Introduction
Bladder cancer is the most common malignancy of the
urinary tract, with an incidence four times higher in men than in
women, and has a high rate of tumor recurrence. Because the
incidence of urinary bladder cancer has continuously increased over
the past two decades, bladder cancer is clearly recognized as a
significant public health issue around the world, especially in
industrialized countries (1,2).
Bladder cancers can be divided into two major subgroups that
possess distinct clinical, pathological and molecular
characteristics. More than 70% of bladder cancer patients have
non-muscle invasive papillary tumors that are managed with
transurethral resection of the tumors followed by intravesical
instillation of anticancer agents, and such patients have a good
prognosis. However, the other 30%, who have muscle-invasive tumors,
have a very poor prognosis, and these tumors can rapidly progress
to become metastatic and can lead to death. Although current
treatments for bladder cancer-surgery, radiation therapy,
chemotherapy or their combination-prolong survival time, bladder
cancer tends to recur and progress (3,4).
Therefore, efforts to develop a novel treatment to combat the
disease with complete efficacy and low toxicity must necessarily be
increased.
Apoptosis, a form of genetically programmed cell
death, plays an important role in the cellular response to
genotoxic stress; hence, loss of apoptotic response in tumor cells
represents an effective mechanism of malignant progression and
resistance to treatment (5,6).
Therefore, searching for agents that can trigger tumor cell
apoptosis has become an attractive strategy in the discovery of
anticancer drugs. Cells undergoing apoptosis are characterized by
membrane blebbing, cytoplasmic shrinkage, DNA fragmentation and
apoptotic body formation. There are two main apoptotic pathways in
mammals: the extrinsic or death receptor-mediated pathway and the
intrinsic or mitochondria-mediated pathway. The former is triggered
by engagement of cell-surface death receptors of the tumor necrosis
factor receptor family with their ligands, leading to the cleavage
of caspase-8. Whereas the latter is activated following
mitochondrial depolarization, release of cytochrome c and
the subsequent activation of caspase-9; the event regulated by
interactions between proteins related to Bcl-2 family (7,8).
Although low physiological levels of reactive oxygen species (ROS)
serve as a signaling messenger to mediate various biological
responses, excessive intracellular ROS, which can induce
depolarization of the mitochondrial membrane potential (MMP, ΔΨm),
are also considered an apoptotic death signal that ultimately
activates the intrinsic apoptotic pathway. Moreover, cancer cells
are more sensitive to fluctuations in ROS levels than normal cells;
therefore, ROS are considered an important target in anticancer
agent research (9–11).
Phytochemicals are regarded as a precious
alternative to modern medicine, and investigations of active
components with anticancer potential and fewer side effects have
opened up new research avenues (12–14).
Among such phytochemicals, sulforaphane (4-methylsulfinylbutyl
isothiocyanate) has been identified as a non-toxic isothiocyanate
of organosulfur compounds that is found in cruciferous vegetables
such as broccoli, Brussels sprouts and cabbage. A body of evidence
shows that this phytochemical is able to inhibit the growth of and
induce apoptosis in many different human cancer cells (15–17).
In the case of bladder cancers, sulforaphane has also shown
anti-bladder cancer activity, the reported mechanisms of which
include modulation of cyclooxygenase-2 expression associated with
p38 mitogen-activated protein kinase activation in T24 bladder
cancer cells (18), inhibition of
4-aminobiphenyl-induced DNA damage in RT4 bladder cancer cells and
in mouse bladder tissue (19) and
inhibition of invasion and metastasis by suppressing the
epithelial-to-mesenchymal transition process (20). We also recently reported that
sulforaphane-induced growth inhibition was associated with a
mitotic arrest and apoptosis of 5637 bladder cancer cells via
ROS-dependent pathway (21).
However, the precise cellular mechanisms of sulforaphane’s effect
on bladder cancer cells are not completely understood. Therefore,
in the present study, we used the T24 human bladder cancer cell
line to further examine the molecular events responsible for
sulforaphane-induced apoptosis. Sulforaphane exhibited significant
growth inhibitory effects and an increase in intrinsic apoptotic
cell death, leading to the regulation of Bcl-2 family proteins.
Furthermore, our data provided strong evidence that ROS generation
was a crucial event in the apoptotic response to sulforaphane in
T24 cells.
Materials and methods
Reagents
Sulforaphane was purchased from Sigma-Aldrich
Chemical Co. (St. Paul, MN), dissolved in dimethyl sulfoxide (DMSO,
Sigma-Aldrich), and then diluted with the medium to the desired
concentration prior to use. RPMI-1640 medium and fetal bovine serum
(FBS) were obtained from Gibco-BRL (Gaithersburg, MD).
3-(4,5-Dimetylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT),
4,6-diamidino-2-phenylindole (DAPI), propidium iodide (PI),
2,7-dichlorodihydrofluorescein diacetate (DCFH-DA),
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylimidacarbocyanine iodide
(JC-1) and N-acetyl L-cysteine (NAC) were obtained from
Sigma-Aldrich. Caspase activity assay kits and an enhanced
chemiluminescence (ECL) kit were purchased from R&D Systems
(Minneapolis, MN) and Amersham Life Science Corp. (Arlington
Heights, IL), respectively. Primary antibodies were purchased from
Santa Cruz Biotechnology (Santa Cruz, CA), Chemicon (Temecula, CA),
and Abcam (Cambridge, UK). Peroxidase-labeled donkey anti-rabbit
and sheep anti-mouse immunoglobulin were purchased from Amersham
Life Science Corp. All other chemicals were purchased from
Sigma-Aldrich.
Cell culture and cytotoxicity assay
T24 cells were purchased from American Type Culture
Collection (Manassas, VA) and maintained in RPMI-1640 medium
supplemented with 10% FBS, 2 μm L-glutamine and
penicillin/streptomycin under a humidified condition of 5%
CO2 at 37°C. In order to measure the inhibition of T24
cell proliferation by sulforaphane, cells were plated in 6-well
culture plates (1×105 cells/well) and allowed to adhere
overnight, and were then treated with different concentrations of
sulforaphane for 24 h. After treatment, cells were incubated with
MTT solution (5 mg/ml) for 3 h, and the medium was then removed.
The formazan precipitate was dissolved in DMSO, and the absorbance
was read at 540 nm in an ELISA reader (Dynatech Laboratories,
Chantilly, VA). Morphological changes were monitored by obtaining
photomicrographs under an inverted phase contrast microscope (Carl
Zeiss, Oberkochen, Germany).
Morphological observation of nuclear
change
After culture with various concentrations of
sulforaphane for 24 h, cells were washed with phosphate-buffered
saline (PBS) and fixed with 3.7% paraformaldehyde in PBS for 10 min
at room temperature. Fixed cells were washed with PBS, and stained
with DAPI solution for 10 min at room temperature. Then, the
nuclear morphology of the cells was examined using a fluorescence
microscope (Carl Zeiss).
Measurement of DNA fragmentation
Cells were harvested, washed with PBS and lysed in a
buffer [50 mM Tris (pH 8.0), 0.5% sarkosyl, 0.5 mg/ml proteinase K
and 1 mM EDTA] at 55°C for 3 h. Lysates were then treated with
RNase A (0.5 mg/ml) for 2 h at 56°C. The lysates were centrifuged
at 10,000 g for 20 min. The genomic DNA in the supernatant was
extracted with an equal volume of neutral phenol/chloroform/isoamyl
alcohol mixture (25/24/1). Approximately 20 μg of DNA was loaded
into each well of a 1.5% agarose gel and separated by
electrophoresis at 50 V for 120 min in Tris-borate/EDTA
electrophoresis buffer (TBE). The DNA was visualized and
photographed under ultraviolet (UV) illumination after staining
with 0.1 μg/ml ethidium bromide (EtBr).
Flow cytometric analysis of DNA content
for apoptosis
After treatment with sulforaphane for 24 h, cells
were harvested, washed with PBS and fixed with 70% ethanol at −20°C
overnight. Cells were washed twice, resuspended in PBS containing
40 μg/ml PI, 0.1 mg/ml RNase A and 0.1% Triton X-100 in a dark room
for 30 min at 37°C, and analyzed by flow cytometry
(Becton-Dickinson, San Jose, CA). The cell cycle distribution and
sub-G1 population (apoptosis) were determined and analyzed.
