Introduction
Laryngeal squamous cell carcinoma (LSCC) is one of
the most common head and neck squamous cell carcinoma (HNSCC)
worldwide. Cancer of the larynx accounts for ~1.2% of new
malignancies worldwide every year, with an incidence of ~151,000
cases per year, ~95% of these tumors have squamous cell carcinoma
as the primary histological type (1,2). It
has been reported that this neoplasia is strongly linked to
cigarette smoking, alcohol ingestion and human papillomavirus
infection (3–5). Treatment options in laryngeal
carcinoma comprise surgery, radiotherapy, chemotherapy or a
combination therapy. Although treatment options have improved, an
epidemiological survey revealed that the survival rate of patients
with LSCC has demonstrated a decreasing trend, although the
survival rates of patients with other types of cancer have been
prolonged (6). Thus, it is
necessary and important to look for new diagnostic methods and new
treatment targets to control this malignancy.
More than 1,000 miRNAs have been estimated in human
genome, only ~200–300 miRNAs have been currently identified in
humans (7). It has been
extensively debated whether miRNAs are just fine-tuning molecules
or they act as key gene switches. Recent studies suggest that both
hypotheses are probably true, depending on the specific biological
context (8). Emerging evidence has
demonstrated that miRNAs have diverse functions in regulating
multiple stages of cancer such as cell proliferation, initiation,
promotion, malignant conversion, progression, angiogenesis,
epithelial-to-mesenchymal transition, invasion and metastasis
(9). Aberrant expressions of
miRNAs have been closely associated with cancer pathogenesis,
>50% of miRNA-encoding loci located in cancer-associated genomic
regions or in fragile sites, some miRNAs have been reported to
function as oncogenes or tumor suppressors (10).
miR-93 is a member of the miR-106b-25 cluster, a
paralog of the miR-17-92 cluster. miR-106b-25 cluster contains
three pre-miRNAs, pre-miR-25, pre-miR-93 and pre-miR-106b, which
all reside in the 13th intron of the MCM7 gene. Previous studies
indicated that miR-93 can repress the tumor suppressor TP53INP1 in
human T-cell leukemia virus 1-transformed human T-cells (11) and FUS-1 in human lung cancer cell
lines (12). Ectopic expression of
miR-93 decreased expression of NRF2, suggested an oncogenic
potential during breast carcinogenesis (13). miR-93 functions as an oncogene by
enhancing tumor cell survival, angiogenesis and metastasis by
targeting integrin-β8 expression (14) and LATS2 expression (15). miR-93 can also function as a new
regulator of PTEN/Akt signaling pathway, and may serve as a
potential target for overcoming CDDP resistance in human ovarian
cancer (16). Consistently,
expression of miR-93 induces mesenchymal-epithelial transition
(MET) and downregulates multiple stem cell regulatory genes in
‘claudin low’ SUM159 cells (17).
Considering all the above it would be interesting and meaningful to
investigate the role of miR-93 and its family members in LSCC
progression.
Cell cycle regulation is the core part in cell
proliferation, which has a close relationship with cell
carcinogenesis. Cyclin G2 is an unconventional cyclin, located on
human chromosome 4q21.1, function as a ubiquitous inhibitor of cell
cycle progression by preventing G1 to S-phase transition. This
inhibition is achieved by interacting with protein phosphatase 2A
(PP2A), the CCNG2-PP2A complex alters PP2A targeting or its
substrate specificity leading to cell cycle arrest. CCNG2 can also
stops cell cycle progression by preventing the phosphorylation of
cyclin-dependent kinase 2 (CDK2), promoting the formation of
unusual nuclear DNA structures suggestive of aberrant mitosis or
cytokinesis (18).
Phosphoinositide 3-kinase and FoxO transcription factors regulate
cyclin G2 expression, thus controling cell cycle progression
(19,20). Ectopic expression of cyclin G2
induces microtubule bundling and resistance to depolymerization,
inhibition of polymer regrowth from microtubule organizing centers
(MTOCs), and a p53-dependent cell cycle arrest (21). These observations suggest that
cyclin G2 is a cell cycle modulator that inhibits cellular
proliferation. However, the role of CCNG2 is not fully
characterized, and the mechanisms of CCNG2 dysregulation in
laryngeal cancer are not defined.
In our previous study, we presented comprehensive
profiling of miRNAs in LSCC through LCM acquisition of homogeneous
samples of cancer cells. Six miRNAs were validated, including
upregulation of miR-21, miR-93, miR-205 and miR-708, downregulation
of miR-125b and miR-145 (22). We
predicted that CCNG2 may be a potential target gene of miR-93. In
the present study, we verified that the expression of miR-93 was
significantly upregulated in 59 cases of LSCC tissues compared with
non-cancerous tissues. Ectopically expressed miR-93 could
accelerate growth and invasion of laryngeal cancer cells.
Furthermore, we found that CCNG2 is a direct and functional target
of miR-93. The expression of CCNG2 is closely correlated with tumor
progression and unfavorable outcome.
Materials and methods
Cell culture
Human embryonic kidney (HEK293) cell line and the
human laryngeal squamous carcinoma cell line Hep-2 was purchased
from the Cell Bank, Shanghai Institute of Life Science, Chinese
Academy of Science (Shanghai, China). The cells were cultured in
RPMI-1640 (HyClone, Novato, CA, USA) supplemented with 10%
fetal-bovine serum (FBS; Gibco-BRL, Gaithersburg, MD, USA),
penicillin G (1×105U/l) and streptomycin (100 mg/l).
These cells were maintained in an incubator at 37°C under an
atmosphere of 5% CO2.
Clinical specimens
Fifty-nine pairs of laryngeal SCC samples and
matched adjacent normal samples were collected from surgical
specimens immediately after primary resection of laryngeal
carcinoma patients in the Department of Otolaryngology-Head and
Neck Surgery, Fudan University Affiliated Eye, Ear, Nose and Throat
Hospital, Shanghai, China. The study was approved by the
institutional review board and written consent was signed by each
patient. None of the patients had received neoadjuvant therapies.
The fresh specimens were immediately taken after the surgery, one
was fixed in 4% paraformaldehyde solution, then embedded in
paraffin for immunohistochemistry, and the other one was stored in
liquid nitrogen for qRT-PCR and western blot assay. Regarding the
pathologic evaluation of surgical specimens, the pathologic
tumor-node-metastasis classification (TNM), and stages of laryngeal
SCC were decided in accordance with the 2002 American Joint
Committee on Cancer - Union Internationale Contre le Cancer
(AJCC-UICC) TNM system. The LSCC clinical information was collected
from patient records, and the details are listed in Tables I and II with the correlation of CCNG2
expression.
 | Table IExpression of CCNG2 in LSCC tissue
and normal laryngeal tissue. |
Table I
Expression of CCNG2 in LSCC tissue
and normal laryngeal tissue.
| | Expression of CCNG2
protein |
|---|
| |
|
|---|
| Group | Case (n) | − | + | ++ | +++ | χ2 | P-value |
|---|
| Cancer tissue | 59 | 38 | 5 | 8 | 8 | 49.015 | <0.001 |
| Normal tissue | 59 | 2 | 19 | 17 | 21 | | |
| Table IIThe relationship between the
expression of cyclin G2 and clinicopathological parameters. |
Table II
The relationship between the
expression of cyclin G2 and clinicopathological parameters.
| | Expression of CCNG2
protein |
|---|
| |
|
|---|
| Case (n) | Negative | Positive
(+~+++) | χ2 | P-value |
|---|
| Age at surgery,
(years) |
| ≤65 | 48 | 17 | 31 | 0.161 | 0.688 |
| >65 | 12 | 5 | 7 | | |
| Gender |
| Male | 54 | 21 | 33 | 0.002 | 0.961 |
| Female | 5 | 2 | 3 | | |
| Clinical stage |
| I–II | 10 | 3 | 7 | 5.51 | 0.019 |
| III–IV | 49 | 34 | 15 | | |
| Site |
| Supraglottic | 24 | 6 | 18 | 0.287 | 0.592 |
| Glottic and
infraglottic | 35 | 11 | 24 | | |
| Lymph node
metastasis |
| Negative | 25 | 12 | 13 | 9.451 | 0.002 |
| Positive | 34 | 29 | 5 | | |
| Histology
grade |
| SCC I–II | 37 | 25 | 12 | 8.984 | 0.003 |
| SCC III | 22 | 6 | 16 | | |
Immunohistochemistry
Paraffin-embedded tissues were cut into 4-μm thick
sections and analyzed with immunohistochemical staining. CCNG2 and
Ki-67 expression was determined based on the percentage of positive
cells, combined with the staining intensity. The proportion of
positive cells was divided into four levels: 0 point, ≤5% of
positive cells; 1 point, 5% ~25%; 2 points, 25–50%, and 3 points,
>50% of positive cells. The intensity of staining was classified
as: 0 point, no staining; 1 point, weak staining (light yellow); 2
points, moderate staining (brown); and 3 points, strong staining
(yellowish brown). The final scores of CCNG2 and Ki-67 expression
was the product of the CCNG2 and Ki-67 expression proportion and
intensity, graded as 0 for negative, + for 1–3 points, ++ for 4–6
points, and +++ for 7–9 points.
In vivo assays for tumor formation
For the in vivo tumor formation assays,
3×106 Hep-2 cells infected with the Lenti-miR-93 or mock
vector were suspended in 200 μl PBS and subcutaneously injected
into the right flank of each nude mouse (female BALB/c-nu/nu,
four-week-old, 6 per group). Tumor volumes were measured using
vernier calipers every 3 days as soon as the tumors were
measurable, and the tumor volumes were calculated: V
(cm3) = width2 (cm2) × length
(cm)/2. On day 32, the mice were sacrificed. The mice were
manipulated and housed according to protocols approved by the
Shanghai Medical Experimental Animal Care Commission.
Lentivirus production and infection
The pri-miR-93 sequence was amplified from normal
genomic DNA and cloned into the pGMLV-PE1 microRNA lentiviral
vector to generate pGMLV-PE1-miR-93. The primer sequences are:
miR-93-F (XhoI): CCGCTCGAGAACCTTCACTGAGAGGGTGGTT and
miR-93-R (BamHI): CGGGATCCGAGGGAGACCAGA CCCTTTTG. The
amplified sequences were inserted into the XhoI and
BamHI restriction sites of the multiple cloning site of
expression vector pGMLV-PE1 and verified by sequencing. The shuttle
vector was co-transfected with virapower packaging mix, containing
packaging plasmid pMDL-gp-RRE, pRSV-Rev, pCMV-VSVG and so on, into
293T cells to produce the lentivirus, using the HG transgene
reagent (Genomeditech Co., Shanghai, China). Virus-containing media
was collected, filtered and added to target Hep-2 cells for
infection in the presence of polybrene (Sigma, St. Louis, MO,
USA).
