Introduction
Neuroblastomas (NB) are tumors that arise from the
embryonic neural crest. In Japan, NBs are the most common childhood
malignancy after leukemia and the most common solid malignant tumor
in childhood. Differentiation and maturation of tumor cells occurs
during the clinical course, and spontaneous tumor regression is
sometimes observed. Prognosis differs with age at onset; while the
prognosis is favorable for children up to 12 months old, the
prognosis is poor for children past the age of 12 months.
Three-year overall survival in the high-risk group is >30%,
based on the Children’s Oncology Group (COG) risk stratification
(1).
Therapeutic strategies for NB have always been based
on risk. For high-risk patients, multidisciplinary treatment is
necessary; in addition to surgical resection, treatment includes
proactive chemotherapy, high-dose chemotherapy + autologous
hematopoietic cell transplantation and radiotherapy (1,2).
However, even with these treatments, 3-year progression-free
survival is low at 20–40% and new, effective treatments are needed
(3–5).
In the 1980s, MYCN gene amplification was reported
to be a cause of NB (6). MYCN gene
amplification, an abnormality observed in ∼10% of NB cases, has
been closely correlated with NB prognosis (7). However, the development of
therapeutic agents targeting this gene is not easy, and no progress
is being made. ALK mutations in NB were reported in 2008 (8–11).
Although research on ALK inhibitors is underway, this research has
not led to the discovery of molecular-targeted drugs (12). Mutations in the CDKN2A gene in NB
cell lines have led to dramatic reductions in the production of
p16INK4a protein (13,14).
Another report confirmed the expression of p16 protein in
neuroblastoma cell lines and cited reduction in 12 of 19 cases
(63%) (15). Non-function of the
cell cycle shutdown system due to absence of p16INK4a at
the G1 checkpoint is also considered a possible cause of NB.
Recently, protein transduction domain systems (PTDs)
have been garnering attention as an effective and safe system for
intracellular drug delivery. PTDs can pass through the membranes of
living cells and are thus useful for introducing molecular-targeted
functional proteins and peptides into cells. Many PTDs have been
reported, such as HIV-1 TAT (16),
pAntp43-58 (17) and polyarginine
(9,18). In 1998, Nagahara et al
created a recombinant fusion of TAT and p27kip1; the
fusion protein was delivered intracellularly, where it induced G1
arrest and cell migration (16).
Kondo et al developed a system for introducing PTDs into
cells in which, rather than directly binding PTDs to functional
peptides or proteins, they synthesized individual functional
peptides and the peptides for introducing them into cells; these
peptides were attached to each other by mixing. The transporter
(Wr-T) is a fusion of a nine D-arginine-residue PTD and a
tryptophan-rich cargo domain to which the functional peptide is
attached. This system is more efficient than the previously
reported Pep-1 and has been used to introduce p16INK4a,
the smallest functional peptide, for its antitumor efficacy in
leukemia, lymphoma, glioma, and renal cell carcinoma (19).
We used Wr-T to introduce p16INK4a and
determined its antitumor efficacy in NB. This method yielded strong
tumor suppression in NB with few side-effects, and is thus a
promising candidate for clinical application in cancer therapy.
Materials and methods
Cells
Human neuroblastoma cell lines SK-N-SH, SK-N-RA,
SK-H-BE, SMS-KAN, and SMS-KCNR (kindly provided by Tsukuba
University), and human cervical cancer cell line HeLa were
maintained in RPMI-1640 containing 10% inactivated fetal bovine
serum (IBL, Gunma, Japan), 100 U/ml penicillin, and 0.1 mg/ml
streptomycin, at 37°C under an atmosphere of 5% CO2.
Peptide synthesis
Wr-T and r9-p16 MIS (minimum inhibitory sequence)
peptides were synthesized at BioGate (Yamagata, Japan). These
peptides were prepared in the HCl form following high-performance
liquid chromatography purification for in vitro and in
vivo applications. Peptide purity was >95%. The identity of
all peptides was confirmed by mass spectrometry. To synthesize
Wr-T, we used the amino acid sequence
KETWWETWWTEWWTEWSQGPGrrrrrrrrr (r, D-enantiomer arginine), as
described by Kondo et al (19). To synthesize p16 MIS, the
FLDTLVVLHR sequence, identified as the MIS of p16 by Fahraeus et
al (22), was defined as the
functional core of the peptide. Since the MIS of p16 is insoluble,
as is the entire p16 molecule (MIS hydrophobicity 69.2%), we fused
r9 to these ten amino acids to make the conjugate less hydrophobic
(hydrophobicity, 40%), thus facilitating transport into cells.
Peptide transduction and measuring
absorbance in vitro
To prepare the peptide mixture for use in
vitro, the Wr-T and r9-p16 MIS peptides were mixed in 10
μl distilled water at room temperature for 60 min (final
concentration: Wr-T, 5 μmol/l; r9-p16 MIS, 8 μmol/l).
The solution was added directly to 190 μl RPMI-1640
containing 5% fetal bovine serum.
Absorbance was measured on a microplate reader at
450 nm with reference at 570 nm. Dunnett’s t-test was used for
statistical analysis.
PCR
Total RNA was extracted from each human
neuroblastoma cell line by using the RNeasy Mini kit (Qiagen
Sciences, MD, USA). cDNA was synthesized using random primers and a
cDNA synthesis kit (High Capacity cDNA RT kit, Applied Biosystems,
Foster City, CA, USA). PCR was performed with AmpliTaq Gold
(Applied Biosystems). Amplification conditions and primer sequences
for p16, CDK4, CDK6 and cyclin D are listed in Table I.
 | Table I.PCR primers and amplification
conditions. |
Table I.