Western blot analysis
For isolation of total protein fractions, cells were
collected, washed twice with cold PBS, and lysed with cell lysis
buffer [20 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100, 2.5 mM
sodium pyrophosphate, 1 mM EDTA, 0.5 g/ml leupeptin, 1%
Na3CO4, 1 mM phenylmethane-sulfonyl
fluoride]. The cell lysates were centrifuged at 13,000 g at 4°C,
and the supernatant was collected for western blot analysis, and
measured for protein concentration by using the Bio-Rad protein
assay kit (Bio-Rad Laboratories, Hercules, CA). In a parallel
experiment, mitochondrial and cytosol cellular fractions were
prepared using a Cytosol/Mitochondria Fractionation kit
(Calbiochem, San Diego, CA) according to the manufacturer’s
protocol. The equal aliquots containing 30–50 μg of each lane were
separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel
electrophoresis followed by electro-transfer onto nitrocellulose
membranes (Schleicher & Schuell, Keene, NH). The membranes were
incubated overnight at 4°C with the primary antibodies and the
corresponding horseradish peroxidase-conjugated secondary
antibodies. The protein bands were visualized using an ECL kit.
Caspase activity assay
Activities of caspases were determined by use of
colorimetric assay kits, which utilize synthetic tetrapeptides
[Asp-Glu-Val-Asp (DEAD) for caspase-3, Ile-Glu-Thr-Asp (IETD) for
caspase-8 and Leu-Glu-His-Asp (LEHD) for caspase-9] labeled with
p-nitroaniline (pNA). Briefly, sulforaphane-treated and untreated
cells were lysed in the supplied lysis buffer. Supernatants were
collected and incubated with the supplied reaction buffer
containing dithiothreitol (DTT) and DEAD-pNA, IETD-pNA or LEHD-pNA
as substrates at 37°C. The reactions were measured by changes in
absorbance at 405 nm using an ELISA reader.
Flow cytometric determination of the
cellular redox state by ROS
The intracellular accumulation of ROS was examined
by flow cytometry after being stained with the cell permeable
fluorescent probe, DCFH-DA. Briefly, the cells were washed,
resuspended in PBS and incubated with 10 μM DCFH-DA for 20 min at
37°C in the dark. The ROS production in the cells was monitored
with a flow cytometer (22).
Flow cytometric detection of MMP
(ΔΨm)
The values of MMP were determined using the
dual-emission potential-sensitive probe JC-1. Briefly, cells were
collected and resuspended in PBS supplemented with 10 μM JC-1 for
30 min at 37°C in the dark. After the JC-1 was removed, the cells
were washed with PBS to remove unbound dye, and the amount of JC-1
retained by cells was immediately analyzed by flow cytometry.
Statistical analysis
Unless otherwise stated, data are expressed as means
