Introduction
Diffuse large B cell lymphoma (DLBCL) is the most
common diagnosed form of non-Hodgkin lymphoma (NHL). While cure
rates have increased, the lack of response to current standard
treatment and/or relapse leaves approximately 50% of patients
incurable (1,2). Many of the cytotoxic drugs used to
treat DLBCL induce caspase-dependent apoptosis, however, a
significant number of patients acquire resistance that is
associated with defective caspase-dependent apoptotic pathways
(3). Upregulation of the
BCL2 gene, due to either the t(14;18) translocation or
genomic BCL2 gain/amplification, results in overexpression
of the Bcl-2 protein and apoptosis resistance (2). The Bcl-2 family is a group of
mitochondrial-associated proteins that are characterized as either
anti-apoptotic or pro-apoptotic. The canonical function of Bcl-2
and other anti-apoptotic proteins, such as Bcl-xL and Mcl-1, is to
prevent mitochondrial outer membrane permeabilization (MOMP)
(4). Pro-apoptotic proteins, such
as BH3-only proteins, bind to and inhibit the function of
anti-apoptotic proteins to induce MOMP (5). There are several BH3 mimetic drugs
that are currently being developed and are in clinical trials
(6). While these drugs are
promising, resistance through the upregulation of Mcl-1 is a
problem (7). Due to their
different binding affinities, a combination of BH3 mimetics that
target different anti-apoptotic proteins would be necessary to
achieve an optimal therapeutic effect (6).
The non-canonical function of Bcl-2 and other
anti-apoptotic proteins is to maintain mitochondrial homeostasis by
regulating mitochondrial membrane potential
(ΔΨm) and/or mitochondrial respiration
(8–10). Regulation of these mitochondrial
processes may or may not contribute to the ability of
anti-apoptotic proteins to prevent apoptosis. In response to
cellular stress, Chen and Pervaiz showed that Bcl-2 alters the
activity of the copper-dependent enzyme cytochrome c oxidase
(CcOX), the terminal subunit of the mitochondrial respiratory chain
(11). Data from their study
suggest that the ability to modulate CcOX activity contributes to
the ability of Bcl-2 to inhibit caspase-dependent cell death. We
recently showed that ATN-224 (choline tetrathiomolybdate), a copper
chelator drug, inhibits CcOX, causing mitochondrial dysfunction.
Treatment with ATN-224 resulted in cell death in parental murine
thymic lymphoma cells and those transfected with Bcl-2 that are
apoptosis resistant (12). The
ability of ATN-224 to induce cell death in an isogenic cell model
overexpressing Bcl-2 led to the hypothesis that ATN-224 treatment
would be effective in DLBCL cells with upregulated Bcl-2.
In this study, we tested whether using ATN-224 to
target the non-canonical function of Bcl-2 and other anti-apoptotic
proteins could induce cell death in DLBCL cells independent of the
level of anti-apoptotic proteins. We show that nanomolar
concentrations of ATN-224 can induce caspase-independent cell death
via release of apoptosis inducing factor (AIF). ATN-224 also
enhanced the overall effect of the BH3 mimetic, ABT-263, in
apoptosis-resistant DLBCL. Taken together these data suggest that
ATN-224 has therapeutic potential as a single agent or as an
adjuvant, specifically in patients with apoptosis-resistant
disease.
Materials and methods
Drug treatments and reagents
ATN-224 was provided by Dr Andrew Mazar
(Northwestern University, Evanston, IL). ABT-263 and ABT-737 were
purchased from ChemieTek (Indianapolis, IN). ZVAD-FMK was purchased
from Enzo Life Sciences (Plymouth Meeting, PA). All other drugs and
chemicals were purchased from Sigma Chemical Co. (St. Louis, MO)
unless otherwise stated.
Cell lines
SUDHL-4, SUDHL-10, U-2932 cells and Granta 519 cells
were obtained from the Arizona Lymphoid Tissue and Blood Repository
(University of Arizona, Tucson, AZ). SUDHL8 and SUDHL4 R2 cells
were obtained from Dr Anthony Letai (Dana-Farber Cancer Institute,
Boston, MA). All cells were maintained in suspension in RPMI-1640
(Cellgro; Mediatech, Manassas, VA) supplemented with 10% fetal
bovine serum (Gemini, Sacramento, CA), 2 mM L-glutamine and 50 U/ml
each of penicillin and streptomycin (all from Invitrogen, Carlsbad,
CA) at 37°C in a 5% CO2 humidified environment. SUDHL-4
R2 cell cultures were supplemented with 5 μg/ml verapamil
and 1 μM ABT-737, as previously described (7).
Cell viability measurements
The number of viable treated cells, relative to
control treated cells, was measured after 72 h of treatment using
the Non-radioactive Cell Proliferation Assay (MTS) according to the
manufacturer’s instructions (Promega Corp., Madison, WI).
Absorbance was read at 490 nm using a Synergy HT plate reader
(BioTek Instruments, Winooski, VT). The MTS assay was used to
determine the estimated ATN-224 concentration needed to decrease
the number of viable cells by 50 (EC50) and 25%
(EC25). Viable cell number was also determined by
propidium iodide (PI) (Molecular Probes, Eugene, OR) uptake, as
previously described (12). PI
uptake was used to determine the effect of drugs alone or in
combination with ATN-224. Caspase 3 activity was measured using
Ac-DEVD-p-nitroanilide (pNA) (Enzo Life Sciences), as previously
described (13).
ΔΨm
The fluorescent probe JC-1 (Molecular Probes) was
used to measure ΔΨm. Cells were incubated
with 2 μg/ml JC-1 for 30 min at 37°C in a 5% CO2
humidified environment. Cells were then washed with PBS,
resuspended in PBS and transferred to a black well plate. JC-1
J-aggregates (Ex: 560/Em: 595) were measured using a Synergy HT
plate reader (Bio Tek Instruments). Fluorescence was normalized to
cellular protein.
Nuclear condensation
Following treatment, cells were transferred to
poly-L-lysine coated chamber slides. Chambers were removed from the
slide, coated with mounting medium containing Dapi (Vectashield;
Vector Laboratories, Burlingame, CA) and cover slips applied.
Slides were visualized using the Olympus Fluorview FV1000 Confocal
Microscope with FV10-ASW software (Olympus America Inc., San Jose,
CA).
Immunoblot analysis
Protein fractions isolated with the Mitochondrial
Isolation kit for Cultured Cells (Thermo Fisher Scientific,
Waltham, MA) or total cell lysates were separated by SDS-PAGE and
transferred to a PVDF membrane using standard protocols. Blots were
probed with antibodies for Mcl-1, AIF, COX IV (Cell Signaling,
Danvers, MA), cyto-chrome c, Bcl-2, Bcl-xL, Bak, Bax, Bim
(BD Pharmingen, San Diego, CA), Bid (Abcam, Cambridge, MA) or Noxa
(Imgenex, San Diego, CA). Proteins were detected with either
horseradish peroxidase-linked anti-rabbit Ig or horse-radish
peroxidase-linked anti-mouse Ig (Cell Signaling), where
appropriate, and visualized by chemiluminescence (Perkin-Elmer,
Waltham, MA). Blots were also probed with anti-β-actin (Abcam) as a
loading control. Restore Western Blot Stripping Buffer (Thermo
Scientific) was used to visualize multiple bands on the same blot.
Film was scanned, and images were cropped to show the bands of
interest then contrast adjustments were made to the cropped image.
Bands were quantified using Image J (NIH).