PCR primers and amplification
conditions.
| Molecule | Sequence | Annealing temperature
(°C) | Fragment size
(bp) |
|---|
| p16 | F:
ATAGTTACGGTCGGAGGCC | 60 | 536 |
| R:
TGGTTACTGCCTCTGGTGC | | |
| Cyclin D | F:
AAAGACAGTTTTTGGGTAATCTTTT | 55 | 126 |
| R:
CCGGAGCATTTTGATACCAG | | |
| CDK4 | F:
CTTCTGGACACTGAGAGGGC | 61 | 110 |
| R:
TGGGAGGGGAATGTCATTAA | | |
| CDK6 | F:
CGGAGAACACCCTTGGTG | 59 | 105 |
| R:
GAGCCTGTCCAGAAGACAGC | | |
| Actin | F:
GTGGGGCGCCCCAGGCACCA | 55 | 539 |
| R:
CTCCTTAATGTCACGCACGATTTC | | |
Western blotting
Cells were lysed with SDS sample buffer and extracts
were separated by SDS-PAGE using SuperSep™Ace 12.5–15% bis-Tris
gradient gels (Wako, Osaka, Japan) and Q-PAGE system 7.5–15%
bis-Tris gradient gels (TEFCO, Tokyo, Japan). Separated proteins
were transferred to a PVDF-membrane (Immobilon-P, Millipore,
Billerica, MA, USA), then sequentially probed with the following
antibodies after blocking with 5% dried milk and 1% normal goat
serum-PBS: mouse monoclonal anti-actin antibody (Clone: AC-74,
Sigma, St. Louis, MO, USA), mouse monoclonal anti-human
p16INK4 (Clone: G175-1239, BD Biosciences Pharmingen,
San Diego, CA, USA), mouse monoclonal anti-RB antibody (Clone:
LM95.1, Oncogene), and rabbit polyclonal anti-phospho-Ser780RB
antibody (Cell Signaling Technology, Beverly, MA, USA). Immune
complexes were visualized with the ECL plus western blotting
dtection system (RPN2132, Amersham Pharmacia Biotech UK Ltd., UK)
according to the manufacturer’s instructions and signals were
visualized and digitally captured using an image analyzer (LAS
1000, Fuji Photo Film Co. Ltd., Tokyo, Japan).
Flow cytometry
Apoptosis assays were performed with the
FITC-Annexin V staining kit (MBL Co. Ltd., Japan) according to the
manufacturer’s instructions followed by FACScan analysis (BD,
Franklin Lakes, NJ, USA).
Cell cycle stage was evaluated by DNA uptake of EdU
(5-ethynyl-2’-deoxyuridine). Staurosporin was used as a positive
control using Click-iT®EdU Imaging kits (Invitrogen,
USA).
Mouse tumor models
Four-week-old NOD/Shi-scid, IL-2Rγnull
female mice were obtained from the Central Institute for
Experimental Animals (Kanagawa, Japan). SMS-KAN human neuroblastoma
cells (6×107) in 100 l RPMI-1640 were subcutaneously
injected into the flanks of each mouse to form a solid tumor
nodule. Treatments were as follows: the Wr-T/r9-p16 MIS peptide mix
(Wr-T, 50 nmol; r9-p16 MIS, 80 nmol) was injected into the heart of
the mouse when the tumor had grown to a diameter of 5 mm (tumor
volume, ∼150 mm3). For control groups, 100 μl PBS
without r9-p16 MIS peptide or Wr-T peptide was injected. Animal
experiments were approved by the Aichi Medical University
Subcommittee on Animal Research. All mouse procedures, euthanasia,
and surgery, including neuroblastoma cell transplantations and
peptide injections, were done pain-lessly or under anesthesia,
within the strict guidelines of the Experimental Animal Facility of
Aichi Medical University. Dunnett’s t-test was used for statistical
analysis.
