Introduction
Prostate cancer is the most frequently diagnosed
non-cutaneous male malignancy and the third leading cause of
cancer-related death in men in most western industrialized
countries (1). Since it is
estimated that around 660,000 men worldwide will be diagnosed with
prostate cancer it will remain a major health problem in coming
years (1). Despite an initial
efficacy of androgen deprivation therapy, most patients with
prostate cancer progress within 2 years from androgen-dependent
status to hormone-refractory prostate cancer, for which there is no
curative therapy. Androgen receptor (AR) signaling plays a key role
in the development of hormone-refractory prostate cancer. AR, a
member of the steroid receptor superfamily, is a ligand-dependent
transcription factor that mediates androgen action in cells. AR is
composed of three major domains: an NH2-terminal transcriptional
activation domain (NTD), a central DNA-binding domain, and a
COOH-terminal ligand-binding domain (LBD) (2,3). AR
is associated with cellular chaperones in the cytosol in its
inactive state (4). After binding
to androgens, such as testosterone and, more potently,
dihydrotestosterone (DHT), AR translocates to the nucleus, binds to
the androgen response elements of AR target gene promoter, and
regulates expression of AR target genes (2,5). AR
hypersensitivity, as a result of AR gene mutation and/or
amplification, overexpression of coactivators, and AR cross-talking
with other signal transduction pathways, often occurs and plays
crucial roles in prostate cancer development, progression, and
androgen-independent growth. In other words, AR and its signaling
axis are the most important targets for therapies against advanced
prostate cancer (5–7). Therefore, finding novel and more
effective inhibitors of AR signaling is of great interest.
Shikonin is an active naphthoquinone compound and
the main component of red pigment extracts from the Chinese
medicinal herb, Lithospermum erythrorhizon Sieb et Zucc.
Shikonin and its analogues can kill cancer cells through a number
of mechanisms, including inhibition of topoisom-erase-I (8), polo-like kinase 1 (PLK1) and protein
tyrosine kinase (PTK) (9);
regulation of phosphorylation-dependent activities of
extracellular-regulated protein kinase (pERK), c-Jun N-terminal
kinase (JNK), and protein kinase Cα PKCα (10); suppression of tumor necrosis factor
receptor-associated protein 1 (TRAP1) expression (11); activation of caspases (12); and inhibition of proteasome
activity (13). In previous
studies, shikonin and its derivatives were shown to exert
anti-proliferative and pro-apoptotic effects against some tumor
cells, including sarcoma 180 (S-180) ascites cells, gastric cancer,
colon adenocarcinoma, and oral cancer (14). A recent report has also shown that
shikonin activates p53 and caspase-9 pathways (15) in human malignant melanoma A375-S2
cells.
In this study, we hypothesized that shikonin may
have a role as an inhibitor of AR signaling and, thus, could serve
as a therapeutic agent for the management of human prostate
cancers. We report strong anti-AR activity of the natural product
shikonin in prostate cancer cells. Shikonin markedly decreased
expression not only of AR, but also PSA, a widely used serologic
marker for prostate cancer burdens and an indicator of therapeutic
efficacy and recurrence (16), and
inhibited growth of AR-positive human prostate cancer cells. We
propose that shikonin is a good candidate for use as an anticancer
drug in chemoprevention and treatment of hormonal-responsive
prostate tumor cells.
Materials and methods
Reagents
RPMI-1640 was purchased from Gibco Technologies,
Inc. (Gaithersburg, MD, USA). Fetal bovine serum (FBS) was obtained
from Hyclone (Logan, UT, USA). Shikonin was purchased from
Sigma-Aldrich (St. Louis, MO, USA). Antibodies for AR, β-actin,
PSA, PCNA, Bcl-2, PARP, and anti-goat peroxidase conjugated
secondary antibody were obtained from Santa Cruz Biotechnology
(Santa Cruz, CA, USA). Anti-mouse and anti-rabbit peroxidase
conjugated secondary antibodies were purchased from Pierce
(Madison, WI, USA). The dual-luciferase reporter assay kit was from
Promega (Madison, WI, USA).
Cell culture
LNCaP and 22RV1 prostate cancer cells were
maintained in RPMI-1640 supplemented with 10% FBS and 1%
penicillin/streptomycin antibiotics.
Immunoblotting
Following reagent treatments, cells were washed
twice with 1X PBS and cell extracts were prepared using RIPA buffer
(1X PBS, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS containing an
additional 100 μl of 10 mg/ml PMSF, and 1 tablet of the
complete mini protease inhibitors). Protein content was quantified
by the Lowry assay. Lysate proteins were resolved by SDS-PAGE and
transferred onto a nitrocellulose membrane. The membrane was
incubated with TBS buffer containing 0.1% Tween-20 and 5% skim
milk, and then exposed to the desired primary antibody. After
treatment with a proper secondary antibody, the immunoreactive
bands were visualized by standard ECL method.