± standard deviation (SD) and analyzed statistically by one-way
analysis of variance. A value of p<0.05 was considered
statistically significant.
Results
Sulforaphane treatment reduces cell
viability and induces apoptosis in T24 cells
Initially, the effects of sulforaphane on
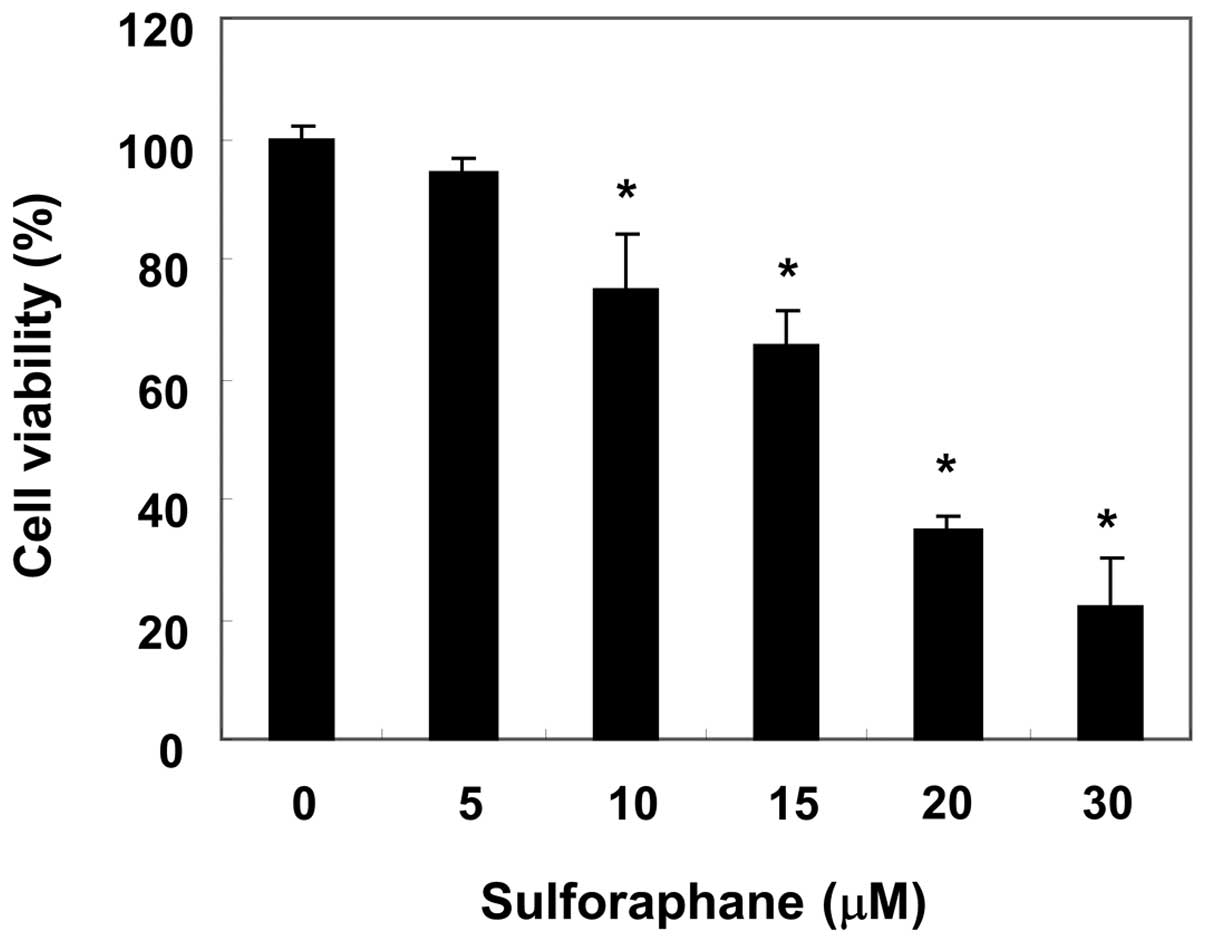
proliferation of T24 cells were measured by an MTT assay. As shown
in Fig. 1, sulforaphane treatment
inhibited the cell viability of T24 cells in a
concentration-dependent manner: sulforaphane at 10 and 20 μM for 24
h inhibited cell proliferation by 25 and 62%, respectively. In
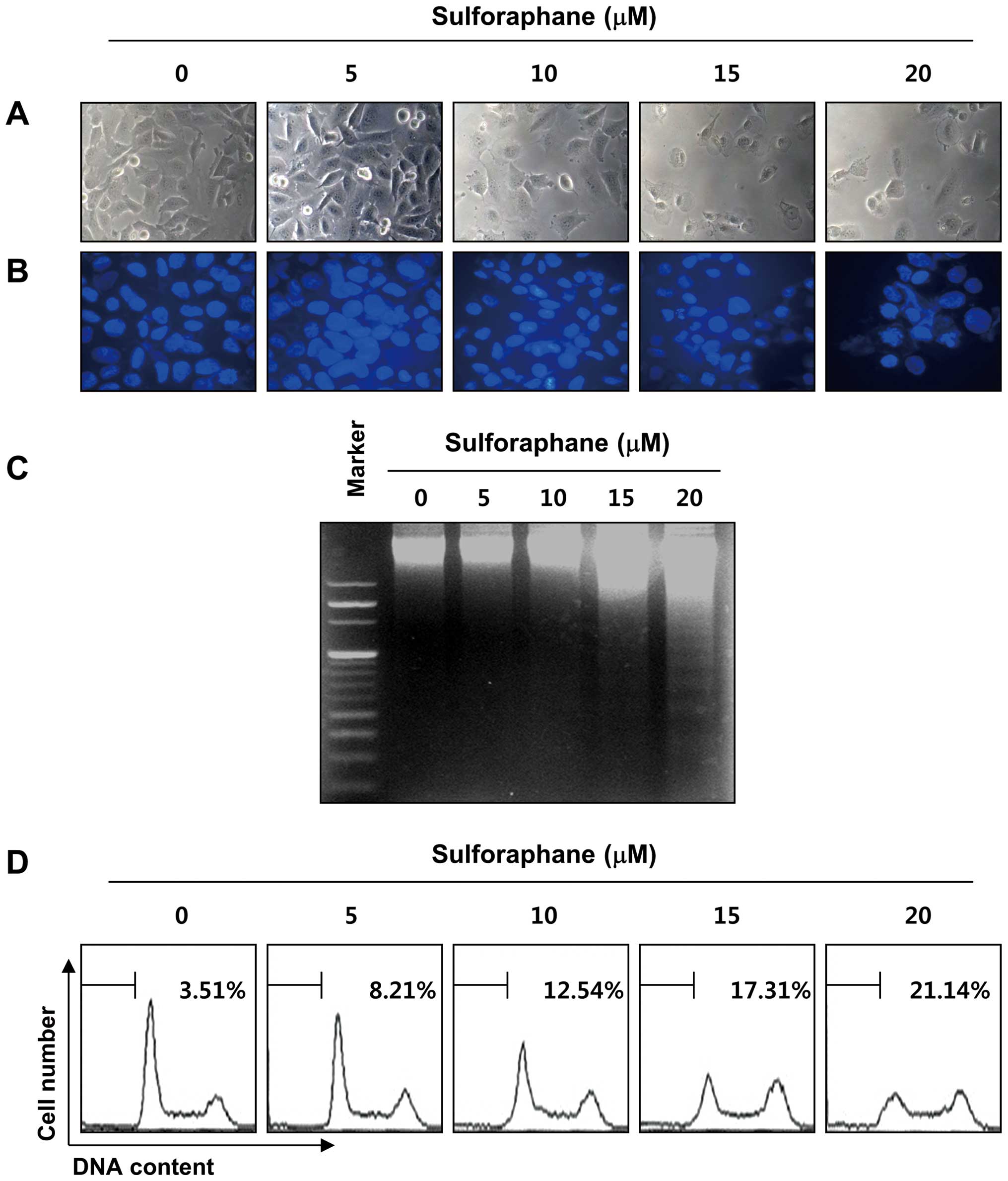
addition, sulforaphane stimulation significantly induced
morphological changes, including extensive cytosolic vacuolization
and the appearance of irregular cell membrane buds (Fig. 2A). To examine whether the
cytotoxicity of sulforaphane was due primarily to apoptosis, we
assessed the apoptosis parameters of T24 cells in response to
sulforaphane treatment. As shown in Fig. 2B, the nuclear structure of the
control cells remained intact, whereas nuclear chromatin
condensation and fragmentation, both of which are characteristics
of apoptosis, were increased in a concentration-dependent manner in
cells treated with sulforaphane, which was associated with
increased DNA fragmentation (Fig.
2C). Under the same conditions, results of flow cytometry
further demonstrated that treatment with sulforaphane induced a
concentration-dependent accumulation of apoptotic sub-G1 population
of T24 cells (Fig. 1D). These
results indicate that the inhibition of cell viability observed in
response to sulforaphane was associated with the induction of
apoptosis.
Sulforaphane treatment activates
caspase-3 and -9 in T24 cells
Several gene products are known to be important in
controlling the apoptotic process. Among them, caspases, a family
of cysteine proteases, play essential roles as important mediators
in apoptosis and as determinants of general apoptotic morphology
through the cleavage of various cellular substrates, including poly
ADP-ribose polymerase (PARP), an endogenous substrate of activated
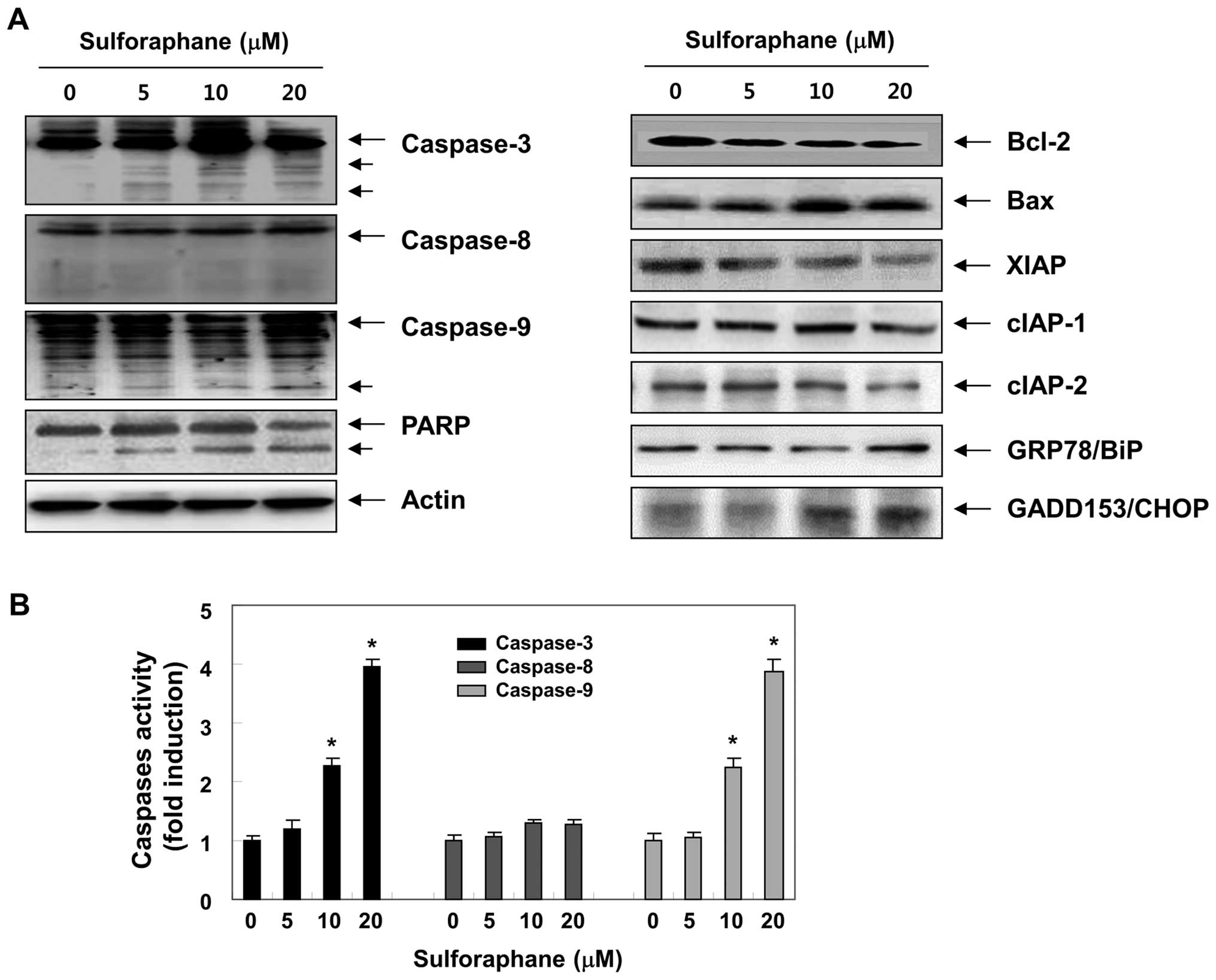
caspase-3 (23,24). To elucidate the molecular
mechanisms of sulforaphane-induced apoptosis, we assessed whether
sulforaphane induces the proteolytic processing of caspases. As
illustrated in Fig. 3A, we
detected a concentration-dependent upregulation of cleaved
caspase-3 and -9, whereas the cleaved forms of caspase-8 were not
detected. In addition, to quantify the proteolytic activation of
the caspases, we evaluated in vitro caspase activities using
fluorogenic substrates (Fig. 3B).