Statistics
Means were compared using Student’s t-test with the
algorithm in Excel (Microsoft Corp., Redmond, WA). Means were
considered significantly different when p≤0.05. When a comparison
required multiple t-tests, the Dunn-Bonferroni method was used to
control for type I error (14).
Results
Apoptosis-resistant cells are sensitive
to ATN-224
To determine the ability of ATN-224 to overcome
apoptosis-resistance, we characterized three DLBCL cell lines for
the anti-apoptotic proteins, Bcl-2, Bcl-xL and Mcl-1. The
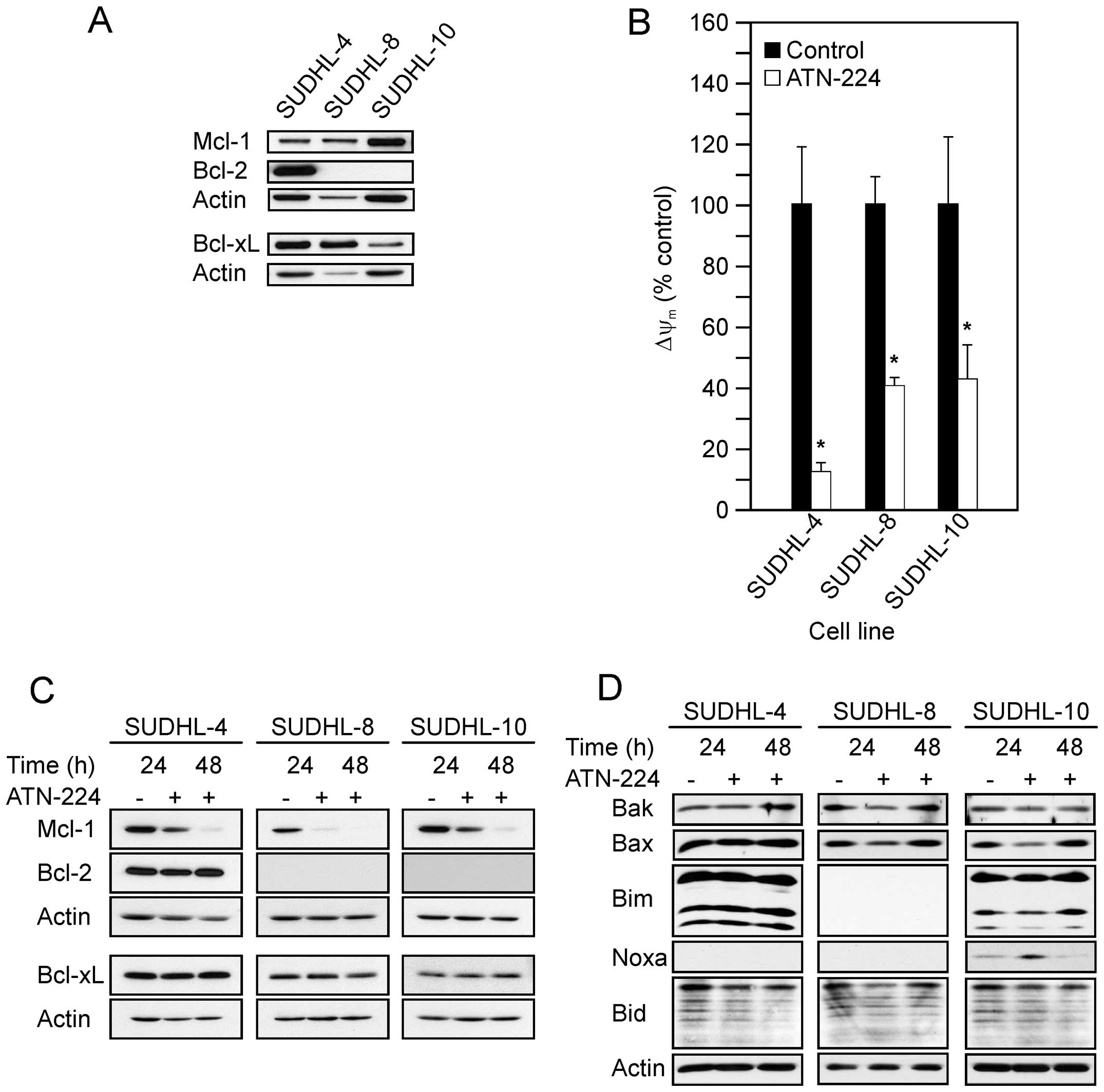
immunoblots in Fig. 1A show the
following: the SUDHL-4 had high levels of Bcl-2 and Bcl-xL, with
moderate levels of Mcl-1; the SUDHL-8 had high levels of Bcl-xL and
moderate levels of Mcl-1; the SUDHL-10 had high levels of Mcl-1 and
moderate levels of Bcl-xL. Taken together all three DLBCL cell
lines displayed various levels of anti-apoptotic proteins, which
contribute to apoptosis-resistance.
To establish whether the DLBCL cells were sensitive
to ATN-224, we measured cell viability following ATN-224 treatment.
In the SUDHL-4, SUDHL-8 and SUDHL-10 cells, nanomolar
concentrations of ATN-224 decreased the number of viable cells
(Table I). Recent studies suggest
that the protective function of Bcl-2, in part, is due to its
ability to regulate mitochondrial respiration (9). In previous studies we have shown that
ATN-224 inhibits CcOX and decreases ΔΨm in
murine thymic lymphoma cells that overexpress Bcl-2 (12). To determine whether ATN-224 is
targeting the mitochondria, we assessed
ΔΨm following ATN-224 treatment. In the
SUDHL-4, SUDHL-8 and SUDHL-10 cells, ATN-224 treatment
significantly decreased ΔΨm at 12 h
(Fig. 1B).
 | Table I.ATN-224 sensitivity. |
Table I.
ATN-224 sensitivity.
| NHL cell line | ATN-224
EC50 (nM) | ATN-224
EC25 (nM) |
|---|
| DLBCL | | |
| SUDHL-4 | 105.61±7.39 | 53.56±4.14 |
| SUDHL-8 | 9.73±0.84 | 4.94±0.64 |
| SUDHL-10 | 32.71±2.04 | 15.16±1.33 |
| U2932 | 29.06±0.22 | 15.24±0.50 |
| MCL | | |
| Granta 519 | 72.77±1.51 | N/A |
In previous studies, we have shown that ATN-224
inhibits superoxide dismutase 1 (SOD1), a copper-dependent enzyme
responsible for the detoxification of superoxide, resulting in
increased levels of superoxide (12). An increase in oxidants causes Bcl-2
degradation through the ubiquitin-proteasomal pathway in other cell
types (15). In response to
ATN-224 treatment, Bcl-2, Bcl-xL and Mcl-1 are unable to maintain
mitochondrial homeostasis, as indicated by the decrease in
ΔΨm, suggesting that they may be indirect
targets of ATN-224. To characterize the effect of ATN-224 on Bcl-2
and other anti-apoptotic proteins, we measured Bcl-2, Bcl-xL and
Mcl-1 protein levels following ATN-224 treatment. In the SUDHL-4,
SUDHL-8 and SUDHL-10 cells, we detected decreases in Mcl-1, but no
change in Bcl-2 or Bcl-xL protein levels (Fig. 1C). Taken together these results
indicate that ATN-224 treatment induces mitochondrial dysfunction,
independent of Bcl-2, Bcl-xL and Mcl-1 status, which may contribute
to the ATN-224 sensitivity of apoptosis-resistant cells.