Cell fractionation
Nuclear and cytoplasmic fractions were prepared
using nuclear and cytoplasmic extraction reagents kit (Fermentas,
St. Leon-Rot, Germany). Cell lysates were spun down for 7 min at
500 × g, and the supernatant was designated the cytoplasmic
fraction. The nuclear pellet was washed three times in wash buffer
and then treated with nuclear lysis buffer for 15 min. The sample
was spun at 20,000 × g for 15 min, and the supernatant designated
the nuclear fraction. PCNA and β-actin were used as markers for
nuclear and cytoplasmic proteins.
Real-time PCR and quantitative real-time
PCR (qRT-PCR)
Total RNA isolation was performed with the use of
the TRIzol (Invitrogen) according to the manufacturer’s protocol.
The cDNA was synthesized with the use of 2 mg of total RNA through
SuperScript reverse transcriptase (Bioneer, Daejeon, Korea) with
oligo dT primers. PCR was done with a specific primer (AR sense
5′-ATGGTGAGCAGAGTGCCCTA-3′; antisense 5′-GTGGTGCTGGAAGCCTCTCCT-3′;
GAPDH sense, 5′-GGCCTCCAAGGAGGAAGACC-3′; and GAPDH antisense,
5′-AGGGGTCTACATGGCAACTG-3′). GAPDH, a non-regulated housekeeping
gene was used as an internal control to normalize input cDNA.
RT-PCR was performed using the LightCycler 480 using SYBR green
master mix (Roche). Each experiment was performed in three
experimental replicates with three technical replicates within each
experiment.
Luciferase reporter assay
LNCaP and 22RV1 cells were transiently
co-transfected with 0.5 μg ARE-, PSA-luciferase plasmid and
0.5 mg pSV-β-galactosidase reporter vector using Lipofectamine 2000
(Invitrogen) transfection reagent. After transfection for 24 h,
cells were treated with shikonin for the described period. Cell
extracts were prepared for the luciferase assays. The luciferase
activity was normalized by β-galactosidase activity.
Cell proliferation and viability
assays
All proliferation assays were based on the MTT
method. Cells were seeded in a 96-well plate, 1×104
cells per well. After 24–72 h, cells were treated with shikonin. At
the end of the experiment, the media was removed and DMSO was added
as MTT solubilization solution. Absorbance was measured at 550
nm.
Results
Shikonin decreases the AR protein level
in LNCaP and 22RV1 cells
In androgen-responsive prostate cancer cells, AR is
required for the initiation of androgen-dependent gene
transcription. We examined the effect of shikonin on the expression
of AR in the androgen-responsive human prostate cancer cell lines
LNCaP and 22RV1. Porstate cancer cells were treated with shikonin
at various concentrations and subjected to western blotting.
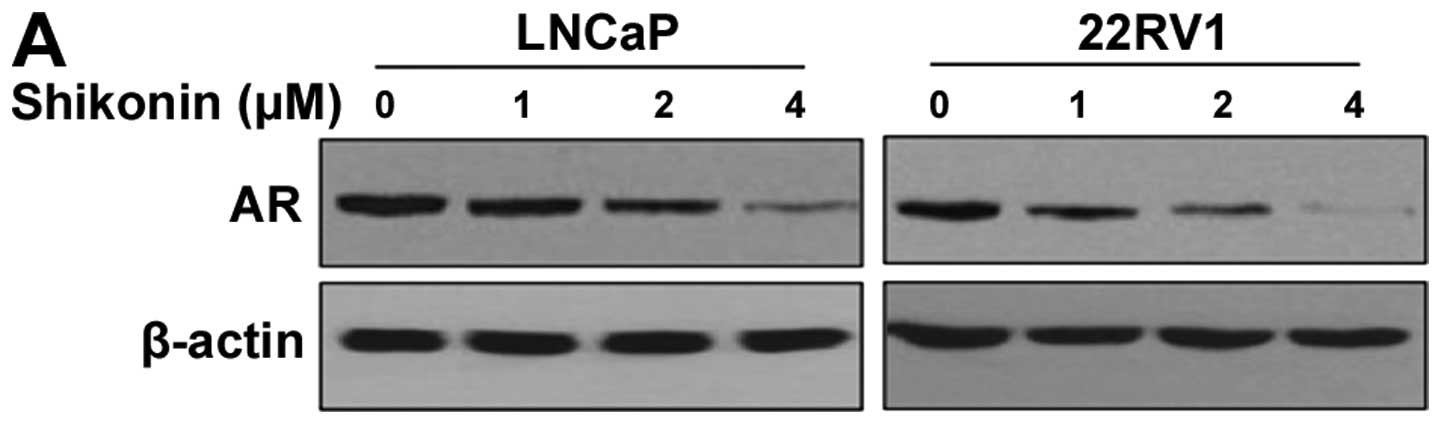
Shikonin decreased the protein level of AR in a dose-dependent
manner in both cell lines (Fig.