Our results indicated that treatment with sulforaphane
significantly increased the activities of caspase-3 and -9 compared
with control cells, whereas a very low level of caspase-8 activity
was detected after the sulforaphane treatment, suggesting the
likely involvement of a mitochondria-dependent cascade for caspase
activation. To further identify the activation of the caspase
cascade, the levels of PARP were examined by western blot analysis.
As shown in Fig. 3A, sulforaphane
induced a concentration-dependent proteolytic cleavage of PARP,
resulting in a reduction of the 116-kDa protein and accumulation of
the 85-kDa fragment. In addition, the levels of inhibitor of
apoptosis proteins (IAP) family members, including XIAP, cIAP-1 and
cIAP-2, were inhibited by sulforaphane treatment in a
concentration-dependent manner (Fig.
3A).
Sulforaphane-induced apoptosis involves
alterations in the intracellular distribution of Bcl-2 family
proteins and cytochrome c in T24 cells
It has been recognized that the Bcl-2 family members
play crucial roles in regulating apoptosis by functioning as
promoters (e.g., Bax) or inhibitors (e.g., Bcl-2) of cell death.
Mitochondria are specialized organelles containing an outer
membrane and an inner membrane separated by an intermembrane space
that contains many proapoptotic proteins, such as cytochrome
c (25,26). We therefore examined the expression
of these molecules after sulforaphane treatment. Western blot
analysis data showed that sulforaphane markedly increased protein
levels of proapoptotic Bax. In contrast to Bax, anti-apoptotic
Bcl-2 expression decreased mildly following sulforaphane treatment.
To further characterize the apoptotic effect of sulforaphane in T24
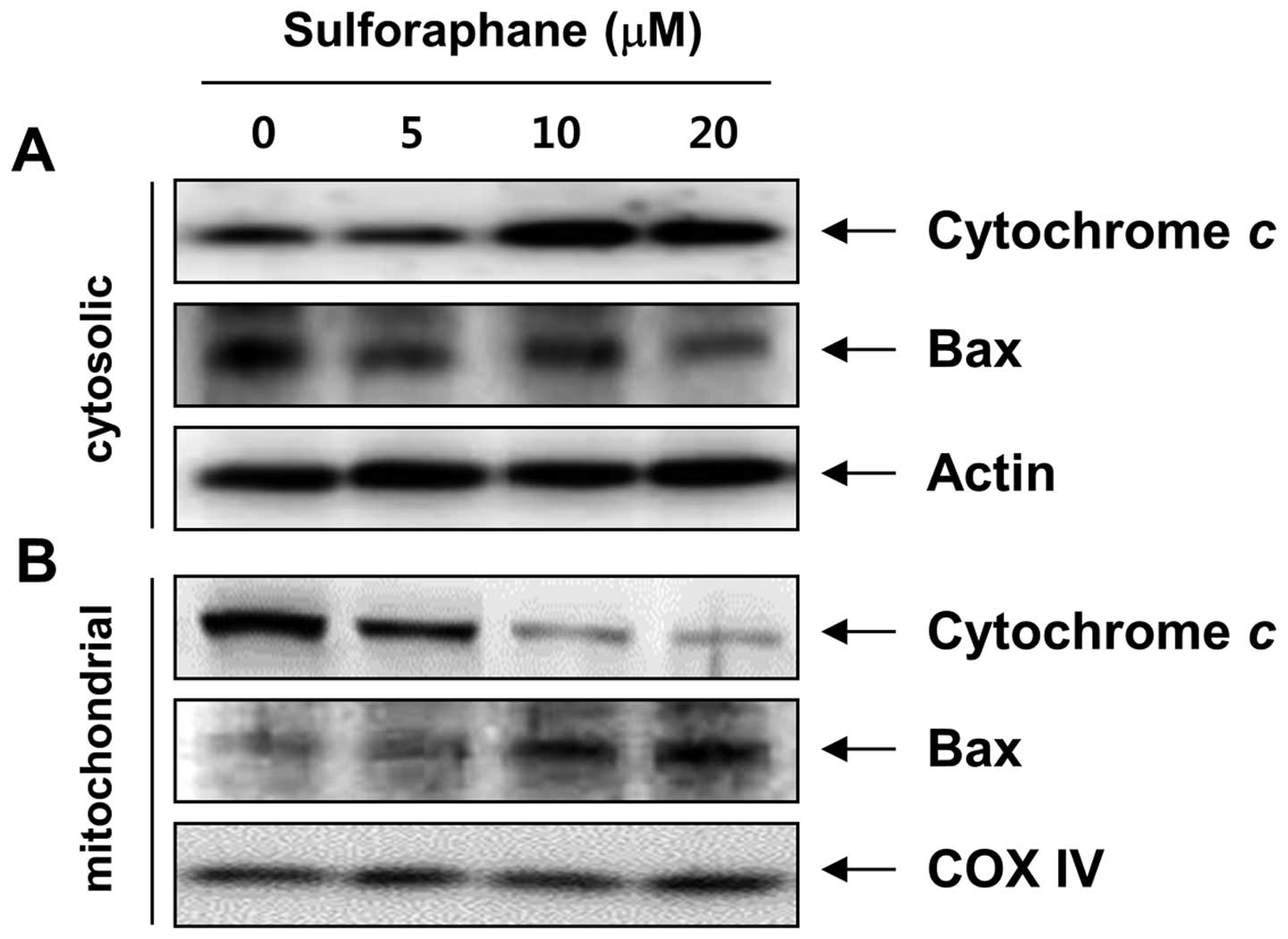
cells, we analyzed the translocation of Bax and the release of
cytochrome c using cytosol and mitochondria fractionation.
Notably, as shown in Fig. 4,
sulforaphane treatment led to a concentration-dependent increase in
cytosolic cytochrome c levels and triggered the
translocation of Bax to the mitochondria. In addition, the MMP,
which is regulated by the Bcl-2 family, was gradually reduced in
the sulforaphane-treated cells compared to the control cells
(Fig. 5B). These results suggest
that sulforaphane-induced apoptosis, which took place by promoting
the translocation of Bax to mitochondria, followed by a loss of MMP
and release of cytochrome c into the cytosol, results in the
activation of caspase-9 and -3, thus leading to apoptosis in the
cells. These observations also suggest that sulforaphane induced
apoptosis in T24 cells via the mitochondrial pathway.
Sulforaphane induces the activation of ER
stress pathway in T24 cells
Evidence has accumulated from many studies that
apoptosis associated with endoplasmic reticulum (ER) stress may be
responsible for cell death induced by antitumor agents (27,28).
ER stress can be characterized by an increase in ER
stress-associated molecules. In particular, glucose-regulated
protein (GRP) 78 and C/EBP-homologous protein (CHOP) have been
considered as vital proteins of ER stress response. To examine
whether ER stress was involved in sulforaphane-induced apoptosis,
we followed the behavior of these two stress markers in response to
sulforaphane treatment. Our data revealed that sulforaphane
exposure increased the expression of GRP78 as well as CHOP compared
with control (Fig. 4A) in a
concentration-dependent manner. These results show that ER stress
was involved in sulforaphane-induced apoptosis in T24 cells, as
evidenced by upregulation of the expression of ER stress-associated
proteins.