Construction of the pcDNA3.1-CCNG2 and
pcDNA3.1-NC expression vectors
The coding region of the CCNG2 mRNA was amplified by
PCR from human genomic DNA. The amplified sequences were inserted
into the EcoRI and BamHI restriction sites of the
multiple cloning site of expression vector pcDNA3.1 and verified by
sequencing. The primers used were: human CCNG2-F (EcoRI):
CCGGAATTCGCCA CCATGAAGGATTTGGGGGCAG, human CCNG2-R (BamHI):
CGGGATCCCTAAGATGGAAAGCACAGTGT TTGT. Empty plasmid was used as
negative control. Cells were transfected with the specified
expression vector pcDNA3.1 using Lipofectamine 2000. After 48 h,
antibiotic-G418 at 800 ng/ml was added to select cells with stably
integrated expression constructs. Stable cell lines were
established within 3–4 weeks.
Cell proliferation and colony formation
assays
The cell proliferation assay was measured using the
Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan).
Approximately 2×103 cells were seeded in each well of a
96-well plate, and 10 μl of CCK-8 was added to 90 μl of the culture
medium at the indicated time. The cells were subsequently incubated
at 37°C for 2 h, and the ultraviolet absorbance at 450 nm was
measured using a BioTek synergy H1 hybrid reader. For the colony
formation assay, 200 cells were plated into each well of a 6-well
plate and incubated at 37°C for two weeks, the culture medium was
changed once every 3 days. The colonies were fixed and stained in a
dye solution containing 1% crystal violet, and the number of
colonies was counted. Colonies were counted using ImageJ software
and differences were assessed by two-tailed t-test. These assays
were replicated three times.
Migration and invasion assays
A 24-well plate containing 8-μm-pore size chamber
inserts (Corning Incorp.) was used to evaluate the migration and
invasion of tumor cells. For the invasion assay, the membrane was
coated with Matrigel to form a matrix barrier, and 2×105
cells were placed in the upper chamber. For the migration assay,
only 5×104 cells were seeded in the upper chamber. Cells
in 200 μl serum-free 1640 medium were gently loaded onto each
filter insert (upper chamber) and in each lower chamber, 600 ml of
1640 medium with 10% FBS was added, then incubated at 37°C for 18
or 24 h, respectively for migration assay and invasion assay. After
that, the filter inserts were removed from the chambers, cells that
migrated or invaded were fixed and stained with haematoxylin for 20
min. The number of cells that had migrated or invaded was counted
and photographed using a Nikon Ti-E inverted microscope.
Cell cycle and cell apoptosis
analysis
Cell Cycle Detection kit was purchased from KeyGen
Biotech Co., Ltd. (Nanjing, China). Approximately 1×106
cells were collected and fixed with ice-cold 70% ethanol in PBS at
−20°C overnight. Cells were treated with RNase A for 30 min and
then labeled for 15 min with PI and immediately analyzed by flow
cytometry. The test was performed in triplicate. The data were
evaluated using ModFit software program and presented as means ± SD
(n=3).
The cell apoptosis assay was performed using flow
cytometry and was detected with the Annexin V-FITC/PI apoptosis
detection kit (KeyGen Biotech) or Annexin V-APC/7-AAD apoptosis
detection kit (KeyGen Biotech). Cells were cultured and treated
with the agents indicated in the figure legends according to the
manufacturer’s instructions. Cells were resuspended in 400 μl
Annexin V binding buffer and subsequently incubated with 5 μl
Annexin V-FITC or Annexin V-APC for 15 min in room temperature;
then, 10 μl of PI or 7-AAD was added. Experimental data were
analyzed using FlowJo software (version 7.6.1) and presented as
means ± SD (n=3).
Real-time quantitative PCR
For qRT-PCR of miRNAs, low-molecular-weight RNA
(<200 nt) was separated from the total RNA using the mirVana
miRNA isolation kit (Ambion, Foster City, CA, USA) according to the
manufacturer’s protocol. Normalization was performed using the
signal of U6 snRNA. cDNA synthesis was performed with 100 ng total
RNA using a Hairpin-it™ MicroRNAs Quantitation PCR kit (GenePharma,
Co., Ltd., Shanghai, China) according to the manufacturer’s
protocol. The sequences of primers specific for individual miR-93
are as follows: forward primer, CGTTATATCCC AAAGTGCTGTTC and
reverse primer, TATGGTTGTTCT CGTCTCCTTCTC. Thermal cycling and
fluorescent monitoring were performed on an ABI ViiA™ 7 Sequence
Detection System. Relative expression (RE) of the sample gene was
calculated using the formula RE = 2−ΔΔCT.
For qRT-PCR of CCNG2 and MMP-9, total RNA was
extracted from clinical specimens or cultured cells using TRIzol
reagent (Invitrogen). The quantity was measured on a BioTek synergy
H1 hybrid reader, and the integrity of the RNA was checked on a 1%
agarose gel. cDNA was synthesized with the PrimeScript™ RT Master
Mix (Takara Bio, Shiga, Japan) using 500 ng of RNA. The real-time
PCR analyses were performed using SYBR® Premix Ex Taq™
(Takara). The levels of CCNG2 transcript were measured by forward
primer, 5′-CAGGATTGAGAAATGCCAAAGT-3′ and reverse primer,
5′-TGACAGCCAGGACAAAAGTT-3′. The levels for MMP-9 were measured by
forward primer, 5′-GCCACTTCCCCTTC ATCTTC-3′ and reverse primer,
5′-GGTCGTCGGTGTCG TAGTTG-3′. β-actin was used as internal controls
and amplified with forward primer, 5′-AGCGAGCATCCCCC AAAGTT-3′ and
reverse primer, 5′-GGGCACGAAGGCTC ATCATT-3′.
Western blots
Cell lysates were prepared using RIPA buffer
(Beyotime Institute of Biotechnology, Shanghai, China). Protein
concentration was determined using the BCA assay (Beyotime
Institute of Biotechnology). For electrophoresis, equal amounts of
cell lysate protein (30 μg) were resolved by SDS-PAGE and
transferred to polyvinylidene difluoride (PVDF) membrane
(Millipore, Billerica, MA, USA). Blots were probed at 4°C overnight
with primary antibodies in 5% milk/TBST. Membranes were blocked and
probed with the following antibodies: mouse anti-CCNG2 (Abcam
BioSciences), mouse anti-GADPH (WeiAo Biotechnology, Shanghai,
China). Bound antibodies were detected with secondary
HRP-conjugated antibodies and visualized by enhanced
chemiluminescent substrate (WeiAo Biotechnology). Western blot data
were quantified by normalizing the signal intensity of each sample
to that of GADPH using ImageJ software.
Luciferase reporter assay
We used a luciferase reporter vector pLUC to
generate luciferase reporter constructs. There are two potential
binding sites for miR-93 in the CCNG2 3′UTR. These two sites are
close to each other, which made the analysis of each site
individually difficult. The segment of the wild-type 3′UTR of CCNG2
(CCNG2-3′UTR) containing the predicted miR-93 target site was
cloned from human genomic DNA. The mutant construct
(CCNG2-3′UTR-mutant), with the part of the target sites
corresponding to the seed sequence of miR-93, CCNG2 3′UTR-186
(5′-CAUUA ACAGUACUUUA-3′) replaced by (5′-CAUUAACAG UACUUUA-3′),
CCNG2 3′UTR-222 (5′-GCACUUU-3′) replaced by (5′-CGUGAAA-3′). The
CCNG2 fragment was cloned with two primers CCNG2 3′UTR-F
XhoI: CACAACT CGAGTCCTGGTTTAGCCCCCATCT, CCNG2 3′UTR-R
BamHI: AAGGATCCATGGCTGTATCACCACACAGA. The PCR product was
digested with XhoI and BamHI, followed by insertion
into a XhoI- and BamHI-opened vector. To generate a
mutant containing a mutation in the second site of the 3′UTR
(leaving the other site active), the primers were used as follows:
CCNG2 3′UTR-MF1: CATTGCGTGAAA ATTTTTCTCGTAGATCTTTAGCTAC, CCNG2
3′UTR-MR1: GAAAAATTTTCACGCAATGTCTAAAGTACTG TTAATGG. To generate a
mutant containing mutations in both sites, the primers used were:
CCNG2 3′UTR-MF2: AAACGTGTTTGTCGTGAAAAGACATTGCGTGAAA ATT, CCNG2
3′UTR-MR2: ATGTCTTTTCACGACAAAC ACGTTTAGAGGCACTACC. For luciferase
assays, HEK 293T cells were seeded in 96-well plates at a density
of 5,000 cells/well. After 24 h, the cells were transiently
transfected with 5 ng of pRL-CMV Renilla luciferase
reporter, 30 ng of either CCNG2-3′UTR or CCNG2-3′UTR-mutant and 5
pmol of miRNA mimics. After 48 h, the luciferase activity was
measured using the dual-luciferase reporter assay system (Promega,
Madison, WI, USA), detected by Tecan Infinite F200/M200 microplate
reader. Firefly luciferase activity was normalized to
Renilla luciferase activity to evaluate the effect of miR-93
mimics.
Transfection of Hep-2 cells with miRNA
mimics, inhibitor or siRNAs
All miRNAs mimics and inhibitors were synthesized by
GenePharma. The siRNAs specific for CCNG2 were synthesized by
Biotend (Shanghai, China). The oligonucleotide transfection was
performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA,
USA).
miRNA mimis (50 nmol), inhibitor (100 nmol) or
siRNAs transfection was performed using Lipofectamine 2000
following the manufacturer’s protocol. In brief, 4×104
cells in 1.5 ml of RPMI-1640 (10% FBS) were plated in each of five
different 35-mm tissue culture dishes and were incubated overnight
at 37°C and 5% CO2 atmosphere. For each dish, 5 μl
mimics or 10 μl inhibitor or 10 μl siRNA was added into 250 μl of
serum-free medium and mixed with 5 μl of Lipofectamine 2000. The
mixture was added to cells and incubated for 6 h before replacing
the medium with complete medium.