Results
The p16INK4a-cyclin
D1/CDK4-pRB pathway (RB pathway) in NB
The RB pathway in cancer cells is the basic pathway
that must be inhibited in order for oncogenic transformation of
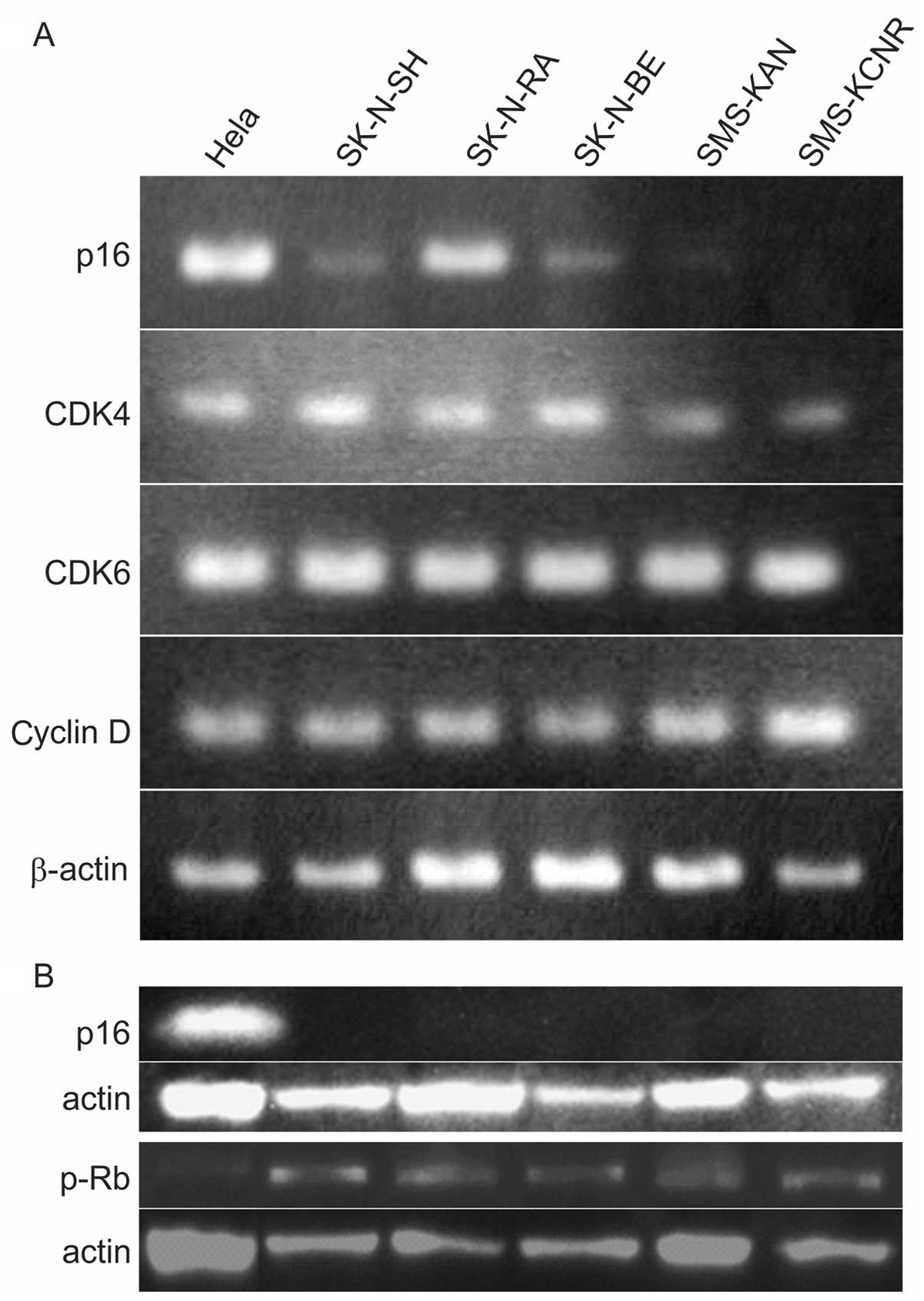
cells to occur. PCR and western blotting were used to verify p16,
cyclin D, CDK4, CDK6, RB, and pRB in five NB cell lines. PCR
revealed no p16 expression in SMS-KAN or SMS-KCNR cells, but
expression was confirmed in SK-NSH, SK-N-RA and SK-N-BE. Expression
of CDK4, CDK6 and cyclin D was confirmed in all cell lines
(Table I and Fig. 1A). p16 protein expression was not
observed in any cell lines and RB phosphorylation, which indicates
cell cycle revolution, was confirmed in all cell lines (Fig. 1B).
Transduction efficiency and tumor
suppression mechanism
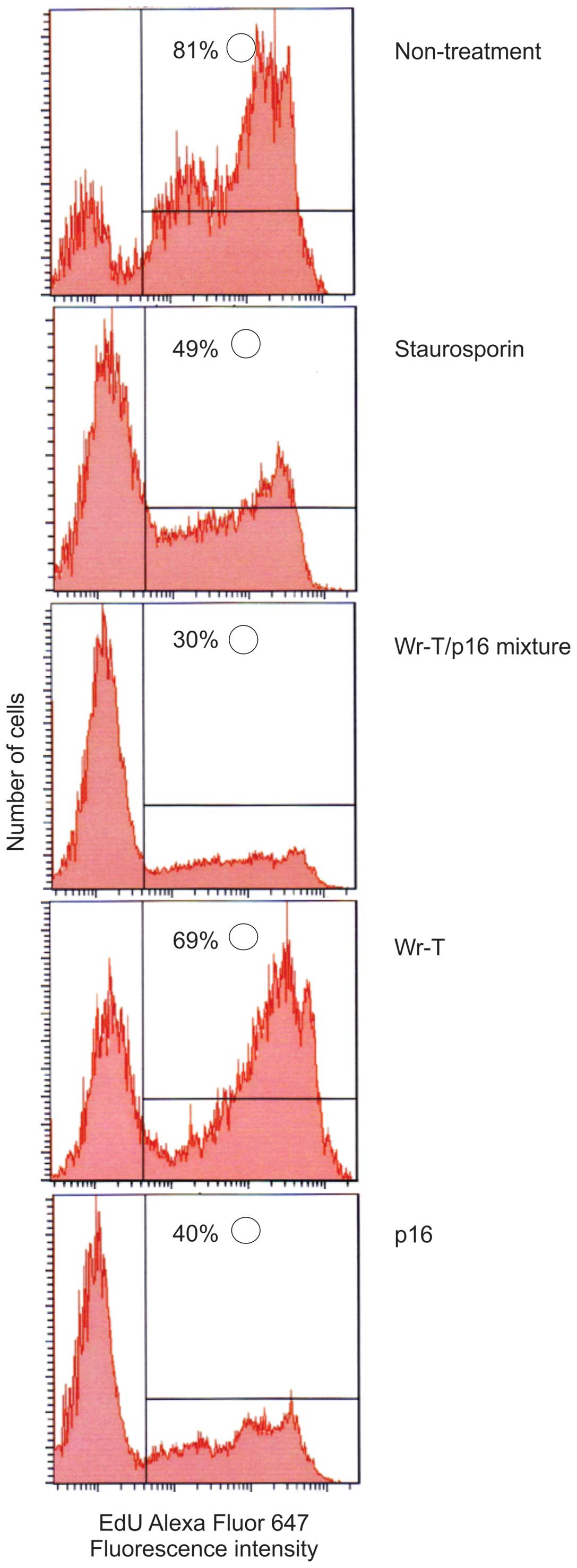
In order to examine the effects of the r9-p16 MIS
peptide on the cell cycle, DNA uptake of EdU was used as a marker
and Staurosporin was used as a positive control. Tumor growth
suppression was verified in the SMS-KAN cell line, which lacks
p16INK4a (Fig. 4).
