Introduction
Renal cell carcinoma (RCC) accounts for
approximately 2% of all cancers worldwide (1) and its incidence has increased by 2–3%
in the last decade with even higher rate in developed countries
(2–6). The underlying mechanisms such as some
environmental and genetic risk factors including smoking, obesity,
acquired cystic kidney disease and inherited susceptibility (von
Hippel-Lindau disease) (3,7,8) have
been indicated, but the etiological and pathological mechanisms of
this disease are still far from fully understood.
Although local renal tumors can be surgically
removed (9–11), distant metastasis is often observed
even if the primary tumor is relatively small (12,13).
Patients with metastatic RCC generally result in extremely poor
outcomes with overall median survival of around 13 months and the 5
year survival rate of <10% (13). For the advanced-stage patients,
systemic therapy including immunotherapy (e.g. IL-2, IFN-α) and/or
molecular-targeted drugs (e.g. sunitinib, bevacizumab, sorafenib,
temsirolimus and everolimus) is recommended (14), but the response rates are not
satisfactory.
To better understand the molecular mechanisms of
renal carcinogenesis and apply the information for the development
of effective treatment and early diagnosis, we performed
genome-wide gene expression profile analysis and identified a small
heat shock protein, HSPB7, whose function in cancer is unknown, to
be downregulated in a great majority of human RCC samples.
In this study, we attempted to address two key
questions, i) whether HSPB7 has growth suppressive function and ii)
how HSPB7 is downregulated in RCCs. We here report for the first
time that HSPB7 is likely to be a tumor suppressor which is
frequently downregulated by DNA methylation in RCCs and is involved
in the p53 pathway.
Materials and methods
Tissue samples and cell lines
Tissue samples used in this study were obtained from
patients with written informed consent at three hospitals: Juntendo
University School of Medicine, Kochi University School of Medicine,
and Kyoto Prefectural University of Medicine. The human RCC cell
lines (Caki-1, Caki-2, 786-O, A-498, ACHN), HEK293 and NCI-H1299
(lung carcinoma, p53-null) were purchased from American Type
Culture Collection (ATCC; Rockville, MD, USA). Colon cancer cell
lines HCT116 p53 wild-type (p53+/+) and its derivative
(p53−/−) were gifts from Dr B. Vogelstein (Johns-Hopkins
University, Baltimore, MD, USA). Normal human renal proximal tubule
epithelial cells (RPTEC) were purchased from Lonza Walkersville
Inc. (Walkersville, MD, USA). All cell lines were grown in
monolayers in appropriate media recommended by suppliers:
Dulbecco’s modified Eagle’s medium (Gibco, Carlsbad, CA, USA) for
HEK293, HCT116 (p53−/−) and HCT116 (p53+/+);
Eagle’s minimal essential medium (Gibco) for A-498; McCoy’s 5A
medium (Gibco) for Caki-1 and Caki-2; RPMI-1640 medium (Gibco) for
ACHN, 786-O and NCT-H1299; in addition, cells were supplemented
with 10% fetal bovine serum (Cell Culture Bioscience, Nichirei
Biosciences, Inc., Tokyo, Japan) except ACHN (5%), and 1%
penicillin-streptomycin-amphotericin B suspension (Wako, Osaka,
Japan). RPTEC were grown in REGM™ BulletKit, purchased from Lonza
Walkersville Inc. (Walkersville, MD, USA). All cells were
maintained at 37°C in humid air with 5% CO2 condition.
Cells were transfected with plasmids using FuGENE 6 transfection
reagent (Roche, Basel, Switzerland) or Lipofectamine LTX and Plus
reagent (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer’s protocols.
cDNA microarray and selection of
candidate genes
We prepared a genome-wide cDNA microarray with
totally 27,648 cDNAs/ESTs selected from the UniGene database of the
National Center for Biotechnology Information (NCBI). This
microarray system was constructed as previously described (15,16).
We analyzed 15 clear cell renal cell carcinomas (RCC) and selected
candidate genes according to the following criteria: i) genes for
which we were able to obtain expression data in more than 50% of
the cancers examined; ii) genes whose expression ratio was <0.2
in more than 50% of informative cases; and iii) the function of the
gene was still unknown. Through these criteria, several candidates
including HSPB7 were further validated. Gene expression data were
deposited in the Gene Expression Omnibus database (accession no.
GSE39364).
Quantitative real-time PCR (qPCR)
We extracted total RNA from the microdissected RCC
clinical samples, microdissected normal renal cortex, 25 different
normal organs (17) and cultured
cells using RNeasy mini kits (Qiagen, Valencia, CA, USA). RNAs from
cell lines were reversely transcribed using the oligo (dT)21 primer
and SuperScript III reverse transcriptase (Invitrogen). RNAs from
tissue samples were treated with DNase I and subjected to two
rounds of RNA amplification using T7-based in vitro
transcription (Epicentre Technologies, Madison, WI, USA), then
amplified RNAs were reversely transcribed to single-stranded cDNAs
using random primer with Superscript II reverse transcriptase
(Invitrogen) according to the manufacturer’s instruction. qPCR was
conducted using the SYBR-Green I Master (Roche) on a LightCycler
480 (Roche). Standard curve method was used for quantification
analysis, and β2 micro-globulin (B2M) served as a control gene. The
qPCR primers for HSPB7 in cell lines were: 5′-ACTTCTCACCTGAAGA
CATCATTG-3′ (forward) and 5′-CATGACAGTGCCG TCAGC-3′ (reverse). The
qPCR primers for HSPB7 in tissues were: 5′-GACCTTCCATCAGCCTTAACC-3′
(forward) and 5′-ATGTGGGAGACGAAACCAAG-3′ (reverse). The qPCR
process was started at 95°C for 5 min, then underwent 45 cycles at
95°C for 10 sec, 55°C for 10 sec and 72°C for 10 sec. Data analysis
including standard curve generation and copy number calculation was
performed automatically. Each reaction was performed in duplicate
and negative controls were included in each experiment.
Immunohistochemistry (IHC)
A kidney tissue array (BioChain Institute, Inc.,
USA) was used to analyze the protein expression of HSPB7 by IHC
staining. This tissue array included 11 cases of RCC with
corresponding normal tissues from the same patients as controls.