Generation of ROS is required as a
mediator for sulforaphane-induced mitochondrial dysfunction in R24
cells
Because oxidative stress and ROS generation are
directly involved in protease cascades, such as induction of
caspases during apoptosis regulation (9,10),
we investigated whether the elevated generation of ROS production
is related to sulforaphane-induced apoptosis following MMP
disruption. To this end, DCFH-DA-based flow cytometric analysis was
utilized to measure the amounts of ROS in control and
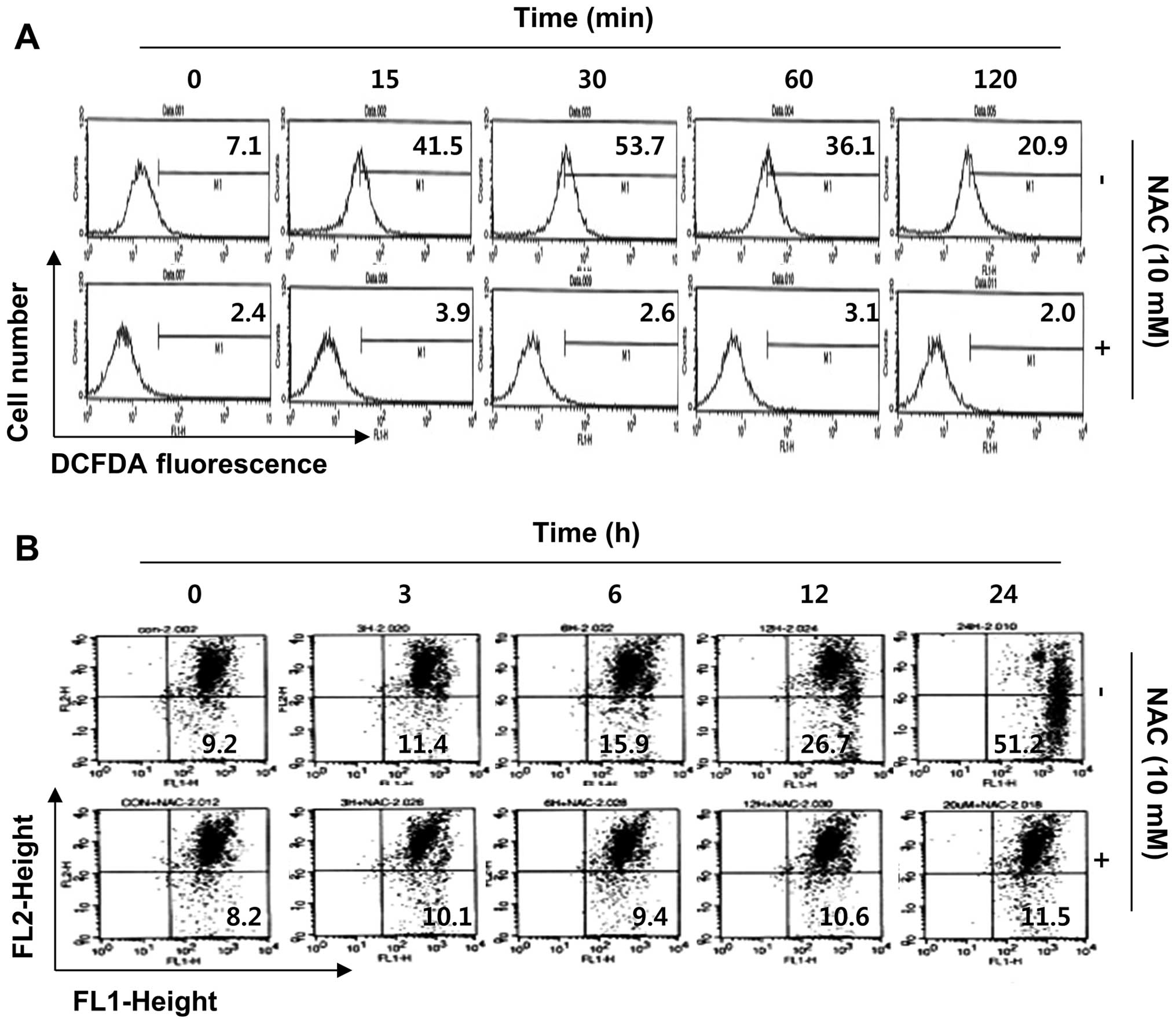
sulforaphane-treated cells. As shown in Fig. 5A, compared with control cells, the
production of ROS in sulforaphane-treated cells dramatically
increased within 15 min of sulforaphane treatment and peaked at 30
min after treatment; thereafter, the production was attenuated
gradually from 1 to 2 h following sulforaphane treatment. In a
parallel experiment, pretreatment of the ROS scavenger NAC, along
with sulforaphane, drastically decreased ROS generation as compared
to the sulforaphane-treated group. Therefore, we proceeded to
investigate whether ROS generation was necessary for
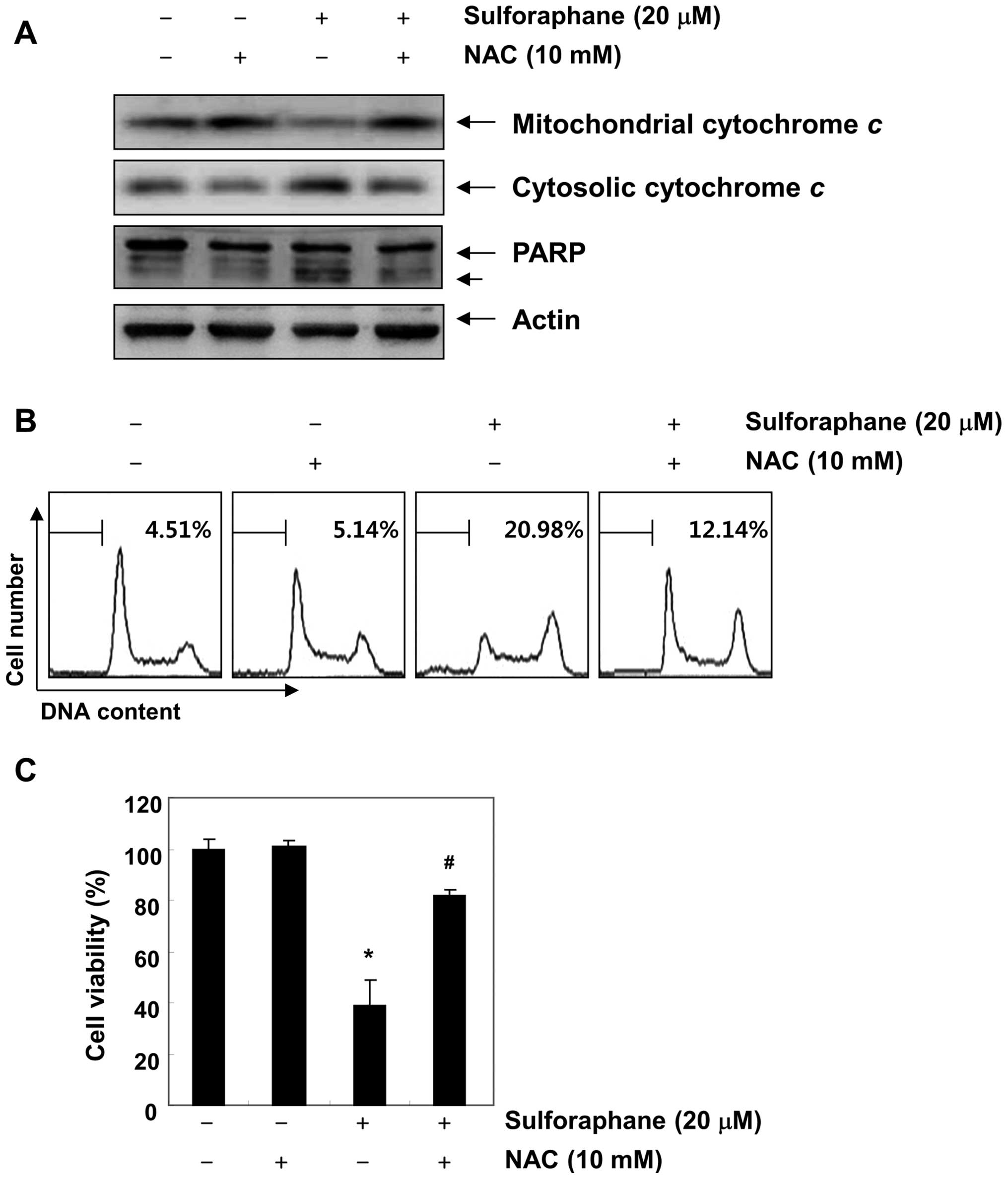
sulforaphane-induced mitochondrial dysfunction. Fig. 5B shows that the scavenging of ROS
by NAC significantly attenuated sulforaphane-induced mitochondrial
membrane permeabilization. Furthermore, blockade of ROS generation
suppressed sulforaphane-induced cytochrome c release, PARP
cleavage, increases of sub-G1 accumulation, and growth inhibition
(Fig. 6). These results indicate
that ROS play a major role in sulforaphane-induced mitochondrial
dysfunction in the T24 cell apoptosis process.