While EdU uptake occurred in 81% of untreated cells, uptake volume
in cells treated with p16 alone and Wr-T/p16 complex was markedly
suppressed (40 and 30%, respectively), indicating that cell
division was suppressed. Tumor growth inhibition was also verified
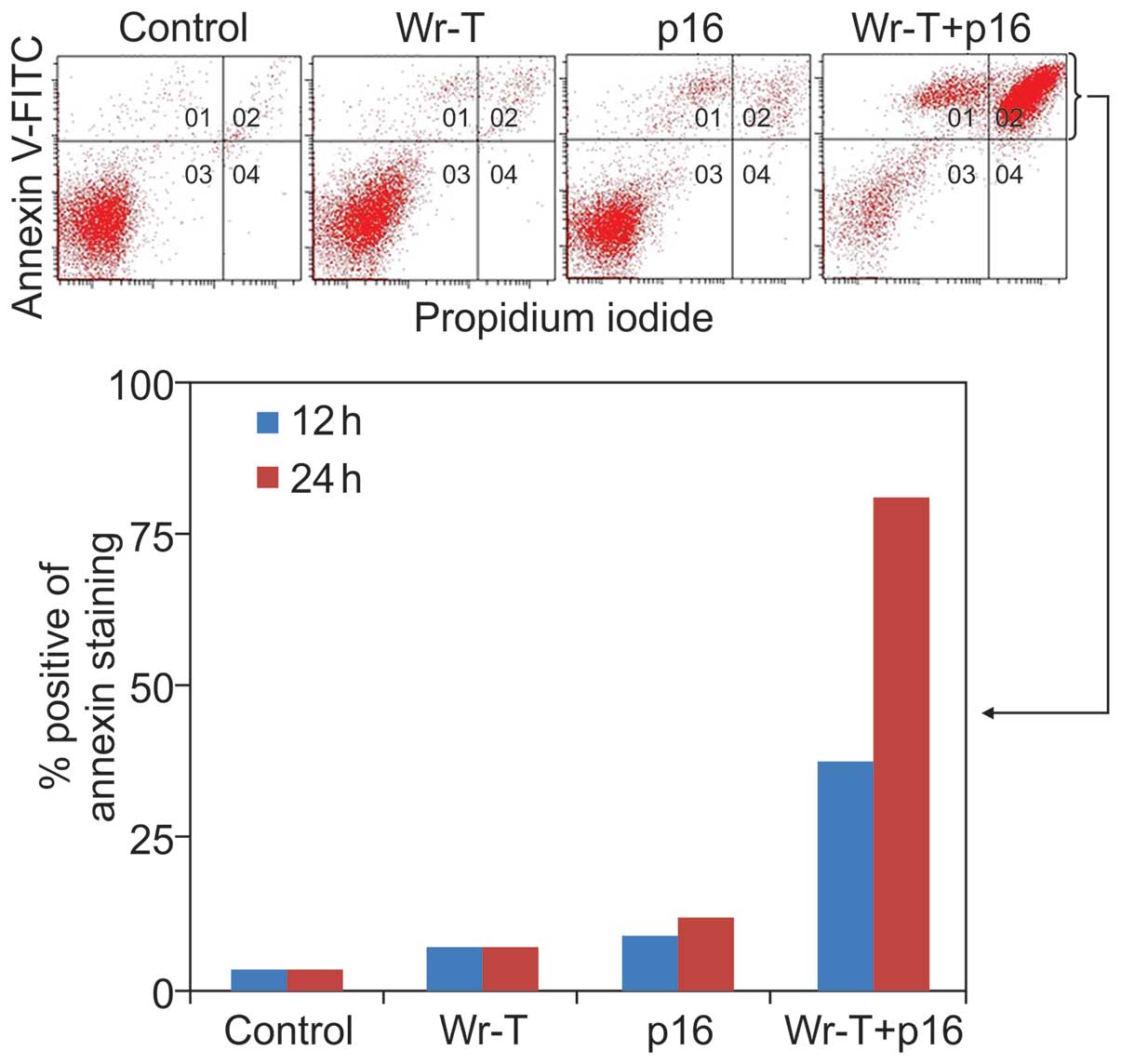
with Annexin V-FITC and propidium iodide (PI) staining using the
SMS-KAN cell line, which lacks p16INK4a expression
(Fig. 5). The mechanism of cell
death due was thought to be apoptosis. Twelve hours after
treatment, the percentage of cells with Annexin
V-positive/PI-negative staining was 2.1% in the control group, 3.6%
in the Wr-T group, 5.8% in the r9-p16 MIS peptide group, and 17.9%
in the Wr-T/r9-p16 MIS peptide mix group. At 24 h, the percentage
of dead cells was 3.4% in the control group, 6.8% in the Wr-T
group, 12% in the r9-p16 MIS peptide group, and 81.3% in the
Wr-T/r9-p16 MIS peptide mix group. A mild increase was observed in
the r9-p16 MIS peptide group, while the Wr-T/r9-p16 MIS peptide mix
group displayed a markedly high value. These results indicate
increased transduction efficiency of r9-p16 MIS peptide due to
Wr-T.