1A). The AR protein level started to decrease with 2 μM
shikonin and almost disappeared with 4 μM shikonin (Fig. 1A). We also examined the
time-dependent effect of shikonin on AR. The AR protein level
decreased in a time-dependent manner in prostate cancer cells
treated with 4 μM shikonin (Fig. 1B). Next, we examined whether
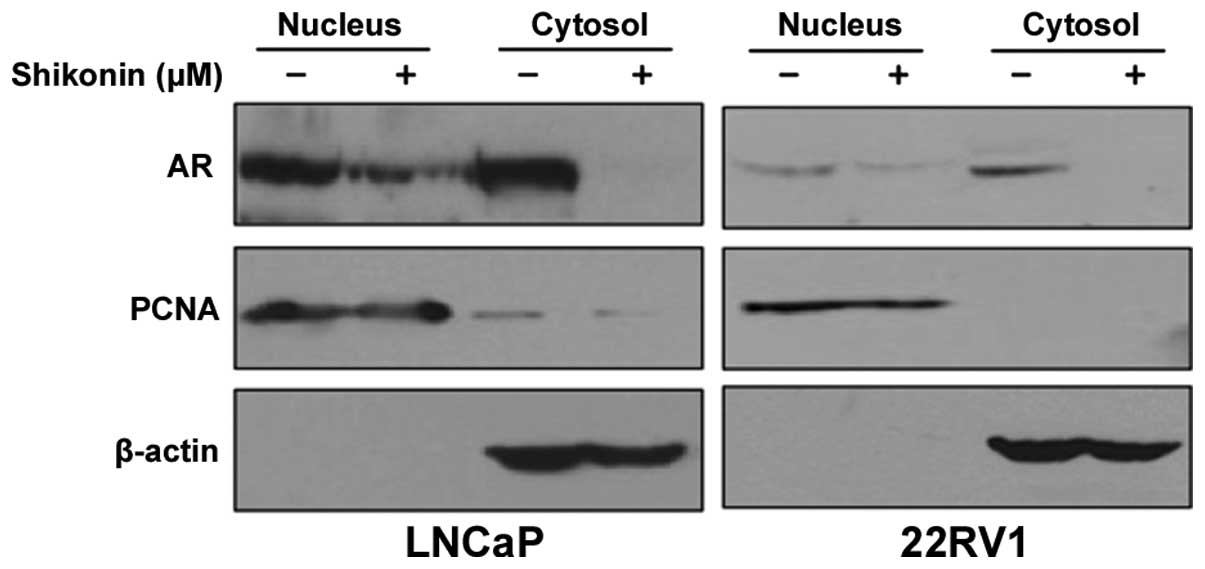
shikonin affects translocation of AR to the nucleus, because
nuclear localization of AR is important for its transcriptional
activity (17). LNCaP and 22RV1
cells were treated with 4 μM shikonin and fractionated into
cytoplasmic and nuclear fractions. Shikonin decreased the AR
protein levels in both the nucleus and cytoplasm (Fig. 2). These results indicated that
shikonin decreases the AR protein level in a dose-, time-dependent
manner and also, effectively block nuclear localization of AR in
LNCaP and 22RV1 prostate cancer cells.