Three siRNAs that were synthesized to target CCNG2
expression were used in this experiment: 5′-3′ Si-1: sense, GGC
UGCUAGAAUAGUUGAAdTdT and antisense, UUCAAC UAUUCUAGCAGCCdTdT; Si-2:
sense, CUCCACAACAG CUACUAUA and antisense, UAUAGUAGCUGUUGUGG AG;
Si-3: sense, CCCGGAGAAUGAUAACACU and anti-sense,
AGUGUUAUCAUUCUCCGGG; Si-NC: sense, uucuc cgaacgugucacguTT and
antisense, acgugacacguucggagaaTT.
Statistical analysis
All statistical analyses were performed using SPSS
20.0 software. For the clinicopathological features, P-values were
calculated using the χ2 test. The results (mean values ±
SD) of all the experiments were subjected to statistical analysis
by the Student’s t-test (two-tailed, with P<0.05 considered
significant). Error bars depict SD. Asterisks were used to
represent statistical significance of P-values in the figures, e.g.
*P<0.05, **P<0.01,
***P<0.001.
Results
Differential expression of CCNG2 in LSCC
and adjacent non-neoplastic tissues
If miR-93 actually regulates the expression of CCNG2
in LSCC, then the expression of these two factors should be
inversely correlated in LSCC. Therefore, we evaluated the
expression of CCNG2 mRNA in corresponding the previous 59 cases of
LSCC and the adjacent non-cancerous laryngeal tissues. CCNG2 mRNA
was downregulated in LSCC tissues compared with their respective
non-cancerous laryngeal tissues. For the immunohistochemical
staining, staining was negative or weak in LSCC tissues, while in
normal laryngeal tissues, CCNG2 staining ranged from light yellow
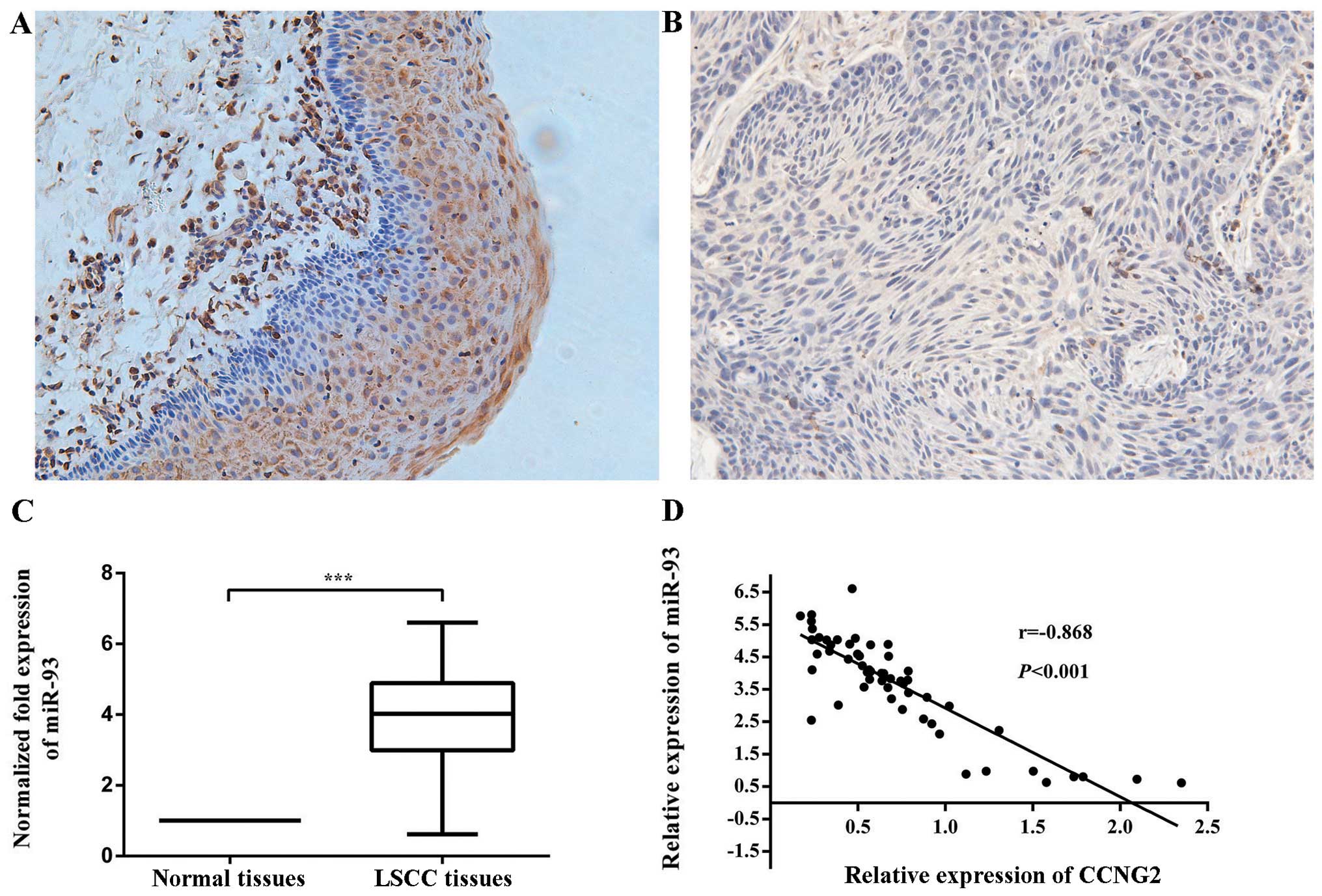
to brown (Fig. 1A and B).
Statistically, CCNG2 was expressed in 35.6% (21/59) of laryngeal
cancer tissues, which was lower than 96.6 (57/59) in normal
tissues. The difference was statistically significant (P<0.001)
(Table I). The expression of CCNG2
was correlated with clinical stages, lymph node metastasis and
pathological differentiation (P<0.05), regardless of age, gender
and tumor site (P>0.05) (Table
II).
CCNG2 mRNA expression in LSCC was
negatively correlated with miR-93 expression
To validate the expression of miR-93 in LSCC, we
detected mature miR-93 in 59 pairs of LSCC and matched
non-cancerous laryngeal tissues by real-time PCR. The results
indicated that miR-93 expression was upregulated in 86.44% of LSCC
tissues compared with the non-cancerous laryngeal tissues, at
3.73-fold higher mean level (Fig.
1C).
Moreover, the downregulation of CCNG2 was inversely
correlated with the upregulation of miR-93 in these LSCC samples.
The Pearson correlation was recorded between CCNG2 and miR-93
expression (r=−0.868, P<0.001) (Fig. 1D). These data suggest that CCNG2
mRNA expression is negatively correlated with miR-93 expression in
LSCC tissues and miR-93 may have an important role in cell cycle
and proliferation.
miR-93 promotes the proliferation and
colony formation in vitro
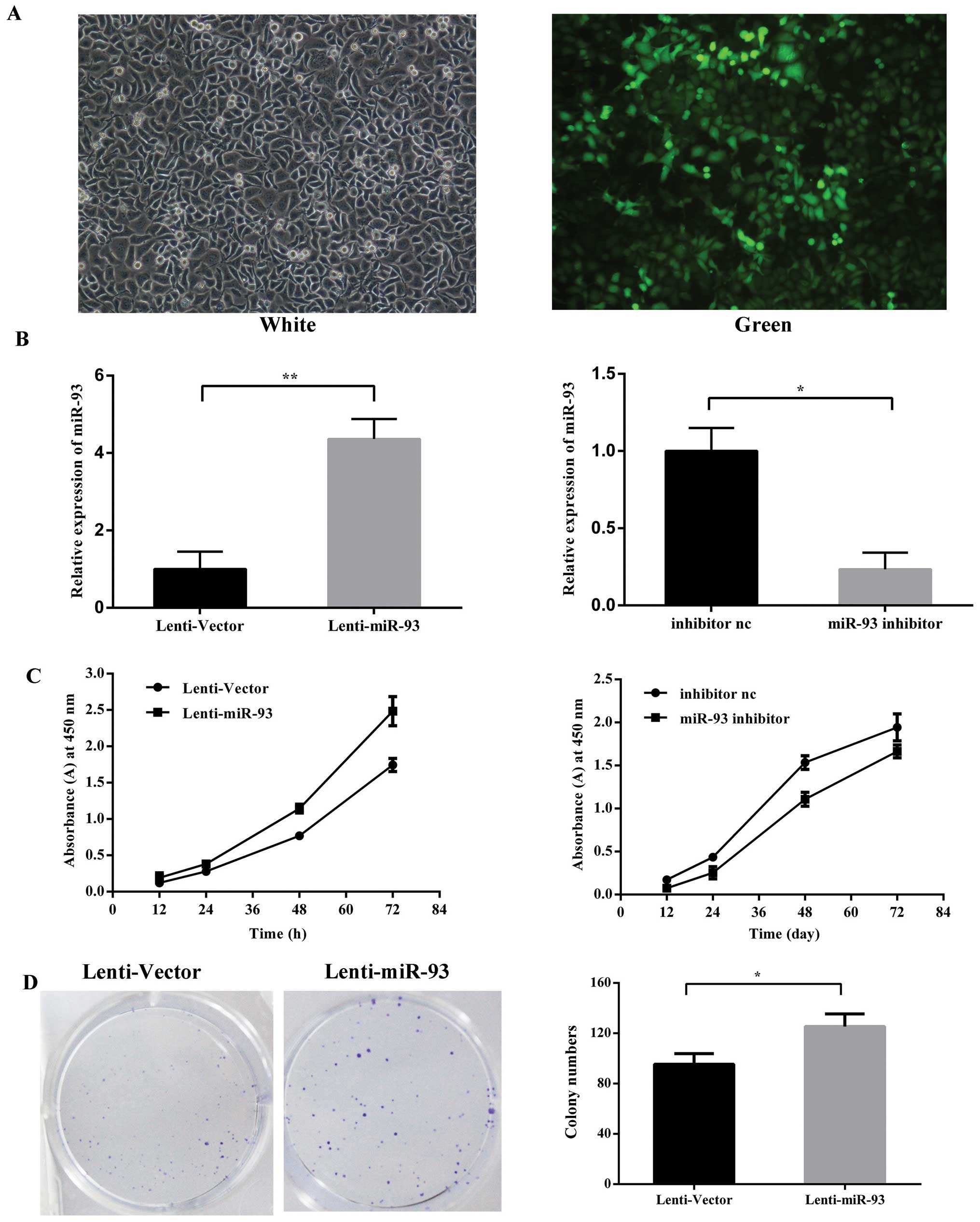
To examine the biological function of miR-93 in the
development of LSCC, a lentivirus vector expressing miR-93 was
constructed and used to infect Hep-2 cells to establish stable cell
lines, denoted as Lenti-miR-93 (Fig.