The decision to undergo apoptosis is influenced by
the balance between the anti- and pro-apoptotic Bcl-2 family
members. To determine whether, in addition to the loss of Mcl-1,
ATN-224 treatment increased pro-apoptotic Bcl-2 family member
proteins we measured the levels of the pro-apoptotic proteins Bak,
Bax, Bim, Noxa and Bid. As shown in Fig. 1D, we did not see a consistent
increase in any of the pro-apoptotic proteins we measured across
the cell types and in several cases there was a decrease, e.g., Bax
in the SUDHL-8 and SUDHL-10 cells. We did see an increase in Noxa
after a 24 h-ATN-224 treatment in the SUDHL-10; Noxa was
undetectable in the other two cell types. These data suggest that
the primary effect of ATN-224 on Bcl-2 family member protein levels
is a decrease in Mcl-1. A decrease in Mcl-1 could tip the balance
toward apoptosis.
ATN-224 induces caspase-independent cell
death in apoptosis-resistant cells
The intrinsic apoptotic pathway involves the
formation of MOMP and the release of cytochrome c from the
mitochondria, which leads to caspase 3 activation and cell death
(5). The upregulation of
anti-apoptotic proteins, such as Bcl-2, Bcl-xL and Mcl-1, prevent
the formation of MOMP (4). To
determine whether ATN-224 treatment results in apoptosis, we
measured cytochrome c release from the mitochondria and
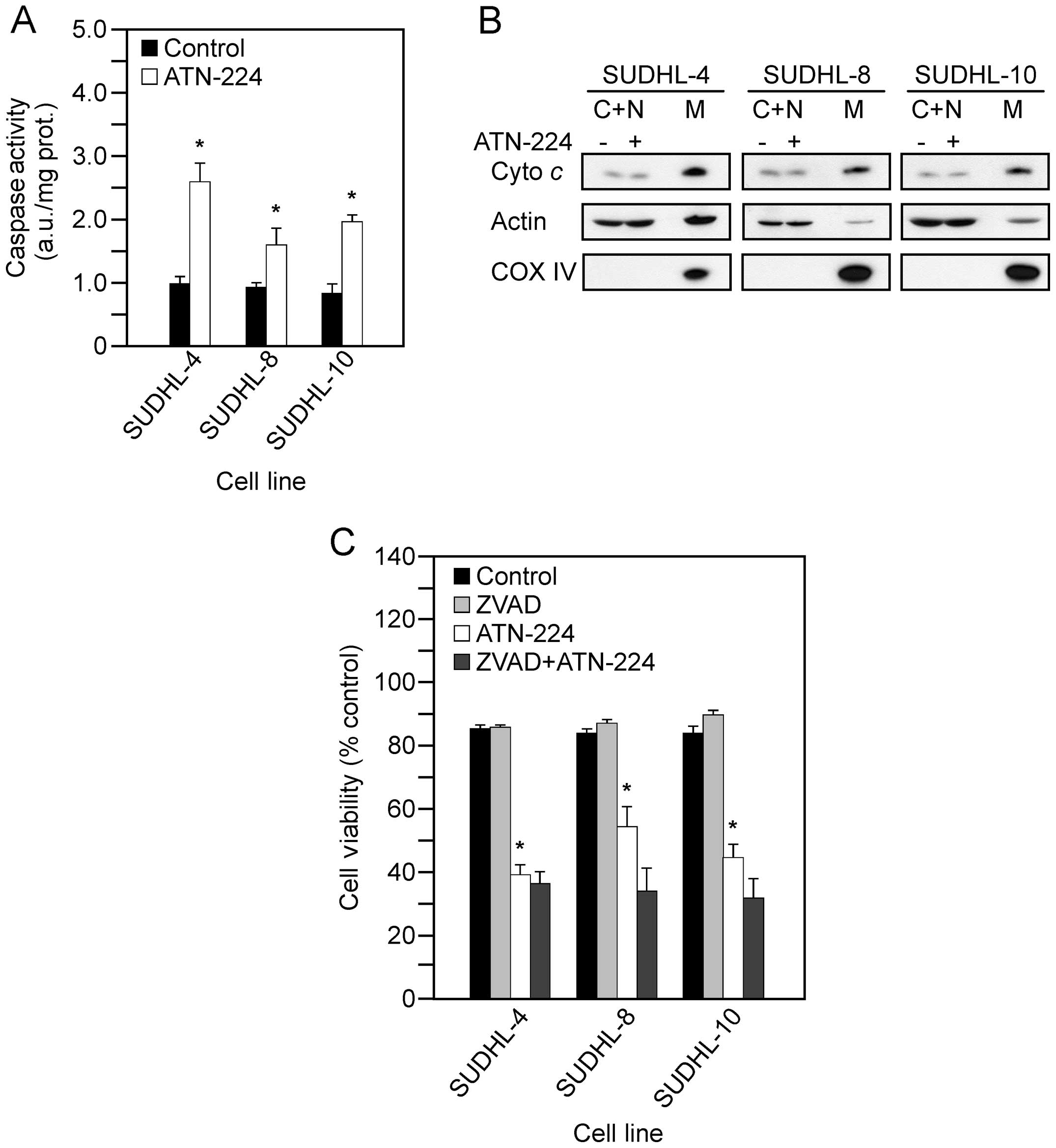
caspase 3 activity following ATN-224 treatment. In the SUDHL-4,
SUDHL-8, SUDHL-10 cells, we measured significant increases in
caspase 3 activity (Fig. 2A), but
detected no significant increase in cytochrome c release
from the mitochondria (Fig. 2B).
To determine whether ATN-224 induced cell death is
caspase-dependent, we used the pan caspase inhibitor, ZVAD-FMK, in
combination with ATN-224 and measured cell viability. In the
SUDHL-4, SUDHL-8 and SUDHL-10 cells, the addition of ZVAD-FMK was
unable to attenuate the effect of ATN-224 (Fig. 2C). These results indicate that
ATN-224 induced cell death is caspase-independent.