Shikonin suppresses the mRNA level of AR
in LNCaP and 22RV1 cells
Since shikonin decreased the AR protein expression,
we examined the effect of shikonin to mRNA level of AR in LNCaP and
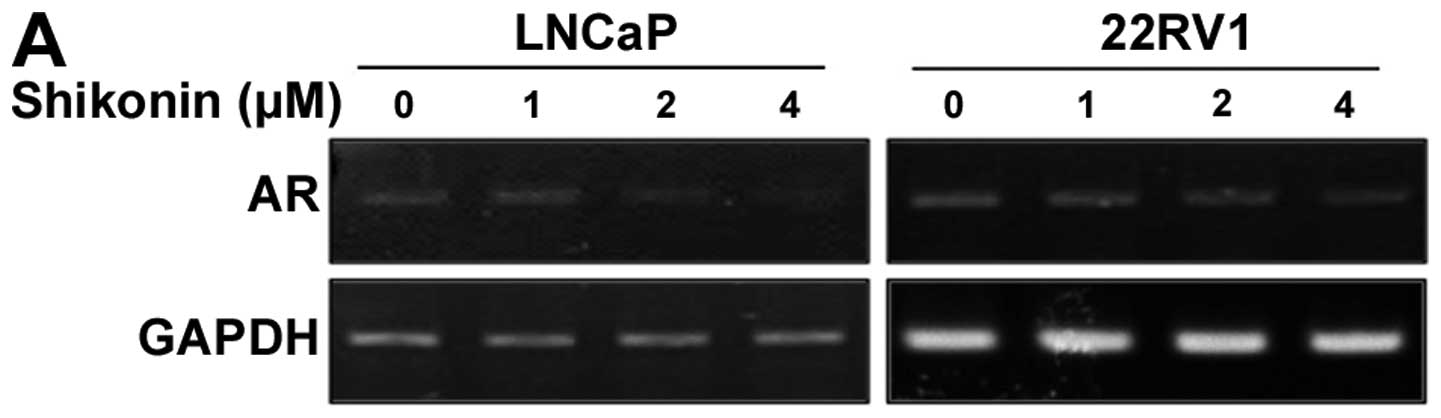
22RV1 cells. The mRNA level of AR was determined using
semi-quantitative RT-PCR. Cells treated with shikonin at various
concentrations for 6 h exhibited a marked and
concentration-dependent decrease in AR mRNA levels (Fig. 3A). The mRNA level of AR in cells
treated with 4 μM shikonin for 6 h was decreased to 10% of
the level compared to without shikonin treatment (Fig. 3A). Shikonin also decreased the AR
mRNA level in a time-dependent manner in LNCaP and 22RV1 cells
(Fig. 3B). As shown in Fig. 3C, shikonin decreased the AR mRNA
expression dose-and time-dependently. These results indicate that
shikonin decreases the expression of AR at the transcriptional
level.
Shikonin decreased the transcriptional
activity of AR
AR is an important transcription factor that
regulates expression of a variety of target genes that harbor the
androgen response element (ARE) in their promoters (17,18).
Therefore, we examined the effect of shikonin on the