Tissue sections were deparaffinized, rehydrated, and processed
under high pressure (125°C, 30 sec) in antigen-retrieval solution
of pH 9.0 (S2367, Dako, Carpinteria, CA, USA). Sections were
blocked with Protein Block Serum Free (Dako) for 1 h at room
temperature, followed by incubation with primary antibody (HSPB7,
1:100, Proteintech, Chicago, IL, USA) overnight at 4°C. At day 2,
endogenous peroxidase activity was blocked by incubation in 3%
hydrogen peroxide for 30 min at room temperature. Sections were
incubated with a secondary antibody (Dako Envision+
system-HRP labeled polymer anti-rabbit K4003) for 30 min at room
temperature, followed by DAB staining (K3468, Dako), counter
stained with hematoxylin QS (H-3404, Vector Laboratories,
Burlingame, CA, USA), dehydrated and mounted. Three independent
investigators semi-quantitatively assessed the HSPB7 positivity
without prior knowledge of clinicopathological data. According to
the intensity of HSPB7 staining, these samples were evaluated as:
negative (−), weakly positive (+), moderate positive (++), and
strong positive (+++). HSPB7 negative or weakly positive (−/+) were
considered low expression, and moderate or strong positive were
considered high expression (++/+++).
5-Aza-2′-deoxycytidine (5-Aza-dC)
treatment
5-Aza-dC (Sigma-Aldrich, St. Louis, MO, USA) was
dissolved in DMSO. For a negative control, 5 RCC cell lines were
treated with DMSO alone for 4 days. For a 5-Aza-dC group, cells
were treated with DMSO for 1 day, following 5-Aza-dC-treatment (1,
3 and 10 μM, respectively) for 3 days. On the fifth day,
total RNAs of all cells were isolated using the RNeasy mini kits
(Qiagen, Valencia, CA, USA), according to the manufacturer’s
directions. qPCR was subsequently performed to detect the
expression of HSPB7. To detect the protein level of HSPB7 in 5 RCC
cell lines after the same treatment (5-Aza-dC 1 μM was used
in 5-Aza-dC group), western blot and immunocyto-chemical (ICC)
analyses were performed.
Bisulfite sequencing
Genomic DNA was extracted from RPTEC, HEK293 and 5
RCC cell lines (Caki-1, Caki-2, 786-O, A-498, ACHN) using the
DNeasy blood and tissue kit (Qiagen). Genomic DNA (3.5 μg
each) were digested at 37°C for 16 h with 35 units of XhoI
(Takara, Tokyo, Japan) and 1X H buffer (Takara) in 50 μl of
reaction volume. After treatment with phenol/chloroform/isoamyl
alcohol (25:24:1, v/v), the DNA was finally dissolved in TE buffer
and denatured in 0.3 N NaOH for 20 min at 37°C, and then the
unmethylated cytosine residues were sulfonated by incubation in
3.12 M of sodium bisulfite (pH 5.0, Sigma-Aldrich) and 0.5 mM of
hydroquinone (Sigma-Aldrich) at 55°C for 16 h. The sulfonated DNA
was recovered using the Nucleospin Extract II (Macherey-Nagel GmbH
and Co. KG, Düren, Germany) according to the manufacturer’s
recommendations. The conversion reaction was completed by
desulfonating in 0.3 N NaOH for 20 min at 37°C. The DNA was ethanol
precipitated, then washed by 70% ethanol and resuspended in TE
buffer. Primers for bisulfite genomic sequencing PCR were designed
by the use of the online program MethPrimer. The primers for region
1 were: 5′-TTT GAAGGGTTTTGGGTTTAATATAT-3′ (forward) and
5′-CTCCTAACTACAAACTATCCAACAC-3′ (reverse). The primers for region 2
were: 5′-GGGTTGGTTTTAAGTTT AGGGATAG-3′ (forward) and
5′-AAAAAAAATTCTA TAACTCATCCAC-3′ (reverse). The primers for region
3 were: 5′-TGTATAT TGATG GAG GAG GTATAGT-3′ (forward) and
5′-AAAAAAAACTAAAAATCTTCTCCC-3′ (reverse). The primers for region 4
were: 5′-TGGAGAAGG TTTTGAGTATGTTTTT-3′ (forward) and 5′-CCACAT
CTATCCCTATAACCACATC-3′ (reverse). The amplification products were
checked by electrophoresis. After gel purification, the PCR
products were cloned into pCR2.1-TOPO vector (Invitrogen), and 10
or more colonies were randomly chosen and sequenced. Methylation
level analysis was performed by using QUMA software (http://quma.cdb.riken.jp/).
Construction of HSPB7 expression
vector
To construct an HSPB7 expression vector, the entire
coding sequence of HSPB7 cDNA (based on NM_014424.4 in Pubmed) was
amplified by PCR using KOD-Plus DNA polymerase (Toyobo, Osaka,
Japan). The primers used for PCR reaction were
5′-AAAGAATTCCGTCCGTGGATGAGCCACAG-3′ (forward) and
5′-TTTCTCGAGGATTTTGATCTCCGTC CGGA-3′ (reverse). The PCR product was
inserted into the EcoRI (Takara) and XhoI (Takara)
sites of pCAGGSnHC expression vector containing the HA tag. The
sequence and protein expression for pCAGGSnHC-HSPB7-HA were
confirmed by DNA sequencing, western blot and ICC analyses.
Western blot analysis
To prepare whole cell extracts, cells were collected
and lysed in chilled radioimmunoprecipitation assay buffer (RIPA)
(50 mM Tris-HCl at pH 8.0, 150 mM sodium chloride, 0.1% SDS, 0.5%
DOC, 1% NP-40), 1 mM phenyl methylsulphonyl fluoride (PMSF), 1 mM
DTT and 0.1% Calbiochem Protease Inhibitor Cocktail Set III,
EDTA-Free (EMD Chemicals Inc., Merck KGaA, Darmstadt, Germany).
Following 15-min ultrasonication and subsequent 30-min incubation
on ice, homogenates were centrifuged for 15 min at 4°C, and the
supernatants were collected and boiled in SDS sample buffer. Each
sample was loaded into a 15% SDS-polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred to a nitrocellulose membrane
(Hybond™ ECL™, Amersham, Piscataway, NJ,
USA). Protein bands on western blots were visualized by
chemiluminescent detection (ECL, Amersham). The primary antibodies
used in this study included rabbit anti-human HSPB7 polyclonal
antibody (Proteintech, diluted 1:500) and goat anti-rabbit IgG-HRP
secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA,
diluted 1:30,000).