Sulforaphane upregulates the antioxidant
response proteins Nrf2 and HO-1 in T24 cells
The transcription factor nuclear factor erythroid
2-related factor 2 (Nrf2) is one of the key regulators in the
antioxidant response in eukaryotic cells. Under normal
physiological conditions, the Nrf2 protein is sequestrated by its
cytoplasmic partner, Kelch-like ECH-associated protein 1 (Keap1).
ROS induce the release of Nrf2 from Keap1 repression with a
subsequent translocation of Nrf2 into the nucleus, where it binds
to the antioxidant response element (ARE) for transcription of
phase II detoxifying and antioxidant enzymes (29–31).
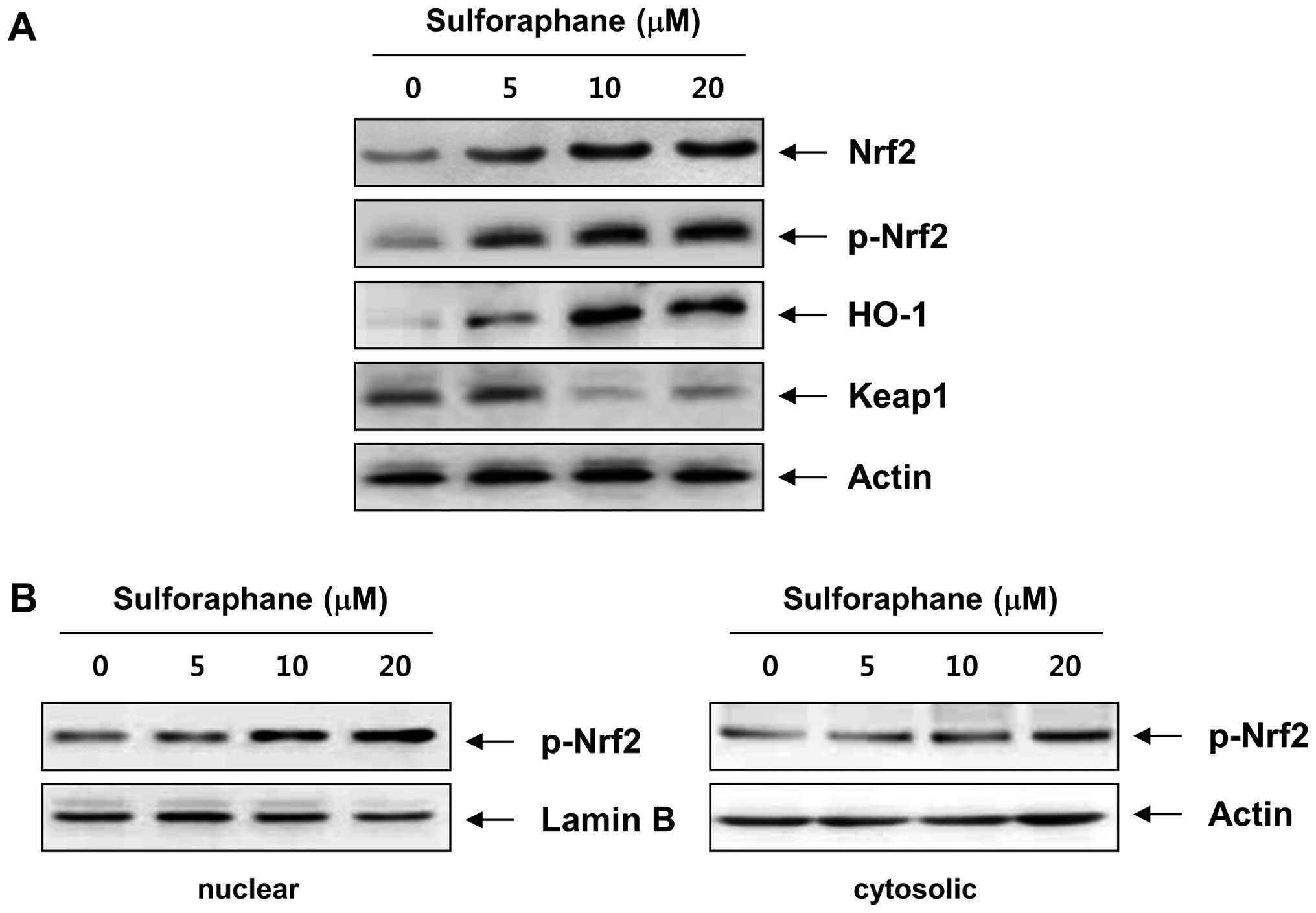
In order to identify the mechanism underlying sulforaphane-induced
apoptosis, we assessed the effect of sulforaphane on the Nrf2
pathway. As shown in Fig. 7A, an
apparent increase in Nrf2 levels and a reduction in Keap1 protein
expression were detected after treatment with sulforaphane. Next,
Nrf2 activation was assessed by the accumulation of phosphorylated
forms of the Nrf2 protein (p-Nrf2) in the nucleus, and we found
that treatment with sulforaphane resulted in a translocation of
p-Nrf2 in the nucleus from cytosol (Fig. 7B). Because one of the downstream
targets of Nrf2 is the heme oxygenase-1 (HO-1) gene, whose gene
product is a sensitive and reliable indicator of cellular stress
(32,33), in the next step, we evaluated the
effect of sulforaphane on HO-1 protein levels following treatment
of T24 cells. As illustrated in Fig.
7A, after treatment of T24 cells with sulforaphane, HO-1 levels
increased steadily in a concentration-dependent manner, clearly
indicating that sulforaphane treatment activated Nrf2 signaling in
T24 cells.
Discussion
Because well-known natural phytochemicals extracted
from plants have been used in an increasing number of cancer
treatment applications, exploring plant-based anticancer agents has
become an effective strategy for chemotherapeutic and anticancer
drug development (12–14). In particular, epidemiological
studies show that there is a low incidence of cancer among people
who eat a lot of cruciferous vegetables. Sulforaphane is a
phytochemical belonging to the family of isothiocyanates found in
cruciferous vegetables, and is well known as a strong antioxidant
and stimulator of natural detoxifying enzymes. Recently, the
results of numerous experimental models (34,35)
have demonstrated the ability of sulforaphane to cause inhibition
of cell growth and induction of apoptosis in cancer cells. Several
studies have also demonstrated that sulforaphane can activate the
extrinsic or intrinsic apoptotic pathways in a variety of cancer
cell lines by altering the expression of apoptosis-associated or
signaling proteins, cell cycle regulatory proteins, and
transcription factors (15–17).
Although sulforaphane may affect different signaling pathways
depending on the cell type or culture conditions used, our previous
results have shown that sulforaphane-induced apoptosis is
correlated with the marked generation of intracellular ROS and loss
of MMP, suggesting that this pro-oxidant function can play a
pivotal role in sulforaphane-induced apoptosis in various human
cancer cell lines (21,36–38).
