Introduction
Lung cancer is the leading cause of cancer-related
deaths worldwide (1). Non-small
cell lung cancer (NSCLC) accounts for ~80% of all lung cancer
cases. Despite recent development in treatment agents, the
prognosis for lung cancer patients remains poor (2).
Overexpression of epidermal growth factor receptor
(EGFR) is observed in various malignancies, including lung cancer
(3). EGFR activation induces many
intracellular signaling pathways, such as the mitogen-activated
protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), and
signal transducer and activator of transcription (STAT) pathways,
which cause tumor cell proliferation and survival (4). The EGFR pathway is an appropriate
target for cancer therapy, and several agents that block this
pathway have been developed. In particular, epidermal growth factor
receptor-tyrosine kinase inhibitors (EGFR-TKIs), such as gefitinib
and erlotinib, demonstrated marked clinical activity against NSCLC
harboring an activating EGFR mutation (5–9).
However, patients develop acquired resistance to EGFR-TKIs almost
without exception (10). A
secondary mutation, resulting in a threonine to methionine change
at codon 790 of EGFR (EGFR T790M), is the major mechanism of
EGFR-TKI resistance (10,11). Additionally, some reports suggest
that the EGFR T790M mutation may not be rare and may exist in a
small population of in tumor cells before TKI treatment (12–14).
Moreover, a pre-existing T790M mutation was associated with shorter
progression-free survival (PFS) in patients receiving TKI treatment
(13,14). At this time, no standard treatment
for EGFR mutant patients with acquired resistance has yet been
established, and novel strategies for overcoming this resistance
issue are required.
Immunotherapy for NSCLC patients is considered to be
a potentially feasible option, because of its high specificity and
low toxicity against normal tissues; indeed, several
tumor-associated antigen (TAA)-targeted phase 2/3 studies are
ongoing (15). However,
unfortunately, the results of a TAA-based vaccine therapy study
were unsatisfactory (16). One
concept for improving the effect of cancer vaccine therapy is to
target mutated antigen-derived epitopes. It has been reported that
various mutated epitopes were recognized by tumor-reactive T cells
(17,18), suggesting that the mutated epitope
was potentially immunogenic and thus might function as an
immunotherapeutic target. There are few studies of immunotherapy
targeting the EGFR T790M mutation. Here, we hypothesized that EGFR
T790M-harboring cancer cells could be targeted by activated immune
cells, and attempted to assess the immunogenicity of the EGFR T790M
mutation-derived antigen in vitro. In the present study, we
identified the human leukocyte antigen (HLA)-A2-restricted EGFR
T790M mutation-derived epitope. Our results suggest that
immunotherapy targeting the EGFR T790M mutation-derived antigen may
be a novel treatment option for NSCLC patients with the T790M
mutation. The combination of immunotherapy and EGFR-TKI therapy
also may be a novel strategy for prevention of T790M-mediated
resistance.
Materials and methods
Cell lines
The human NSCLC cell line H1975 was provided by
Professor Seiji Yano (Kanazawa University, Ishikawa, Japan).
H1975-A2 (H1975 transfected with HLA-A2) was provided by Dr Tetsuro
Sasada (Kurume University, Fukuoka, Japan). Artificial APC-A2
(aAPC-A2) cells, which were generated by transduction of
HLA-A*02:01, CD80, and CD83 molecules into K562 cells,
were provided by Dr Naoto Hirano (Dana-Farber Cancer Institute,
Boston, MA, USA). T2 cells (HLA-A*02:01,
TAP−) and human NSCLC cell line 11–18 were purchased
from Riken (Saitama, Japan). These cell lines were cultured in
RPMI-1640 (Sigma Chemical Co., St. Louis, MO, USA), supplemented
with 10% FBS (Gibco-BRL, Carlsbad, CA, USA), 100 U/ml penicillin,
and 100 μg/ml streptomycin in a humidified atmosphere containing 5%
CO2.
PBMC collection
Peripheral blood samples were collected from four
HLA-A*02:01-positive healthy donors, after informed
consent was obtained. Peripheral blood mononuclear cells (PBMCs)
were isolated by density centrifugation using Ficoll-Hypaque
(Pharmacia, Uppsala, Sweden) and frozen in liquid nitrogen until
use.