Immunocytochemistry (ICC)
Five RCC cell lines were seeded on Lab-Tek II
chamber slide system (Nalge Nunc International). At day 5 after the
5-Aza-2′-dC-treatment, the cells were fixed with 4%
paraformaldehyde in PBS for 10 min and permeabilized with 0.2%
Triton X-100 in PBS for 5 min at room temperature. Cells were
covered with blocking solution (3% BSA in PBS contained 0.2% Triton
X-100) for 60 min at room temperature. Then the cells were
incubated with rabbit anti-human HSPB7 polyclonal antibody
(Proteintech, diluted 1:250) overnight at 4°C, following an Alexa
Fluor 488 goat anti-rabbit IgG antibody (Molecular Probes, Eugene,
OR, USA, diluted 1:1,000) for 1 h at room temperature. PBS or 0.2%
Triton X-100 in PBS was used for washing after each step. Then
cells were stained with DAPI (Vector) and viewed with a laser
scanning spectral confocal microscope (Leica TCS SP2).
Colony formation assay
Cells were plated in a 6-well plate and transfected
with pCAGGSnHC-HSPB7-HA or empty vector using FuGENE 6 (ACHN and
Caki-1) or lipofectamine LTX (Caki-2, A498 and 786-O) transfection
reagent (Roche). After 48 h of transfection, cells were selected
with G418 (Gibco) for 14–21 days. Colonies (>1 mm diameter) were
counted using the Image J software after fixed with methanol and
stained with 0.1% crystal violet. The experiment was carried out
twice in duplicate wells.
DNA-damaging treatments
When cells reached 60–70% confluence in the culture
dish, HCT116 (p53−/−) and HCT116 (p53+/+)
cells were incubated with adriamycin for 2 h at the indicated
concentration. The cells were harvested at different time points
after cell-damaging treatment as indicated in the figure legends.
Replication-deficient recombinant adenovirus encoding p53 (Ad-p53)
or LacZ (Ad-LacZ) was generated and purified as previously
described (18,19). NCI-H1299 lung cancer cells were
infected with viral solutions at an indicated multiplicity of
infection (MOI) and incubated at 37°C until harvest.
p53-binding site screening by Luciferase
assay
Two DNA fragments including candidate p53-binding
sites of HSPB7 were amplified by PCR, digested with MluI and
BglII and cloned into pGL3-Promoter vector (Promega,
Madison, WI, USA). Primer sequences (including MluI and
