Introduction
Exposure to asbestos (i.e., chrysotile, crocidolite
or amosite) causes malignant mesothelioma (MM) and serious social
problems (1–4). Therefore, early diagnosis of
asbestos-related MM is required for early medical treatment because
MM has an extremely poor prognosis. We reported previously that
exposure to chrysotile asbestos causes immunological abnormalities
in immunocompetent cells, such as T cells and NK cells, following
research concerning identification of an early diagnosis indicator
(5–10). In particular, the establishment of
an in vitro experimental model of asbestos exposure using a
human T-cell leukemia virus type-1 (HTLV-1)-immortalized human
polyclonal T cell line, MT-2 (11), enabled analysis of immunological
abnormalities induced by asbestos exposure and the identification
of target molecules related to antitumor immunity (12–16).
Actually, people exposed to asbestos usually have been receiving
exposure from work or environmental circumstances extrinsically or
from remaining fibers inhaled intrinsically. To establish an in
vitro model of asbestos-exposure to immunocompetent cells, we
have been using original MT-2 (MT-2Org) cells and examined
short-term exposure as reported previously (13,14,17).
The culturing of MT-2Org cells (1×105/2 ml of medium)
with 0, 2.5, 5, 12.5 and 25 μg/cm2 of chrysotile fibers
was used to examine their growth features and the appearance of
apoptosis measured by the TUNEL method. Growth inhibition and the
appearance of apoptosis were dependent on dose and time (1–3 days)
and resulted in the production of ROS, activation of pro-apoptotic
MAPK signaling molecules such as p38 and JNK, and activation of the
mitochondrial apoptotic pathway. However, relatively low doses (2.5
and 5 μg/ml) induced apoptosis in less than half of the cells.
Thus, we continuously added these doses of chrysotile fibers to
establish an in vitro cell line model of long-term exposure
for more than eight months. Although short-term, high-dose exposure
to asbestos causes apoptosis via a caspase-dependent pathway in
original MT-2 cells, long-term (more than eight months) and
low-dose (5 μg/cm2) exposure to chrysotile-B (CB)
results in resistance to apoptosis. Acquisition of resistance to CB
occurs by increased Src family kinase-mediated interleukin-10
(IL-10) production, with subsequent activation of signal transducer
and activator of transcription 3 (STAT3), and overexpression of
anti-apoptotic protein Bcl-2 located down-stream of STAT3,
resulting in the establishment of a CB-induced apoptosis-resistant
subline (MT-2Rst) (14). Moreover,
bcl2 mRNA expression increases in peripheral CD4+
T cells from MM patients, suggesting that MT-2Rst cells are useful
as a model for chronic CB exposure. Recently, we found reduced
expression of cell surface chemokine receptor CXCR3 in MT-2Rst
cells, and reported that the CXCR3 expression is decreased in
peripheral CD4+ T cells from patients with
asbestos-related diseases such as pleural plaque or MM (18,19).
Therefore, it is important to examine the cellular features of the
MT-2Rst cell line to determine whether this subline has modified
characteristics as immunocompetent cells which affect tumor
immunity. On the other hand, MT-2Org cells are a regulatory T
(Treg) cell-like cell line, as previously reported (20). Treg cells produce anti-inflammatory
cytokine IL-10 and transforming growth factor-β1 (TGF-β1), which
suppress antitumor immune function by inhibition of proliferation
and differentiation of various immunocompetent cells (21,22).
In this study, we have shown that long-term exposure
of MT-2Org cells to CB promotes a remarkable production of TGF-β1
through activation of chronic p38 mitogen-activated protein kinase
(MAPK). In addition, MT-2Rst cells acquire resistance to
TGF-β1-mediated growth inhibition. Our findings may indicate that
increased production of IL-10 and TGF-β1 by chronic exposure to
asbestos, and alteration of immunocompetent cells, may contribute
to the development of asbestos-related MM by suppression of an
antitumor immune system.
Materials and methods
Reagents
Recombinant human TGF-β1 was purchased from
Peprotech, London, UK. PD98059 was obtained from Cell Signaling
Technology, Inc., Danvers, MA, USA. SB203580 was acquired from
Calbiochem, Madison, WI, USA. SP600125 was obtained from
SABiosciences, Frederick, MD, USA.
Cell culture and asbestos
MT-2Org cells were kindly provided as a gift by the
Cell Biology Institute, Research Center, Hayashibara Biochemical
Laboratories, Inc. Okayama, Japan. MT-2Org cells were seeded in
RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS),
streptomycin and penicillin. MT-2Rst cells were established from
the MT-2Org cells by continuous exposure to CB (5
μg/cm2) for more than eight months as previously
described (12,14). 293FT cells were cultured in
Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA, USA)
supplemented with 10% FBS. The UICC (the International Union
Against Cancer) standard of CB was kindly provided by the
Department of Occupational Health, National Institute for
Occupational Health, South Africa (23). Chrysotile asbestos is composed of
Mg3Si2O5 (OH)4.
Chrysotile-A from Zimbabwe contains 2% fibrous anthophyllite,
although CB from Canada does not contain any fibrous
impurities.
Real-time RT-PCR analysis
Total RNA was extracted from cells using the RNeasy
mini kit (Qiagen, Hilden, Germany), and cDNAs were synthesized
using the PrimeScript II® 1st strand cDNA Synthesis kit
(Takara, Shiga, Japan), according to the manufacturer’s
instructions. Real-time RT-PCR was performed using Brilliant II
Fast SYBR® Green QPCR Master Mix (Stratagene, La Jolla,
CA, USA) with the Mx3000P QPCR System (Agilent Technologies Inc.),
according to the manufacturer’s instructions. Real-time RT-PCR of
the TGF-β1 primers were 5′-TTCAACACATCAGAGCTCCG-3′ (forward) and
5′-ATAACCACTCTGGCGAGTCG-3′ (reverse), for TGF-βRI:
5′-TAATTCCTCGAGATAGGCCG-3′ (forward) and 5′-TCGATGGTGAATGACAGTGC-3′
(reverse), for TGF-βRII: 5′-CAGCAGAAGCTGAGTTCAACC-3′ (forward) and
5′-GTGTTCTGCTTCAGCTTGGC-3′ (reverse), for SMAD2:
5′-GGAATTTGCTGCTCTTCTGG-3′ (forward) and 5′-TCTGCCTTCGGTATTCTGCT-3′
(reverse), for SMAD3: 5′-CCCCAGAGCAATATTCCAGA-3′ (forward) and
5′-GGCTCGCAGTAGGTAACTGG-3′ (reverse), for GAPDH:
5′-GAGTCAACGGATTTGGTCGT-3′ (forward) and 5′-TTGATTTTGGAGGGATCTCG-3′
(reverse). The relative gene expression was calculated by the ΔΔCt
method using an endogenous control (GAPDH) as 1.0. The formula is
expressed as follows: 2−ΔΔCt = 2− (ΔCt for target
gene − ΔCt for GAPDH).
ELISA
Cultured cells were purified using the Ficoll-Paque
method to remove CB. Cells (2×105/ml) were cultured in
24-well plates in RPMI-1640 medium supplemented with 10% 1X Serum
Replacement 1 (Sigma-Aldrich) for 3 days. Levels of TGF-β were
quantified by immunoassay using Quantikine ELISA kits (R&D
Systems, Minneapolis, MN, USA) according to the manufacturer’s
instructions.
Flow cytometry
Cell surface proteins were stained with anti-human
latency associated peptide (LAP) (TGF-β1)-PE antibody (27232) or
anti-human TGF-βRII-PE antibody (FAB241P) (R&D Systems).
Analysis was performed by a flow cytometer (FACSCalibur™; BD
Biosciences, Franklin Lakes, NJ, USA). For intracellular staining
of TGF-β1, cell surface TGF-β1 was blocked with 2.5 μg/ml of
anti-human TGF-β1 antibody (9016) (R&D Systems) for 30 min at
room temperature. Cells were fixed and permeabilized using a
fixation/permeabilization solution (BD Biosciences) for 20 min at
4°C. After washing twice in BD Perm/Wash buffer (BD Biosciences),
cells were stained with anti-human TGF-β1-PE (9016) (R&D
Systems) for 30 min at 4°C. After washing with BD Perm/Wash buffer,
cells were resuspended in PBS and analyzed on a flow cytometer.