Epitope prediction and synthesis
The epitope prediction software BIMAS (http://www-bimas.cit.nih.gov/molbio/hla_bind/) was
used to predict peptides that could bind to HLA-A2. EGFR T790M
mutation-derived peptides (purity >95%) were purchased from
Scrum, Inc. (Tokyo, Japan). H-2 Kb-restricted ovalbumin (OVA)
(257–264) (SIINFEKL) peptide (AnaSpec, Inc., Fremont, CA, USA) was
used as a negative control in the peptide-binding assay.
HLA-A2-restricted cytomegalovirus (CMV) (495–503) (NLV PMVATV)
peptide was used as a positive control peptide, and an
HLA-A2-restricted HIV-gag (77–85) (SLYNTYATL) peptide (American
Peptide Company, Sunnyvale, CA, USA) as an irrelevant peptide in
cytotoxic T lymphocyte (CTL) assays.
Peptide-binding assay
After incubation in culture medium at 26°C
overnight, T2 cells were washed with PBS and suspended in 1 ml
Opti-MEM (Invitrogen Life Technologies, Carlsbad, CA, USA) with
peptide (100 μg/ml), followed by incubation at 26°C for 3 h and
then at 37°C for 2.5 h. After washing with PBS, HLA-A2 expression
was measured using a BD FACSCanto II flow cytometer (BD
Biosciences, San Jose, CA, USA) using a FITC-conjugated HLA-A2 (MBL
Co., Ltd., Aichi, Japan)-specific monoclonal antibody. Mean
fluorescence intensity (MFI) was analyzed using the FlowJo software
(Tomy Digital Biology Co., Ltd., Tokyo, Japan). An OVA peptide was
used as a negative control. A CMV peptide was used as a positive
control peptide.
Generation of DCs
CD14+ cells were isolated from PBMCs
using human CD14 microbeads (Miltenyi Biotec GmbH, Bergisch
Gladbach, Germany). Immature dendritic cells (DCs) were generated
from CD14+ cells using IL-4 (10 ng/ml; PeproTech, Inc.,
Rocky Hill, NJ, USA) and granulocyte-macrophage colony-stimulating
factor (GM-CSF) (10 ng/ml; PeproTech, Inc.) in RPMI-1640
supplemented with 10% FBS. Maturation of DCs was induced by
prostaglandin E2 (PGE2) (1 μg/ml; Sigma Chemical Co.) and tumor
necrosis factor-α (TNF-α) (10 ng/ml; PeproTech, Inc.).
Induction of peptide-specific CTLs
CD8+ cells were isolated using human CD8
microbeads (Miltenyi Biotec GmbH) from PBMCs. CD8+ cells
(2×106 cells/well) were stimulated with peptide-pulsed
(10 μg/ml) 100-Gy-irradiated autologous mature DCs
(1×105 cells/well) in RPMI-1640 containing 10%
heat-inactivated human AB serum. After 1 week, these cells were
stimulated twice weekly with peptide-pulsed (10 μg/ml)
200-Gy-irradiated aAPC-A2 cells (1×105 cells/well).
Supplementation with 10 IU/ml IL-2 (Proleukin; Novartis, Basel,
Switzerland) and 10 ng/ml IL-15 (PeproTech, Inc.) was performed at
3–4-day intervals between stimulations.
IFN-γ ELISPOT assay
Specific secretion of interferon-γ (IFN-γ) from
human CTLs in response to stimulator cells was assayed using the
IFN-γ enzyme-linked immuno spot (ELISPOT) kit (BD Biosciences),
according to the manufacturer’s instructions. Stimulator cells were
pulsed with peptide for 2 h at room temperature and then washed
three times. Responder cells were incubated with stimulator cells
for 20 h. The resulting spots were counted using an ELIPHOTO
counter (Minerva Tech, Tokyo, Japan).
CD107a assay and generation of a CTL
line
CD8+ cells isolated using human CD8
microbeads from cultured cells were incubated with peptide-pulsed
T2 cells at a ratio of 2:1 for 3.5 h at 37°C. CD107a-specific
antibodies (BD Biosciences) were included in the mixture during the
incubation period. CD8+ CD107a+ cells were
sorted using a FACSAria II cell sorter (BD Biosciences). Sorted
CTLs were stimulated, and the CTL line was established as described
previously (19).