BglII site) for p53-binding sites of HSPB7 were: region 1
forward, 5′-AAAACGCGTTCCAAGGTCACACAGCAGAG-3′; and reverse,
5′-TTTAGATCTGCTTCAAACCGGTCATCCT-3′; and region 2 forward,
5′-AAAACGCGTTGAGCAGGAGCA GTCAGAGA-3′; and reverse,
5′-TTTAGATCTAGCCCCAAG AGGACAAAGTT-3′.
H1299 cells were seeded in 12-well plates
(5×104 cells per well). Twenty-four hours later, cells
were co-transfected with i) 25 ng of the pRL-CMV vector (Promega)
(for internal control); ii) 125 ng of either pcDNA3.1(+)-wild-type
p53 or pcDNA3.1(+) empty vector; and iii) 125 ng of pGL3-promoter
vector with either the p21 promoter region corresponding to
p53-binding site (for positive control) (20), that with p53-binding site 1 of
HSPB7, that with p53-binding site 2 of HSPB7, or pGL3-Promoter mock
vector (for negative control) by using FuGENE 6 transfection
reagent (Roche). After 36-h incubation, luciferase activity was
measured using the Dual Luciferase Assay System (Promega) (21).
Statistical analysis
All statistical analyses including t-test and
Fisher’s exact test were carried out by using the SPSS software
(version 17). Data are shown as mean ± SD. All tests were 2-sided
and p-value of <0.05 was considered to indicate a statistically
significant difference.
Results
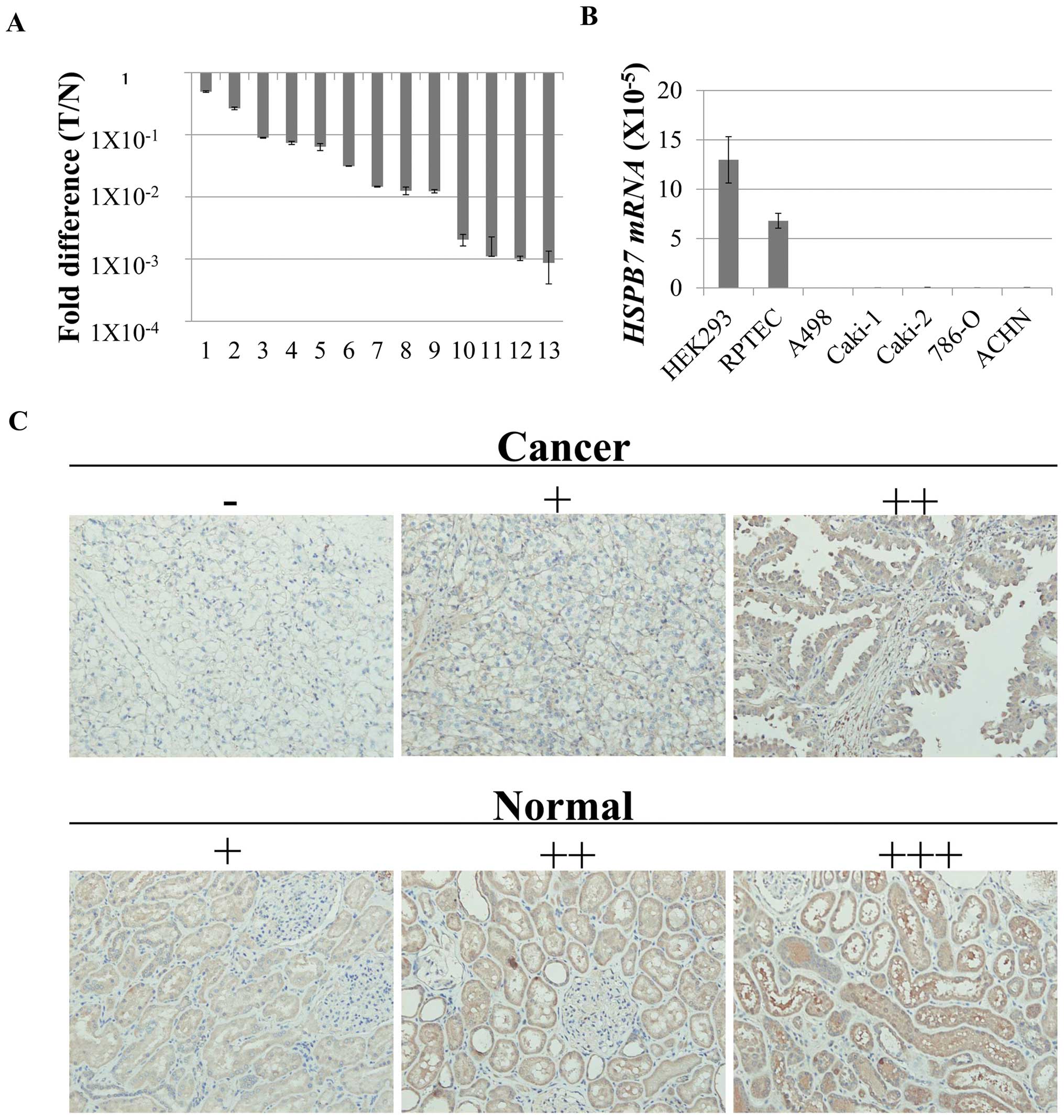
Downregulation of HSPB7 in RCC
Based on the analysis of microarray data of 15 clear
cell renal cell carcinomas, we found HSPB7 to be significantly and
commonly downregulated in RCC. qPCR experiment confirmed its
downregulation in 11 (85%) of 13 RCC tissues and in all of the five
RCC cell lines (Fig. 1A and B),
compared with their corresponding normal controls. IHC analysis of
a tissue array consisting of 11 pairs of human RCC sample revealed
that the expression of HSPB7 was significantly higher in normal
kidney tissues than that in RCC tissues (Fig. 1C and Table I). We also detected HSPB7
expression mainly in the cytoplasm of normal renal tubular
epithelial cells. To explore the expression patterns of HSPB7 in
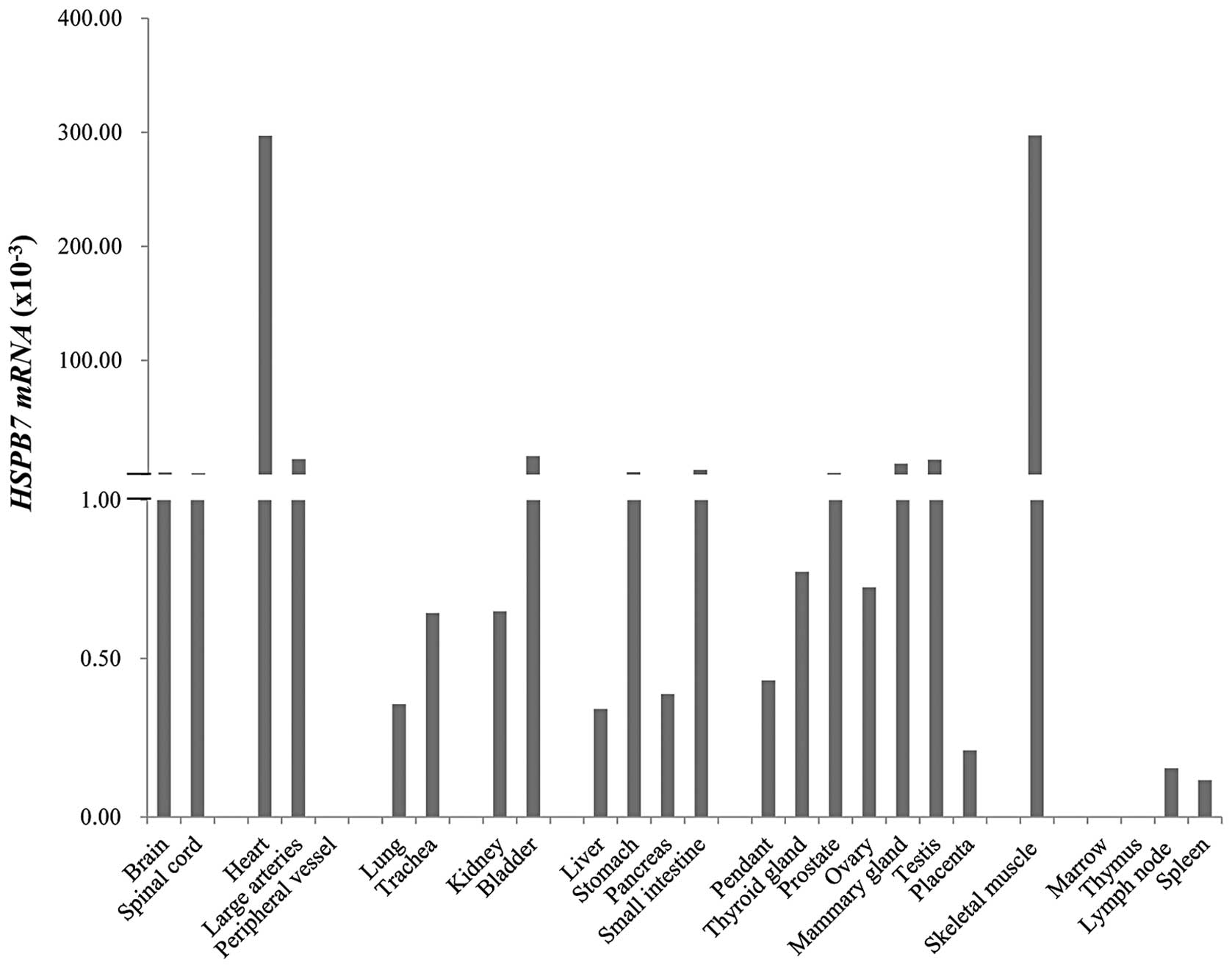
other normal tissues, we performed qPCR analysis using mRNAs
isolated from 25 normal tissues. HSPB7 expression was detected
ubiquitously in human tissues (Fig.
2).
 | Table I.Immunohistochemical expression of
HSPB7 in RCC tissue array. |
Table I.