ATN-224 induces AIF release and nuclear
condensation in apoptosis-resistant cells
We have shown that ATN-224 targets the mitochondria
and induces caspase-independent cell death. The mitochondria
contain other death inducing proteins, such as apoptosis inducing
factor (AIF). The release of AIF from the mitochondria results in
the activation of caspases and nuclear condensation, however,
AIF-induced cell death is caspase-independent (16). To determine whether AIF is involved
in ATN-224-induced cell death, we measured AIF release from the
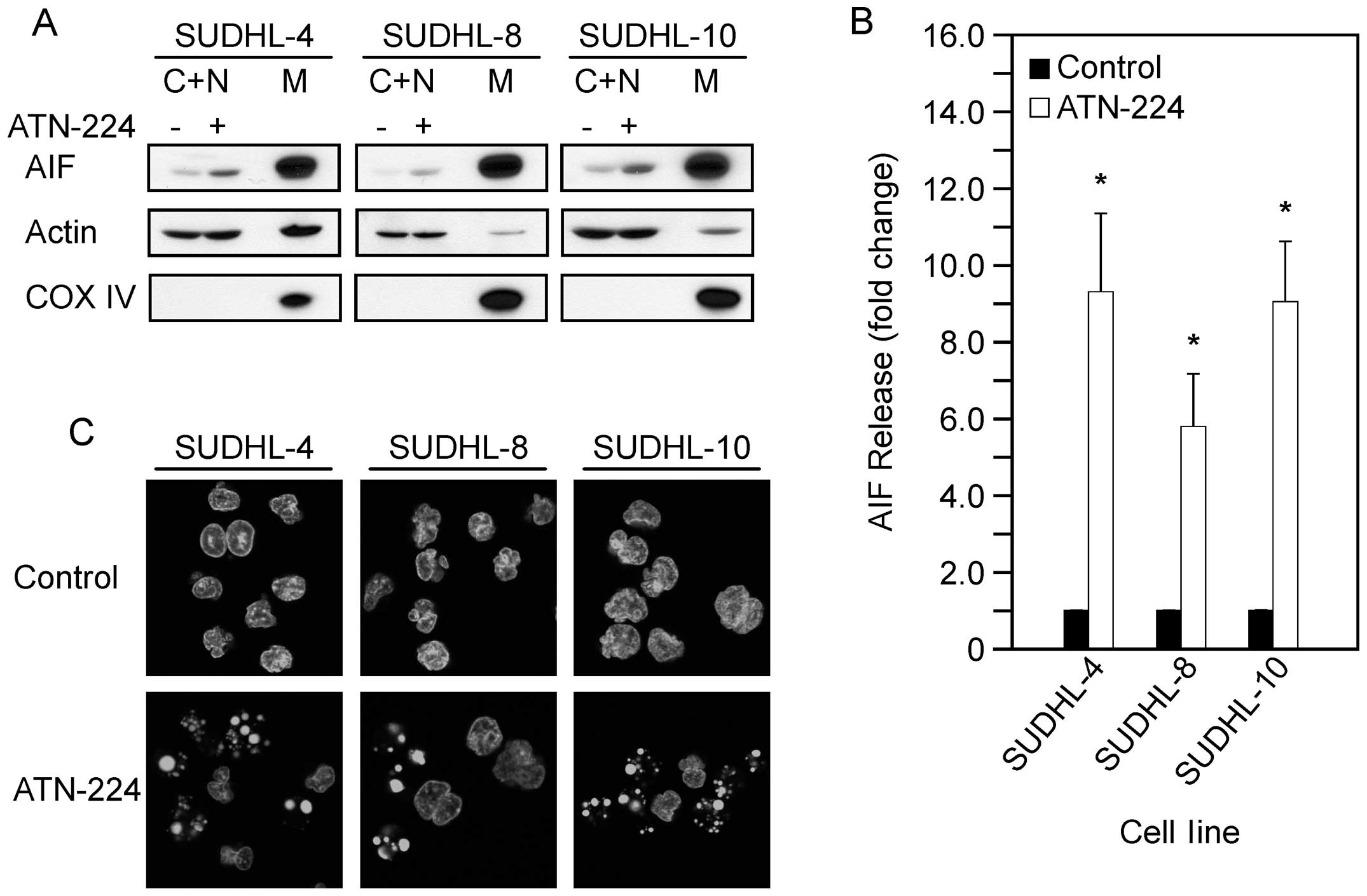
mitochondria following ATN-224 treatment. In the SUDHL-4, SUDHL-8
and SUDHL-10 cells, we detected a significant increase in AIF
release from the mitochondria (Fig. 3A
and B). To further assess the involvement of AIF, we looked for
nuclear condensation. In the SUDHL-4, SUDHL-8 and SUDHL-10 cells,
we detected nuclear condensation following ATN-224 treatment
(Fig. 3C). These results indicate
that ATN-224 induces AIF release and nuclear condensation. These
data suggest that ATN-224 induces cell death via a mechanism that
circumvents apoptosis resistance.
ATN-224 induces cell death in aggressive
lymphomas
The t(14;18) translocation or genomic
gain/amplification causes BCL2 upregulation. BCL2
translocations are commonly associated with follicular lymphoma and
the DLBCL germinal center B-cell-like (GBC) subtype (17). BCL2 gain/amplification are
commonly associated with the DLBCL activated B-cell-like (ABC)
subtype and mantle cell lymphoma (MCL), two aggressive types of NHL
(18,19). We have shown that ATN-224 treatment
circumvents Bcl-2 overexpression and induces death in the SUDHL-4
cells, which have the t(14;18) translocation (20). To determine whether our findings
extend to aggressive NHLs that have BCL2 gain/amplification,
we used the U-2932 and Granta 519, a DLBCL and an MCL cell line,
respectively, which have BCL2 gain/amplification (21,22).
We first measured cell viability to establish whether the U-2932
and Granta 519 cells were sensitive to ATN-224 (Table I). To determine whether ATN-224
treatment had an effect in the U-2932 and Granta 519 cells similar
to the effect in the SUDHL-4, SUDHL-8 and SUDHL-10 cells, we
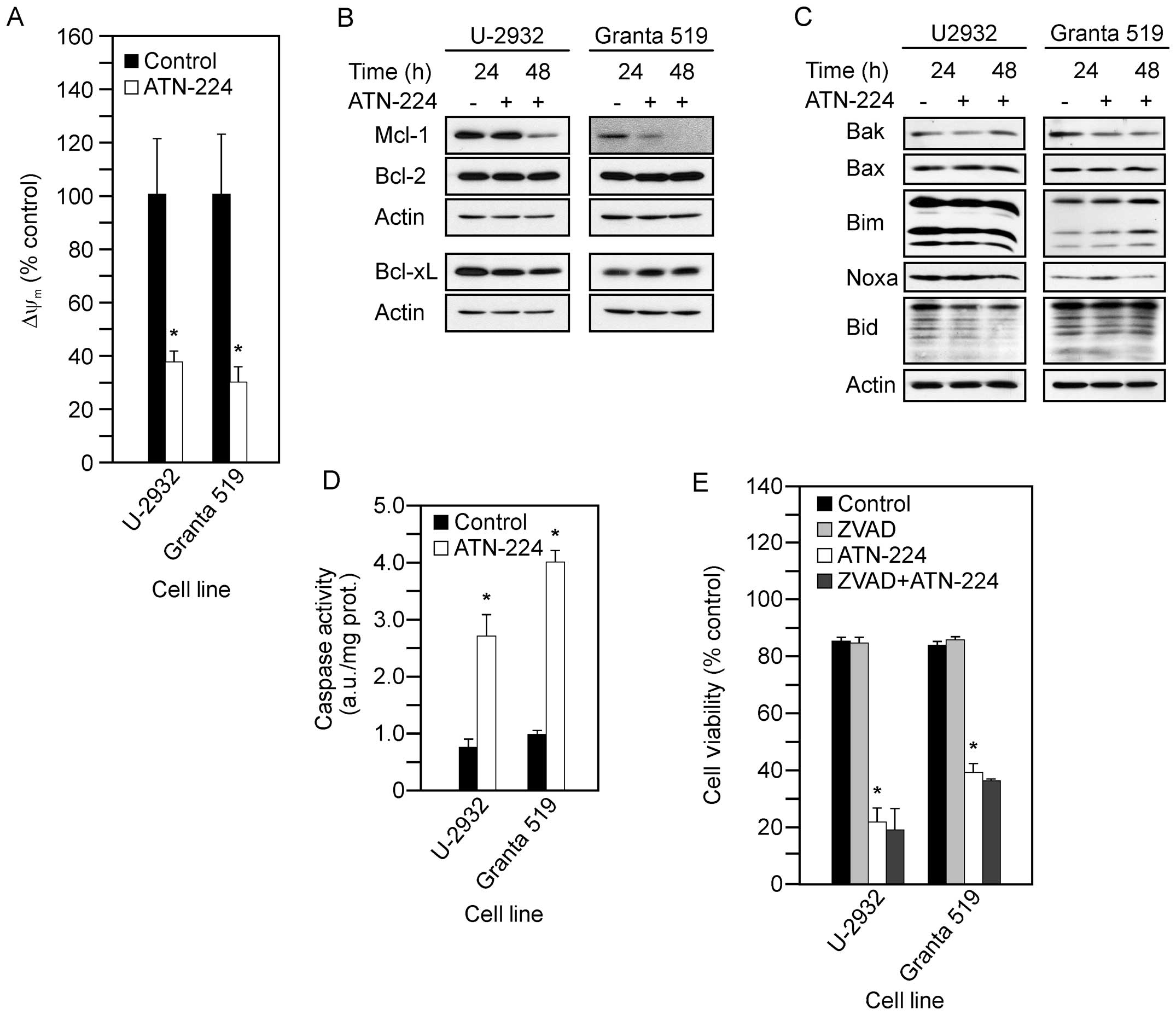
measured the following: ΔΨm; Mcl-1, Bcl-2 and
Bcl-xL protein levels; pro-apoptotic Bcl-2 family member protein
levels; and caspase 3 activity. Following ATN-224 treatment in the
U-2932 and Granta 519 cells, we detected: i) decreases in
ΔΨm at 12 h (Fig.