2A), the negative control as Lenti-Vector after lentivirus
transduction. The silencing of miR-93 was via the tansfection of
Hep-2 cells with miR-93 inhibitor and the inhibitor negative
control. The relative expression levels of mature miR-93 were
detected using qRT-PCR (Fig. 2B).
Cell proliferation assays revealed that the ectopic expression of
miR-93 in these stable cancer cells resulted in a significant
increase in cell proliferation. In contrast, the silencing of
miR-93 reduced the cell proliferation rates compared with the
negative control (Fig. 2C). To
further examine the effect of miR-93 on the selfrenewing capacity
in vitro, Lenti-miR-93 cells stably expressing miR-93 or the
vector control were plated at a density of 200 cells/well in 6-well
cell cultrue cluster, Lenti-miR-93 cells generated a greater number
of tumor spheres than the vector control (95.33±8.5 and
125.30±10.0, respectively) (Fig.
2D). miR-93 overexpression in laryngeal cancer cells
significantly promotes long-term cell growth as measured by colony
formation assay.
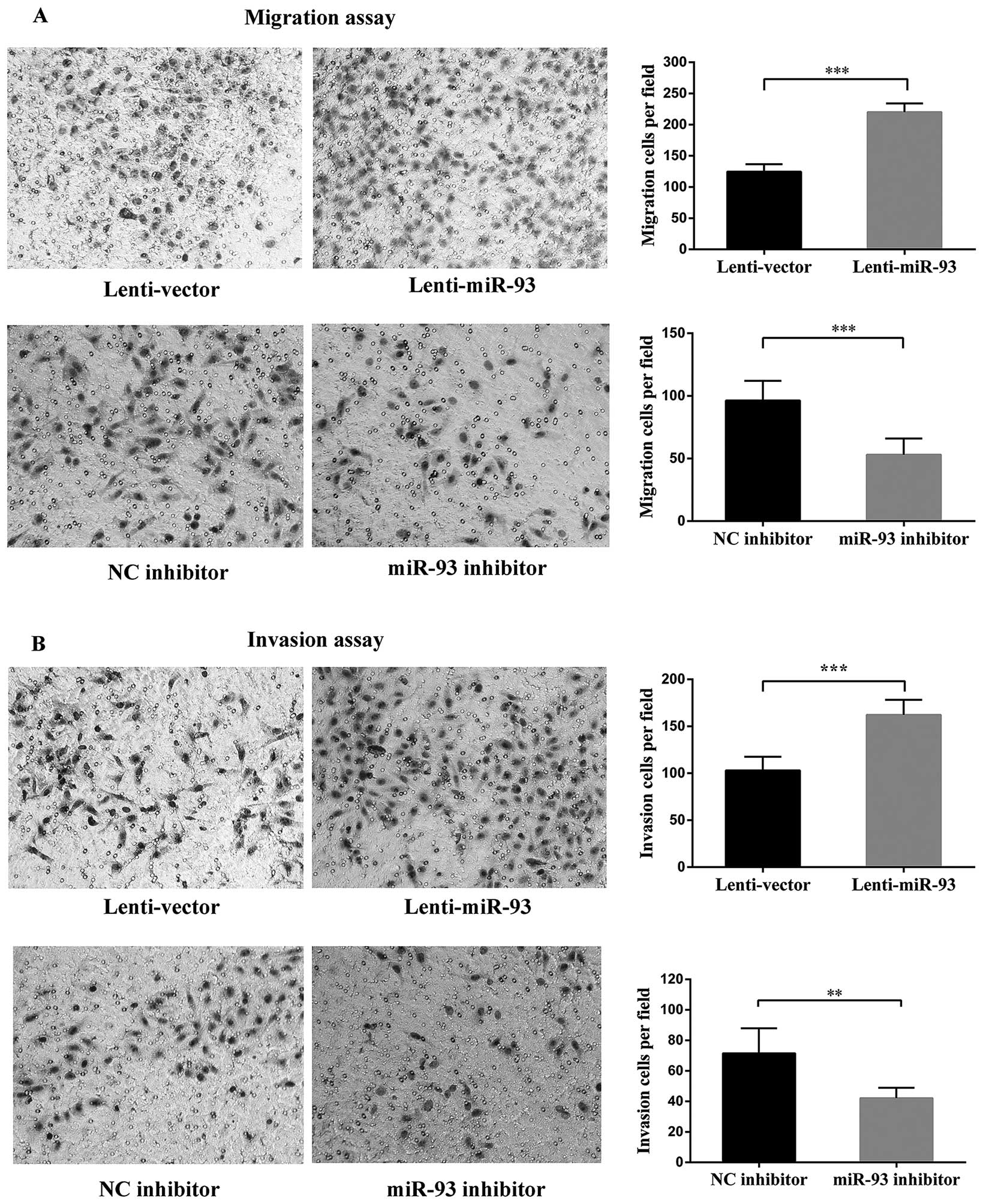
miR-93 promotess cancer cell invasion and
metastasis in vitro
miR-93 was recently reported to promote the
metastasis of breast cancer cells in vitro (15), thus, prompting us to investigate
whether miR-93 could also promote cell invasion and metastasis in
LSCC. Transwell assays without or with Matrigel demonstrated that
the cells transfected with miR-93 displayed higher levels of
migration and invasion than the mock-transfected cells (Fig. 3). In contrast, the silencing of
endogenous miR-93 in Hep-2 cells decreased cell migration and
invasion (Fig. 3). Notably, the
incubation times for the migration and invasion assays were 18 and
24 h, respectively, and at those time-points, the cell growth was
not significantly affected by miR-93. The migrated and invasive
cells were stained and counted in six randomly selected fields
under a light microscope. In contrast, the mRNA level of MMP-9 was
upregulated in the Lenti-miR-93 cells, the fold change was 22.9
compared with Lenti-Vector cells. Taken together, these findings
indicated that miR-93 promotes cancer cell migration and invasion
in vitro.
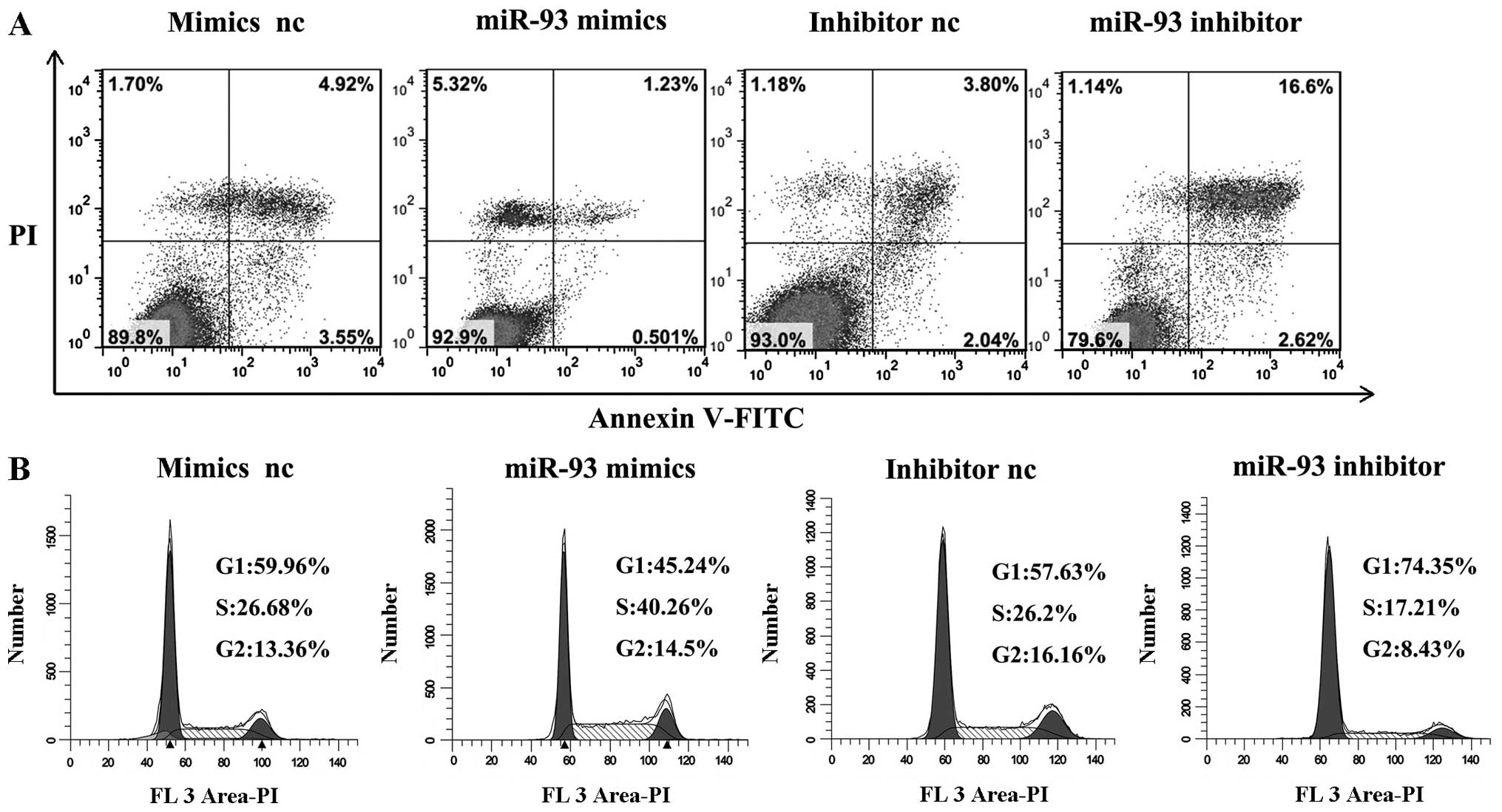
miR-93 inhibits LSCC cell apoptosis in
vitro and promotes cell cycle progression of LSCC cells
Given that miR-93 may function as an oncogene in
LSCC, we considered whether miR-93 might have an important role in
LSCC cell apoptosis. The flow cytometry assay demonstrated that the
apoptosis rates of Hep-2 cells transfected with miR-93 mimics were
significantly decreased compared with those of negative control
miRNA-transfected cells (P<0.01). When transfected with miR-93
inhibitor, the apoptosis rates increased (P<0.01) (Fig. 4A). These results indicated that
miR-93 inhibited the cellular apoptosis of laryngeal cancer
cells.