Immunohistochemical expression of
HSPB7 in RCC tissue array.
| Total | Low (−/+) | High (++/+++) | Fisher’s t-test |
|---|
| Clear cell | | | | |
| Cancer | 9 | 7 | 2 | P=0.015 |
| Normal | 9 | 1 | 8 | |
| Papillary | | | | |
| Cancer | 2 | 1 | 1 | - |
| Normal | 2 | 0 | 2 | |
| Total | | | | |
| Cancer | 11 | 8 | 3 | P=0.008 |
| Normal | 11 | 1 | 10 | |
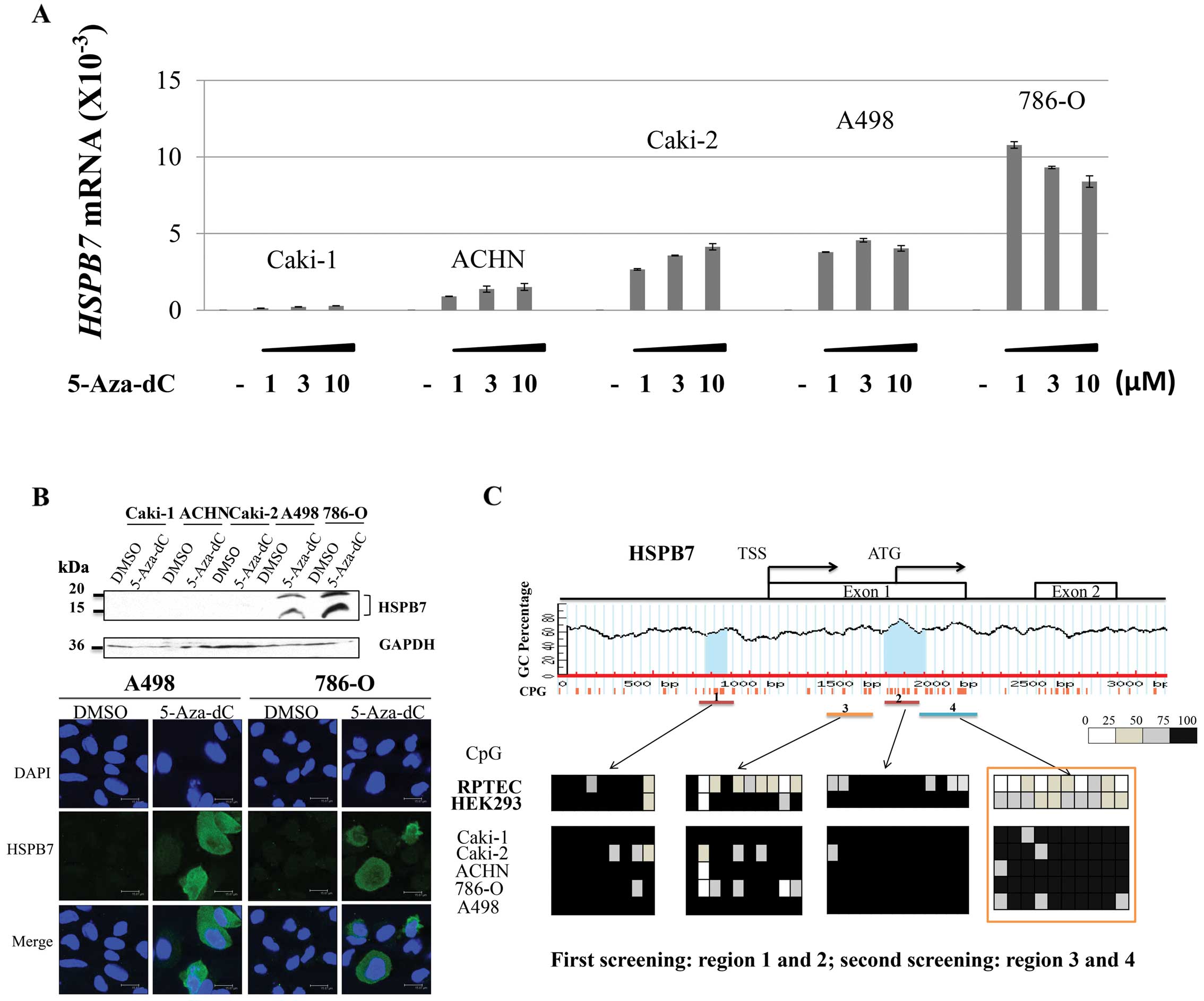
5-Aza-dC treatment restores HSPB7
expression in RCC cell lines
To investigate whether the methylation status of the
HSPB7 gene could affect HSPB7 expression in RCCs, 5 RCC cell lines,
Caki-1, Caki-2, ACHN, 786-O and A498 were treated with a
demethylating agent 5-Aza-dC, and then the expression levels of
HSPB7 were analyzed by qPCR, western blot and IHC analysis. We
found that HSPB7 mRNA expression were restored in all the 5 RCC
cell lines by the treatment with 5-Aza-dC (Fig. 3A), and the HSPB7 protein expression
could also be detected in two cell lines, 786-O and A498, in which
mRNA expression was most highly induced (Fig. 3B), indicating suppression of HSPB7
in RCC was caused probably by DNA hypermethylation. We performed
exon sequencing of HSPB7 in these five RCC cell lines, but no
mutation or deletion/insertion was detected (data not shown).
Hypermethylation of HSPB7 in RCC
To confirm the methylation status of the HSPB7 gene,
bisulfite sequencing was performed for the 5 RCC cell lines Caki-1,
Caki-2, ACHN, 786-O and A498 as well as 2 normal renal cell lines
RPTEC and HEK293. We first screened two CpG islands, regions 1 and
2 shown in Fig. 3C, but no
significant difference of methylation status was found in these two
regions in normal and cancer cell lines. Then, we performed the
second screening for regions 3 and 4 (Fig. 3C) (we also screened the other
regions in normal cells, but data are not shown). In region 4, we
found significantly higher levels of methylation in the 5 RCC cell
lines than in the 2 normal renal cell lines.