transcriptional activity of AR. LNCaP and 22RV1 cells were
transfected with an ARE-containing promoter reporter gene. AR
transactivation was determined using a luciferase assay in cells
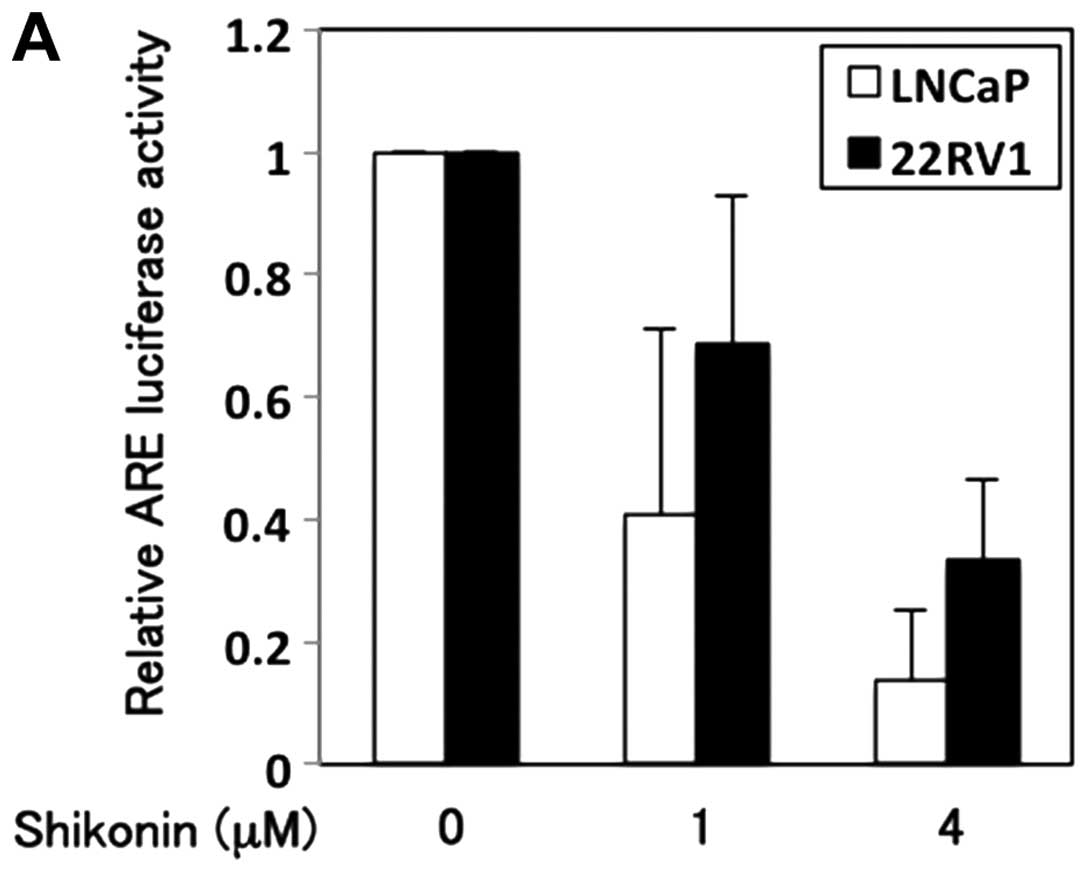
treated with or without shikonin. Shikonin decreased the luciferase
activity in a dose-dependent manner (Fig. 4A). Cells treated with 4 μM
shikonin showed ∼5-fold and 2.5-fold decrease in the AR
transcriptional activity in LNCaP and 22RV1 cells, respectively. To
further determine the inhibitory effects of shikonin on PSA gene
transcription, the PSA promoter-driven luciferase reporter
activity, was assessed in prostate cancer cells transfected with
pGL3-PSA-luc. PSA promoter activity was reduced when the cells were
treated with shikonin (Fig. 4B).
These results indicated that the shikonin mediated decrease in AR
protein level correlated with repression of its promoter
activity.
Inhibition of AR transcriptional activity
by shikonin leads to suppression of AR-induced gene expression
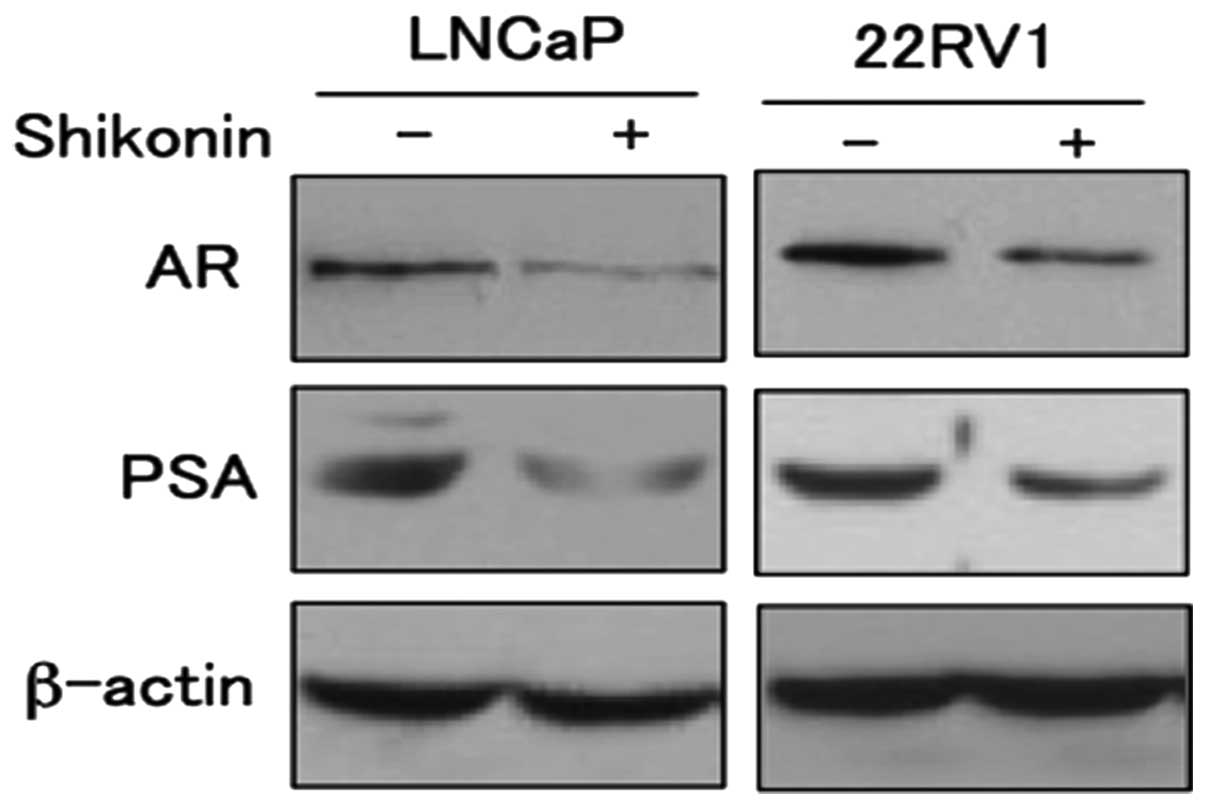
To determine the effect of decreased AR
transcriptional activity by shikonin, we examined the expressions
of the AR target gene PSA (Fig.