4A); ii) decreases in Mcl-1 and no change in Bcl-2 or Bcl-xL
protein levels (Fig. 4B); iii) no
consistent change in pro-apoptotic Bcl-2 family member protein
levels, although similar to the SUDHL-10 cells, Granta 519 cells
showed a slight increase in Noxa after 24 h (Fig. 4C); and significant increases in
caspase 3 activity (Fig. 4D). To
confirm that the ATN-224-induced cell death was
caspase-independent, we measured cell viability using the
pan-caspase inhibitor, ZVAD-FMK, in combination with ATN-224. In
both the U-2932 and Granta 519 cells, the addition of ZVAD-FMK was
unable to attenuate the effect of ATN-224 (Fig. 4E). Taken together, these data
suggest ATN-224 circumvents the overexpression of Bcl-2, regardless
of the mechanism by which BCL2 is upregulated, and has
therapeutic potential in the treatment of aggressive NHL.
ATN-224 induces cell death in
ABT-737-resistant cells and enhances the effect of ABT-263
Drugs targeting Bcl-2 and Bcl-xL, such as ABT-737
and ABT-263 derivatives, are currently in clinical trials. Yecies
et al showed that SUDHL-4 R2 cells, selected for resistance
to ABT-737, upregulate Mcl-1 (7).
The ability of ATN-224 to degrade Mcl-1 suggests that the SUDHL-4
R2 cells may be sensitive to ATN-224. To determine the effect of
ATN-224 on SUDHL-4 R2 cells, we measured cell viability following
treatment with various concentrations of ATN-224. In the SUDHL-4
and SUDHL4-R2 cells, we measured a significant decrease in the
number of viable cells, which appears to be concentration-dependent
(Fig. 5A). We also detected
decreases in Mcl-1 protein levels (Fig. 5B). These data suggest ATN-224 has
the potential to overcome Mcl-1 resistance, thus sensitizing cells
to drugs that target Bcl-2 and Bcl-xL.
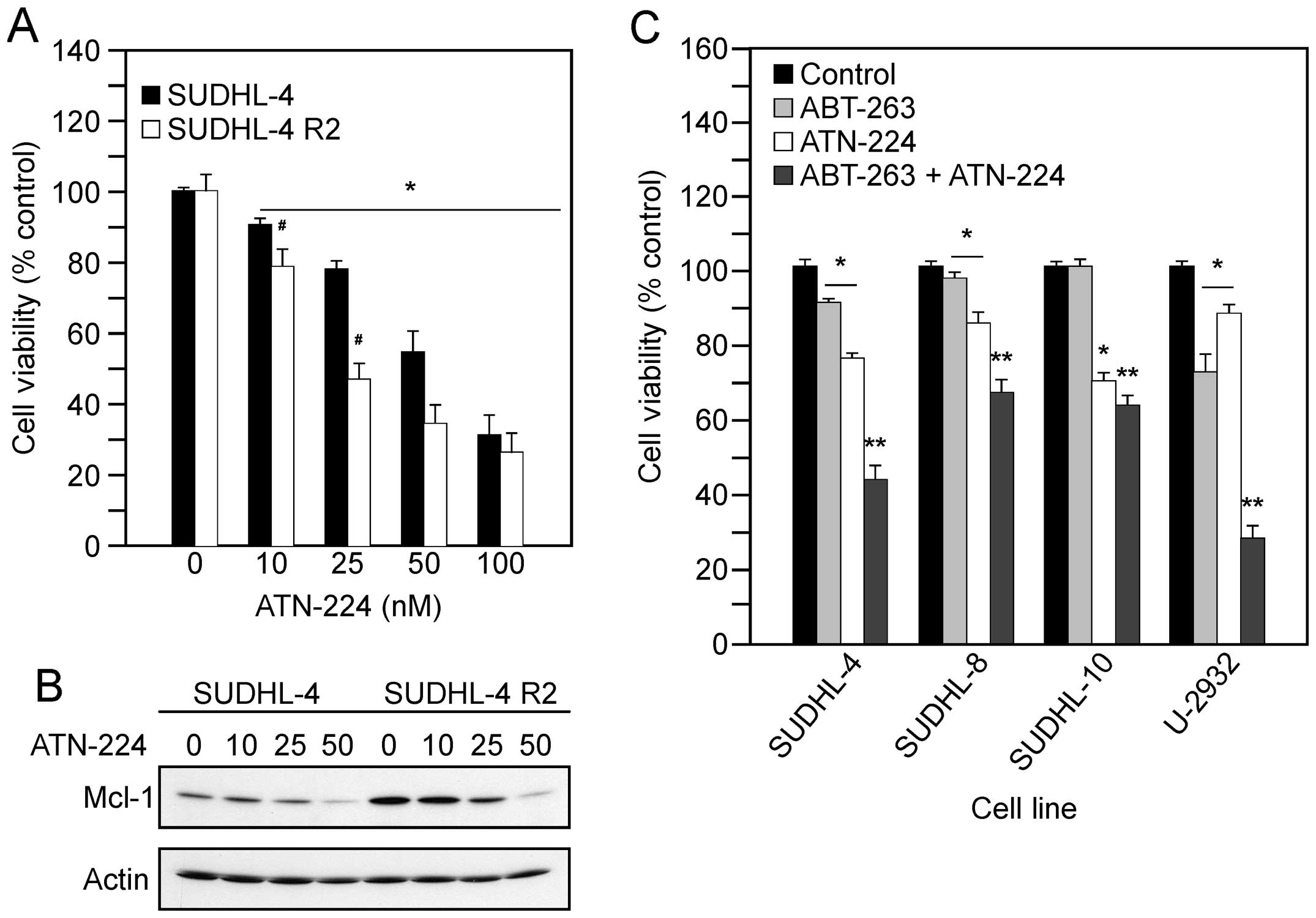 | Figure 5.ATN-224 enhances the effect of BH3
mimetics. (A) Cell viability in SUDHL-4 and SUDHL-4 R2 cells
treated with vehicle or ATN-224 (10, 25, 50 and 100 nM) for 72 h.
(B) Immunoblot showing Mcl-1 protein levels in SUDHL-4 and SUDHL-4
R2 cells treated with vehicle or ATN-224 (10, 25 and 50 nM) for 24
h. Immunoblot showing actin protein levels to demonstrate similar
loading. All immunoblots are representative blots from three
independent sample collections. (C) Cell viability in SUDHL-4,
SUDHL-8, SUDHL-10 and U-2932 cells treated with vehicle, 250 nM
ABT-263, ATN-224 (EC25) or a combination of ABT-263 and
ATN-224 for 72 h. All values are mean ± SEM (n ≥3).