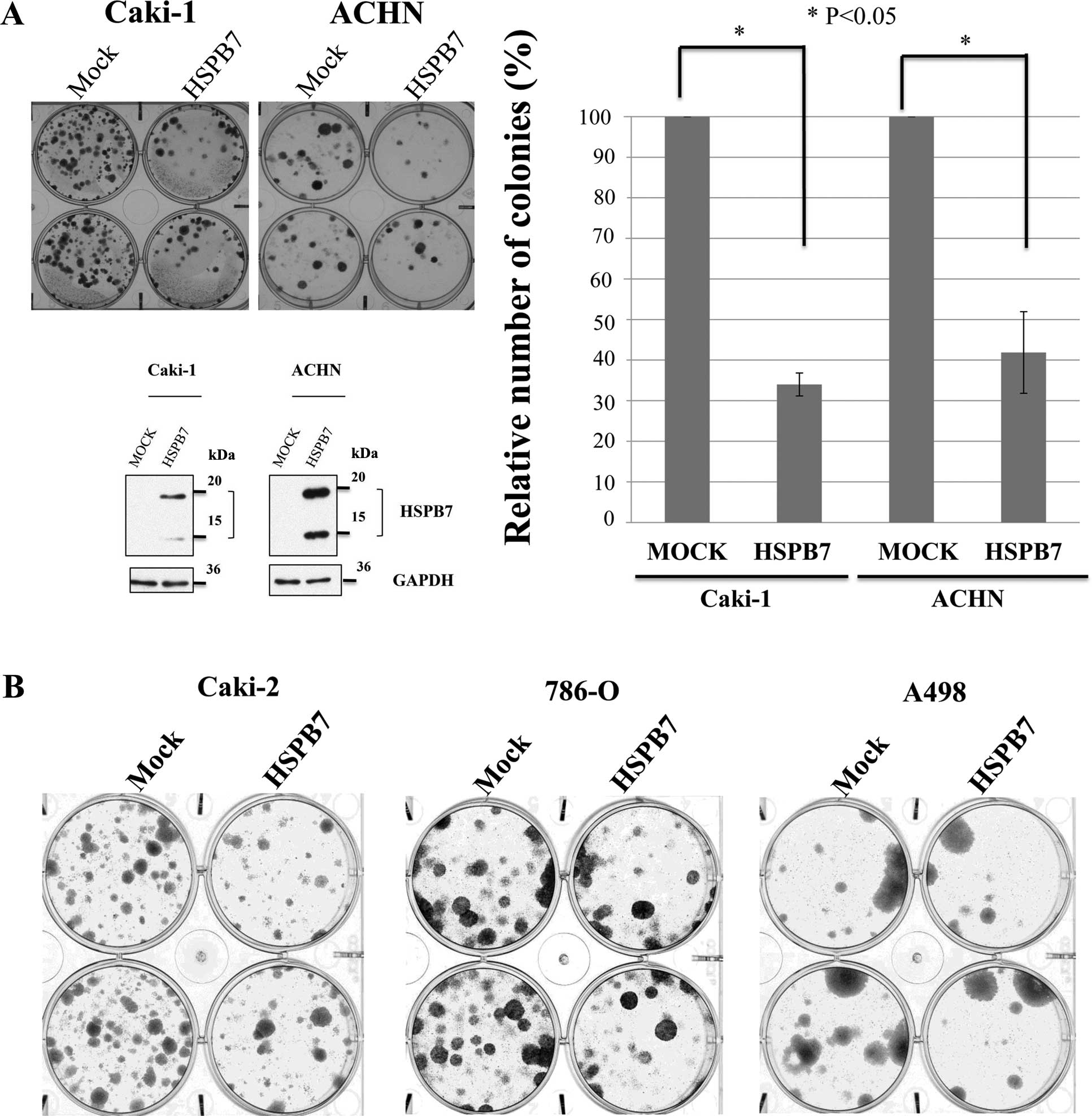
Ectopic HSPB7 expression suppresses RCC
cell clonogenicity
To study the effect of HSPB7 expression on tumor
growth, Caki-1 and ACHN cells were transfected with HSPB7
expression vector, pCAGGSnHC-HSPB7-HA. Introduction of HSPB7 into
these two cancer cell lines caused significant decrease in the
number of colonies, compared with corresponding mock-transfected
controls (Fig. 4A). We also
performed colony formation assay in 3 other RCC cell lines (Caki-2,
A498 and 786-O) using the same vectors, and confirmed similar
growth-suppressive effects (Fig.
4B), implying that HSPB7 may function as a tumor suppressor
gene.
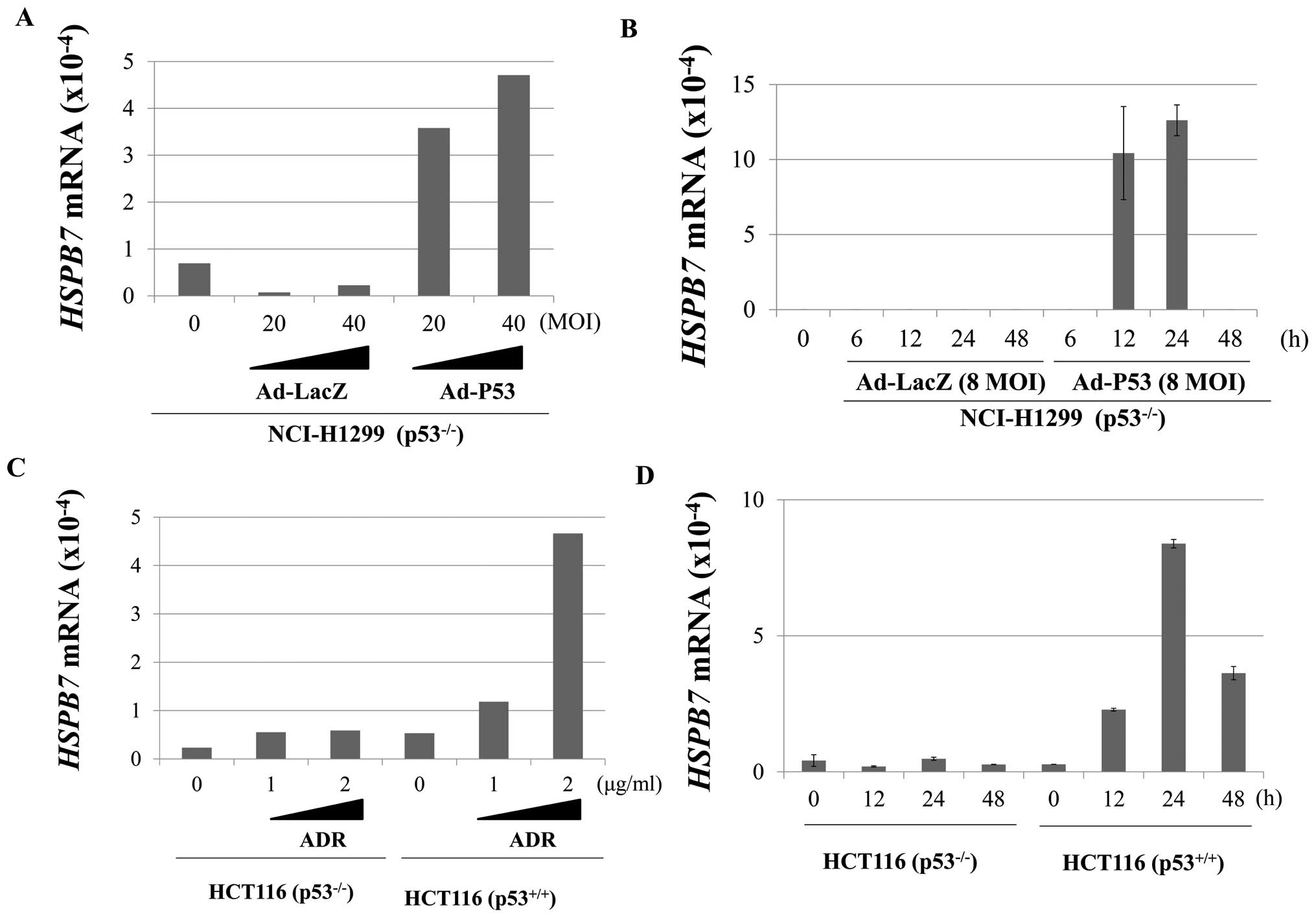
HSPB7 is regulated by p53
To further elucidate the biological significance, we
first investigated its possible involvement in the p53-pathway
because α B-crystallin, one of the small heat shock protein family
members, was reported to be induced by p53 (22,23).
