills numerous
functions, the best characterized one being related to its role in
homologous recombination during DNA repair, chromatin remodeling,
DNA decatenation, transcriptional regulation of the estrogen
receptor, cell cycle checkpoint control and ubiquitylation
(20,21). BRCA1 mutations increase the
breast cancer risk (22).
PARPs are members of a large family of
multifunctional enzymes that play a key role in DNA single-strand
break repair through BER (23). In
the current study, we established that a BRCAl-deficient rather
than a BRCAl-proficient cell line was sensitive to AZD2281.
Mechanisms to explain this observation include defects in DNA
repair pathways involved in HR. BRCA1 germ-line mutation
carriers commonly develop DNA repair defects and, therefore, their
cells are sensitive to PARP inhibitors (6). However, BRCAl-deficient breast tumors
are rare (24), which might
restrict the application of PARP inhibitor monotherapy. Because
PARP plays a major role in the BER, we hypothesized that the
inhibition of another target, which is indispensable in HR, may act
syner-gistically with PARP inhibitors.
CDK1, a protein that is essential for multiple steps
in yeast HR (25), acts as a core
component of the cell cycle machinery and forms complexes with
cyclins to promote cell cycle progression (12). Genetic ablation of all interphase
CDKs (CDK2, CDK4 and CDK6) does not result in cell cycle defects in
most cell types, while CDK1 deletion causes cell cycle arrest and
prevents embryos from developing beyond the two-cell stage
(26). Johnson et al
(16) reported that CDK1 is
essential for cell division and CDK1 inhibition in lung cancer
cells reduces the formation of BRCA1 foci and sensitizes cancer
cells to DNA damaging treatments. It is likely that the reduced
CDK1 activity may also sensitize cells to PARP inhibition by
disrupting BRCA1 function in other cancer cells.
We observed that when PARP was inhibited alone, the
GI50 of AZD2281 was over 100 μM in MDA-MB-231 cells, a
finding supported by Lehmann et al (27). However, PARP and CDK1 inhibition
together reduced the GI50 dramatically. To our knowledge, studies
on the molecular mechanisms of PARP inhibition are limited
(28), because most studies
focused on characterizing the DNA damage response defects and on
measuring PARP activity (29,30).
No published reports have shown that PARP inhibition leads to cell
death through apoptosis in BRCA-proficient breast cancer cells. To
gain more insight into the exact molecular mechanisms involved in
decreasing viability following treatment, we investigated several
representative apoptosis markers and found that PARP inhibition
causes apoptosis. The G2/M phase was associated with DNA synthesis
and the mitotic preparation period, which plays a crucial role in
cell cycle progression and the accumulation of cells in the G2/M
phase results in cell death. Consistent with Inbar-Rozensal et
al (31), who showed that PARP
inhibition promotes cell cycle arrest at the G2/M phase in breast
cancer cell lines lacking BRCA1 mutations (MCF-7 and
MDA-MB-231), we observed that AZD2281 caused G2/M phase arrest.
This can be explained by an inhibitory effect that PARP exerts on
kinase (ERK)-dependent kinase cascades regulated by extracellular
signals, and the resulting decrease in the proportion of putative
cancer stem cells (27) and the
inhibition of signal transduction pathways involving cell cycle
proteins (cyclins, p21, CDK1). On the other hand, by using the CDK1
inhibitor RO3306, we also showed that G2/M phase arrest and
apoptosis occur in MDA-MB-231 cells, the finding was supported by
Payton et al (32) who
found that CDK1 expression was required for osteosarcoma and breast
tumor cell proliferation and CDK1 suppression decreased the S phase
while markedly increasing the G2/M phase, thus tumor cells treated
with CDK1 inhibitors showed an overall decrease in cell
proliferation and apoptosis.
We then investigated the underlying mechanisms by
which CDK1 inhibition sensitized cells to PARP inhibition. First,
MTT showed that RO3306 followed by AZD2281 treatment inhibited the
growth of MDA-MB-231 cells in a time- and dose-dependent manner,
with the combination effect synergistic. We subsequently treated
MDA-MB-231 cells with the two compounds in sequential combination
for flow cytometry and examined not only the dysregulation of the
G2/M phase arrest but also the increased proportion of apoptotic
cells. The mitochondria-initiated cell death pathway plays an
important role in triggering apoptosis in response to stimuli
(33) and our data confirmed this.
Autophagy is a lysosomal degradation pathway that is essential for
survival, development and homeostasis (34). It was previously reported that
autophagic cell death has biochemical and morphological features
distinguishing it from apoptosis and that some cancer cells could
undergo autophagy following cancer therapy (35). We explored both RO3306- and
AZD2281-induced autophagy, which were consistent with the prior
studies, although the exact mechanism requires further
investigation. Furthermore, AZD2281 combined with RO3306 caused
dramatic DNA damage refracted by Rad51 and γ-H2AX. The experiments
above taken together suggested that the cytotoxic effects of the
combined inhibition were achieved through the accumulation of DNA
DSBs, thereby blocking cells in G2/M and leading to cell death.