Cytotoxicity assay
Cytotoxic capacity was analyzed using the Terascan
VPC system (Minerva Tech). The CTL line was used as the effector
cell type. Target cells were labeled in calcein-AM (Dojindo
Molecular Technologies, Inc., Kumamoto, Japan) solution for 30 min
at 37°C. The labeled cells were then co-cultured with the effector
cells for 4–6 h. Fluorescence intensity was measured before and
after the culture period, and specific cytotoxic activity was
calculated using the following formula: % cytotoxicity = {1 −
[(average fluorescence of the sample wells − average fluorescence
of the maximal release control wells)/(average fluorescence of the
minimal release control wells − average fluorescence of the maximal
release control wells)]} × 100%.
Results
Assessment of EGFR T790M-derived peptide
binding to HLA-A*02:01 molecules
As the candidates of
HLA-A*02:01-restricted EGFR T790M-derived CTL epitopes,
we selected five 9- or 10-mer peptides with high predicted
HLA-A*02:01-binding scores, calculated using BIMAS
software. Three of the five EGFR T790M-derived peptides had higher
binding scores than the corresponding wild-type peptides. Some
studies have reported that modified peptides with single amino acid
substitutions exhibit improved affinity for HLA molecules and
enhanced immunogenicity (20–22);
thus, we also designed two modified peptides. These modified
peptides with a substitution of Cys for Val (T790M-D) or Leu
(T790M-E) at codon 797 showed higher binding scores (Table I).
 | Table IPredicted EGFR T790M-derived peptides
binding to HLA-A2. |
Table I
Predicted EGFR T790M-derived peptides
binding to HLA-A2.
| Peptide name | Position | Length | Sequence | BIMAS scorea |
|---|