To study whether miR-93 promoted the proliferation
of LSCC carcinoma cells by affecting their cell cycle, cell cycle
analysis was performed. Cell cycle analysis showed that after
transfection with miR-93 mimics, the cell cycle was arrested at the
S phase compared with cells transfected with mimics negative
control in Hep-2 cells. When transfection was carried out with
miR-93 inhibitor, the cell cycle was arrested at G1 phase compared
with cells transfected with inhibitor negative control in Hep-2
cells (Fig. 4B). These results
suggested that miR-93 promoted the progression through the G1-S
phase of the cell cycle.
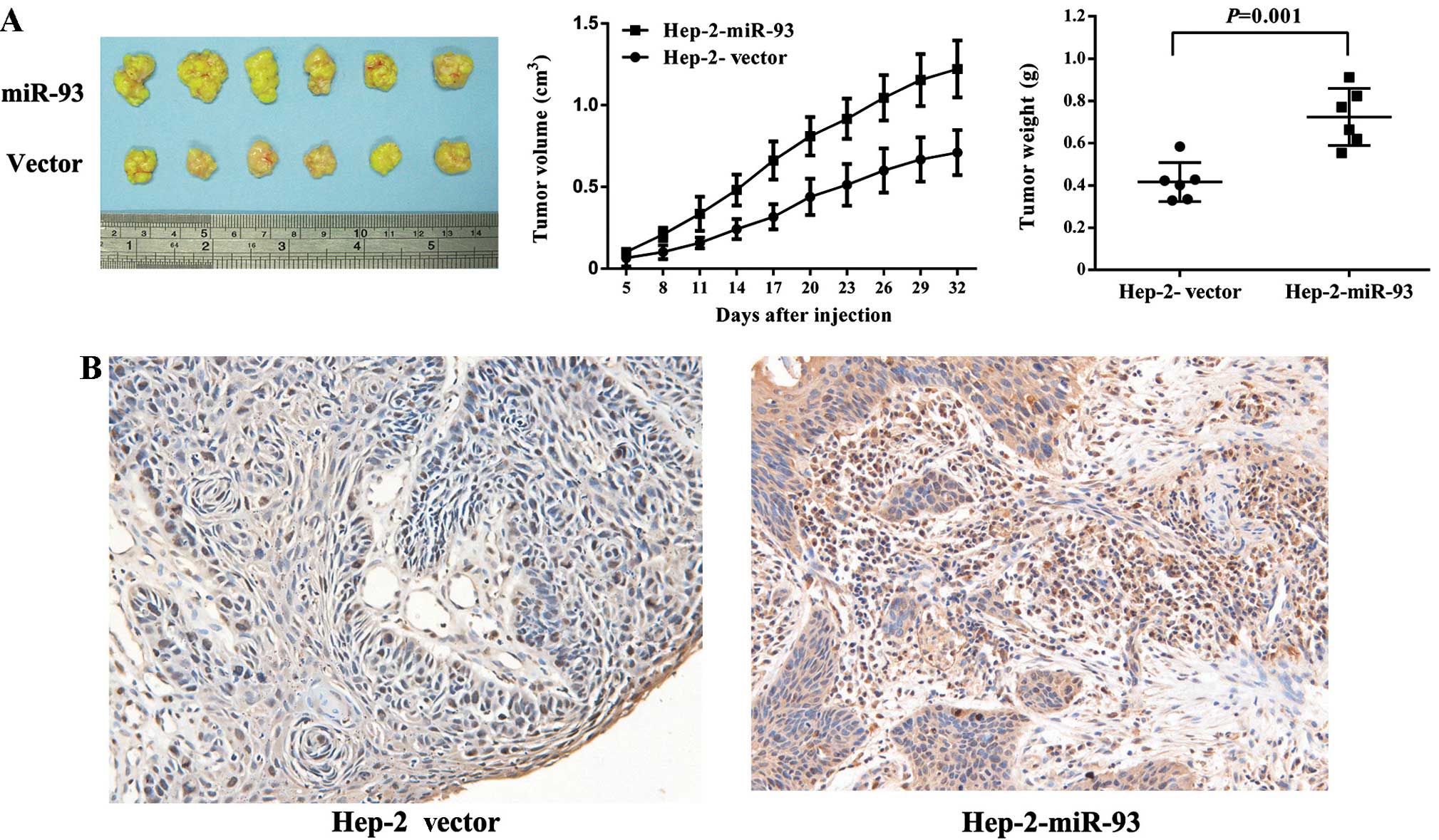
miR-93 overexpression promotes tumor
growth in laryngeal xenografts
To further characterize the oncogenic function of
miR-93 in vivo, we generated subcutaneous xenografts using
Hep-2 cells stably transfected with either miR-93 or a control
construct. As shown in Fig. 5A, we
found tumors in the mice injected with the Lenti-miR-93 cells, and
significantly increased tumor growth in the nude mice compared to
the Lenti-Vector cells. Fig. 5B
shows that miR-93 overexpression increased expression of Ki-67
protein, a marker of proliferation, in tumor samples. Collectively,
these findings demonstrated that miR-93 promotes LSCC cancer cell
proliferation in vivo.
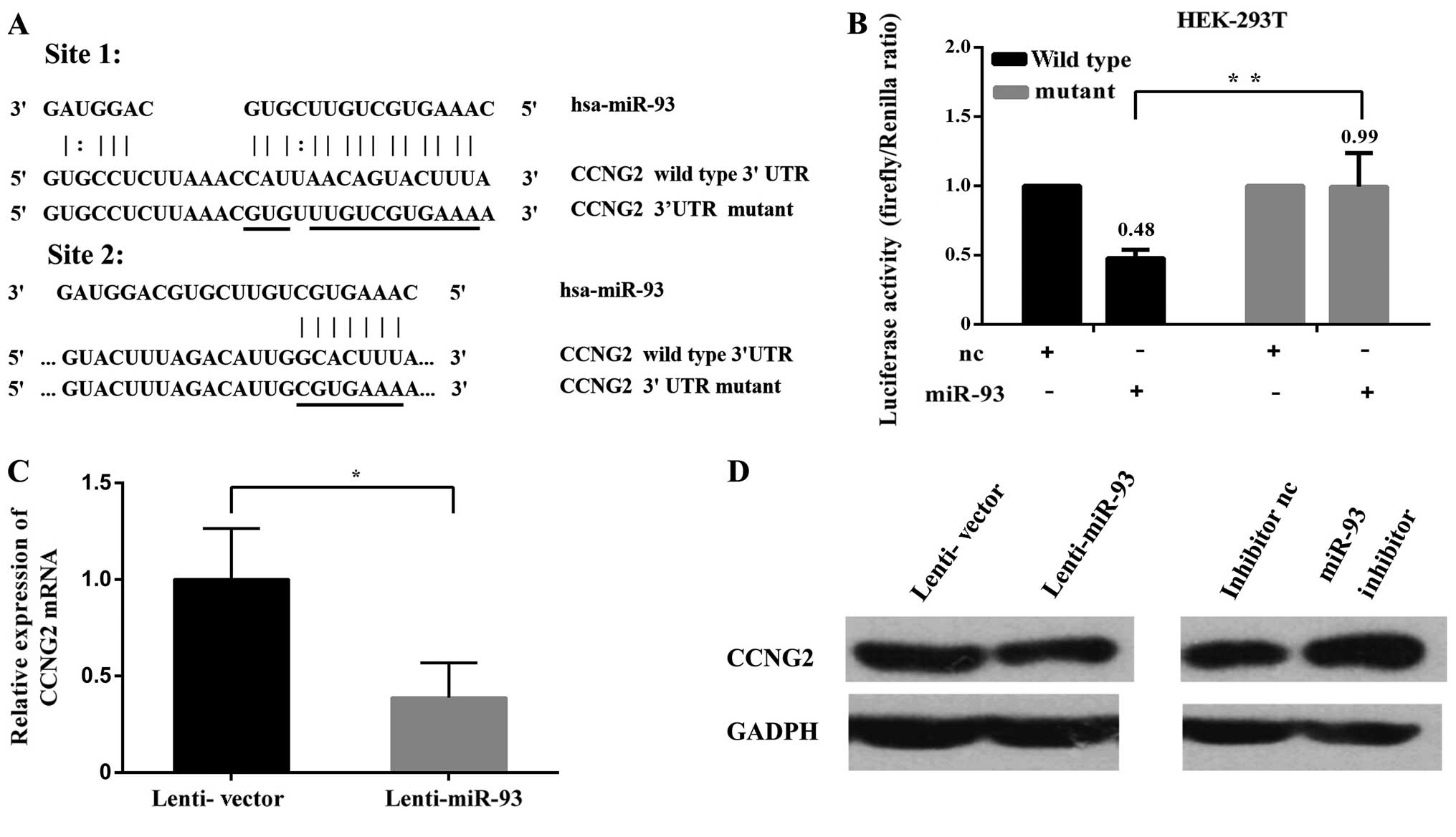
miR-93 represses CCNG2 expression by
directly targeting its 3′UTR
We investigated the target of miR-93 in mediating
the observed effects focusing on tumor suppressors. The CCNG2 was
identified as a potential target of miR-93. The 3′UTR of CCNG2
harbored two typical target sequences for miR-93 at nucleotides
186–214 and 222–229 (Fig. 6A). To
obtain direct evidence that the 3′UTR of CCNG2 was a target of
miR-93, we generated luciferase expression constructs harboring
fragments of the CCNG2 3′UTR containing the miR-93 target sites,
which produced the wild-type constructs CCNG2-186 and CCNG2-222.
Two mutant constructs CCNG2-186-mutant and CCNG2-222-mutant were
also generated (Fig. 6A). As
expected, expression of CCNG2-186 and CCNG2-222 showed that
luciferase activities were significantly repressed when the
constructs were cotransfected with miR-93 mimics. Mutations of the
miR-93 target sites abolished the effects of miR-93 (Fig. 6B). The mRNA levels of CCNG2 were
determined by qRT-PCR in Hep-2 cells stably expressing miR-93 or
the negative control (Fig. 6C). In
addition, western blot analyses showed that overexpression of
miR-93 could lead to decreased CCNG2 protein level in Hep-2 cells.