Cell growth
Cultured MT-2Org cells, MT-2Rst cells, MT-2Org
control cells, MT-2Rst control cells, and TGF-β1-knockdown in
MT-2Rst cells were purified using the Ficoll density gradient
method to remove CB completely, and cells were then cultured for
2–4 days in the absence of CB. Cells (2×104/100 μl) were
cultured in 96-well U-bottom plates in RPMI-1640 medium
supplemented with 10% 1X Serum Replacement 1 (Sigma-Aldrich) in the
presence or absence of TGF-β1. The proliferation was evaluated on
day 3 based on [3H]-thymidine incorporation. After 2
days of culture, 3.7 kBq (0.1 μCi) [3H]-thymidine (10
μl) (GE Healthcare UK Ltd., Buckinghamshire, UK) was added to each
well. After 16 h of culture, [3H]-thymidine
incorporation was measured using a liquid scintillation counter
(LSC-5100, Aloka, Japan).
Western blot analysis
Cells were lysed in 50 mM Tris-HCl (pH 7.2) buffer
containing 150 mM NaCl, 1% Nonidet P-40, 1% deoxycholic acid, 0.05%
sodium dodecyl sulfate (SDS), 1X protease inhibitor cocktail
(Sigma-Aldrich, St. Louis, MO, USA), and 1X Halt Phosphatase
Inhibitor Cocktail (Thermo Fisher Scientific Inc., Rockford, IL,
USA). Proteins were quantified using the BCA assay kit (Thermo
Fisher Scientific Inc.), and 10 μg of protein was resolved on 10%
SDS-PAGE under reducing conditions with 5% 2-mercaptoethanol and
transferred to a PVDF membrane. Proteins were probed with the
following antibodies: p-SMAD2, p-SMAD3, SMAD2/3 (Cell Signaling
Technology, Inc.), p-ERK, p-JNK, ERK1, JNK1/3, p38α, (Santa Cruz
Biotechnology, Santa Cruz, CA, USA), p-p38 (BD Biosciences, San
Jose, CA, USA), and GAPDH (Millipore Corp. Headquarters, Billerica,
MA, USA), and incubated with HRP-conjugated anti-mouse IgG or
anti-rabbit IgG (Santa Cruz Biotechnology). Proteins were detected
with ECL Plus Western Blotting Detection Reagents (GE Healthcare UK
Ltd.). The intensity of western blotting was quantified with
Dolphon-View2 Band Tool (Kurabo Industries Ltd, Osaka, Japan).
RNA interference
Three kinds of double-stranded oligonucleotides
5′-GATCCCCGGAGGTCACCCGCGTGCTATTC A
AGAGATAGCACGCGGGTGACCTCCTTTTTGGAAA-3′
(no. 1), 5′-GATCCCCGTTCAAGCAGAGTACACACTTCAAG
AGAGTGTGTACTCTGCTTGAACTTTTTGGAAA-3′
(no. 2) and 5′-GATCCCCGTGGACATCAACGGGTTCATTCAAG
AGATGAACCCGTTGATGTCCACTTTTTGGAAA
(no. 3) were subcloned into pSUPER digested by
BglII-HindIII (24).
Resulting constructs were digested with BamHI/SalI,
and short hairpin RNA (shRNA) containing human H1 RNA polymerase
III promoter subcloned into the BamHI-SalI site of
pRDI292 as described previously (25).
Lentiviral vector production and viral
infection
The vesicular stomatitis virus G protein
(VSV-G)-pseudotyped HIV-1-based vector system was generated as
described previously (26). The
Replication-defective lentiviral vector particles were produced by
transient cotransfection of the second-generation packaging
construct pCMV-ΔR8.91 (27), the
VSV-G envelope plasmid pMDG2 and the lentiviral vector into 293FT
cells with FuGENE6 (Roche Diagnostics, Mannheim, Germany). The
supernatant containing the virus was collected 48 and 72 h after
transfection. The lentivirus-containing supernatants were subjected
to MT-2Org and MT-2Rst cells (0.5×105 in 2 ml of medium)
in a 6-well plate. After 3 days, cells were treated with 1 μg/ml of
puromycin to select stable clones expressing the shRNA.
Analysis of apoptosis by Annexin V
staining
Cells (1×105/ml) were cultured in the
absence or presence of 5, 12.5 or 25 μg/cm2 CB in
24-well plates for 24 h. Apoptotic cells were detected by staining
with Annexin V-FITC and propidium iodide (PI) (Roche Applied
Science, Indianapolis, IN, USA) according to the manufacturer’s
protocol, and stained cells were analyzed using a flow
cytometer.
Statistical analysis
A t-test and a Fisher’s parametric least significant
difference (PLSD) were performed to determine statistical
differences between the experimental groups.
Results
Enhancement of TGF-β1 production in
MT-2Rst cells by continuous exposure to CB
Long-term exposure to CB results in CB-dependent
resistance to apoptosis and upregulates IL-10 production in MT-2Org
cells, as previously described (14). Given that MT-2Org cells are known
to have a Treg cell-like suppressive function (20), in this study we investigated the
production of an anti-inflammatory cytokine, TGF-β1, in MT-2Rst
cells by long-term exposure to CB. The production of TGF-β1 in
culture supernatants was augmented large in MT-2Rst compared to
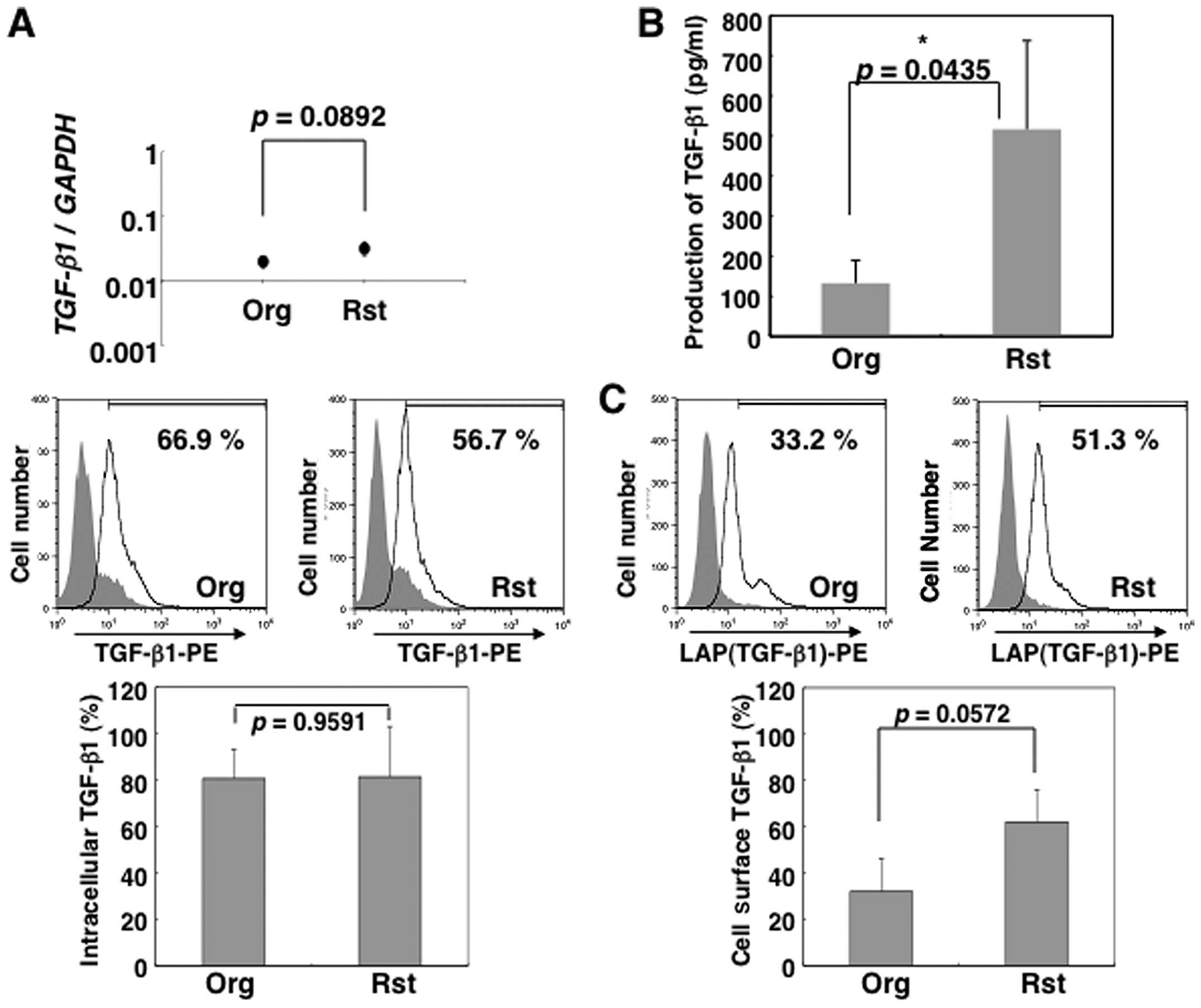
MT-2Org cells (Fig. 1B). Besides
FACS analysis showed that intracellular TGF-β1 was strongly
expressed in both MT-2Org and MT-2Rst cells as shown in the middle
and bottom panels of Fig. 1A, but
there were no significant differences such as those revealed by
real-time RT-PCR (top panel of Fig.