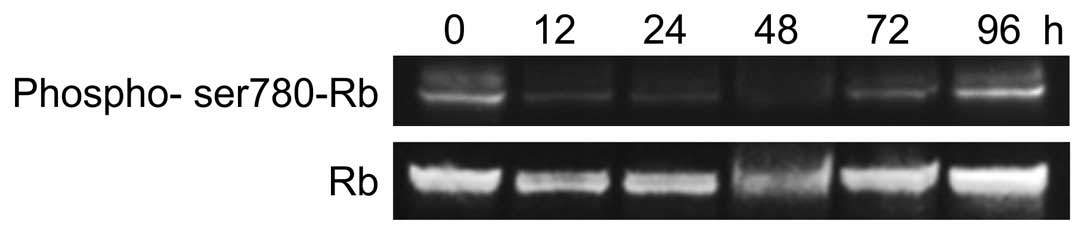
We also observed the effect of r9-p16 MIS peptide on
endogenous RB phosphorylation in SMS-KAN cells by western blotting
(Fig. 3). A clear quantitative
reduction in pRB was observed at 12 h (Fig. 3). The r9-p16 MIS peptide, which was
transported efficiently into nuclei due to Wr-T, likely suppressed
RB phosphorylation and arrested the cell cycle at G1 by replacing
the absent p16INK4a in the RB pathway. Quantitative
reduction reached its peak at 48 h after introduction, while tumor
re-enlargement was observed by 96 h. While the Wr-T/r9-p16 MIS
peptide mix suppressed cell proliferation through G1 arrest, the
complex degrades over time, allowing RB phosphorylation to progress
again.
In vitro tumor cell suppression
An examination of cell growth suppression in each
cell line at 96 h after administration yielded no differences
between the control and Wr-T groups. In the r9-p16 MIS peptide
group, growth suppression rates in the SK-N-SH, SK-N-RA, SK-N-BE,
SMS-KAN, and SMS-KCNR cell lines were 40.4, 28.1, 23.1, 15.1 and
33.5%, respectively. In the Wr-T/r9-p16 MIS peptide mix group,
growth suppression rates were 83.1 (p<0.0005), 85.4
(p<0.0005), 79.6 (p=0.004), 91 (p=0.025), and 91.7% (p=0.005),
respectively; significant differences were observed in all cell
lines. The r9-p16 MIS peptide group exhibited a marked tumor
inhibition effect (Fig. 2).