| T790M-A | 789–797 | 9 | IMQLMPFGC | 35.378 |
| T790M-B | 790–799 | 10 | MQLMPFGCL | 51.77 |
| T790M-C | 788–797 | 10 | LIMQLMPFGC | 24.921 |
| T790M-D | 789–797b | 9 | IMQLMPFGV | 495.288 |
| T790M-E | 789–797c | 9 | IMQLMPFGL | 152.124 |
| T790M-Awt | 789–797 | 9 | ITQLMPFGC | 0.68 |
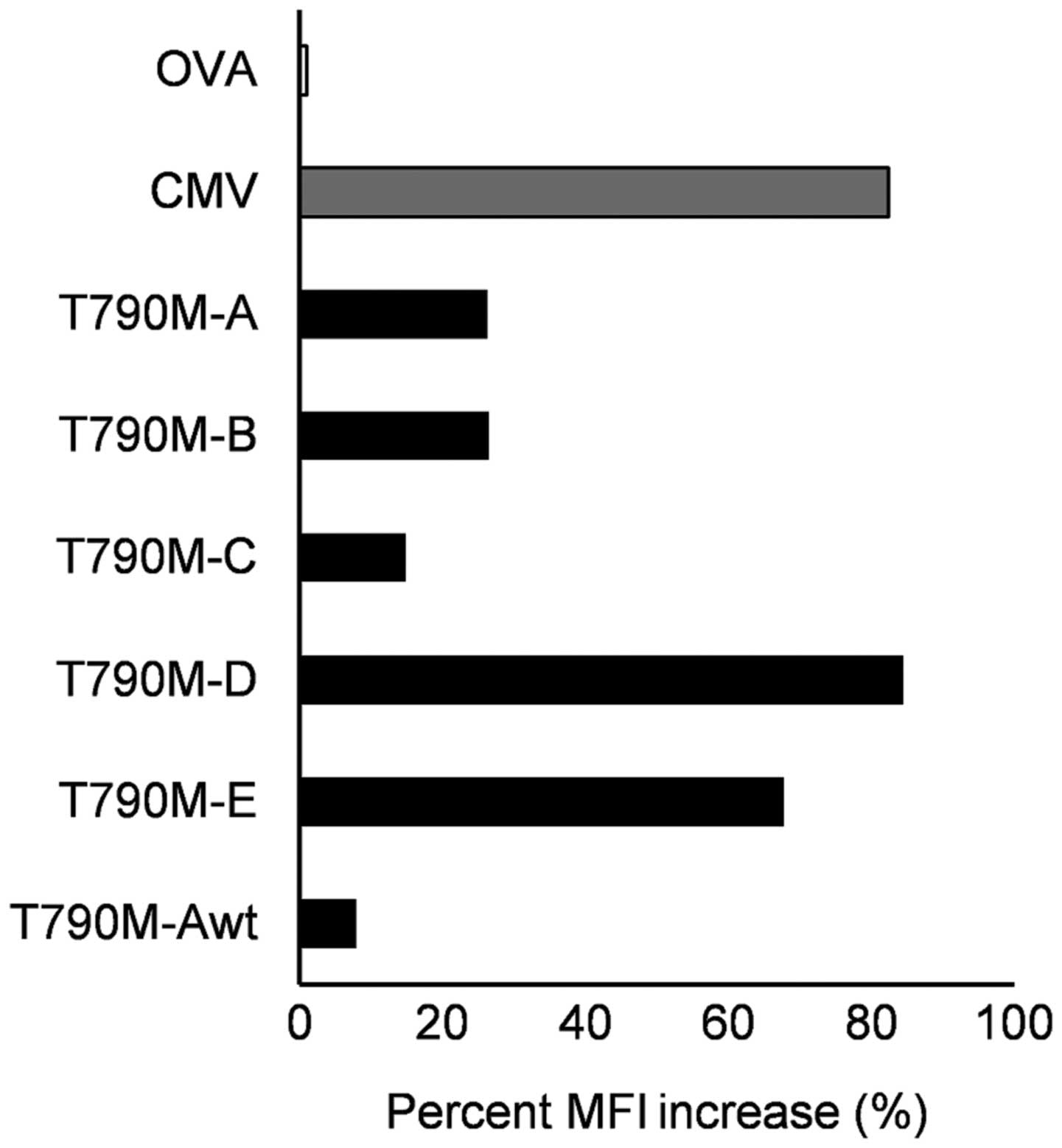
Using the HLA-A2 TAP-deficient T2 cell line, the
binding affinity of the five synthetic peptides to HLA-A2 was
assessed. A peptide-binding assay showed that three EGFR
T790M-derived peptides were able to bind to HLA-A*02:01
molecules. In particular, the binding capability of the T790M-A
peptide to HLA-A*02:01 molecules was higher than that of
the corresponding wild-type peptide. This result suggests that the
single amino acid substitution at codon 790 improved the binding
affinity for HLA-A*02:01 molecules. The binding
affinities of two mutated peptides (T790M-D and -E) to
HLA-A*02:01 were equivalent to that of the CMV peptide
used as a positive control (Fig.
1).
Induction of EGFR T790M-derived
peptide-specific CTLs from human PBMCs
To evaluate the immunogenic potential of the five
predicted HLA-A*02:01-binding peptides derived from EGFR
T790M, we attempted to induce peptide-specific CTLs from human
PBMCs obtained from four healthy donors. Several reports have shown
the usefulness of artificial antigen-presenting cells (aAPCs) for
the induction and expansion of peptide-specific CTLs from PBMCs
(23,24). Thus, we attempted to induce such
CTLs using aAPCs. CD8+ cells were isolated from human
PBMCs using human CD8 microbeads, and then stimulated with
peptide-pulsed DCs for 1 week and subsequently, stimulated twice
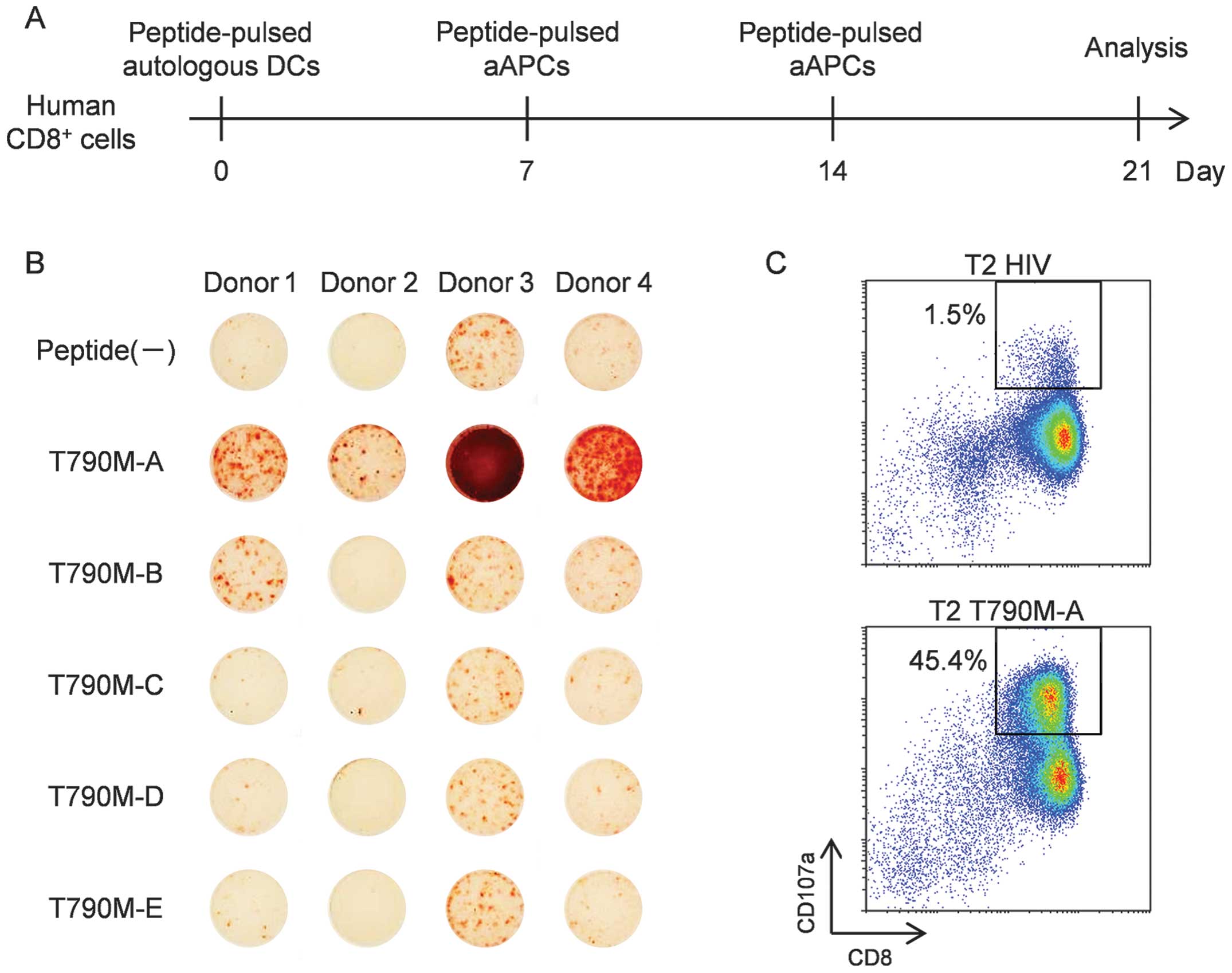