*p≤0.05, significantly different from vehicle treated
control cells. **p≤0.01, significantly different from
ABT-263 or ATN-224 treated cells. #p≤0.05, significantly
different from SUDHL-4 ATN-224 treated cells. |
The ability of ATN-224 to induce cell death
independent of Bcl-2/Bcl-xL status and degrade Mcl-1 suggests that
ATN-224 has potential as an adjuvant to ABT-263 treatment. To
determine whether ATN-224 enhances the effect of ABT-263, we
combined low concentrations of ATN-224 with a low concentration of
ABT-263. The combination of ATN-224 with ABT-263 resulted in an
enhanced effect, in comparison to either drug alone, especially in
those with high levels of Bcl-2 (Fig.
5C). These results suggest ATN-224 has potential as an adjuvant
to drugs that target Bcl-2 and Bcl-xL.
Discussion
Our data suggest that use of a copper chelator drug
to target the mitochondria has potential as a therapeutic strategy
to overcome apoptosis resistance and induce caspase-independent
cell death in DLBCL. In cells with high levels of Bcl-2, Bcl-xL or
Mcl-1, ATN-224 treatment causes mitochondrial dysfunction and
induces the release of AIF from the mitochondria, resulting in
nuclear condensation. ATN-224 treatment enhances the effect of
ABT-263, which may be attributed to the ability of ATN-224 to
degrade Mcl-1. These data suggest that ATN-224 has potential as an
adjuvant with drugs that target Bcl-2 and Bcl-xL. The ability of
ATN-224 to trigger caspase-independent cell death is an attractive
alternative approach, either as a single agent or as an adjuvant,
in the treatment of patients with defective caspase-dependent
apoptotic pathways.
Targeting the non-canoncial function of Bcl-2 and
other anti-apoptotic proteins is a successful strategy for
circumventing apoptosis-resistance. Chen and Pervaiz showed that in
response to cellular stress, cells with upregulated Bcl-2 maintain
mitochondrial homeostasis, in comparison to cells without
upregulated Bcl-2, by regulating the activity of CcOX (11). Other anti-apoptotic proteins, such
as Bcl-xL and Mcl-1, have also been shown to maintain mitochondrial
homeostasis by regulating mitochondrial respiration (8). The data suggest that the ability of
these proteins to prevent cell death may be attributed to their
ability to maintain mitochondrial homeostasis. CcOX, the terminal
subunit of the mitochondrial respiratory chain, tightly controls
ΔΨm and is a target of ATN-224 (13,23,24).
Here we show that ATN-224 treatment decreases
ΔΨm, independent of Bcl-2, Bcl-xL or Mcl-1
status, thus affecting their ability to maintain homeostasis.
Recently, Ni Chonghaile et al showed that decreases in
ΔΨm, or mitochondrial ‘priming’, describes the
proximity to death and correlates with better response and outcome
(25). Our data suggest that the
ability of ATN-224 to target the mitochondria and induce
mitochondrial dysfunction is an attractive alternative approach to
circumvent apoptosis-resistance and enhance the efficacy of
cytotoxic agents, such as doxorubicin, vincristine or etoptoside
(25).
Our data suggest that ATN-224 does not target the
canonical function of the anti-apoptotic Bcl-2 family members.
ATN-224 treatment results in loss of Mcl-1 either via inhibiting
synthesis or promoting degradation via induction of Noxa, as seen
in two of the cell types; Noxa specifically binds Mcl-1 resulting
in degradation of the Mcl-1/Noxa complex (26). However, the release of cytochrome
c or caspase dependence of the cell death mechanism have not
been reported. Release of cytochrome c into the cytoplasm is
a two step process (27). It
requires release of cytochrome c from the outer face of the
inner mitochondrial membrane into the intermembrane space and
movement of cytochrome c into the cytoplasm through a pore
formed in the outer membrane. Although the outer membrane may be
compromised by ATN-224 treatment, our data on the reactive oxygen
species-dependence of ATN-224-induced cell death suggest a
mechanism by which cytochrome c would not be released into
the intermembrane space. Release of cytochrome c from the
inner membrane requires the peroxidatic activity of cytochrome
c in the presence of H2O2 (28). ATN-224 inhibits SOD1, which
converts superoxide to H2O2, resulting in
increased superoxide and a decrease in H2O2
(data not shown). We have shown that the increased superoxide forms
peroxynitrite and that ATN-224 induced cell death is
peroxynitrite-dependent (12).
Peroxynitrite forms nitrotyrosine residues on target proteins. In
the presence of H2O2, cytochrome c is
nitrated on Tyr74 which results in increased peroxidatic activity
and triggers cytochrome c release and apoptosome formation
(29). Under conditions of low
H2O2, nitration occurs on alternate tyrosine
residues resulting in cytochrome c that is unable to trigger
downstream apoptotic events (29)
and may quench the peroxidatic activity of cytochrome c
(30).
Lack of cytochrome c release combined with
the caspase-independence of cell death indicates that ATN-224
triggers cell death by a mechanism other than traditional
apoptosis. The inability of cathepsin B, D and L inhibitors
[indicators of lysosomal-induced cell death pathways (31)] or calpain inhibitors to attenuate
cell death (data not shown) combined with release of AIF suggests
that ATN-224 induces an alternative form of cell death. AIF is a
mitochondrial-localized flavoprotein that translocates to the
nucleus where it induces chromatin condensation and DNA degradation
in a caspase-independent manner (32). AIF release can increase caspase
activity; however, the AIF-induced cell death is
caspase-independent (16). The
expression of AIF is relatively high in DLBCL and is associated
with a more favorable overall survival (OS) in patients treated
with CHOP-like therapy (33). In
this study, we found that ATN-224-induced cell death involves the
release of AIF from the mitochondria. Our data suggest a mechanism
by which ATN-224 could trigger AIF release. The mitochondrial
permeability transition (MPT) pore is susceptible to oxidation by
peroxynitrite, which could lead to the opening of the pore,
allowing the release of AIF (34).
The upregulation of anti-apoptotic proteins results in defective
caspase-dependent apoptotic pathways. Many of the cytotoxic drugs
used to treat DLBCL induce caspase-dependent apoptosis (3). The ability of ATN-224 to induce cell
death via the release of AIF from the mitochondria suggests that
patients with tumors that have defective caspase-dependent pathways
could benefit from ATN-224 treatment.
Gene-deletion studies have demonstrated that the
expression of Mcl-1 is critical to survival (35) and has been shown to correlate with
high-grade follicular lymphomas, mantle cell lymphoma and DLBCL,
predominately the ABC subtype (36–38).
BH3 mimetics that target anti-apoptotic proteins are currently
being developed and are in clinical trials (39). While those that target Bcl-2 and
Bcl-xL have shown promise, resistance through the upregulation of
Mcl-1 is a problem (7). BH3
mimetics that target Mcl-1 have been less successful due to their
different binding affinities (6).
Taken together, these data suggest that an alternative approach to
target Mcl-1 is needed. In cells, Mcl-1 is tightly regulated and
inactivated by different mechanisms that depend on the stimulus.