In summary, we have used targeted kinase inhibition
to inactivate BRCA1, impair the homologous recombination DNA
repair, and selectively improve the BRCA-proficient breast cancer
cell line MDA-MB-231 to PARP inhibition. Analysis of CDK1-mediated
phosphorylation of BRCA1 suggests that a reduction of the CDK1
activity by small molecule inhibitors improved the response to PARP
inhibition in vitro, and serves as a guide to translate this
to substantial antitumor activity in vivo. The data add
substantially to our understanding of the roles that CDK1 and PARP
play, and support the clinical development of the combined
inhibition. This approach avoids the use of DNA-damaging
chemo-therapeutic drugs with cellular toxicity, thereby providing
the potential to extend well-tolerated PARP inhibition to treating
BRCA-proficient breast cancers in the near future.
Abbreviations:
|
TNBC
|
triple-negative breast cancer;
|
|
DSB
|
double-strand break;
|
|
HR
|
homologous recombination;
|
|
NER
|
nucleotide excision repair;
|
|
BER
|
base excision repair;
|
|
PARP
|
poly (ADP-ribose) polymerase;
|
|
CDK1
|
cyclin-dependent kinase 1;
|
|
GI50
|
50% growth inhibition;
|
|
CI
|
combination index;
|
|
PI
|
propidium iodide
|
Acknowledgements
This study was supported by the
National-Eleventh Five technology major project (grant no.
2008ZX09312-002) and the Research Award Fund for Outstanding Young
researchers at the Sun Yat-sen Cancer Center.
References
|
1.
|
Gucalp A and Traina TA: Triple-negative
breast cancer: adjuvant therapeutic options. Chemother Res Pract.
2011:6962082011.PubMed/NCBI
|
|
2.
|
Moynahan ME, Chiu JW, Koller BH and Jasin
M: Brca1 controls homology-directed DNA repair. Mol Cell.
4:511–518. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Zhang J and Powell SN: The role of the
BRCA1 tumor suppressor in DNA double-strand break repair. Mol
Cancer Res. 3:531–539. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Hartman AR and Ford JM: BRCA1 induces DNA
damage recognition factors and enhances nucleotide excision repair.
Nat Genet. 32:180–184. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Alli E, Sharma VB, Sunderesakumar P and
Ford JM: Defective repair of oxidative DNA damage in
triple-negative breast cancer confers sensitivity to inhibition of
poly(ADP-ribose) polymerase. Cancer Res. 69:3589–3596. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
McCabe N, Turner NC, Lord CJ, Kluzek K,
Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka
MZ, Smith GC and Ashworth A: Deficiency in the repair of DNA damage
by homologous recombination and sensitivity to poly(ADP-ribose)
polymerase inhibition. Cancer Res. 66:8109–8115. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Ashworth A: A synthetic lethal therapeutic
approach: poly(ADP) ribose polymerase inhibitors for the treatment
of cancers deficient in DNA double-strand break repair. J Clin
Oncol. 26:3785–3790. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Tutt A, Robson M, Garber JE, Domchek SM,
Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler
RK, Wardley A, Mitchell G, Earl H, Wickens M and Carmichael J: Oral
poly(ADP-ribose) polymerase inhibitor olaparib in patients with
BRCA1 or BRCA2 mutations and advanced breast cancer: a
proof-of-concept trial. Lancet. 376:235–244. 2010. View Article : Google Scholar
|
|
9.
|
Fong PC, Boss DS, Yap TA, Tutt A, Wu P,
Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ,
Ashworth A, Carmichael J, Kaye SB, Schellens JH and de Bono JS:
Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA
mutation carriers. N Engl J Med. 361:123–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Wooster R and Weber BL: Breast and ovarian
cancer. N Engl J Med. 348:2339–2347. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: a changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Morgan DO: Principles of CDK regulation.