5). LNCaP and 22RV1 cells were treated with shikonin and
subjected to western blotting. The protein expression levels of PSA
decreased in response to shikonin in a dose-dependent manner (data
not shown). The expression of prostate specific antigen (PSA) has
been used extensively as a marker of prostate cancer growth and is
a well-known target gene of AR. These results indicate that
shikonin decrease the transcriptional activity of AR leading to
suppression of the target protein PSA expression.
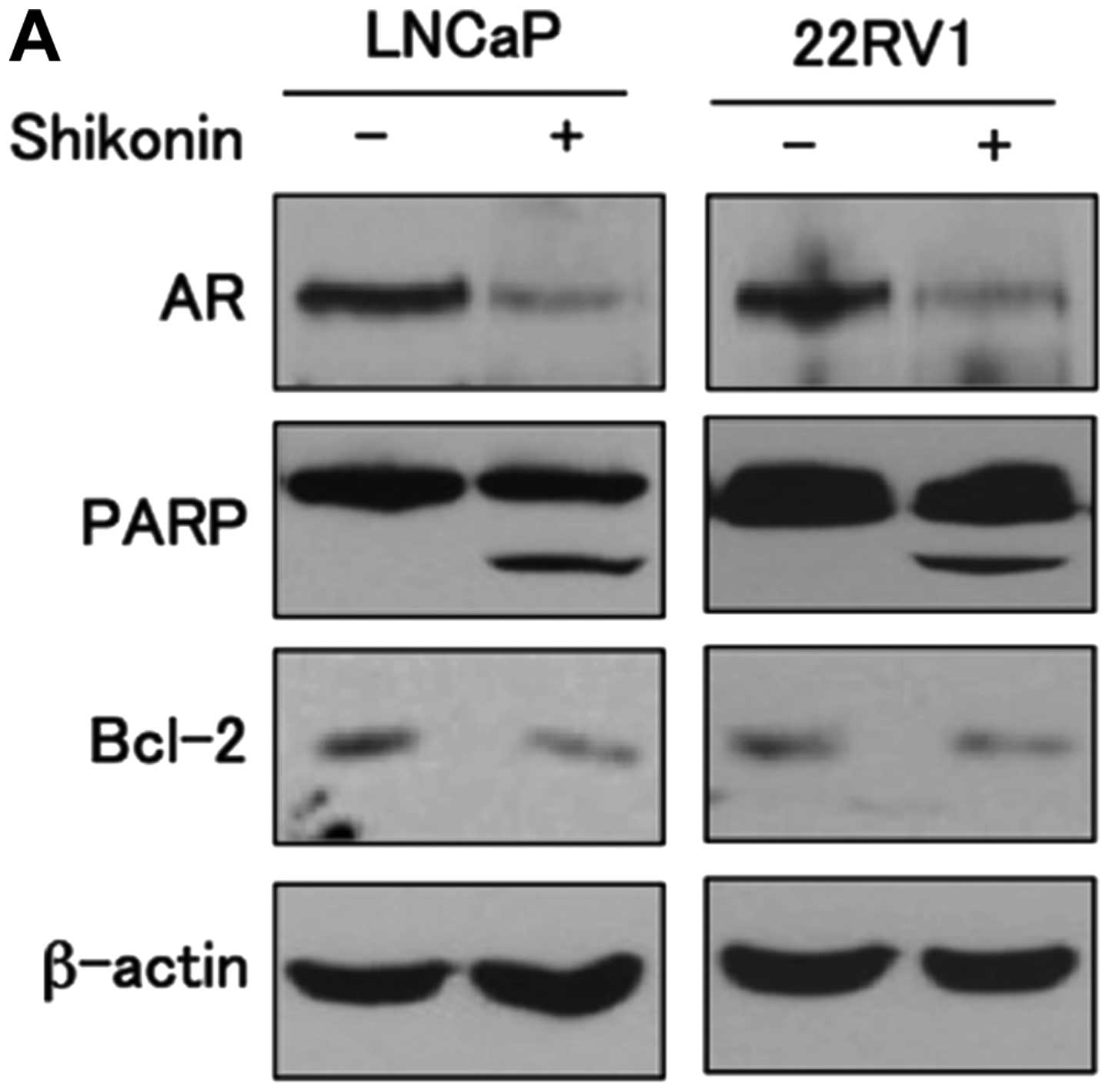
Shikonin inhibits the growth of LNCaP and
22RV1 cells
AR is an important regulator of cellular growth in
androgen-dependent prostate cancer cells. Shikonin suppresses AR
expression at the transcriptional level and subsequently inhibits
the transcriptional activity of AR, resulting in inhibition of cell
proliferation in androgen-positive prostate cancer cells.