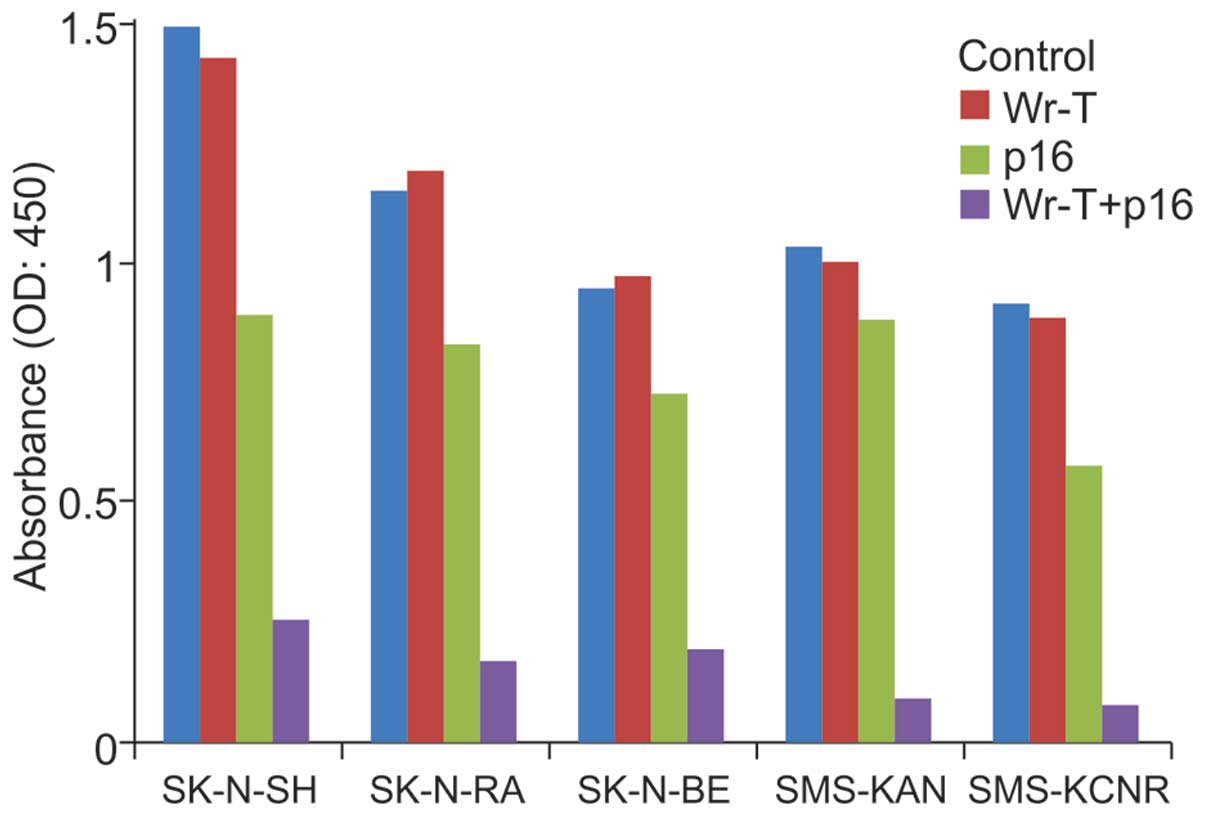 | Figure 2.Growth suppression by p16. Wr-T alone,
p16 peptide alone, Wr-T+p16 complex, and non-treatment control
groups in all cell lines were assessed by WST-1 assays for cell
proliferation after 96-h culture. There were no differences between
the control group and the Wr-T group in any cell lines. In the p16
group, growth suppression rates compared to the control group in
SK-N-SH, SK-N-RA, SK-N-BE, SMS-KAN and SMS-KCNR were 40.4, 28.1,
23.1, 15.1 and 33.5%, respectively. In the Wr-T+p16 complex group,
a marked growth suppression was observed with growth suppression
rates of 83.1, 85.4, 79.6, 91 and 91.7%, respectively. |
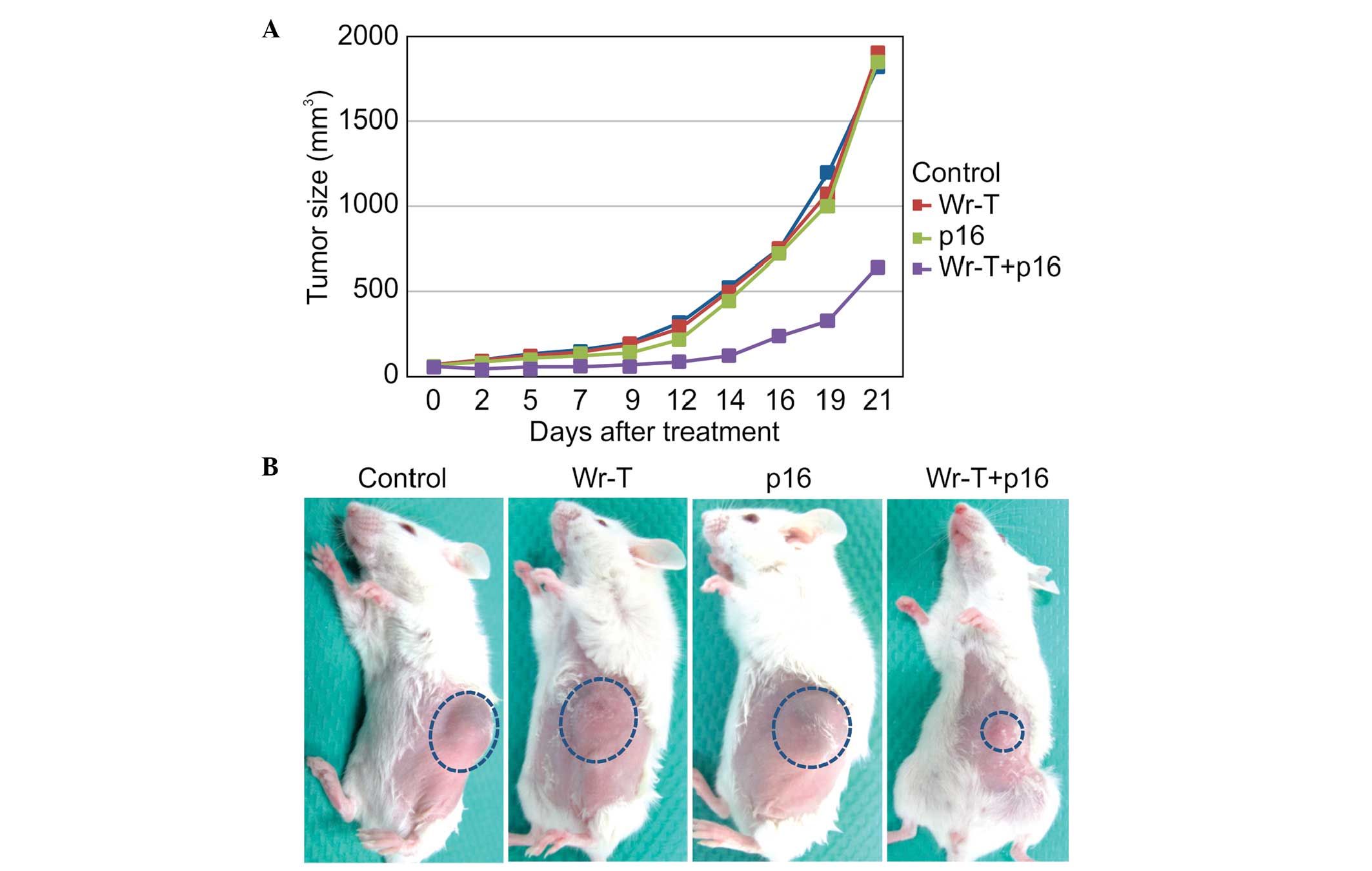
In vivo tumor suppression
Based on the in vitro results, SMS-KAN cells
were injected into four-week-old NOD/Shi-scid,
IL-2Rγnull female mice. At 14 days after tumor
inoculation, tumor volumes in the control, Wr-T, r9-p16 MIS
peptide, and Wr-T/r9-p16 MIS peptide mix groups were 523, 500.2,
439.3 and 127.3 mm3, respectively. Growth in the
Wr-T/r9-p16 MIS peptide mix group was inhibited by 75.6% compared
to the control group (p<0.0005) (Fig. 6A). The image shows tumors at 14
days after administration of each peptide. While similar tumors
were observed in the single peptide administration groups and the
non-treatment control group, only a small tumor was observed in the
Wr-T/p16 complex administration group (Fig. 6B).