In contrast, miR-93 inhibitor increased the protein level of CCNG2
(Fig. 6D). Collectively, these
results indicated that miR-93 could regulate CCNG2 expression by
directly binding to its 3′UTR.
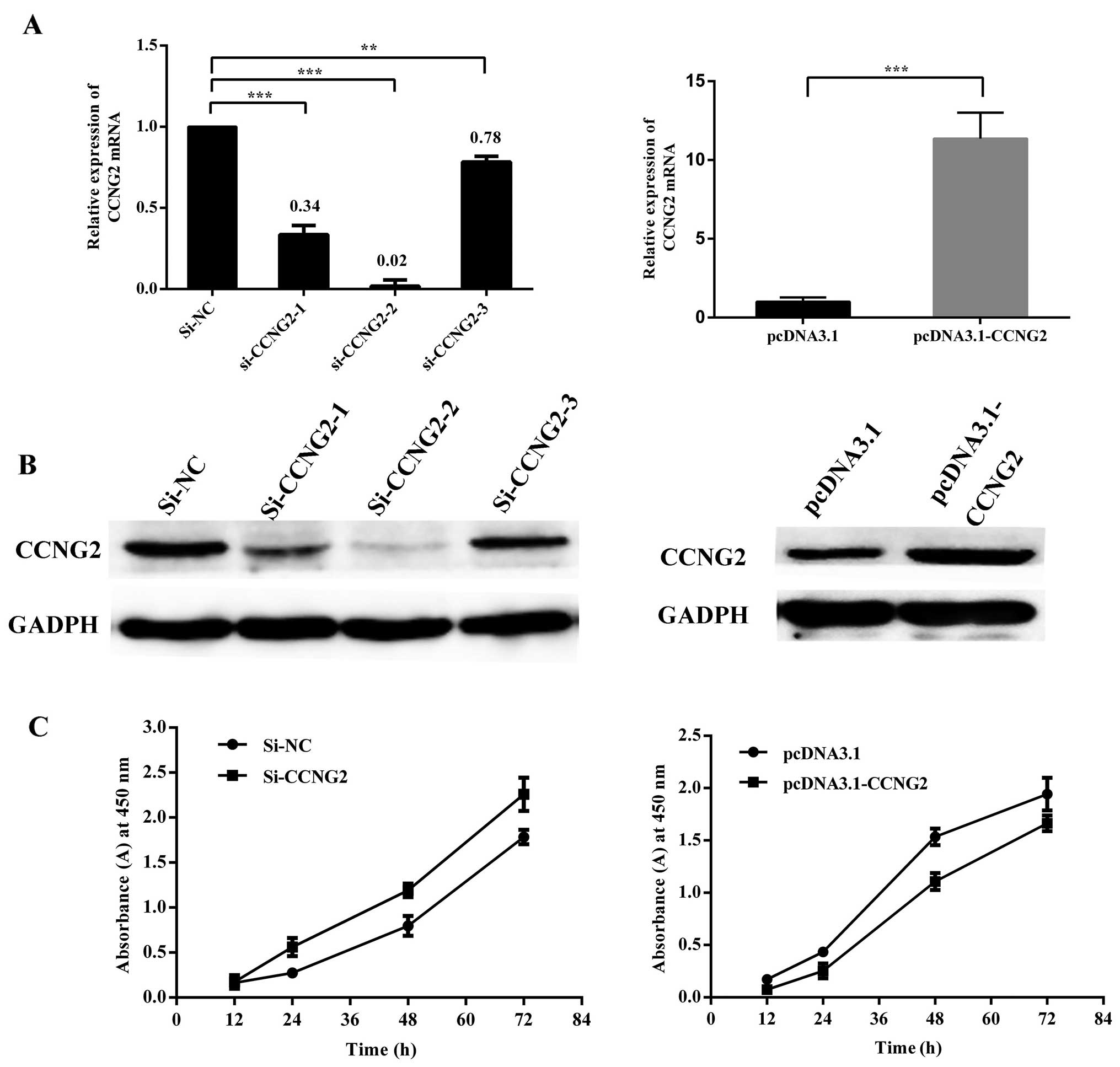
CCNG2 acts as a tumor suppressor in
laryngeal carcinoma
To confirm that CCNG2 played an important role in
mediating the miR-93 effects in carcinogenesis, we investigated the
role of CCNG2 overexpression and knockdown in cultured Hep-2 cells.
An expression plasmid vector pcDNA3.1 was used for overexpression,
three siRNAs targeting CCNG2 were maintained in serum-free medium.
Western blot assays showed that CCNG2 protein levels were
significantly increased by transfection of a CCNG2 expression
construct, and were reduced by transfection of specific siRNA
against CCNG2. As shown in Fig. 7A and
B, since all siRNAs appeared to have functioned efficiently,
one of them, the siRNA-2, was used for further analysis. CCNG2
overexpression significantly reduced cell proliferation as measured
by CCK8, and after transfection with CCNG2-siRNA, cell
proliferation of Hep-2 cells had increased markedly as compared
with cells transfected with the control oliogo (Fig. 7C). To examine if ectopic expression
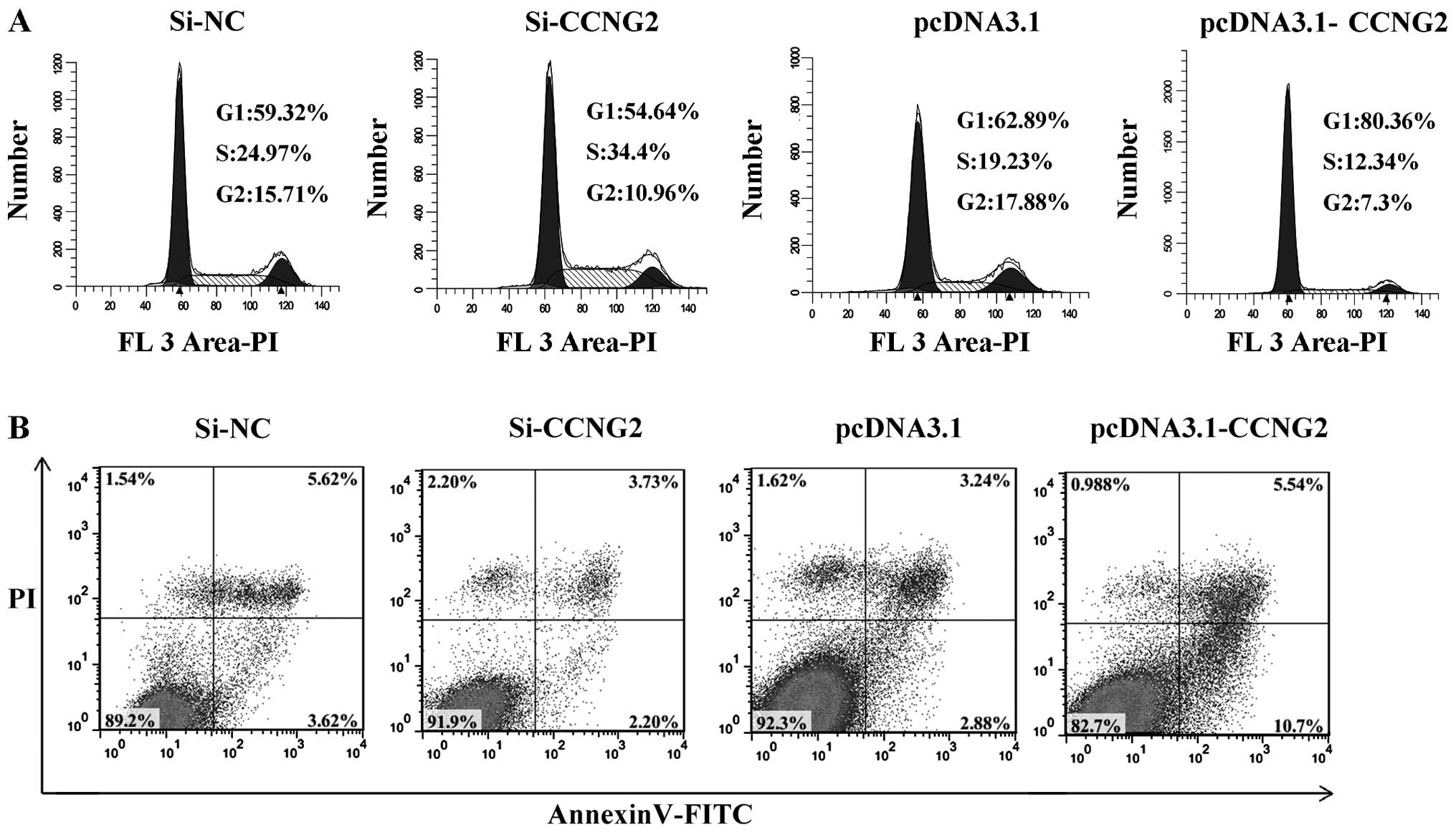
of CCNG2 altered the cell cycle profile in Hep-2 cells, we
determined the cell cycle positions. Compared with control
transfectants, CCNG2-overexpression cells showed a decrease in S
phase and an increase in G1 phase. Knockdown of CCNG2 expression
leads to an increase in S phase, the cell cycle was arrested at S
phase (Fig. 8A). To examine if
ectopic expression of CCNG2 induced apoptosis, we measured
percentage of apoptotic cell population by Annexin V-FITC/PI
analysis. Consitent with the observation of the proliferation and
cell cycle, we observed CCNG2-overexpression significantly induced
apoptosis compared with null plasmid transfection (P<0.01)
(Fig. 8B). These results indicate
that CCNG2 inhibits growth of LSCC cells by inducing cell arrest at
the G1/S checkpoint of the cell cycle, the downregulation of CCNG2
promotes the proliferation and cell cycle progress of LSCC cells.
These data support the growth inhibitory function of CCNG2, CCNG2
acted as a tumor suppressor in laryngeal carcinoma.