1A). Additionally, the cell surface TGF-β1 expression was
increased in MT-2Rst cells, although there were no significant
differences between MT-2Org and MT-2Rst cells (Fig. 1C). MT-2 cells are
HTLV-1-immortalized human polyclonal T cell line and express Tax
protein which represses TGF-β1 signaling in human T cells (28). Although we compared the gene
expression level of Tax1 between MT-2Org and MT-2Rst cells, there
were no differences in the gene expression level (data not shown).
These results suggested that the upregulation of TGF-β1 production
in MT-2Rst cells induced by long-term exposure to CB leads to an
enhancement of Treg-like phenotypes in a Tax-independent
manner.
MT-2Rst cells acquire resistance to the
growth inhibitory effect of TGF-β1
Although TGF-β1 inhibits the proliferation of T
cells and NK cells, TGF-β1 does not inhibit the growth of
TGF-β1-producing Treg cells (29).
Therefore, we examined whether TGF-β1 inhibits the proliferation of
MT-2Rst cells. As shown in Fig.
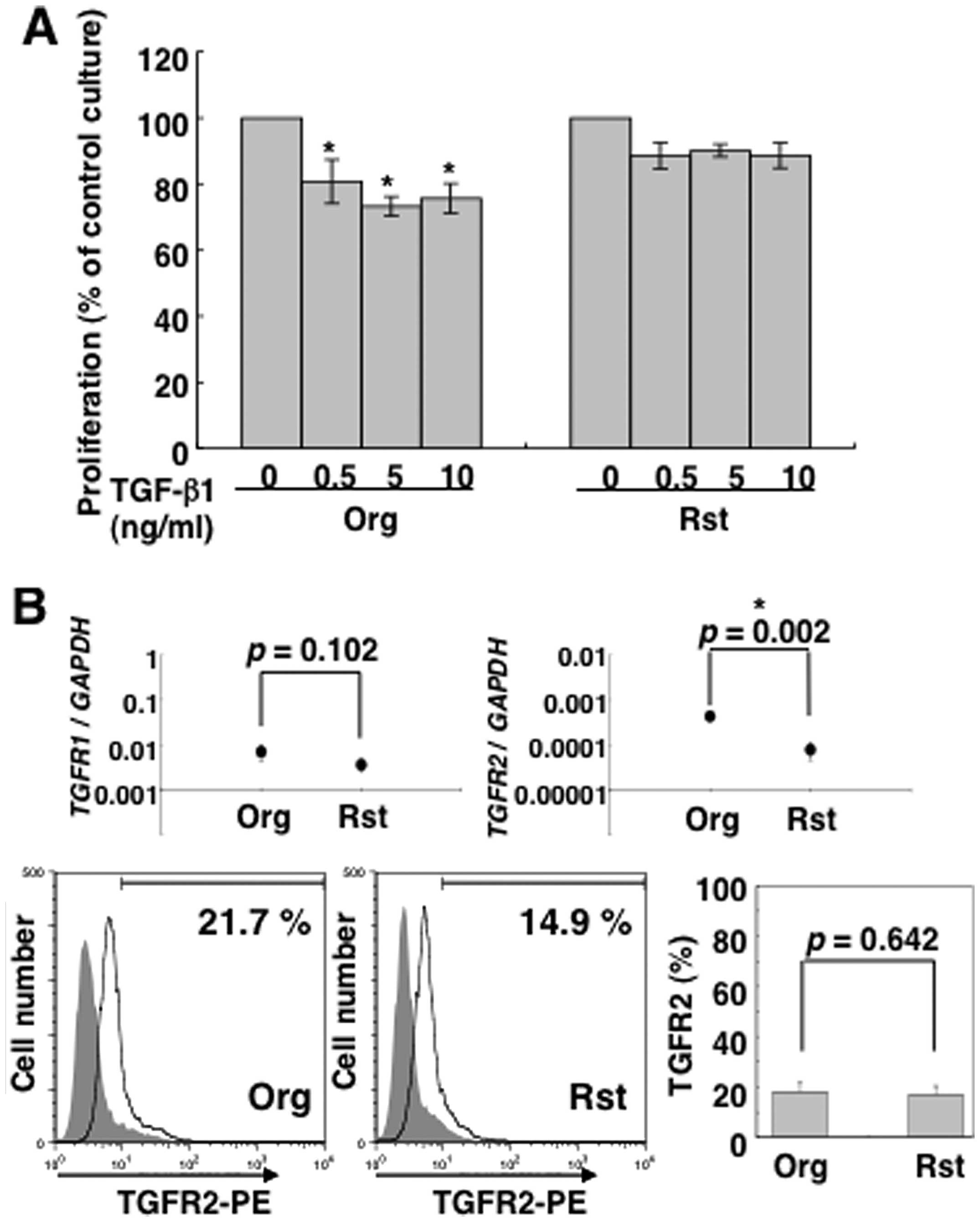
2A, the proliferation of MT-2Org cells was significantly
inhibited by TGF-β1, whereas MT-2Rst cells were not inhibited. In
order to confirm whether the resistance to TGF-β1-mediated growth
inhibition depends on TGF-β1 production, TGF-β1 knockdown in
MT-2Rst cells was generated using lentiviral vector-mediated RNA
interference. The knockdown efficiency of three TGF-β1 shRNA
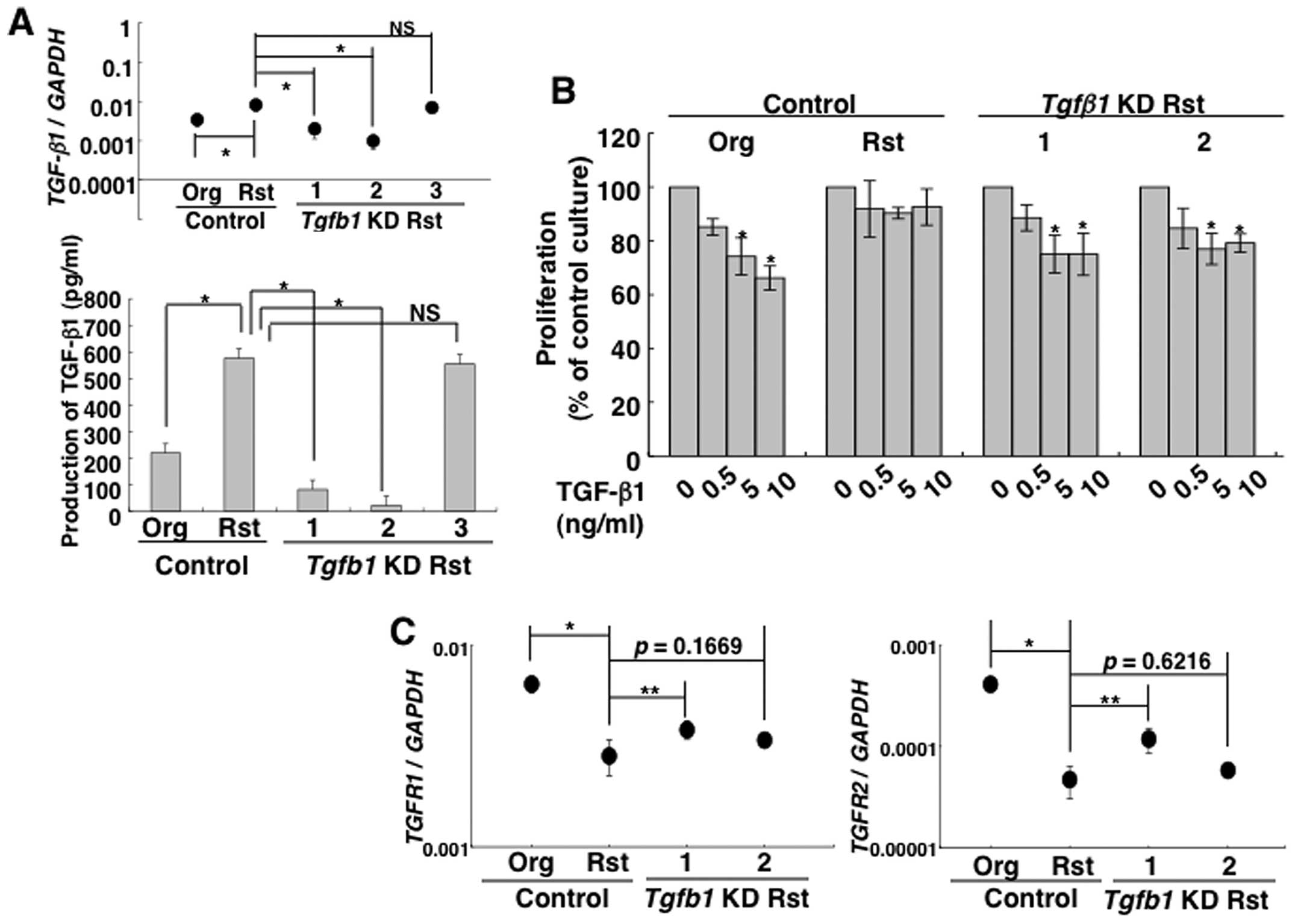
constructs was examined by real-time RT-PCR and ELISA (Fig. 3A), and the results confirmed that
the expression of TGF-β1 mRNA and production of TGF-β1
significantly decreased in construct nos. 1 and 2. Therefore, these
cell clones (construct nos. 1 and 2) were employed in subsequent
experiments. Interestingly, TGF-β1-knockdown cells significantly
inhibited their proliferation by TGF-β1 treatment in a manner
similar to the MT-2Org control transduced with a control lentiviral
vector, although these growth suppressions due to TGF-β1 were not
observed in MT-2Rst control cells transduced with a control
lentiviral vector (Fig. 3B). These
results suggested that MT-2Rst cells acquired the resistance to
TGF-β1-mediated growth inhibition through upregulation of TGF-β1
production.