Discussion
The failure of cell cycle regulation, which
functions properly in normal cells, is the cause of abnormal
proliferation in cancer cells. For example, cyclin-D
overexpression, caused by translocation or amplification of the
PRAD1 gene, promotes cell cycle revolution. The absence of cyclin
inhibitors such as p16INK4a and p21cip1 also
promote cell cycle revolution. There are few reports on the
expression of cyclin inhibitors such as p16INK4a and
p21cip1 in NB; however, Diccianni et al reported
that, while alterations of p16INK4a at the genetic level
are rare, they confirmed the expression of p16 protein in
neuroblastoma cell lines and cited reduction in 12 of 19 cases
(63%) (15).
We analyzed p16INK4a expression in five
NB cell lines. Although p16INK4a expression was
confirmed by PCR in three of the five cell lines, western blotting
did not indicate p16INK4a protein expression (Fig. 1). Kondo et al reported that
introduction of p16INK4a abrogated phosphorylation of
retinoblastoma (RB), thereby suppressing cancer cell growth
(19). We conclude that
p16INK4a induces RB expression; RB phosphorylation
requires normal expression of CDK4, CDK6 and cyclin D, which
mediate the G1 checkpoint pathway. RB, pRB, CDK4, CDK6, and cyclin
D expression was observed by PCR and western blotting in all five
cell lines in which p16INK4a protein expression was
absent (Fig. 1). We believe the
evidence of pRB expression indicates a loss of cyclin inhibition in
NB, thus producing the observed loss of growth suppression.
Therefore, introduction of a cyclin inhibitor may suppress tumor
growth if it inhibits RB phosphorylation. This method may have
clinical therapeutic utility and differs from current therapies for
NB (chemotherapy and autologous stem cell transplantation).
We synthesized the smallest functional sequence of
p16INK4a and introduced it into NB cell lines using a
system for transporting peptides/proteins that was previously
established by our group (19–21).
This system produced significant growth suppression in all five
cell lines (Fig. 2). We used the
same nine-arginine sequence attached to the p16INK4a
peptide as described in the PTD system and observed a much greater
effect with the peptide/protein transporter system complex than
with the p16INK4a peptide alone. p16INK4a
inhibits the function of complexes that induce RB phosphorylation
(cyclin D, CDK4, and CDK6), thereby suppressing RB phosphorylation.
Indeed, RB phosphorylation was suppressed in p16INK4a
peptide-treated cells; we conclude the introduced peptide
functioned in place of the original p16INK4a molecule.
In a previous report on B-cell lymphoma, G1 arrest and apoptosis
occurred in p16INK4 peptide-treated cells (19). S-phase cells are also reduced in
NB, indicating that G1 arrest is occurring. While Annexin
V-positive cells increase over time, no Annexin
V-negative/PI-positive cells were observed in this study. We
concluded that p16INK4a peptide not only inhibits cell
growth but also induces tumor cell apoptosis (Figs. 4 and 5).
In vitro experiments have suggested the
feasibility of treating NB with peptides. We tested this treatment
in nude mouse models of transplanted human tumors derived from
SMS-KAN cell lines. Administration of a p16INK4a
peptide/transporter complex resulted in a high tumor suppression
effect at 14 days (75.6% inhibition vs. controls, p<0.0005).
However, on day 21, despite significant suppression in comparison
to controls, the effect size was reduced (65% inhibition compared
to the control group, p=0.024). Although extremely strong tumor
suppression was observed after only a single administration, the
tumor began to grow again; this suggests therapeutic efficacy is
limited after peptide inoculation. In a report on renal cell
carcinoma, multiple administrations produced prolonged therapeutic
efficacy and tumor regression. In a report on gliomas, combined
administration of p14 and p16INK4a produced a strong
antitumor effect (20). Based on
these reports, we believe it is feasible to strive for tumor
regression by optimizing combined therapies and number of
administrations.
The p16INK4a functional
peptide/transporter peptide complex described here was not
cell-specific. Its introduction into normal and tumor cells makes
toxicity a particular concern. In a report on B-cell lymphoma,
peptides were introduced into normal lymphocytes, but apoptosis was
not induced in these cells. In an experiment on gliomas and renal
cell carcinoma in mice, no H&E staining abnormalities were
observed in normal tissue surrounding the transplanted tumor or
other normal organs. We also observed no abnormalities in H&E
staining in the normal subcutaneous tissue surrounding the
transplanted tumor or other normal tissue (brain, lungs, heart,
liver, kidneys and spleen).
The introduction of a functional peptide using the
transporter developed by Kondo et al (19) was effective in NB treatment and had
almost no effect on normal cells, thus showing its therapeutic
promise for further development and clinical trials.