Confirmation of the targeting effects by
rescue experiments
Rescue experiments were performed to further confirm
that miR-93 promoted cell proliferation and migration by targeting
CCNG2. Hep-2 cells stably transfected with miR-93 were transfected
with pcDNA3.1-CCNG2 plasmid or a control plasmid. Western blot
analysis was used as confirmation of the expression for the protein
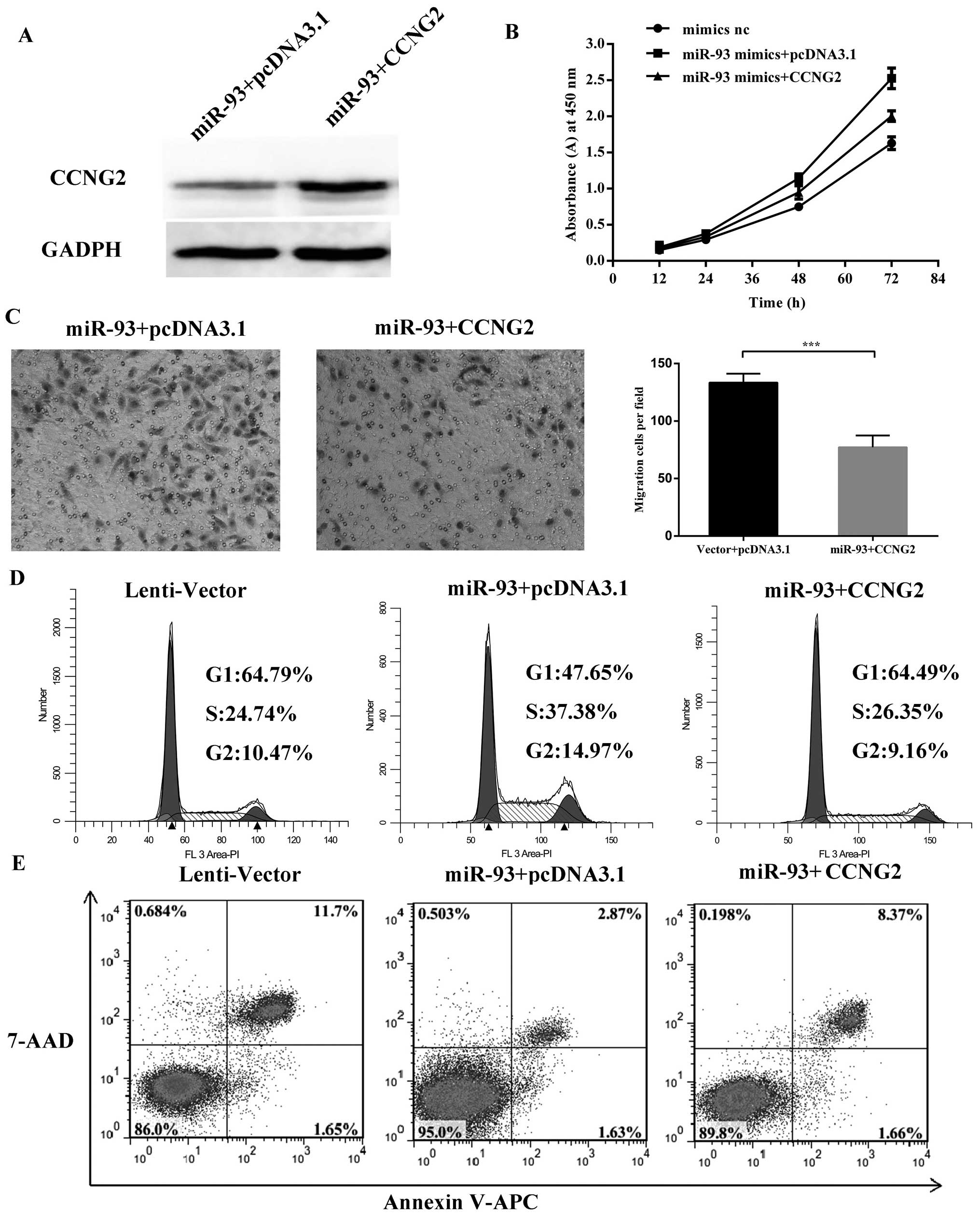
gel blot probed with anti-CCNG2 antibody (Fig. 9A). Cell proliferation assay showed
that reintroduction of CCNG2 into the miR-93-expressing cells
reversed the effect of miR-93 on cell proliferation (Fig. 9B) and failed to promote the cell
cycle process (Fig. 9D). Decreased
migration was found in cells transfected with CCNG2 (Fig. 9C). The flow cytometry data showed
that overexpression of CCNG2 in miR-93 cells can attenuate the
anti-apoptotic effect of miR-93 (Fig.
9E). Thus, re-expression of CCNG2 was sufficient to cause cell
death and decrease both cell proliferation and migration. This
suggested that the effects of miR-93 on enhanced proliferation and
metastasis were at least partly taking place through repression of
CCNG2 expression.
Discussion
The role of miR-93 in cancer is controversial
because reports have indicated that it can be tumor suppressive or
tumor promoting, depending on context. miR-93 is upregulated in a
subset of human tumors and leads to deregulation of important
cancer-related genes (23–27) whereas, in some types of cancer,
miR-93 represses proliferation paradoxically (28). Despite increasing evidence pointing
to a role for miR-93 as a tumor oncomiR, the tumor promotion effect
of miR-93 has not been fully elucidated in LSCC. In the present
study, we provide the first integrated investigation of the
function and probable underlying mechanisms of miR-93 and CCNG2 in
LSCC at both cellular and clinical levels.
Previous data showed that as a key negative
regulator of cell cycle proliferation, apoptosis, cell death
signaling, and even carcinogenesis, suggesting that the decreased
expression levels of CCNG2 significantly correlates with tumor
progression and unfavorable prognosis, such as studies in
colorectal carcinoma, kidney, esophageal, oral cancer, gastric
carcinoma, prostate cancer and papillary carcinoma of the thyroid
(29–35). In the present study, we found that
CCNG2 expression was markedly decreased in the LSCC tissues
compared with the adjacent normal tissues and lower expression of
CCNG2 tended to have more advanced clinical stage. The expression
of CCNG2 in laryngeal carcinoma tissues without lymph node
metastasis or with high differentiation presented to be higher than
laryngeal carcinoma tissues with lymph node metastasis or with poor
differentiation, suggesting that downregulation of CCNG2 expression
in LSCC is associated with malignant degree and development
tendency. We further demonstrate that CCNG2 functions as a potent
repressor of cell proliferation and invasion at cellular
levels.
The downregulation of CCNG2 expresion in cancer
cells may be caused by multiple mechanisms, as we show here,
including through miRNA regulation. We verified that miR-93
regulates CCNG2 expression by directly targeting the 3′UTR of CCNG2
mRNA. There was a significant negative correlation between CCNG2
expression and miR-93 levels in laryngeal cancer specimens,
confirming the role of miR-93 in regulating CCNG2 expression. After
validation experiments, we found that the aberrant overexpression
of miR-93 markedly attenuated CCNG2 expression at the mRNA and
protein level in the Hep-2 cells. Data of luciferase reporter assay
showed that miR-93 could bind to the wild-type target sequence but
not the mutant target sequence. Therefore, to a certain extent,
this result supports our hypothesis that CCNG2 is a direct target
gene of miR-93 in Hep-2 cells, miR-93 has the primary role in
determining CCNG2 expression in laryngeal cancer. Silencing of
CCNG2 expression by small interfering RNAs indicated that CCNG2
plays crucial roles in cell proliferation. In addition, rescue
experiments demonstrated that CCNG2 re-expression can only partly
reverse the function of miR-93, which may suggest that CCNG2 was
not the only target of miR-93 in laryngeal carcinoma and thus there
may be some other targets of miR-93.
It is well recognized that enhanced cell
proliferation, cell cycle arrest, resistance to apoptosis and the
migration state of LSCC cells plays key roles in the progression of
LSCC (36–39). Another important finding of our
study is that miR-93 acted as an oncomiR affecting the
proliferation, cell cycle progress, apoptosis and migration state
of LSCC cells. Firstly, we found that elevated miR-93 levels
promoted the growth of Hep-2 cells in vitro and in
vivo which was associated with the inhibition of apoptosis and
induction of cell cycle arrest. In vivo tumor formation
assays, the Lenti-miR-93 group displayed stronger Ki-67
immunoreactivity, an indication of extensive cell proliferation,
than Lenti-Vector group. Secondly, miR-93 appeared to play roles in
migraton and invasion of Hep-2 cells, cellular migration and
invasion was enhanced following ectopic expression of miR-93 in the
Hep-2 cells. To further confirm whether silencing of miR-93
inhibits proliferation, clonability, migratory and invasion
potential, we silenced miR-93 in Hep-2 cells using miR-93 inhibitor
and all the assays discussed above were carried out. On the
contrary, knocking down the expression of miR-93 performed the
reverse function. All of the above indicated an oncogenic role of
miR-93 in LSCC carcinogenesis.
The dysregulation of cell cycle control and
apoptosis state is emerging as a central theme of carcinogenesis.
Cyclin G2 is an unconventional cyclin highly expressed in cells
undergoing apoptosis, its expressiom is upregulated as cells
undergo cell cycle arrest or apoptosis in response to inhibitory
stimuli independent of P53 (40,41).
In lymphocytes, cyclin G2 expression oscillating in late S/early G2
phase of cell cycle (42).
Correspondingly, CCNG2 has a potential pro-apoptotic role; however,
the underlying mechanisms remain unclear. However, it has been
shown that there are two different mechanisms associated with cell
apoptosis: the extrinsic receptor-mediated pathway and the
intrinsic mitochondrial-dependent pathway (43). Our results revealed that the
overexpression of miR-93 markedly downregulated CCNG2 expression,
and finally induced cell cycle S phase blockage in the Hep-2 cells.
Our results also indicated that the anti-apoptotic effect of miR-93
in Hep-2 cells is partially dependent on CCNG2-mediated
mitochondrial apoptotic signaling.
To initiate the metastatic process, cancer cells
must first penetrate the epithelial basement membrane. The
degradation of extracellular matrix (ECM) is associated with cell
invasion. Among matrix metalloproteinases (MMPs), MMP-9 digest type
IV collagen, a major compotent of basement membrane. Thus, MMP-9
plays a central role in cancer cell invasion and is frequently
upregulated in cancer cells (44,45).
In the present study, the miR-93 Hep-2 cells were able to penetrate
through the Matrigel as compared with the control cells. It
appeared that the miR-93 Hep-2 cells were able to digest the matrix
molecules, which facilitated cell invasion and led to metastasis.
The mRNA level of MMP-9 were upregulated in the miR-93-transfected
cells, which significantly stimulated the migratory capability of
Hep-2 cells, supporting that miR-93 might promote cell migration
through the CCNG2-MMP-9 pathway, partly due to its effect on MMP-9.
The specific molecular mechanisms of this pathway remain to explore
further.