Next, we investigated the expression of TGF-β
receptor (R) I and TGF-βRII in MT-2Org and MT-2Rst cells to
elucidate a mechanism in which TGF-β1 did not inhibit the growth of
TGF-β1-producing MT-2Rst cells. Real-time RT-PCR revealed that both
cells expressed TGF-βR1 and TGF-βR2 mRNA, and that
the latter expression was significantly decreased in MT-2Rst cells
(Fig. 2B). However, FACS analysis
showed that the expression of cell surface TGF-βRII was low in both
cells and there were no significant differences (Fig. 2B). Additionally, expression of
TGF-βR1 and TGF-βR2 mRNA decreased in MT-2Rst control
cells was enhanced in TGF-β1-knockdown cells, and was particularly
remarkable in construct no. 1 when compared with the MT-2Rst
control cells (Fig. 3C). These
results suggested that the acquisition of the resistance to
TGF-β1-mediated growth inhibition might be partly associated with
the expression level of TGF-βRI/II.
TGF-β1 production in MT-2Rst cells via
p38 MAP kinase activation
To elucidate the molecular mechanism involved in
overexpression and over-production of TGF-β1 in MT-2Rst cells, we
investigated the importance of these aspects and the activation of
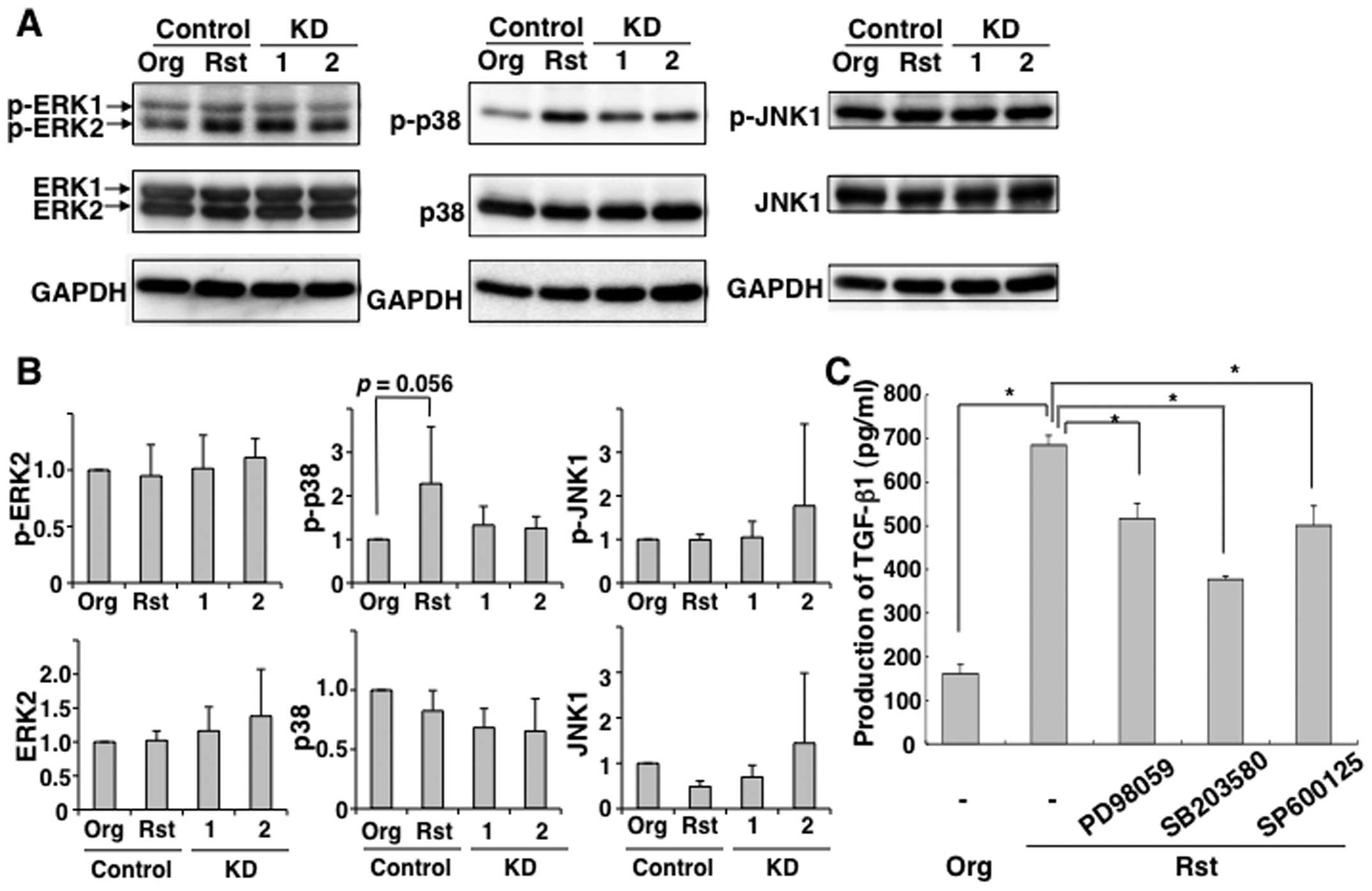
MAPKs, such as ERK1/2, p38 and JNK1. Given that we previously
reported that exposure to chrysotile-A induces apoptosis via
phosphorylation of p38 and JNK (13), we examined the activation of MAPKs
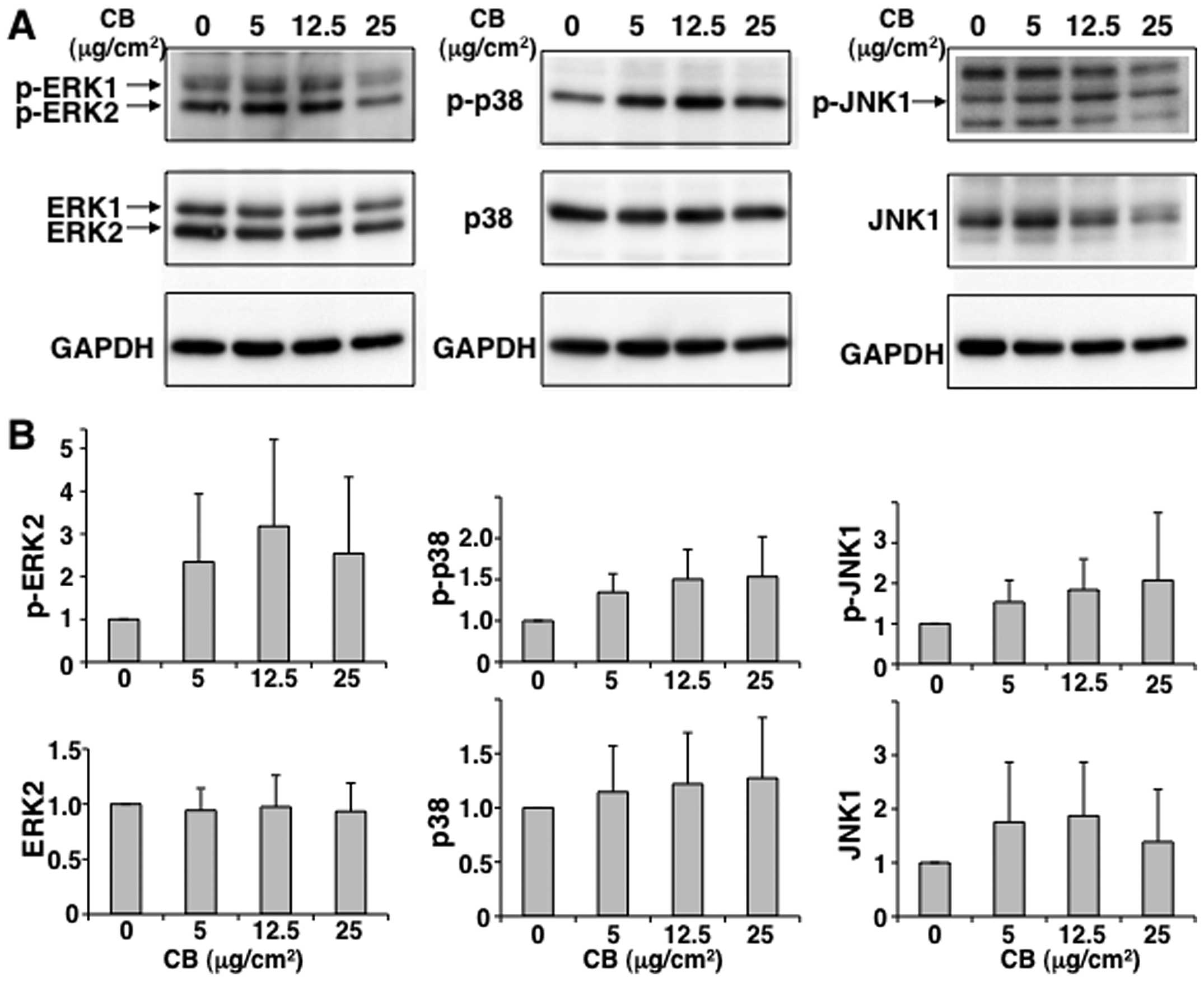
in MT-2Org cells subjected to short-term exposure to CB. As shown
in Fig. 4, ERK1/2, p38 and JNK1
were phosphorylated when MT-2Org cells were exposed to 5 or 12.5
μg/cm2 of CB, although there were no significant
differences between the treated and control groups. Furthermore, as