For example, in response to oxidative stress, phosphorylation by
JNK results in the loss of survival function; in dying cells,
caspase-mediated cleavage results in the generation of a potent
pro-apoptotic protein; and in response to genotoxic stress,
poly-ubiquitylation results in proteasome-dependent degradation
(40,41). Induction of Noxa is also a strategy
to increase Mcl-1 degradation (26). While the mechanism by which ATN-224
treatment degrades Mcl-1 remains to be tested, our data suggest
that ATN-224 could prove effective in tumors with increased
Mcl-1.
In addition to BCL2, the upregulation of many
other oncongenes, such as NF-κB, MYC and BCL6, occur
in DLBCL and are associated with poor clinical outcome (42–45).
The constitutive activation of NF-κB occurs in the more aggressive
DLBCL ABC subtype (2). NF-κB is a
redox sensitive transcription factor with known anti-apoptotic
target genes, such as BCL2 and BCLXL (46). A study by Pan et al has
shown that ammonium tetrathiomolybdate treatment suppresses NF-κB
transcription and decreases nuclear protein binding to the κB
sequence (27). This suggests that
NF-κB may be a target of ATN-224, however, this remains to be
tested. The upregulation of BCL2 can present with other
complex karyotypes, which include upregulated MYC and/or
BCL6, resulting in what is referred to as ‘double-hit’ and
‘triple-hit’ DLBCL (47). Double
hit DLBCL with upregulated BCL2 and MYC are
characterized by highly aggressive clinical behavior and poor
response to therapy (48). While
double hit and triple hit DLBCL are rare, one retrospective study
reported median survival of 6 and 4 months, respectively (49). Recently Quentmeier et al
(21) reported that the U-2932
cells are actually two clones in one cell line. While both clones
overexpress BCL2, one clone overexpresses MYC and the
other BCL6 (21). The
sensitivity of the U-2932 cells to ATN-224 suggests that ATN-224
may also prove effective in tumors with increased MYC and/or
BCL6.
In conclusion, our data indicate that ATN-224 has
potential for the treatment of DLBCL. ATN-224 induces cell death in
DLBCL cell lines that represent phenotypes that show
characteristics of drug resistance and correlate with poor clinical
outcome. The mechanism of action is different from many of the
currently used therapies, which suggests that it could be used as a
single agent or as an adjuvant in refractory disease.
Tetrathiomolybdate has been used in clinical trials for the
treatment of Wilson disease, a copper transport disorder (50), and for several tumor types other
than lymphoma [(51–54) and references therein]. Our data
combined with the safety data in the published clinical trials
supports pursuing ATN-224 as a potential DLBCL
chemotherapeutic.
Acknowledgements
We thank Amanda Bahe for technical
assistance and Dr Anthony Letai (Dana-Farber Cancer Institute) for
the SUDHL-8 and SUDHL-4R2 cells. This study was supported by the
National Cancer Institute Grants CA09213 (K.L.), CA71768 (M.M.B.),
CA130805 (K.L., M.M.B., M.E.T.) and CA023074 (M.E.T.). A.P.M. was
supported by U01 CA151461-02, P50 HL 107186-01 and H Foundation
Funds. A.P.M. is a consultant to Wilson Therapeutics AB who is
developing ATN-224 for Wilson disease and has a small amount of
equity in the company. All other authors declare no conflict of
interest.
References
|
1.
|
Coiffier B: State-of-the-art therapeutics:
diffuse large B-cell lymphoma. J Clin Oncol. 23:6387–6393. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Lenz G and Staudt LM: Aggressive
lymphomas. N Engl J Med. 362:1417–1429. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Bosch R, Dieguez-Gonzalez R, Cespedes MV,
et al: A novel inhibitor of focal adhesion signaling induces
caspase-independent cell death in diffuse large B-cell lymphoma.
Blood. 118:4411–4420. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Low IC, Kang J and Pervaiz S: Bcl-2: a
prime regulator of mitochondrial redox metabolism in cancer cells.
Antioxid Redox Signal. 15:2975–2987. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Elkholi R, Floros KV and Chipuk JE: The
role of BH3-only proteins in tumor cell development, signaling, and
treatment. Genes Cancer. 2:523–537. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Kang MH and Reynolds CP: Bcl-2 inhibitors:
targeting mitochondrial apoptotic pathways in cancer therapy. Clin
Cancer Res. 15:1126–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Yecies D, Carlson NE, Deng J and Letai A:
Acquired resistance to ABT-737 in lymphoma cells that up-regulate
MCL-1 and BFL-1. Blood. 115:3304–3313. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Andersen JL and Kornbluth S: Mcl-1 rescues
a glitch in the matrix. Nat Cell Biol. 14:563–565. 2012. View Article : Google Scholar
|
|
9.
|
Krishna S, Low IC and Pervaiz S:
Regulation of mitochondrial metabolism: yet another facet in the
biology of the oncoprotein Bcl-2. Biochem J. 435:545–551. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Vander Heiden MG, Chandel NS, Williamson
EK, Schumacker PT and Thompson CB: Bcl-xL regulates the membrane
potential and volume homeostasis of mitochondria. Cell. 91:627–637.
1997.PubMed/NCBI
|
|
11.
|
Chen ZX and Pervaiz S: Involvement of
cytochrome c oxidase subunits Va and Vb in the regulation of cancer
cell metabolism by Bcl-2. Cell Death Differ. 17:408–420. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Lee K, Briehl MM, Mazar AP, et al: The
copper chelator ATN-224 induces peroxynitrite-dependent cell death
in hematological malignancies. Free Radic Biol Med. 60:157–167.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Jaramillo MC, Frye JB, Crapo JD, Briehl MM
and Tome ME: Increased manganese superoxide dismutase expression or
treatment with manganese porphyrin potentiates
dexamethasone-induced apoptosis in lymphoma cells. Cancer Res.
69:5450–5457. 2009. View Article : Google Scholar
|
|
14.
|
Myers JL and Well AD: Research design and
statistical analysis. 2nd edition. Lawrence Erlbaum Associates;
Mahwah, NJ: pp. 244–246. 2003
|
|
15.
|
Azad N, Iyer AK, Manosroi A, Wang L and
Rojanasakul Y: Superoxide-mediated proteasomal degradation of Bcl-2
determines cell susceptibility to Cr(VI)-induced apoptosis.
Carcinogenesis. 29:1538–1545. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Cregan SP, Dawson VL and Slack RS: Role of
AIF in caspase-dependent and caspase-independent cell death.
Oncogene. 23:2785–2796. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Shaffer AL, Rosenwald A and Staudt LM:
Lymphoid malignancies: the dark side of B-cell differentiation. Nat
Rev Immunol. 2:920–932. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Lenz G, Wright GW, Emre NC, et al:
Molecular subtypes of diffuse large B-cell lymphoma arise by
distinct genetic pathways. Proc Natl Acad Sci USA. 105:13520–13525.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Bea S, Salaverria I, Armengol L, et al:
Uniparental disomies, homozygous deletions, amplifications, and
target genes in mantle cell lymphoma revealed by integrative
high-resolution whole-genome profiling. Blood. 113:3059–3069. 2009.
View Article : Google Scholar
|
|
20.
|
Robetorye RS, Bohling SD, Morgan JW,
Fillmore GC, Lim MS and Elenitoba-Johnson KS: Microarray analysis
of B-cell lymphoma cell lines with the t(14;18). J Mol Diagn.
4:123–136. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Quentmeier H, Amini RM, Berglund M, et al:
U-2932: two clones in one cell line, a tool for the study of clonal
evolution. Leukemia. 27:1155–1164. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Pfreundschuh M, Trumper L, Kloess M, et
al: Two-weekly or 3-weekly CHOP chemotherapy with or without
etoposide for the treatment of elderly patients with aggressive
lymphomas: results of the NHL-B2 trial of the DSHNHL. Blood.