Nature. 374:131–134. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Vassilev LT, Tovar C, Chen S, Knezevic D,
Zhao X, Sun H, Heimbrook DC and Chen L: Selective small-molecule
inhibitor reveals critical mitotic functions of human CDK1. Proc
Natl Acad Sci USA. 103:10660–10665. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Goga A, Yang D, Tward AD, Morgan DO and
Bishop JM: Inhibition of CDK1 as a potential therapy for tumors
over-expressing MYC. Nat Med. 13:820–827. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Kojima K, Shimanuki M, Shikami M, Andreeff
M and Nakakuma H: Cyclin-dependent kinase 1 inhibitor RO-3306
enhances p53-mediated Bax activation and mitochondrial apoptosis in
AML. Cancer Sci. 100:1128–1136. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Johnson N, Cai D, Kennedy RD, Pathania S,
Arora M, Li YC, D’Andrea AD, Parvin JD and Shapiro GI: Cdkl
participates in BRCA1-dependent S phase checkpoint control in
response to DNA damage. Mol Cell. 35:327–339. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Johnson N, Li YC, Walton ZE, Cheng KA, Li
D, Rodig SJ, Moreau LA, Unitt C, Bronson RT, Thomas HD, Newell DR,
D’Andrea AD, Curtin NJ, Wong KK and Shapiro GI: Compromised CDK1
activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat
Med. 17:875–882. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Gartner EM, Burger AM and Lorusso PM:
Poly(adp-ribose) polymerase inhibitors: a novel drug class with a
promising future. Cancer J. 16:83–90. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Paull TT, Rogakou EP, Yamazaki V,
Kirchgessner CU, Gellert M and Bonner WM: A critical role for
histone H2AX in recruitment of repair factors to nuclear foci after
DNA damage. Curr Biol. 10:886–895. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Turner NC and Reis-Filho JS: Basal-like
breast cancer and the BRCA1 phenotype. Oncogene. 25:5846–5853.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Gudmundsdottir K and Ashworth A: The roles
of BRCA1 and BRCA2 and associated proteins in the maintenance of
genomic stability. Oncogene. 25:5864–5874. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Miki Y, Swensen J, Shattuck-Eidens D,
Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM
and Ding W: A strong candidate for the breast and ovarian cancer
susceptibility gene BRCA1. Science. 266:66–71. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Amé JC, Spenlehauer C and de Murcia G: The
PARP super-family. Bioessays. 26:882–893. 2004.
|
|
24.
|
Turner N, Tutt A and Ashworth A: Hallmarks
of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 4:814–819.
2004.
|
|
25.
|
Ira G, Pellicioli A, Balijja A, Wang X,
Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth
NM, Haber JE and Foiani M: DNA end resection, homologous
recombination and DNA damage checkpoint activation require CDK1.
Nature. 431:1011–1017. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Santamaría D, Barrière C, Cerqueira A,
Hunt S, Tardy C, Newton K, Cáceres JF, Dubus P, Malumbres M and
Barbacid M: Cdk1 is sufficient to drive the mammalian cell cycle.
Nature. 448:811–815. 2007.PubMed/NCBI
|
|
27.
|
Lehmann BD, Bauer JA, Chen X, Sanders ME,
Chakravarthy AB, Shyr Y and Pietenpol JA: Identification of human
triple-negative breast cancer subtypes and preclinical models for
selection of targeted therapies. J Clin Invest. 121:2750–2767.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Kimbung S, Biskup E, Johansson I, Aaltonen
K, Ottosson-Wadlund A, Gruvberger-Saal S, Cunliffe H, Fadeel B,
Loman N, Berglund P and Hedenfalk I: Co-targeting of the PI3K
pathway improves the response of BRCA1 deficient breast cancer
cells to PARP1 inhibition. Cancer Lett. 319:232–241. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Drew Y, Mulligan EA, Vong WT, Thomas HD,
Kahn S, Kyle S, Mukhopadhyay A, Los G, Hostomsky Z, Plummer ER,
Edmondson RJ and Curtin NJ: Therapeutic potential of
poly(ADP-ribose) polymerase inhibito AG014699 in human cancers with
mutated or methylated BRCA1 or BRCA2. J Natl Cancer Inst.
103:334–346. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Mendes-Pereira AM, Martin SA, Brough R,
McCarthy A, Taylor JR, Kim JS, Waldman T, Lord CJ and Ashworth A:
Synthetic lethal targeting of PTEN mutant cells with PARP
inhibitors. EMBO Mol Med. 1:315–322. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Inbar-Rozensal D, Castiel A, Visochek L,
Castel D, Dantzer F, Izraeli S and Cohen-Armon M: A selective
eradication of human nonhereditary breast cancer cells by
phenanthridine-derived polyADP-ribose polymerase inhibitors. Breast
Cancer Res. 11:R782009. View Article : Google Scholar
|
|
32.
|
Payton M, Chung G, Yakowec P, Wong A,
Powers D, Xiong L, Zhang N, Leal J, Bush TL, Santora V, Askew B,
Tasker A, Radinsky R, Kendall R and Coats S: Discovery and
evaluation of dual CDK1 and CDK2 inhibitors. Cancer Res.
66:4299–4308. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Gao LL, Li FR, Jiao P, Yang MF, Zhou XJ,
Si YH, Jiang WJ and Zheng TT: Paris chinensis dioscin induces G2/M
cell cycle arrest and apoptosis in human gastric cancer SGC-7901
cells. World J Gastroenterol. 17:4389–4395. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Levine B and Klionsky DJ: Development by
self-digestion: molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Cuervo AM: Autophagy: in sickness and in
health. Trends Cell Biol. 14:70–77. 2004. View Article : Google Scholar : PubMed/NCBI
|