shown in Fig. 5A and B,
phosphorylated p38 in MT-2Rst control was decreased by knockdown of
TGF-β1. To confirm the association between TGF-β1 production and
p38 MAP kinase activation, MT-2Rst cells were treated with ERK
inhibitor PD98059, p38 MAP kinase inhibitor SB203580, or JNK
inhibitor SP600125. As shown in Fig.
5C, TGF-β1 production was largely reduced by inhibition of p38
phosphorylation, while decreased TGF-β1 production was also induced
by inhibition of ERK and JNK phosphorylation involved in
proliferation of cells (30).
These results suggested that long-term exposure to CB upregulates
TGF-β1 production via constitutive activation of the
phosphorylation of p38.
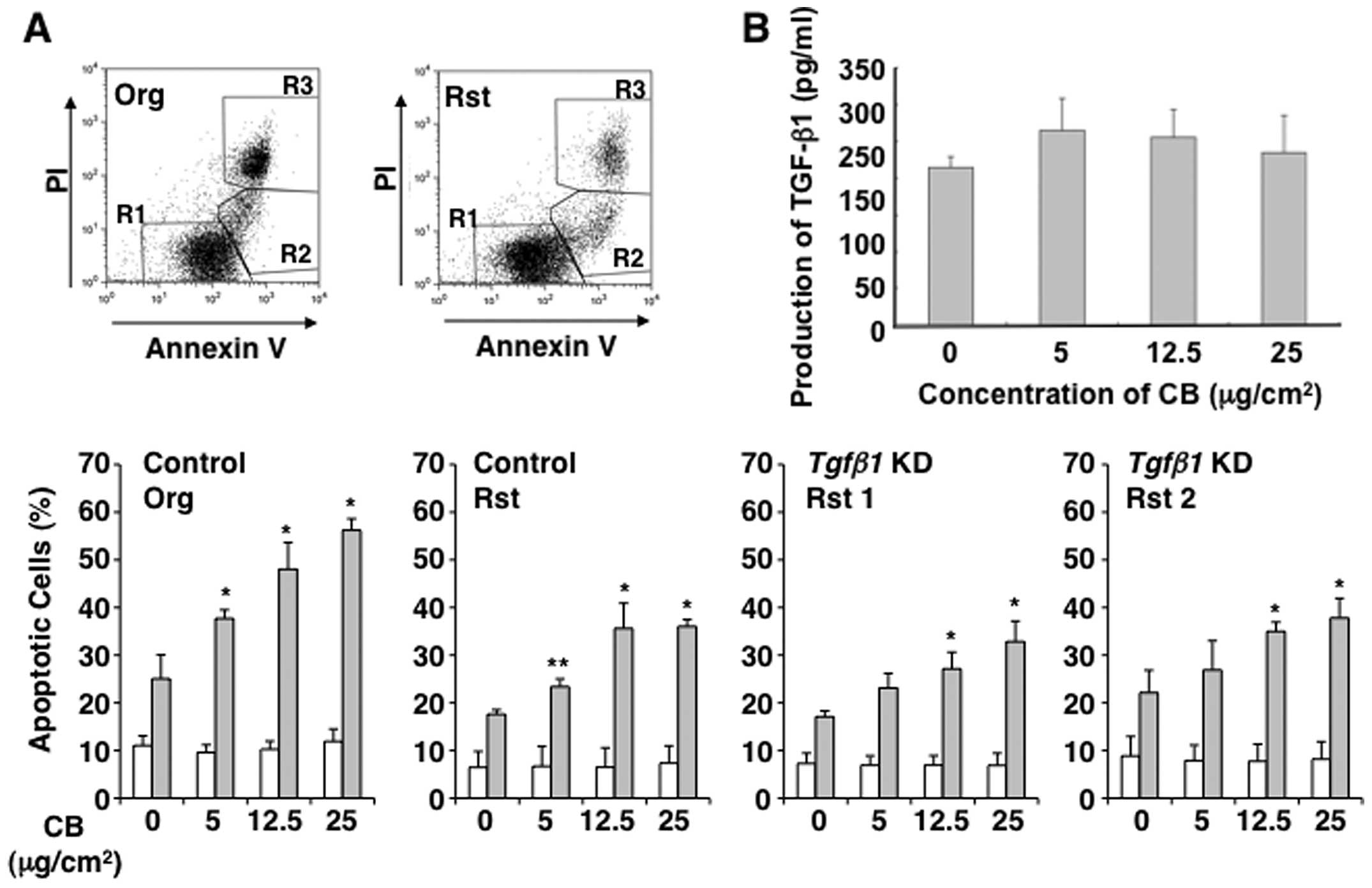
Apoptosis induced by CB occurs
independently of increased TGF-β1 production
To determine whether MT-2Rst cells acquire
resistance to asbestos-induced apoptosis depending on the
modification of TGF-β1 production, we examined the occurrence of
apoptosis caused by co-culturing with CB in TGF-β1-knockdown cells
using the Annexin V method. As shown in Fig. 6A, the short-term and high-dose
exposure to CB did not induce apoptosis in TGF-β1-knockdown cells
as in the MT-2Rst control cells, not knocked down, when compared
with the appearance of apoptosis in MT-2Org cells. The short-term
and high-dose exposure to CB in MT-2Org cells slightly increased in
TGF-β1 production via apoptosis, although there were no significant
differences between the treated and control groups (Fig. 6B). These results indicated that
acquisition of resistance to CB-induced apoptosis in MT-2Rst cells
occurs independently of enhanced TGF-β1 production by long-term
exposure to CB.