104:634–641. 2004. View Article : Google Scholar
|
|
23.
|
Pacelli C, Latorre D, Cocco T, Capuano F,
Kukat C, Seibel P and Villani G: Tight control of mitochondrial
membrane potential by cytochrome c oxidase. Mitochondrion.
11:334–341. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Juarez JC, Betancourt O Jr, Pirie-Shepherd
SR, et al: Copper binding by tetrathiomolybdate attenuates
angiogenesis and tumor cell proliferation through the inhibition of
superoxide dismutase 1. Clin Cancer Res. 12:4974–4982. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Ni Chonghaile T, Sarosiek KA, Vo TT, et
al: Pretreatment mitochondrial priming correlates with clinical
response to cytotoxic chemotherapy. Science. 334:1129–1133.
2011.PubMed/NCBI
|
|
26.
|
Weber A, Auslander D and Hacker G: Mouse
Noxa uses only the C-terminal BH3-domain to inactivate Mcl-1.
Apoptosis. 18:1093–1105. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Pan Q, Kleer CG, van Golen KL, et al:
Copper deficiency induced by tetrathiomolybdate suppresses tumor
growth and angiogenesis. Cancer Res. 62:4854–4859. 2002.PubMed/NCBI
|
|
28.
|
Ott M, Robertson JD, Gogvadze V,
Zhivotovsky B and Orrenius S: Cytochrome c release from
mitochondria proceeds by a two-step process. Proc Natl Acad Sci
USA. 99:1259–1263. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Radi R: Protein tyrosine nitration:
biochemical mechanisms and structural basis of functional effects.
Acc Chem Res. 46:550–559. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Vlasova II, Tyurin VA, Kapralov AA, et al:
Nitric oxide inhibits peroxidase activity of cytochrome c:
cardiolipin complex and blocks cardiolipin oxidation. J Biol Chem.
281:14554–14562. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Aits S and Jaattela M: Lysosomal cell
death at a glance. J Cell Sci. 126:1905–1912. 2013. View Article : Google Scholar
|
|
32.
|
Modjtahedi N, Giordanetto F, Madeo F and
Kroemer G: Apoptosis-inducing factor: vital and lethal. Trends Cell
Biol. 16:264–272. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Troutaud D, Petit B, Bellanger C, et al:
Prognostic significance of BAD and AIF apoptotic pathways in
diffuse large B-cell lymphoma. Clin Lymphoma Myeloma Leuk.
10:118–124. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Szabo C, Ischiropoulos H and Radi R:
Peroxynitrite: biochemistry, pathophysiology and development of
therapeutics. Nat Rev Drug Discov. 6:662–680. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Perciavalle RM, Stewart DP, Koss B, et al:
Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and
couples mitochondrial fusion to respiration. Nat Cell Biol.
14:575–583. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Cho-Vega JH, Rassidakis GZ, Admirand JH,
et al: MCL-1 expression in B-cell non-Hodgkin’s lymphomas. Hum
Pathol. 35:1095–1100. 2004.
|
|
37.
|
Wenzel SS, Grau M, Mavis C, et al: MCL1 is
deregulated in subgroups of diffuse large B-cell lymphoma.
Leukemia. 27:1381–1390. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Khoury JD, Medeiros LJ, Rassidakis GZ,
McDonnell TJ, Abruzzo LV and Lai R: Expression of Mcl-1 in mantle
cell lymphoma is associated with high-grade morphology, a high
proliferative state, and p53 overexpression. J Pathol. 199:90–97.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Merino D, Khaw SL, Glaser SP, et al:
Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737
and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood.
119:5807–5816. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Opferman JT: Unraveling MCL-1 degradation.
Cell Death Differ. 13:1260–1262. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Zhong Q, Gao W, Du F and Wang X:
Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the
polyubiquitination of Mcl-1 and regulates apoptosis. Cell.
121:1085–1095. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Bavi P, Uddin S, Bu R, et al: The
biological and clinical impact of inhibition of NF-kappaB-initiated
apoptosis in diffuse large B cell lymphoma (DLBCL). J Pathol.
224:355–366. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Savage KJ, Johnson NA, Ben Neriah S, et
al: MYC gene rearrangements are associated with a poor prognosis in
diffuse large B-cell lymphoma patients treated with R-CHOP
chemotherapy. Blood. 114:3533–3537. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Akasaka T, Ueda C, Kurata M, Akasaka H,
Yamabe H, Uchiyama T and Ohno H: Nonimmunoglobulin (non-Ig)/BCL6
gene fusion in diffuse large B-cell lymphoma results in worse
prognosis than Ig/BCL6. Blood. 96:2907–2909. 2000.PubMed/NCBI
|
|
45.
|
Shustik J, Han G, Farinha P, et al:
Correlations between BCL6 rearrangement and outcome in patients
with diffuse large B-cell lymphoma treated with CHOP or R-CHOP.
Haematologica. 95:96–101. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Davis RE, Brown KD, Siebenlist U and
Staudt LM: Constitutive nuclear factor kappaB activity is required
for survival of activated B cell-like diffuse large B cell lymphoma
cells. J Exp Med. 194:1861–1874. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Thieblemont C and Briere J: MYC, BCL2,
BCL6 in DLBCL: impact for clinics in the future? Blood.
121:2165–2166. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Snuderl M, Kolman OK, Chen YB, et al:
B-cell lymphomas with concurrent IGH-BCL2 and MYC rearrangements
are aggressive neoplasms with clinical and pathologic features
distinct from Burkitt lymphoma and diffuse large B-cell lymphoma.
Am J Surg Pathol. 34:327–340. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Tomita N, Tokunaka M, Nakamura N, et al:
Clinicopathological features of lymphoma/leukemia patients carrying
both BCL2 and MYC translocations. Haematologica. 94:935–943. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Brewer GJ, Askari F, Lorincz MT, et al:
Treatment of Wilson disease with ammonium tetrathiomolybdate: IV.
Comparison of tetrathiomolybdate and trientine in a double-blind
study of treatment of the neurologic presentation of Wilson
disease. Arch Neurol. 63:521–527. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Crowe A, Jackaman C, Beddoes KM, Ricciardo
B and Nelson DJ: Rapid copper acquisition by developing murine
mesothelioma: decreasing bioavailable copper slows tumor growth,
normalizes vessels and promotes T cell infiltration. PLoS One.
8:e736842013. View Article : Google Scholar
|
|
52.
|
Jain S, Cohen J, Ward MM, et al:
Tetrathiomolybdate-associated copper depletion decreases
circulating endothelial progenitor cells in women with breast
cancer at high risk of relapse. Ann Oncol. 24:1491–1498. 2013.
View Article : Google Scholar
|
|
53.
|
Schneider BJ, Lee JS, Hayman JA, et al:
Pre-operative chemoradiation followed by post-operative adjuvant
therapy with tetrathiomolybdate, a novel copper chelator, for
patients with resectable esophageal cancer. Invest New Drugs.
31:435–442. 2013. View Article : Google Scholar
|
|
54.
|
Lin J, Zahurak M, Beer TM, et al: A
non-comparative randomized phase II study of 2 doses of ATN-224, a
copper/zinc superoxide dismutase inhibitor, in patients with
biochemically recurrent hormone-naive prostate cancer. Urol Oncol.
31:581–588. 2013. View Article : Google Scholar
|