Therefore, we next examined whether shikonin affects the growth of
prostate cancer cells. LNCaP and 22RV1 cells were treated with or
without 4 μM shikonin for 6 h and the levels of proteins
involved in apoptosis were examined. Cells treated with shikonin
increased cleavage of poly(ADP-ribose) polymerase (PARP) cleavage
and decreased expression of the anti-apoptotic molecule Bcl-2
compared to cells without shikonin in both cell lines (Fig. 6A). We next examined the effect of
shikonin on the growth of LNCaP and 22RV1 cells. Cell proliferation
assays were determined using MTT analysis. As shown in Fig. 6B, the viability of prostate cancer
cells treated with 4 μM shikonin for 24 h was reduced in a
time-dependent manner to ∼50% of the viability of control cells.
These data are consistent with the induction of apoptosis by the
shikonin as indicated by the PARP cleavage in both LNCaP and 22RV1
cells treated with shikonin. Taken together, these results indicate
that shikonin led to growth inhibition of AR-positive prostate
cancer cells thereby suppressing the transcriptional and
translational level of AR.
Discussion
Prostate cancer is a leading cause of cancer-related
deaths among men in the United States (19). The mechanism underlying the
pathogenesis of prostate cancer is not fully understood, but age,
race, dietary habits, and androgen secretion and metabolism are
some of the risk factors associated with this malignancy (20). AR, a ligand-activated transcription
factor belonging to the steroid receptor super-family, is
critically involved in prostate cancer progression as well as
maintenance of the male reproductive organ (17). Ligand-free AR predominantly resides
in the cytoplasm complexed with chaperone proteins, including
Hsp90, but in a conformational state responsive to ligand binding
(17). Ligand-activated regulation
of AR leads to its subsequence events such as nuclear
translocation, dimerization, and binding to androgen response
elements in the DNA of target genes (21). Moreover, AR is assumed to be a
major molecule in the transition from hormone-sensitive to
androgen-independent prostate cancer (22). It is hormone ablation therapy that
is the main treatment for early-stage prostate cancer. However,
this treatment leads to incurable and acute hormone refractory
disease. Therefore, it is necessary for novel strategies to
effectively eliminate AR signaling from prostate cancer for the
clinical control of this lethal disease. Herein, the
shikonin-mediated down-modulation in AR protein level correlates
with a reduction in AR message as presented by reverse
transcription-PCR and inhibition of AR promoter activity as
revealed by the luciferase reporter assay, which is supported by
the following observations: i) nuclear level of AR is markedly
suppressed in the presence of shikonin in both LNCaP and 22RV1 cell
lines; ii) shikonin treatment results in a critical decrease in
expression levels of the AR-regulated target gene PSA; and iii)
shikonin inhibits growth of LNCaP and 22RV1 cells in association
with apoptosis induction.
Shikonin is the main component of Chinese herbal
medicine Zi Cao (gromwell) that has antitumor activity
(23). Although several in
vitro molecular targets were found to be associated with
shikonin-induced apoptotic cell death (14,24),
the cellular target of shikonin is still unknown. Natural compounds
might have multiple cellular targets in order to achieve their
biological beneficial effects such as tumor growth inhibition
(25).
In this study, we firstly describe that the AR is
one of the targets of shikonin in vitro, inhibition of which
leads to cell death in human prostate tumor cells, shikonin is
highly effective in reducing the protein level of AR and the
transcriptional activity of AR. Consequently, we suggest that
shikonin has great potential to be used clinically for treatment of
human prostate cancers.
Acknowledgements
This research was supported by the
grant from Basic Science Research Program through the National
Research Foundation of Korea grant funded by the Ministry of
Education, Science and Technology of the Korea government
(2011-0010430).
References
|
1.
|
Jemal A, Murray T, Ward E, et al: Cancer
statistics, 2005. CA Cancer J Clin. 55:10–30. 2005. View Article : Google Scholar
|
|
2.
|
Gelmann EP: Molecular biology of the
androgen receptor. J Clin Oncol. 20:3001–3015. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Culig Z, Klocker H, Bartsch G and Hobisch
A: Androgen receptors in prostate cancer. Endocr Relat Cancer.