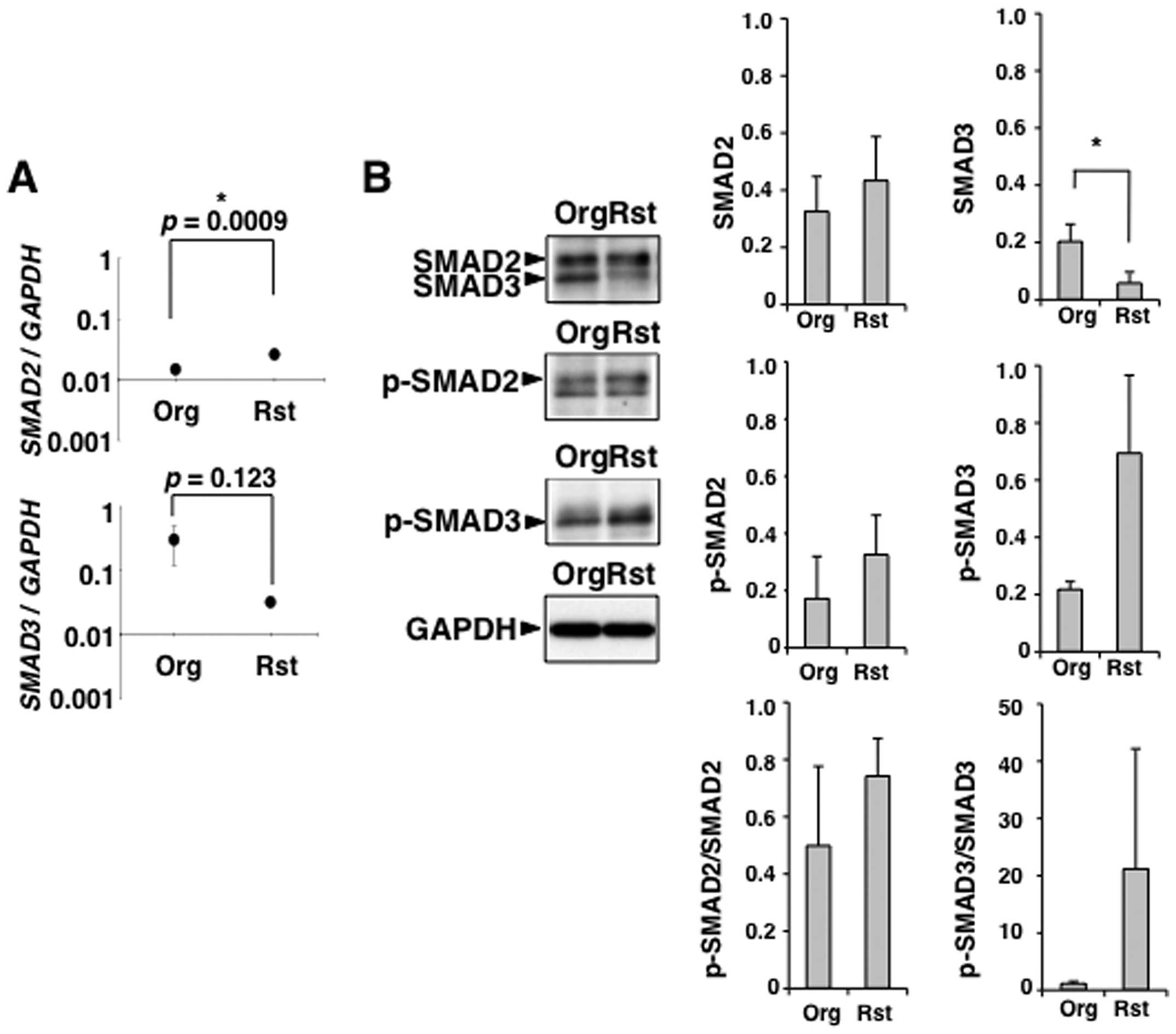
Regulation of Smad-dependent TGF-β1
signaling in MT-2Rst cells
TGF-β1-Smad pathway is involved in the inhibition of
T cell proliferation (31–34). We examined that the expression and
phophorylation of SMAD2/3 in TGF-β1 producing MT-2Rst cells. SMAD2
was highly expressed at mRNA and protein levels in MT-2Org and
MT-2Rst cells, and the level of SMAD2 mRNA was significantly
higher in MT-2Rst than MT-2Org cells (Fig. 7A). Similarly, in Fig. 8, there were no significant
differences in the protein level of SMAD2 arising from the level of
SMAD2 mRNA between MT-2Org control and MT-2Rst control
cells, suggesting that SMAD2 is degraded by proteasome to maintain
a certain amount of SMAD2 protein (35). On the other hand, mRNA and protein
expression levels of SMAD3 were lower in MT-2Rst than MT-2Org
cells, and the protein level of SMAD3 was significantly decreased
in MT-2Rst compared with MT-2Org cells (Fig. 7). Similarly, the mRNA and protein
level of SMAD3 were significantly decreased in MT-2Rst cells
(Fig. 8). SMAD2/3 was highly
phosphorylated in MT-2Rst cells compared with MT-2Org cells
(Fig. 7B), although the level of
phosphorylated SMAD3 was not enhanced in MT-2Rst cells (Fig. 8). The level of phosphorylated SMAD2
(p-SMAD2) and SMAD3 (p-SMAD3) were normalized to the protein
expression level of SMAD2 and SMAD3, respectively. The levels of
p-SMAD2/SMAD2 and p-SMAD3/SMAD3 were increased in MT-2Rst cells
compared with MT-2Org cells (Figs.
7B and 8), suggesting that
Smad-dependent TGF-β1 signaling through the activation of
TGF-βRI/II stimulated by autocrine TGF-β1 from MT-2Rst cells was
performed normally in MT-2Rst cells.
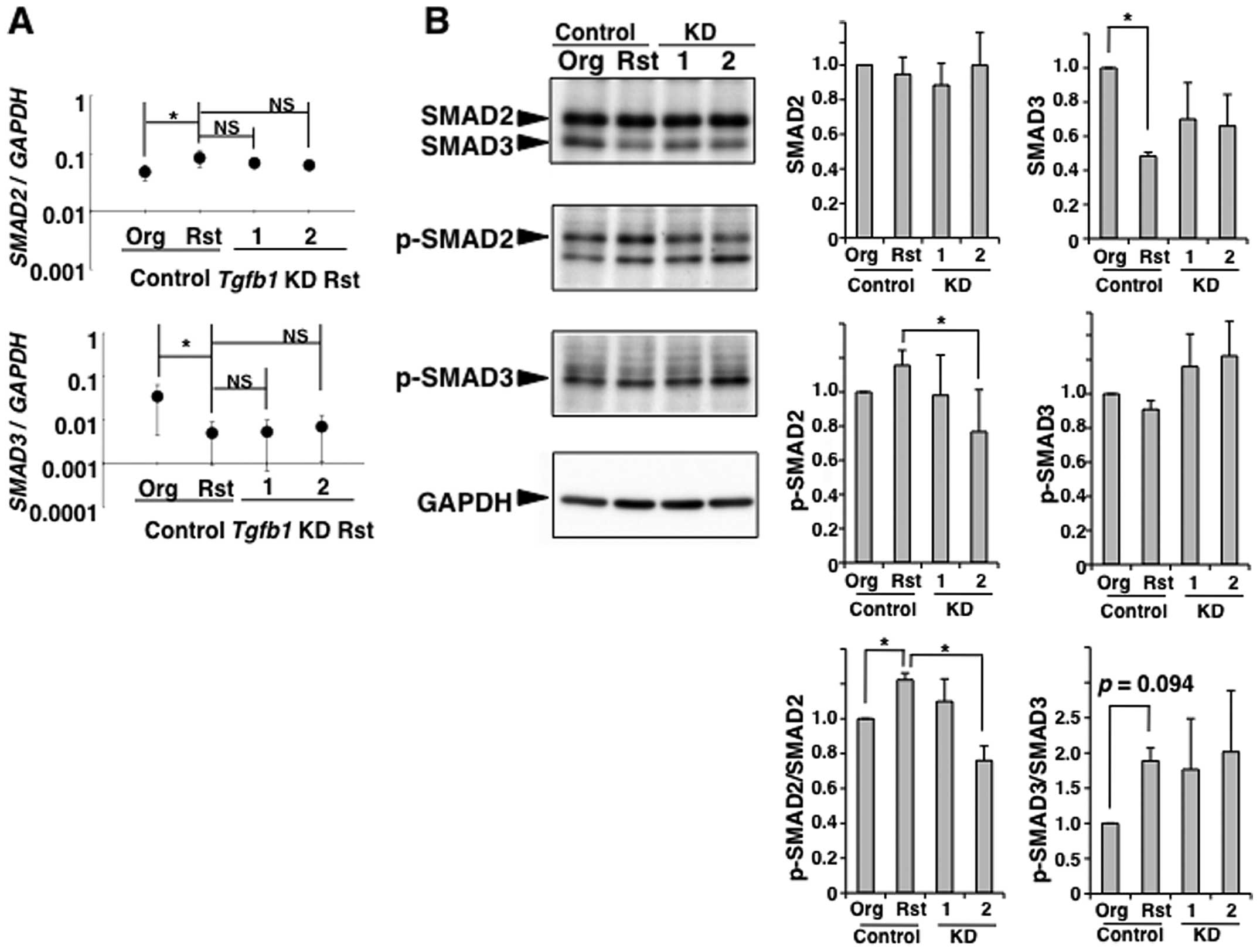
However, the results from TGF-β1-knockdown in
MT-2Rst cells showed that there were no significant differences in
mRNA expression of SMAD2 and SMAD3 among MT-2Rst
control, TGF-β1-knockdown nos. 1 and 2 (Fig. 8). Whereas the level of
p-SMAD2/SMAD2 was decreased in TGF-β1-knockdown cells, and was
particularly remarkable in construct no. 2 when compared with the
MT-2Rst control cells (Fig. 8B).