We applied qPCR analysis to evaluate the expression of HSPB7 in
NCI-H1299 (p53 null) cell lines with or without introduction of p53
using the adenovirus system. After the infection of Ad-p53, we
observed induction of HSPB7 in a dose- and time-dependent manner
(Fig. 5A and B), while no
induction was observed in the control cells. After the 48-hour
treatment with 40 MOI of Ad-p53, the expression level of HSPB7
became nearly 5 times higher than the control cells (Fig. 5A). Induction of HSPB7 was also
confirmed under the treatment with relative lower dose of Ad-p53 (8
MOI) at different time points. Concordantly, DNA damage by
adriamycin treatment induced HSPB7 expression in HCT116 cells with
wild-type p53, but not in HCT116 cells without wild-type p53
(Fig. 5C and D), indicating that
HSPB7 expression is regulated by wild-type p53. To further
investigate whether HSPB7 is directly regulated by p53, we screened
two possible p53-binding sites indicated by the p53-binding site
search software developed by us, but neither of these two candidate
sites was confirmed to be a direct p53-binding site (data not
shown). Although there might be another site(s) that p53 binds to,
we are unable to conclude whether HSPB7 is directly or indirectly
regulated by p53, it is certain that HSPB7 expression is inducible
by wild-type p53.
Discussion
Scarce knownledge exists on the biological function
of HSPB7, a member of the small heat shock protein family that is
characterized by possessing a conserved α-crystallin domain. HSPB7
has been shown to interact with the cytoskeletal protein α-filamin
(24) as well as other small heat
shock proteins (25). HSPB7
belongs to a non-canonical HSPB protein that prevents the
aggregation of polyQ proteins in an active autophagy machinery, but
overexpression of HSPB7 alone did not affect the autophagy event
(26). Several genome-wide
association studies found that SNPs in the HSPB7 gene were strongly
associated with idiopathic cardiomyopathies and heart failure
(27–31). Recently, HSPB7 was suggested to
regulate early developmental steps in cardiac morphogenesis
(32). However, the involvement of
HSPB7 in carcinogenesis has not been described.
Through the genome-wide expression analysis in RCCs,
we identified HSPB7 as a candidate tumor suppressor gene because of
its common and significant downregulation in RCCs. Subsequent
functional analysis revealed that HSPB7 was downregulated in cancer
cells by hypermethylation. Bisulfite sequencing of genomic regions
of HSPB7 confirmed hypermethylation in RCC cell lines. Although
region 4 (Fig. 3C) contained no
CpG Island, we observed significantly higher level of methylation
in RCC cell lines than normal cell lines. Consistently, restoration
of HSPB7 expression was observed by the treatment of cancer cells
with 5-Aza-dC. In addition, since no somatic changes in coding
regions of the HSPB7 gene were found in our sequence analysis of
RCC cell lines or in the COSMIC database, HSPB7 in RCC is
considered to be downregulated mostly by hypermethylation.
The second key finding in this study is that HSPB7
showed growth suppressive effect in cancer cells. Ectopic
expression of HSPB7 significantly impaired colony-forming ability
for 5 RCC cell lines, indicating that HSPB7 may function as a tumor
suppressor gene. Similarly α B-crystallin, one of the small heat
shock protein family members, was also indicated to function as a
tumor suppressor in nasopharyngeal carcinoma cells (33). Furthermore, the region on
chromosome 1p36.23-p34.3, where HSPB7 is located, showed frequent
loss of heterozygosity in many types of solid tumors (34). However, further studies are needed
to clarify the detailed tumor suppressor function of HSPB7 in
RCC.
The third important finding in this study is that
HSPB7 was likely to be involved in the p53 pathway. The expression
of HSPB7 was significantly induced in p53-dependent manner that was
clearly demonstrated by two experiments, i) that introduction of
adeno-p53 in p53-negative cancer cells showed strong induction of
HSPB7 and ii) that DNA-damage-dependent introduction of HSPB7 was
observed in HCT116 cells with wild-type p53, but not in those
lacking p53. Although we failed to identify the p53-binding site in
or near the HSPB7 gene, these two pieces of evidence strongly imply
a critical role of HSPB7 as the direct/indirect p53-signal
transducer and its downregulation may be involved in the
development of various types of cancer including RCC.
In conclusion, we carried out a genome-wide gene
expression analysis and identified HSPB7 to be a candidate tumor
suppressor gene in RCC. We confirmed downregulation of this gene
caused by DNA hypermethylation, its growth suppressive effect in
RCC cell lines and its p53-dependent expression, indicating the
important roles of HSPB7 in renal carcinogenesis. Our finding could
contribute to better understanding of the novel function of HSPB7
in cancer.
Acknowledgements
We thank Ryuji Hamamoto, Daechun Kang
and Takashi Fujitomo for technological support of IHC. We also
thank Jinichi Mori for making cDNA template after Adriamycin
treatment. We thank Satomi Takahashi for providing general
reagents, materials and sequencing.
References
|
1.
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
3.
|
Naito S, Tomita Y, Rha SY, et al: Kidney
Cancer Working Group report. Jpn J Clin Oncol. 40(Suppl 1):
i51–i56. 2010. View Article : Google Scholar
|
|
4.
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
5.
|
Chow WH, Devesa SS, Warren JL and Fraumeni
JF Jr: Rising incidence of renal cell cancer in the United States.