In summary, miR-93, as an oncomiR, promotes
proliferation, inhibits apoptosis, induces cell cycle arrest and
promotes the migration and invasion of LSCC cells by targeting
CCNG2. Intriguingly, relatively high levels of CCNG2 predicted a
better outcome in LSCC patients and function as a prognostic factor
in LSCC. We also found that miR-93 and CCNG2 expression were
inversely associated in LSCC samples. The associations of CCNG2
levels with clinical stages, lymph node metastasis, and
histological grade further demonstrate the relevance of the
miR-93/CCNG2 pathway in LSCC tumorigenesis. Given the increasing
appreciation of miRNAs in cancer therapeutics and diagnostics, this
novel oncogenic pathway clearly warrants further investigation to
determine whether new therapeutic tools and prognostic biomarkers
for laryngeal cancer can be developed.
Acknowledgements
The present study was financially supported by the
Key Project of Shanghai Science and Technology Committee (grant no.
12JC1402100).
References
|
1
|
Ferlay J, Shin HR, Bray F, et al:
Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int
J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marioni G, Marchese-Ragona R, Cartei G, et
al: Current opinion in diagnosis and treatment of laryngeal
carcinoma. Cancer Treat Rev. 32:504–515. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Talamini R, Bosetti C, La Vecchia C, et
al: Combined effect of tobacco and alcohol on laryngeal cancer
risk: a case-control study. Cancer Causes Control. 13:957–964.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Menach P, Oburra HO and Patel A: Cigarette
smoking and alcohol ingestion as risk factors for laryngeal
squamous cell carcinoma at Kenyatta National Hospital, Kenya. Clin
Med Insights Ear Nose Throat. 5:17–24. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mohamadian Roshan N, Jafarian A,
Ayatollahi H, et al: Correlation of laryngeal squamous cell
carcinoma and infections with either HHV-8 or HPV-16/18. Pathol Res
Pract. 210:205–209. 2014.PubMed/NCBI
|
|
6
|
Hoffman HT, Porter K, Karnell LH, et al:
Laryngeal cancer in the United States: changes in demographics,
patterns of care, and survival. Laryngoscope. 116(Suppl 111): 1–13.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang B, Pan X, Cobb GP, et al: microRNAs
as oncogenes and tumor suppressors. Dev Biol. 302:1–12. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shah MY and Calin GA: MicroRNAs as
therapeutic targets in human cancers. Wiley Interdiscip Rev RNA.
5:537–548. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kong YW, Ferland-McCollough D, Jackson TJ,
et al: microRNAs in cancer management. Lancet Oncol. 13:e249–258.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Calin GA, Sevignani C, Dumitru CD, et al:
Human microRNA genes are frequently located at fragile sites and
genomic regions involved in cancers. Proc Natl Acad Sci USA.
101:2999–3004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yeung ML, Yasunaga J, Bennasser Y, et al:
Roles for microRNAs, miR-93 and miR-130b, and tumor protein
53-induced nuclear protein 1 tumor suppressor in cell growth
dysregulation by human T-cell lymphotrophic virus 1. Cancer Res.
68:8976–8985. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Du L, Schageman JJ, Subauste MC, et al:
miR-93, miR-98, and miR-197 regulate expression of tumor suppressor
gene FUS1. Mol Cancer Res. 7:1234–1243. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Singh B, Ronghe AM, Chatterjee A, et al:
MicroRNA-93 regulates NRF2 expression and is associated with breast
carcinogenesis. Carcinogenesis. 34:1165–1172. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fang L, Deng Z, Shatseva T, et al:
MicroRNA miR-93 promotes tumor growth and angiogenesis by targeting
integrin-β8. Oncogene. 30:806–821. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fang L, Du WW, Yang W, et al: MiR-93
enhances angiogenesis and metastasis by targeting LATS2. Cell
Cycle. 11:4352–4365. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fu X, Tian J, Zhang L, et al: Involvement
of microRNA-93, a new regulator of PTEN/Akt signaling pathway, in
regulation of chemotherapeutic drug cisplatin chemosensitivity in
ovarian cancer cells. FEBS Lett. 586:1279–1286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu S, Patel SH, Ginestier C, et al:
MicroRNA93 regulates proliferation and differentiation of normal
and malignant breast stem cells. PLoS Genet. 8:e10027512012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bennin DA, Don AS, Brake T, et al: Cyclin
G2 associates with protein phosphatase 2A catalytic and regulatory
B’ subunits in active complexes and induces nuclear aberrations and
a G1/S phase cell cycle arrest. J Biol Chem. 277:27449–27467. 2002.
View Article : Google Scholar
|
|
19
|
Burgering BM and Kops GJ: Cell cycle and
death control: long live Forkheads. Trends Biochem Sci. 27:352–360.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Martínez-Gac L, Marqués M, García Z, et
al: Control of cyclin G2 mRNA expression by forkhead transcription
factors: novel mechanism for cell cycle control by phosphoinositide
3-kinase and forkhead. Mol Cell Biol. 24:2181–2189. 2004.PubMed/NCBI
|
|
21
|
Arachchige Don AS, Dallapiazza RF, Bennin
DA, et al: Cyclin G2 is a centrosome-associated nucleocytoplasmic
shuttling protein that influences microtubule stability and induces
a p53-dependent cell cycle arrest. Exp Cell Res. 312:4181–4204.
2006.
|
|
22
|
Cao P, Zhou L, Zhang J, et al:
Comprehensive expression profiling of microRNAs in laryngeal
squamous cell carcinoma. Head Neck. 35:720–728. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang L, Wang Q, Li HL, et al: Expression
of MiR200a, miR93, metastasis-related gene RECK and MMP2/MMP9 in
human cervical carcinoma--relationship with prognosis. Asian Pac J
Cancer Prev. 14:2113–2118. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim BH, Hong SW, Kim A, et al: Prognostic
implications for high expression of oncogenic microRNAs in advanced
gastric carcinoma. J Surg Oncol. 107:505–510. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu D, He XX, Chang Y, et al:
Downregulation of MiR-93 expression reduces cell proliferation and
clonogenicity of HepG2 cells. Hepatogastroenterology. 59:2367–2373.
2012.PubMed/NCBI
|
|
26
|
Chen L, Jiang M, Yuan W, et al: Prognostic
value of miR-93 overexpression in resectable gastric
adenocarcinomas. Acta Gastroenterol Belg. 75:22–27. 2012.PubMed/NCBI
|
|
27
|
Montanini L, Lasagna L, Barili V, et al:
MicroRNA cloning and sequencing in osteosarcoma cell lines:
differential role of miR-93. Cell Oncol (Dordr). 35:29–41. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu XF, Zou J, Bao ZJ, et al: miR-93
suppresses proliferation and colony formation of human colon cancer
stem cells. World J Gastroenterol. 17:4711–4717. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun GG, Zhang J and Hu WN: CCNG2
expression is down-regulated in colorectal carcinoma and its
clinical significance. Tumour Biol. 35:3339–3346. 2014. View Article : Google Scholar
|
|
30
|
Cui DW, Sun GG and Cheng YJ: Change in
expression of cyclin G2 in kidney cancer cell and its significance.
Tumour Biol. 35:3177–3183. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen JQ, Liu CJ, Wen HX, et al: Changes in
the expression of cyclin G2 in esophageal cancer cell and its
significance. Tumour Biol. 35:3355–3362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim Y, Shintani S, Kohno Y, et al: Cyclin
G2 dysregulation in human oral cancer. Cancer Res. 64:8980–8986.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sun GG, Hu WN, Cui DW, et al: Decreased
expression of CCNG2 is significantly linked to the malignant
transformation of gastric carcinoma. Tumour Biol. 35:2631–2639.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cui DW, Cheng YJ, Jing SW, et al: Effect
of cyclin G2 on proliferative ability of prostate cancer PC-3 cell.
Tumour Biol. 35:3017–3024. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ito Y, Yoshida H, Uruno T, et al:
Decreased expression of cyclin G2 is significantly linked to the
malignant transformation of papillary carcinoma of the thyroid.
Anticancer Res. 23:2335–2338. 2003.PubMed/NCBI
|
|
36
|
Jiao J, Qin Z, Li S, et al: Potential role
of Notch1 signaling pathway in laryngeal squamous cell carcinoma
cell line Hep-2 involving proliferation inhibition, cell cycle
arrest, cell apoptosis, and cell migration. Oncol Rep. 22:815–823.
2009.PubMed/NCBI
|
|
37
|
Ren J, Zhu D, Liu M, et al: Downregulation
of miR-21 modulates Ras expression to promote apoptosis and
suppress invasion of Laryngeal squamous cell carcinoma. Eur J
Cancer. 46:3409–3416. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Che XH, Chen H, Xu ZM, et al:
14-3-3epsilon contributes to tumour suppression in laryngeal
carcinoma by affecting apoptosis and invasion. BMC Cancer.
10:3062010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tian L, Li M, Ge J, et al: MiR-203 is
downregulated in laryngeal squamous cell carcinoma and can suppress
proliferation and induce apoptosis of tumours. Tumour Biol.
35:5953–5963. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Horne MC, Goolsby GL, Donaldson KL, et al:
Cyclin G1 and cyclin G2 comprise a new family of cyclins with
contrasting tissue-specific and cell cycle-regulated expression. J
Biol Chem. 271:6050–6061. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Horne MC, Donaldson KL, Goolsby GL, et al:
Cyclin G2 is up-regulated during growth inhibition and B cell
antigen receptor-mediated cell cycle arrest. J Biol Chem.
272:12650–12661. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bates S, Rowan S and Vousden KH:
Characterisation of human cyclin G1 and G2: DNA damage inducible
genes. Oncogene. 13:1103–1109. 1996.PubMed/NCBI
|
|
43
|
Wang DH, Hu JR, Wang LY, et al: The
apoptotic function analysis of p53, Apaf1, Caspase3 and Caspase7
during the spermatogenesis of the Chinese fire-bellied newt
Cynops orientalis. PLoS One. 7:e399202012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Johansson N, Ahonen M and Kähäri VM:
Matrix metalloproteinases in tumor invasion. Cell Mol Life Sci.
57:5–15. 2000. View Article : Google Scholar
|
|
45
|
Fink K and Boratyński J: The role of
metalloproteinases in modification of extracellular matrix in
invasive tumor growth, metastasis and angiogenesis. Postepy Hig Med
Dosw (Online). 66:609–628. 2012.(In Polish).
|