The SMAD3 protein expression decreased in the MT-2Rst control was
recovered slightly by TGF-β1-knockdown, although there were no
differences in p-SMAD3/SMAD3 among MT-2Rst control,
TGF-β1-knockdown nos. 1 and 2. Thus, phosphorylation of SMAD2 and
SMAD3 may be coordinated with the expression of SMAD2 and SMAD3.
These results were insufficient to understand that long-term
exposure of MT-2Org cells to CB induces TGF-β1 production, and
results in acquisition of the resistance to TGF-β1-mediated growth
inhibition.
Discussion
In this study, it was revealed that MT-2Rst cells,
which were established from MT-2Org cells by continuous exposure to
CB, produced high levels of TGF-β1 through phosphorylation of p38
MAPK, and acquire resistance to inhibition of cell growth by
TGF-β1. Moreover, it was suggested that continuous exposure of the
CD4+CD25+ HTLV-1 immortalized T cell line
(MT-2Org cells) to CB induces modification of cellular phenotypes
and makes these cells resemble Treg cells.
TGF-β1 inhibits proliferation and differentiation of
various immunocompetent cells, resulting in suppression of
antitumor immune function (22,29).
On the other hand, TGF-β1 has contributed to the development of
induced Treg cells (36).
Therefore, induced Treg cells that have the ability to produce
TGF-β1 do not exhibit inhibited cell growth by TGF-β1. Given that
TGF-β1 is produced not only from Treg cells but also tumor cells,
including MM cells (37), the
tumor microenvironment is rich in TGF-β1 (38) and results in the proliferation of
MM cells by TGF-β1 (39).
Consequently, TGF-β1 derived from Treg cells and tumor cells, which
inhibits the antitumor function of immune cells, induces an
immunosuppressive microenvironment surrounding the tumor and
promotes tumor growth. Plasma from patients with MM has high TGF-β1
levels, as previously reported (40). Our findings suggest that long-term
exposure to asbestos induces T cells that exhibit resistance to the
inhibitory effect against T cell proliferation by TGF-β1 derived
from tumor cells, resulting in TGF-β1 development of Treg cells,
suppression of antitumor immune function, and enhancement of tumor
growth. We need additional investigations of Treg cells in MM
patients to elucidate the immunosuppressive state induced by
TGF-β1.
TGF-β1 signaling depends on a heteromeric complex of
two types of transmembrane serine/threonine kinase receptors
(41). TGF-β1 binds to the
receptor complex, which activates TGF-βRII kinase to phosphorylate
and activate TGF-βRI kinase. The activated TGF-βRI phosphorylates
SMAD2 and SMAD3. Once SMAD2 or SMAD3 has been phosphorylated, it
interacts with SMAD4, and the complex translocates to the nucleus,
where it associates with other transcription factors to activate
transcription of target genes (42). It is known that mutations of
SMAD2/3 are involved with the progression of cancer (43,44).
In this study, Smad-dependent TGF-β1 signaling operated correctly
in MT-2Rst cells. Therefore, it seemed that the acquisition of
resistance to TGF-β1 in MT-2Rst cells caused by the long-term
exposure to CB due to reduced mRNA expression of TGF-β1 receptors
in a Smad-independent manner. Furthermore, it was observed that the
acquisition of resistance to the cell-proliferation inhibition
effects of TGF-β1 through increased TGF-β1 production in MT-2Rst
cells might not directly participate in the acquisition of
resistance to apoptosis induced by CB exposure.
It is known that apoptotic cells secrete TGF-β1
(45). Given that induced Treg
cells were developed by TGF-β1, we examined the relation between
TGF-β1 production and apoptosis in MT-2 cells. However, the results
suggested that TGF-β1 production was not related to apoptosis by
exposure to asbestos. On the other hand, it has been reported that
the conversion of CD4+CD25− T cells into
induced Treg cells is mediated by activation of p38 MAPK (46). Interestingly, phosphorylation of
p38 in MT-2Rst cells increased markedly, which was decreased by
TGF-β1 knockdown. Furthermore, TGF-β1 production in MT-2Rst cells
decreased by treatment with the p38 inhibitor, suggesting that
MT-2Org cells secrete TGF-β1 through constitutive phosphorylation
of p38 due to chronic exposure to CB. Finally, MT-2Rst cells became
much more similar to the Treg-like cell phenotype.
It is thought that MT-2Org cells are Treg-like
cells, since MT-2 cells possess a high level of forkhead box P3
(Foxp3) and exhibit suppressive activity in relation to T cell
proliferation (20). Our findings
have shown that long-term exposure of MT-2Org cells to CB enhanced
increased production of anti-inflammatory cytokine IL-10 and
TGF-β1. Therefore, it would be necessary to analyze expression of
Treg cell-related molecules [Foxp3, cytotoxic T-lymphocyte antigen
4 (CTLA-4), glucocorticoid-induced TNF-receptor (GITR)], and the
suppressive function of T cell proliferation in MT-2Rst cells
(21). In fact, Italian group has
recently reported that CTLA-4 had been used as a target for
treatment of advanced malignant mesothelioma (47). Furthermore, given that we have
found that the TGF-β1 production in MT-2Rst cells was induced by
chronic exposure to chrysotile-A and crocidolite, it would be
interesting to determine whether immunocompetent cells are affected
depending on the asbestos character (data not shown).
Taken together, these results may indicate the
possibility of using TGF-β1, TGF-βRI/II and SMAD2/3 as target
molecules in CD4+ T cells for the diagnosis and
treatment of asbestos-related MM.
Acknowledgements
We thank Dr Yasuo Ariumi for the VSV-G-pseudotyped
HIV-1-based vector system (pCMVΔR8.91 and pMDG2), pSUPER, pRDI292,
and 293FT cells. We also thank Misao Kuroki for technical
assistance. The authors thank the former members of our department,
Dr Yoshie Miura, Shuko Murakami, Fuminori Hyodoh, Akiko
Takata-Tomokuni and Ayako Ueki, for their contribution to the
establishment of the fundamental concepts of this investigation. We
also thank Ms. Tamayo Hatayama, Minako Kato, Naomi Miyahara, Shoko
Yamamoto, Keiko Kimura, Tomoko Sueishi and Yoshiko Yamashita for
their technical assistance. This study was supported in part by
Special Coordination Funds for Promoting Science and Technology
(H18-1-3-3-1, Comprehensive approach on asbestos-related diseases),
Takeda Science Foundation, grants from the Ministry of Education,
Culture, Sports, Science and Technology of Japan (20390178 and
22700933), Ryobi Teien Memory Foundation, The Promotion and Mutual
Aid Corporation for Private Schools of Japan, Kawasaki Medical
School Project Grants (21-201), Program to disseminate tenure
tracking system (FY 2011-2013) from the Ministry of Education,
Culture, Sports, Science and Technology of Japan, and Strategic
Research Foundation Grant-aided Project for Private Universities
from Ministry of Education, Culture, Sport, Science, and
Technology, Japan.
Abbreviations:
|
CB
|
chrysotile-B
|
|
Org
|
original
|
|
Rst
|
resistant
|
|
MM
|
malignant mesothelioma
|
References
|
1
|
Pan XL, Day HW, Wang W, Beckett LA and
Schenker MB: Residential proximity to naturally occurring asbestos
and mesothelioma risk in California. Am J Respir Crit Care Med.
172:1019–1025. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kurumatani N and Kumagai S: Mapping the
risk of mesothelioma due to neighborhood asbestos exposure. Am J
Respir Crit Care Med. 178:624–629. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heintz NH, Janssen-Heininger YM and
Mossman BT: Asbestos, lung cancers, and mesotheliomas: from
molecular approaches to targeting tumor survival pathways. Am J
Respir Cell Mol Biol. 42:133–139. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
LaDou J, Castleman B, Frank A, et al: The
case for a global ban on asbestos. Environ Health Perspect.
118:897–901. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ueki A, Yamaguchi M, Ueki H, et al:
Polyclonal human T-cell activation by silicate in vitro.
Immunology. 82:332–335. 1994.PubMed/NCBI
|
|
6
|
Aikoh T, Tomokuni A, Matsukii T, et al:
Activation-induced cell death in human peripheral blood lymphocytes
after stimulation with silicate in vitro. Int J Oncol.