In accordance with these results, we observed a significant
decrease in cell viability as well as an early increase in ROS
levels after sulforaphane treatment (Fig. 5A). Moreover, the effects of
sulforaphane on the depolarization of the MMP and apoptotic events
were abrogated in the presence of NAC (Figs. 5B and 6). This result may reflect a
chemotherapeutic potential of sulforaphane to induce oxidative
injury in T24 cells. Furthermore, the apoptosis induced by the
sulforaphane in T24 cells was related to the increase in the
Bax/Bcl-2 ratio, translocation of Bax from cytosol to mitochondria,
downregulation of IAP family proteins, release of cytochrome
c from the mitochondria, and activation of caspase-9 and -3,
but not caspase-8 (Figs. 3 and
4). These observations suggest
that sulforaphane induces apoptosis in T24 cells via the
ROS-mediated intrinsic pathway.
In addition to the ROS-mediated apoptosis induction,
known cellular responses in sulforaphane-induced apoptosis include
changes in the ER and Nrf2-ARE signaling pathways. ER is an
important organelle involved in calcium signaling and the
synthesis, folding and processing of proteins. Evidence is emerging
that the impaired function of ER leads to ER stress, which is
caused by oxidative stress, changes in Ca2+ homeostasis,
and accumulation of unfolded or misfolded proteins, indicating that
ER stress also plays a crucial role in the response to oxidative
stress. The ER stress pathway is also another possible signaling
pathway involved in anticancer agent-induced apoptosis in cancer
cells (27,28). A central regulator of ER function
is GRP78, also referred to as a binding immunoglobulin protein
(BiP), due to its roles in protein folding and assembly, targeting
misfolded protein for degradation, ER Ca2+-binding and
controlling the activation of transmembrane ER stress sensors. Due
to its anti-apoptotic property, stress induction of GRP78
represents an important prosurvival component of the unfolded
protein response (39,40). In addition, one of the components
of ER stress-mediated apoptosis is the transcription factor CHOP,
also known as the growth arrest- and DNA damage-inducible gene 153
(GADD153), which is expressed at low levels under normal
physiological conditions, but is strongly induced in response to ER
stress. Subsequent upregulation of certain CHOP target genes
promotes induction of ER stress-mediated apoptosis (41,42).
In the present study, we found that sulforaphane promoted the
expression of GRP78 and CHOP (Fig.
3), suggesting that sulforaphane induced apoptosis in part
through ER stress.
The Nrf2-ARE pathway is also involved in the
modulation of oxidative and ER stresses and plays a cytoprotective
role in response to these stresses. It is well known that the
activation of the Nrf2-ARE pathway plays an important role in the
antioxidant activity of sulforaphane (42,43).
Nrf2 is a transcription factor that binds to the cis-acting element
in the genome, termed ARE in the regulatory regions of target
genes. Under normal physiological conditions, Nrf2 is complexed
with Keap1 in the cytoplasm and constantly degraded in the
cytoplasm. However, upon exposure to oxidants and electrophiles,
degradation of Nrf2 protein is halted, which makes it stabilized
and phosphorylated, and free to translocate into the nucleus,
thereby activating target phase II cytoprotective genes including
HO-1 by binding to ARE (29,31).
Moreover, ROS can induce the liberation of Nrf2 from Keap1
repression with a subsequent translocation of Nrf2 into the
nucleus. Furthermore, Nrf2 activation has been implicated in the
promotion of cell survival following ER stress, and the ER stress
signaling pathway can also activate an antioxidant program by
preferentially inducing the expression of the mRNA encoding
activating transcription factor 4 (ATF4) and by phosphorylation of
Nrf2 (44,45). In accordance with the previous data
(46–52), we found activation of Nrf2-ARE
signaling pathway by elevated levels of Nrf2 and HO-1, which was
strongly correlated with an accumulation of phosphorylated Nrf2 in
the nucleus and downregulation of Keap1 in sulforaphane-treated T24
cells (Fig. 7). Based on these
observations, it is somewhat reasonable to conclude that Nrf2
activation mediates sulforaphane-induced apoptosis and ER stress in
T24 cells. However, further studies are required to elucidate the
function of Nrf2 target genes involved in the modulation of ER
stress by sulforaphane.
Taken together, our findings demonstrate that
sulforaphane induces apoptosis in T24 cells through the
ROS-mediated intrinsic pathway, by increasing ROS production and
inducing mitochondrial oxidative damage, MMP depolarization, and
release of cytochrome c, as well as inducing an imbalance
between Bax and Bcl-2, downregulation of IAP family proteins,
activation of caspase-9 and -3, and the cleavage of PARP. Moreover,
treatment with sulforaphane resulted in an increase of ER
stress-associated proteins and accumulation of p-Nrf2 in the
nucleus, indicating that sulforaphane-induced apoptosis is possibly
related to the activation of the ER and Nrf2-ARE signaling
pathways. However, the detailed mechanism by which sulforaphane
affects the molecular connections between the two pathways remains
unclear. Studies regarding this issue are ongoing.
Acknowledgements
This research was supported by Grants from the
Globalization of Korean Foods R&D Program (912001-1), funded by
the Ministry of Food, Agriculture, Forestry and Fisheries and the
National Research Foundation of Korea (NRF) Grant funded by the
Korea government (2008-0062611), Republic of Korea.
References
|
1
|
Cheung G, Sahai A, Billia M, Dasgupta P
and Khan MS: Recent advances in the diagnosis and treatment of
bladder cancer. BMC Med. 11:132013. View Article : Google Scholar
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
3
|
Pinto-Leite R, Arantes-Rodrigues R,
Palmeira C, Colaço B, Lopes C, Colaço A, Costa C, da Silva VM,
Oliveira P and Santos L: Everolimus combined with cisplatin has a
potential role in treatment of urothelial bladder cancer. Biomed
Pharmacother. 67:116–121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Black PC and Dinney CP: Bladder cancer
angiogenesis and metastasis-translation from murine model to
clinical trial. Cancer Metastasis Rev. 26:623–634. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chung KM and Yu SW: Interplay between
autophagy and programmed cell death in mammalian neural stem cells.
BMB Rep. 46:383–390. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jin Z and El-Deiry WS: Overview of cell
death signaling pathways. Cancer Biol Ther. 4:139–163.
2005.PubMed/NCBI
|
|
8
|
Burz C, Berindan-Neagoe I, Balacescu O and
Irimie A: Apoptosis in cancer: key molecular signaling pathways and
therapy targets. Acta Oncol. 48:811–821. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fleury C, Mignotte B and Vayssière JL:
Mitochondrial reactive oxygen species in cell death signaling.
Biochimie. 84:131–141. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fruehauf JP and Meyskens FL Jr: Reactive
oxygen species: a breath of life or death? Clin Cancer Res.
13:789–794. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Saeidnia S and Abdollahi M: Toxicological
and pharmacological concerns on oxidative stress and related
diseases. Toxicol Appl Pharmacol. 273:442–455. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dorai T and Aggarwal BB: Role of
chemopreventive agents in cancer therapy. Cancer Lett. 215:129–140.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shu L, Cheung KL, Khor TO, Chen C and Kong
AN: Phytochemicals: cancer chemoprevention and suppression of tumor
onset and metastasis. Cancer Metastasis Rev. 29:483–502. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vinod BS, Maliekal TT and Anto RJ:
Phytochemicals as chemosensitizers: from molecular mechanism to
clinical significance. Antioxid Redox Signal. 18:1307–1348. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fimognari C and Hrelia P: Sulforaphane as
a promising molecule for fighting cancer. Mutat Res. 635:90–104.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Clarke JD, Dashwood RH and Ho E:
Multi-targeted prevention of cancer by sulforaphane. Cancer Lett.
269:291–304. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheung KL and Kong AN: Molecular targets
of dietary phenethyl isothiocyanate and sulforaphane for cancer
chemoprevention. AAPS J. 12:87–97. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shan Y, Wang X, Wang W, He C and Bao Y:
p38 MAPK plays a distinct role in sulforaphane-induced
up-regulation of ARE-dependent enzymes and down-regulation of COX-2
in human bladder cancer cells. Oncol Rep. 23:1133–1138.
2010.PubMed/NCBI
|
|
19
|
Ding Y, Paonessa JD, Randall KL, Argoti D,
Chen L, Vouros P and Zhang Y: Sulforaphane inhibits
4-aminobiphenyl-induced DNA damage in bladder cells and tissues.