JAMA. 281:1628–1631. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Hock LM, Lynch J and Balaji KC: Increasing
incidence of all stages of kidney cancer in the last 2 decades in
the United States: An analysis of Surveillance, Epidemiology and
End Results program data. J Urol. 167:57–60. 2002. View Article : Google Scholar
|
|
7.
|
Lindblad P: Epidemiology of renal cell
carcinoma. Scand J Surg. 93:88–96. 2004.
|
|
8.
|
Murai M and Oya M: Renal cell carcinoma:
etiology, incidence and epidemiology. Curr Opin Urol. 14:229–233.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Motzer RJ, Agarwal N, Beard C, et al: NCCN
clinical practice guidelines in oncology: kidney cancer. J Natl
Compr Canc Netw. 7:618–630. 2009.PubMed/NCBI
|
|
10.
|
Ljungberg B, Cowan NC, Hanbury DC, et al:
EAU guidelines on renal cell carcinoma: the 2010 update. Eur Urol.
58:398–406. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Escudier B, Eisen T, Porta C, et al: Renal
cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis,
treatment and follow-up. Ann Oncol. 23(Suppl 7): vii65–vii71. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Lam JS, Leppert JT, Belldegrun AS and
Figlin RA: Novel approaches in the therapy of metastatic renal cell
carcinoma. World J Urol. 23:202–212. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Cairns P: Renal cell carcinoma. Cancer
Biomark. 9:461–473. 2010.
|
|
14.
|
Molina AM and Motzer RJ: Current
algorithms and prognostic factors in the treatment of metastatic
renal cell carcinoma. Clin Genitourin Cancer. 6(Suppl 1): S7–S13.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Ono K, Tanaka T, Tsunoda T, et al:
Identification by cDNA microarray of genes involved in ovarian
carcinogenesis. Cancer Res. 60:5007–5011. 2000.PubMed/NCBI
|
|
16.
|
Hirota E, Yan L, Tsunoda T, et al:
Genome-wide gene expression profiles of clear cell renal cell
carcinoma: Identification of molecular targets for treatment of
renal cell carcinoma. Int J Oncol. 29:799–827. 2006.PubMed/NCBI
|
|
17.
|
Saito-Hisaminato A, Katagiri T, Kakiuchi
S, Nakamura T, Tsunoda T and Nakamura Y: Genome-wide profiling of
gene expression in 29 normal human tissues with a cDNA microarray.
DNA Res. 9:35–45. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Oda K, Arakawa H, Tanaka T, et al:
p53AIP1, a potential mediator of p53-dependent apoptosis, and its
regulation by Ser-46-phosphorylated p53. Cell. 102:849–862. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Matsuda K, Yoshida K, Taya Y, Nakamura K,
Nakamura Y and Arakawa H: p53AIP1 regulates the mitochondrial
apoptotic pathway. Cancer Res. 62:2883–2889. 2002.PubMed/NCBI
|
|
20.
|
Tanikawa C, Ueda K, Nakagawa H, Yoshida N,
Nakamura Y and Matsuda K: Regulation of Protein Citrullination
through p53/PAD14 Network in DNA Damage Response. Cancer Res.
69:8761–8769. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Tanikawa C, Matsuda K, Fukuda S, Nakamura
Y and Arakawa H: p53RDL1 regulates p53-dependent apoptosis. Nat
Cell Biol. 5:216–223. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Watanabe G, Kato S, Nakata H, Ishida T,
Ohuchi N and Ishioka C: alphaB-crystallin: a novel p53-target gene
required for p53-dependent apoptosis. Cancer Sci. 100:2368–2375.
2009. View Article : Google Scholar
|
|
23.
|
Evans JR, Bosman JD, Brown-Endres L,
Yehiely F and Cryns VL: Induction of the small heat shock protein
alphaB-crystallin by genotoxic stress is mediated by p53 and p73.
Breast Cancer Res Treat. 122:159–168. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Krief S, Faivre JF, Robert P, et al:
Identification and characterization of cvHsp. A novel human small
stress protein selectively expressed in cardiovascular and
insulin-sensitive tissues. J Biol Chem. 274:36592–36600. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Sun X, Fontaine JM, Rest JS, Shelden EA,
Welsh MJ and Benndorf R: Interaction of human HSP22 (HSPB8) with
other small heat shock proteins. J Biol Chem. 279:2394–2402. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Vos MJ, Zijlstra MP, Kanon B, et al: HSPB7
is the most potent polyQ aggregation suppressor within the HSPB
family of molecular chaperones. Hum Mol Genet. 19:4677–4693. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Cappola TP, Li M, He J, et al: Common
variants in HSPB7 and FRMD4B associated with advanced heart
failure. Circ Cardiovasc Genet. 3:147–154. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Matkovich SJ, Van Booven DJ, Hindes A, et
al: Cardiac signaling genes exhibit unexpected sequence diversity
in sporadic cardiomyopathy, revealing HSPB7 polymorphisms
associated with disease. J Clin Invest. 120:280–289. 2010.
View Article : Google Scholar
|
|
29.
|
Stark K, Esslinger UB and Reinhard W:
Genetic association study identifies HSPB7 as a risk gene for
idiopathic dilated cardiomyopathy. PLoS Genet. 6:e10011672010.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Villard E, Perret C, Gary F, et al: A
genome-wide association study identifies two loci associated with
heart failure due to dilated cardiomyopathy. Eur Heart J.
32:1065–1076. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Li XP, Luo R and Hua W and Hua W:
Polymorphisms of Hspb7 gene associate with idiopathic dilated
cardiomyopathy susceptibility in a Chinese population. Heart.
98:E47. 2012. View Article : Google Scholar
|
|
32.
|
Rosenfeld GE, Mercer EJ, Mason CE and
Evans T: Small heat shock proteins Hspb7 and Hspb12 regulate early
steps of cardiac morphogenesis. Dev Biol. 381:389–400. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Huang Z, Cheng Y, Chiu PM, et al: Tumor
suppressor Alpha B-crystallin (CRYAB) associates with the
cadherin/catenin adherens junction and impairs NPC
progression-associated properties. Oncogene. 31:3709–3720. 2012.
View Article : Google Scholar
|
|
34.
|
Ragnarsson G, Eiriksdottir G,
Johannsdottir JT, Jonasson JG, Egilsson V and Ingvarsson S: Loss of
heterozygosity at chromosome 1p in different solid human tumours:
association with survival. Br J Cancer. 79:1468–1474. 1999.
View Article : Google Scholar : PubMed/NCBI
|