12:1355–1359. 1998.
|
|
7
|
Nishimura Y, Miura Y, Maeda M, et al:
Impairment in cytotoxicity and expression of NK cell- activating
receptors on human NK cells following exposure to asbestos fibers.
Int J Immunopathol Pharmacol. 22:579–590. 2009.
|
|
8
|
Nishimura Y, Maeda M, Kumagai N, Hayashi
H, Miura Y and Otsuki T: Decrease in phosphorylation of ERK
following decreased expression of NK cell-activating receptors in
human NK cell line exposed to asbestos. Int J Immunopathol
Pharmacol. 22:879–888. 2009.PubMed/NCBI
|
|
9
|
Kumagai-Takei N, Nishimura Y, Maeda M, et
al: Effect of asbestos exposure on differentiation of cytotoxic T
lymphocytes in mixed lymphocyte reaction of human peripheral blood
mononuclear cells. Am J Respir Cell Mol Biol. 49:28–36. 2013.
View Article : Google Scholar
|
|
10
|
Nishimura Y, Maeda M, Kumagai-Takei N, et
al: Altered functions of alveolar macrophages and NK cells involved
in asbestos-related diseases. Environ Health Prev Med. 18:198–204.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miyoshi I, Kubonishi I, Yoshimoto S, et
al: Type C virus particles in a cord T-cell line derived by
co-cultivating normal human cord leukocytes and human leukaemic T
cells. Nature. 294:770–771. 1981. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maeda M, Miura Y, Nishimura Y, et al:
Immunological changes in mesothelioma patients and their
experimental detection. Clin Med Circ Respirat Pulm Med. 2:11–17.
2008.PubMed/NCBI
|
|
13
|
Hyodoh F, Takata-Tomokuni A, Miura Y, et
al: Inhibitory effects of anti-oxidants on apoptosis of a human
polyclonal T-cell line, MT-2, induced by an asbestos, chrysotile-A.
Scand J Immunol. 61:442–448. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Miura Y, Nishimura Y, Katsuyama H, et al:
Involvement of IL-10 and Bcl-2 in resistance against an
asbestos-induced apoptosis of T cells. Apoptosis. 11:1825–1835.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nishimura Y, Miura Y, Maeda M, et al:
Expression of the T cell receptor Vbeta repertoire in a human T
cell resistant to asbestos-induced apoptosis and peripheral blood T
cells from patients with silica and asbestos-related diseases. Int
J Immunopathol Pharmacol. 19:795–805. 2006.
|
|
16
|
Maeda M, Chen Y, Kumagai-Takei N, et al:
Alteration of cytoskeletal molecules in a human T cell line caused
by continuous exposure to chrysotile asbestos. Immunobiology.
218:1184–1191. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maeda M, Yamamoto S, Chen Y, et al:
Resistance to asbestos-induced apoptosis with continuous exposure
to crocidolite on a human T cell. Sci Total Environ. 429:174–182.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maeda M, Nishimura Y, Hayashi H, et al:
Reduction of CXC chemokine receptor 3 in an in vitro model of
continuous exposure to asbestos in a human T-cell line, MT-2. Am J
Respir Cell Mol Biol. 45:470–479. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Maeda M, Nishimura Y, Hayashi H, et al:
Decreased CXCR3 expression in CD4+ T cells exposed to
asbestos or derived from asbestos-exposed patients. Am J Respir
Cell Mol Biol. 795–803. 2011.
|
|
20
|
Chen S, Ishii N, Ine S, et al: Regulatory
T cell-like activity of Foxp3+ adult T cell leukemia
cells. Int Immunol. 18:269–277. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sakaguchi S, Miyara M, Costantino CM and
Hafler DA: FOXP3+ regulatory T cells in the human immune
system. Nat Rev Immunol. 10:490–500. 2010.
|
|
22
|
Flavell RA, Sanjabi S, Wrzesinski SH and
Licona-Limon P: The polarization of immune cells in the tumour
environment by TGFbeta. Nat Rev Immunol. 554–567. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kohyama N, Shinohara Y and Suzuki Y:
Mineral phases and some reexamined characteristics of the
International Union Against Cancer standard asbestos samples. Am J
Ind Med. 30:515–528. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brummelkamp TR, Bernards R and Agami R: A
system for stable expression of short interfering RNAs in mammalian
cells. Science. 296:550–553. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bridge AJ, Pebernard S, Ducraux A,
Nicoulaz AL and Iggo R: Induction of an interferon response by RNAi
vectors in mammalian cells. Nat Genet. 34:263–264. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Naldini L, Blomer U, Gallay P, et al: In
vivo gene delivery and stable transduction of nondividing cells by
a lentiviral vector. Science. 272:263–267. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zufferey R, Nagy D, Mandel RJ, Naldini L
and Trono D: Multiply attenuated lentiviral vector achieves
efficient gene delivery in vivo. Nat Biotechnol. 15:871–875. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Arnulf B, Villemain A, Nicot C, et al:
Human T-cell lymphotropic virus oncoprotein Tax represses TGF-beta
1 signaling in human T cells via c-Jun activation: a potential
mechanism of HTLV-I leukemogenesis. Blood. 100:4129–4138. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li MO, Wan YY, Sanjabi S, Robertson AK and
Flavell RA: Transforming growth factor-beta regulation of immune
responses. Annu Rev Immunol. 24:99–146. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang W and Liu HT: MAPK signal pathways
in the regulation of cell proliferation in mammalian cells. Cell
Res. 12:9–18. 2002. View Article : Google Scholar
|
|
31
|
Letterio JJ: TGF-beta signaling in T
cells: roles in lymphoid and epithelial neoplasia. Oncogene.
24:5701–5712. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li L, Iwamoto Y, Berezovskaya A and
Boussiotis VA: A pathway regulated by cell cycle inhibitor
p27Kip1 and checkpoint inhibitor Smad3 is involved in
the induction of T cell tolerance. Nat Immunol. 7:1157–1165. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Delisle JS, Giroux M, Boucher G, et al:
The TGF-beta-Smad3 pathway inhibits CD28-dependenT cell growth and
proliferation of CD4 T cells. Genes Immun. 14:115–126. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yoshimura A, Wakabayashi Y and Mori T:
Cellular and molecular basis for the regulation of inflammation by
TGF-beta. J Biochem. 147:781–792. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhu H, Kavsak P, Abdollah S, Wrana JL and
Thomsen GH: A SMAD ubiquitin ligase targets the BMP pathway and
affects embryonic pattern formation. Nature. 400:687–693. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wrzesinski SH, Wan YY and Flavell RA:
Transforming growth factor-beta and the immune response:
implications for anticancer therapy. Clin Cancer Res. 13:5262–5270.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Suzuki E, Kim S, Cheung HK, et al: A novel
small-molecule inhibitor of transforming growth factor beta type I
receptor kinase (SM16) inhibits murine mesothelioma tumor growth in
vivo and prevents tumor recurrence after surgical resection. Cancer
Res. 67:2351–2359. 2007. View Article : Google Scholar
|
|
38
|
Zou W: Regulatory T cells, tumour immunity
and immunotherapy. Nat Rev Immunol. 6:295–307. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fujii M, Toyoda T, Nakanishi H, et al:
TGF-beta synergizes with defects in the Hippo pathway to stimulate
human malignant mesothelioma growth. J Exp Med. 209:479–494. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Murakami S, Nishimura Y, Maeda M, et al:
Cytokine alteration and speculated immunological pathophysiology in
silicosis and asbestos-related diseases. Environ Health Prev Med.
14:216–222. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Brown KA, Pietenpol JA and Moses HL: A
tale of two proteins: differential roles and regulation of Smad2
and Smad3 in TGF-beta signaling. J Cell Biochem. 101:9–33. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Siegel PM and Massague J: Cytostatic and
apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev
Cancer. 3:807–821. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Levy L and Hill CS: Alterations in
components of the TGF-beta superfamily signaling pathways in human
cancer. Cytokine Growth Factor Rev. 17:41–58. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xiao YQ, Freire-de-Lima CG, Schiemann WP,
Bratton DL, Vandivier RW and Henson PM: Transcriptional and
translational regulation of TGF-beta production in response to
apoptotic cells. J Immunol. 181:3575–3585. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Huber S, Schrader J, Fritz G, et al: P38
MAP kinase signaling is required for the conversion of
CD4+CD25− T cells into iTreg. PLoS One.
3:e33022008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Calabro L, Morra A, Fonsatti E, et al:
Tremelimumab for patients with chemotherapy-resistant advanced
malignant mesothelioma: an open-label, single-arm, phase 2 trial.
Lancet Oncol. 14:1104–1111. 2013. View Article : Google Scholar : PubMed/NCBI
|