Carcinogenesis. 31:1999–2003. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shan Y, Zhang L, Bao Y, Li B, He C, Gao M,
Feng X, Xu W, Zhang X and Wang S: Epithelial-mesenchymal
transition, a novel target of sulforaphane via COX-2/MMP2, 9/Snail,
ZEB1 and miR-200c/ZEB1 pathways in human bladder cancer cells. J
Nutr Biochem. 24:1062–1069. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Park HS, Han MH, Kim GY, Moon SK, Kim WJ,
Hwang HJ, Park KY and Choi YH: Sulforaphane induces reactive oxygen
species-mediated mitotic arrest and subsequent apoptosis in human
bladder cancer 5637 cells. Food Chem Toxicol. 64:157–165. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim NH, Hong BK, Choi SY, Moo Kwon H, Cho
CS, Yi EC and Kim WU: Reactive oxygen species regulate
context-dependent inhibition of NFAT5 target genes. Exp Mol Med.
45:e322013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
MacKenzie SH and Clark AC: Targeting cell
death in tumors by activating caspases. Curr Cancer Drug Targets.
8:98–109. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wen X, Lin ZQ, Liu B and Wei YQ:
Caspase-mediated programmed cell death pathways as potential
therapeutic targets in cancer. Cell Prolif. 45:217–224. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Susnow N, Zeng L, Margineantu D and
Hockenbery DM: Bcl-2 family proteins as regulators of oxidative
stress. Semin Cancer Biol. 19:42–49. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shore GC, Papa FR and Oakes SA: Signaling
cell death from the endoplasmic reticulum stress response. Curr
Opin Cell Biol. 23:143–149. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Logue SE, Cleary P, Saveljeva S and Samali
A: New directions in ER stress-induced cell death. Apoptosis.
18:537–546. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li N and Nel AE: Role of the Nrf2-mediated
signaling pathway as a negative regulator of inflammation:
implications for the impact of particulate pollutants on asthma.
Antioxid Redox Signal. 8:88–98. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Keum YS: Regulation of the Keap1/Nrf2
system by chemopreventive sulforaphane: implications of
posttranslational modifications. Ann N Y Acad Sci. 1229:184–189.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kansanen E, Kuosmanen SM, Leinonen H and
Levonen AL: The Keap1-Nrf2 pathway: mechanisms of activation and
dysregulation in cancer. Redox Biol. 1:45–49. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Paine A, Eiz-Vesper B, Blasczyk R and
Immenschuh S: Signaling to heme oxygenase-1 and its
anti-inflammatory therapeutic potential. Biochem Pharmacol.
80:1895–1903. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Na HK and Surh YJ: Oncogenic potential of
Nrf2 and its principal target protein heme oxygenase-1. Free Radic
Biol Med. 67C:353–365. 2014.PubMed/NCBI
|
|
34
|
Roy SK, Srivastava RK and Shankar S:
Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of
FOXO transcription factor, leading to cell cycle arrest and
apoptosis in pancreatic cancer. J Mol Signal. 5:102010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bryant CS, Kumar S, Chamala S, Shah J, Pal
J, Haider M, Seward S, Qazi AM, Morris R, Semaan A, Shammas MA,
Steffes C, Potti RB, Prasad M, Weaver DW and Batchu RB:
Sulforaphane induces cell cycle arrest by protecting RB-E2F-1
complex in epithelial ovarian cancer cells. Mol Cancer. 9:472010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Choi WY, Choi BT, Lee WH and Choi YH:
Sulforaphane generates reactive oxygen species leading to
mitochondrial perturbation for apoptosis in human leukemia U937
cells. Biomed Pharmacother. 62:637–644. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Moon DO, Kang SH, Kim KC, Kim MO, Choi YH
and Kim GY: Sulforaphane decreases viability and telomerase
activity in hepatocellular carcinoma Hep3B cells through the
reactive oxygen species-dependent pathway. Cancer Lett.
295:260–266. 2010. View Article : Google Scholar
|
|
38
|
Moon DO, Kim MO, Kang SH, Choi YH and Kim
GY: Sulforaphane suppresses TNF-alpha-mediated activation of
NF-kappaB and induces apoptosis through activation of reactive
oxygen species-dependent caspase-3. Cancer Lett. 274:132–142. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li J and Lee AS: Stress induction of
GRP78/BiP and its role in cancer. Curr Mol Med. 6:45–54. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Roller C and Maddalo D: The molecular
chaperone GRP78/BiP in the development of chemoresistance:
mechanism and possible treatment. Front Pharmacol. 4:102013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sano R and Reed JC: ER stress-induced cell
death mechanisms. Biochim Biophys Acta. 1833:3460–3470. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cullinan SB and Diehl JA: Coordination of
ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway.
Int J Biochem Cell Biol. 38:317–332. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xu H, Zhou YL, Zhang XY, Lu P and Li GS:
Activation of PERK signaling through fluoride-mediated endoplasmic
reticulum stress in OS732 cells. Toxicology. 277:1–5. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Miyamoto N, Izumi H, Miyamoto R, Bin H,
Kondo H, Tawara A, Sasaguri Y and Kohno K: Transcriptional
regulation of activating transcription factor 4 under oxidative
stress in retinal pigment epithelial ARPE-19/HPV-16 cells. Invest
Ophthalmol Vis Sci. 52:1226–1234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jeong WS, Keum YS, Chen C, Jain MR, Shen
G, Kim JH, Li W and Kong AN: Differential expression and stability
of endogenous nuclear factor E2-related factor 2 (Nrf2) by natural
chemopreventive compounds in HepG2 human hepatoma cells. J Biochem
Mol Biol. 38:167–176. 2005. View Article : Google Scholar
|
|
47
|
Keum YS, Yu S, Chang PP, Yuan X, Kim JH,
Xu C, Han J, Agarwal A and Kong AN: Mechanism of action of
sulforaphane: inhibition of p38 mitogen-activated protein kinase
isoforms contributing to the induction of antioxidant response
element-mediated heme oxygenase-1 in human hepatoma HepG2 cells.
Cancer Res. 66:8804–8813. 2006. View Article : Google Scholar
|
|
48
|
Ping Z, Liu W, Kang Z, Cai J, Wang Q,
Cheng N, Wang S, Wang S, Zhang JH and Sun X: Sulforaphane protects
brains against hypoxic-ischemic injury through induction of
Nrf2-dependent phase 2 enzyme. Brain Res. 1343:178–185. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lee YJ, Jeong HY, Kim YB, Lee YJ, Won SY,
Shim JH, Cho MK, Nam HS and Lee SH: Reactive oxygen species and
PI3K/Akt signaling play key roles in the induction of Nrf2-driven
heme oxygenase-1 expression in sulforaphane-treated human
mesothelioma MSTO-211H cells. Food Chem Toxicol. 50:116–123. 2012.
View Article : Google Scholar
|
|
50
|
Oh CJ, Kim JY, Min AK, Park KG, Harris RA,
Kim HJ and Lee IK: Sulforaphane attenuates hepatic fibrosis via
NF-E2-related factor 2-mediated inhibition of transforming growth
factor-β/Smad signaling. Free Radic Biol Med. 52:671–682.
2012.PubMed/NCBI
|
|
51
|
Alfieri A, Srivastava S, Siow RC, Cash D,
Modo M, Duchen MR, Fraser PA, Williams SC and Mann GE: Sulforaphane
preconditioning of the Nrf2/HO-1 defense pathway protects the
cerebral vasculature against blood-brain barrier disruption and
neurological deficits in stroke. Free Radic Biol Med. 65:1012–1022.
2013. View Article : Google Scholar
|
|
52
|
Kleszczyński K, Ernst IM, Wagner AE, Kruse
N, Zillikens D, Rimbach G and Fischer TW: Sulforaphane and
phenylethyl isothiocyanate protect human skin against UVR-induced
oxidative stress and apoptosis: role of Nrf2-dependent gene
expression and antioxidant enzymes. Pharmacol Res. 78:28–40.
2013.PubMed/NCBI
|