References
|
1.
|
Brodeur GM and Maris JM: Neuroblastoma.
Principle and Practice of Pediatric Oncology. 5th edition. Pizzo PA
and Poplack DG: Lippincott Williams & Wilkins; Philadelphia,
PA: pp. 993–970. 2006
|
|
2.
|
Matthay KK, Reynolds CP, Seeger RC, et al:
Long-term results for children with high-risk neuroblastoma treated
on a randomized trial of myeloablative therapy followed by
13-cis-retinoic acid: a children’s oncology group study. J Clin
Oncol. 27:1007–1013. 2009.PubMed/NCBI
|
|
3.
|
Matthay KK, Villablanca JG, Seeger RC, et
al: Treatment of high-risk neuroblastoma with intensive
chemotherapy, radiotherapy, autologous bone marrow transplantation,
and 13-cis-retinoic acid. N Engl J Med. 341:1165–1173. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Berthold F, Boos J, Burdach S, et al:
Myeloablative megatherapy with autologous stem-cell rescue versus
oral maintenance chemotherapy as consolidation treatment in
patients with high-risk neuroblastoma: a randomized controlled
trial. Lancet Oncol. 6:649–658. 2005. View Article : Google Scholar
|
|
5.
|
Kaneko M, Tsuchida Y, Mugishima H, et al:
Intensified chemotherapy increases the survival rate in patients
with stage 4 neuroblastoma with MYCN amplification. J Pediatr
Hematol Oncol. 24:613–621. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Brodeur GM, Seeger RC, Schwab M, Varmus HE
and Bishop JM: Amplification of N-myc in untreated human
neuroblastomas correlates with advanced disease stage. Science.
224:1121–1123. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Maris JM, Hogarty MD, Bagatell R and Cohn
SL: Neuroblastoma. Lancet. 369:2106–2120. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Chen Y, Takita J, Choi YL, et al:
Oncogenic mutations of ALK kinase in neuroblastoma. Nature.
455:971–974. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
George RE, Sanda T, Hanna M, et al:
Activating mutations in ALK provide a therapeutic target in
neuroblastoma. Nature. 455:975–978. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Janoueix-Lerosey I, Lequin D, Brugières L,
et al: Somatic and germline activating mutations of the ALK kinase
receptor in neuroblastoma. Nature. 455:967–970. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Mossé YP, Laudenslager M, Longo L, et al:
Identification of ALK as a major familial neuroblastoma
predisposition gene. Nature. 455:930–935. 2008.PubMed/NCBI
|
|
12.
|
Ogawa S: Shinkeigashu no genomu kaiseki ni
yoru ALK idenshi heni no dotei (Identification of ALK gene mutation
by neuroblastoma genome analysis). Exp Med. 27:60–68. 2009.
|
|
13.
|
Ghiorzo P, Gargiulo S, Pastorino L, et al:
Impact of E27X, a novel CDKN2A germ line mutation, on p16 and
p14ARF expression in Italian melanoma families displaying
pancreatic cancer and neuroblastoma. Hum Mol Genet. 15:2682–2689.
2006. View Article : Google Scholar
|
|
14.
|
Bassi CL, Martelli L, Cipolotti R,
Scrideli CA, Defávery R and Tone LG: Lack of evidence for mutations
or deletions in the CDKN2A/p16 and CDKN2B/p15 genes of Brazilian
neuroblastoma patients. Braz J Med Biol Res. 37:1683–1687. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Diccianni MB, Omura-Minamisawa M, Batova
A, Le T, Bridgeman L and Yu AL: Frequent deregulation of p16 and
the p16/G1 cell cycle-regulatory pathway in neuroblastoma. Int J
Cancer. 80:145–154. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Nagahara H, Vocero-Akbani AM, Snyder EL,
et al: Transduction of full-length TAT fusion proteins into
mammalian cells: TAT-p27Kip1 induces cell migration. Nat
Med. 4:1449–1452. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Derossi D, Jolint AH, Chassaing G, et al:
The third helix of the antennapedia homeodomain translocations
through biological membranes. J Biol Chem. 269:10444–10450.
1994.PubMed/NCBI
|
|
18.
|
Futaki S, Suzuki T, Ohashi W, et al:
Arginine-rich peptides, An abundant source of membrane-permeable
peptides having potential as carriers for intracellular protein
delivery. J Biol Chem. 276:5836–5840. 2001.
|
|
19.
|
Kondo E, Seto M, Yoshikawa K and Yoshino
T: Highly efficient delivery of p16 antitumor peptide into
aggressive leukemia/ lymphoma cells using a novel transporter
system. Mol Cancer Ther. 3:1623–1630. 2004.PubMed/NCBI
|
|
20.
|
Kondo E, Tanaka T, Miyake T, et al: Potent
synergy of dual antitumor peptides for growth suppression of human
glioblastoma cell lines. Mol Cancer Ther. 6:1461–1471. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Yoshikawa K, Kondo E, Seto M, et al:
Transport system for the biologically active peptides/proteins into
mammalian cells with transporter peptide. Cytometry Res. 16:25–32.
2006.
|
|
22.
|
Fahraeus R, Lain S, Ball KL, et al:
Characterization of the cyclin-dependent kinase inhibitory domain
of the INK4 family as a model for a synthetic tumour suppresspr
molecule. Oncogene. 16:587–596. 1998. View Article : Google Scholar : PubMed/NCBI
|