9:155–170. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Prescott J and Coetzee GA: Molecular
chaperones throughout the life cycle of the androgen receptor.
Cancer Lett. 231:12–19. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Heinlein CA and Chang C: Androgen receptor
in prostate cancer. Endocr Rev. 25:276–308. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Chen CD, Welsbie DS, Tran C, et al:
Molecular determinants of resistance to antiandrogen therapy. Nat
Med. 10:33–39. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Isaacs JT and Isaacs WB: Androgen receptor
outwits prostate cancer drugs. Nat Med. 10:26–27. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Masuda Y, Nishida A, Hori K, et al:
Beta-hydroxyisovalerylshikonin induces apoptosis in human leukemia
cells by inhibiting the activity of a polo-like kinase 1 (PLK1).
Oncogene. 22:1012–1023. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Kim SH, Kang IC, Yoon TJ, et al: Antitumor
activities of a newly synthesized shikonin derivative,
2-hyim-DMNQ-S-33. Cancer Lett. 172:171–175. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Masuda Y, Shima G, Aiuchi T, et al:
Involvement of tumor necrosis factor receptor-associated protein 1
(TRAP1) in apoptosis induced by beta-hydroxyisovalerylshikonin. J
Biol Chem. 279:42503–42515. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Yoon Y, Kim YO, Lim NY, Jeon WK and Sung
HJ: Shikonin, an ingredient of Lithospermum erythrorhizon
induced apoptosis in HL60 human premyelocytic leukemia cell line.
Planta Med. 65:532–535. 1999.
|
|
12.
|
Yang H, Zhou P, Huang H, et al: Shikonin
exerts antitumor activity via proteasome inhibition and cell death
induction in vitro and in vivo. Int J Cancer. 124:2450–2459. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Gaddipati JP, Mani H, Shefali, et al:
Inhibition of growth and regulation of IGFs and VEGF in human
prostate cancer cell lines by shikonin analogue 93/637 (SA).
Anticancer Res. 20:2547–2552. 2000.PubMed/NCBI
|
|
14.
|
Wu Z, Wu L, Li L, Tashiro S, Onodera S and
Ikejima T: p53-mediated cell cycle arrest and apoptosis induced by
shikonin via a caspase-9-dependent mechanism in human malignant
melanoma A375-S2 cells. J Pharmacol Sci. 94:166–176. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Chung TW, Moon SK, Chang YC, et al: Novel
and therapeutic effect of caffeic acid and caffeic acid phenyl
ester on hepatocarcinoma cells: complete regression of hepatoma
growth and metastasis by dual mechanism. FASEB J. 18:1670–1681.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Balk SP, Ko YJ and Bubley GJ: Biology of
prostate-specific antigen. J Clin Oncol. 21:383–391. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Burnstein KL: Regulation of androgen
receptor levels: implications for prostate cancer progression and
therapy. J Cell Biochem. 95:657–669. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Rosenfeld MG and Glass CK: Coregulator
codes of transcriptional regulation by nuclear receptors. J Biol
Chem. 276:36865–36868. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Jemal A, Siegel R, Ward E, Murray T, Xu J
and Thun MJ: Cancer statistics, 2007. CA Cancer J Clin. 57:43–66.
2007. View Article : Google Scholar
|
|
20.
|
Nelson WG, De Marzo AM and Isaacs WB:
Prostate cancer. N Engl J Med. 349:366–381. 2003. View Article : Google Scholar
|
|
21.
|
Richter E, Srivastava S and Dobi A:
Androgen receptor and prostate cancer. Prostate Cancer Prostatic
Dis. 10:114–118. 2007. View Article : Google Scholar
|
|
22.
|
Tamura K, Furihata M, Tsunoda T, et al:
Molecular features of hormone-refractory prostate cancer cells by
genome-wide gene expression profiles. Cancer Res. 67:5117–5125.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Sankawa U, Ebizuka Y, Miyazaki T, Isomura
Y and Otsuka H: Antitumor activity of shikonin and its derivatives.
Chem Pharm Bull. 25:2392–2395. 1977. View Article : Google Scholar
|
|
24.
|
Han W, Li L, Qiu S, et al: Shikonin
circumvents cancer drug resistance by induction of a necroptotic
death. Mol Cancer Ther. 6:1641–1649. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Cucciolla V, Borriello A, Oliva A,
Galletti P, Zappia V and Della Ragione F: Resveratrol: from basic
science to the clinic. Cell Cycle. 6:2495–2510. 2007. View Article : Google Scholar : PubMed/NCBI
|