weekly with peptide-pulsed aAPC-A2 (Fig. 2A). As shown in Fig. 2B, ELISPOT assays revealed that
T790M-A (789–797) (IMQLMPFGC)-specific CTLs were induced from PBMCs
from all four donors. Also, induction of T790M-B (790–799)
(MQLMPFGCLL)-specific CTLs were induced from PBMCs from two of the
four healthy donors. However, stimulation with three other
peptides, including modified peptides, did not induce
peptide-specific CTLs. These results suggest that T790M-A.(789–797)
and T790M-B (790–799) have immunogenic potential and that CTLs
specific for these peptides can be induced from human PBMCs. Given
the effective induction of T790M-A.(789–797) peptide-specific CTLs,
we performed further analysis of the T790M-A peptide.
Generation of EGFR T790M-A-specific CTL
line from human PBMCs
Next, we attempted to generate a purified T790M-A
(789–797)-specific CTL line. Because the surface mobilization of
CD107a is useful for identifying and isolating functional
tumor-reactive T cells (25), we
performed a CD107a assay to generate a purified T790M-A
(789–797)-specific CTL line. Cultured cells stimulated by T790M-A
peptide-pulsed DCs and aAPC-A2 in vitro were incubated with
peptide-pulsed T2 cells at a ratio of 2:1 for 3.5 h at 37°C in the
presence of an anti-CD107a antibody. More frequent
CD107a+ cells were observed when CTLs were co-cultured
with T790M-A peptide-pulsed T2 cells compared to HIV-peptide-pulsed
T2 cells, and CD8+ CD107a+ cells were sorted
as a purified, peptide-specific CTL line using a FACSAria II cell
sorter (Fig. 2C). A purified
T790M-A-specific CTL line was established from healthy donor 3.
Cross-reactivity of the T790M-A-specific
CTL line with other EGFR T790M-derived peptides
To assess its cross-reactivity with other EGFR
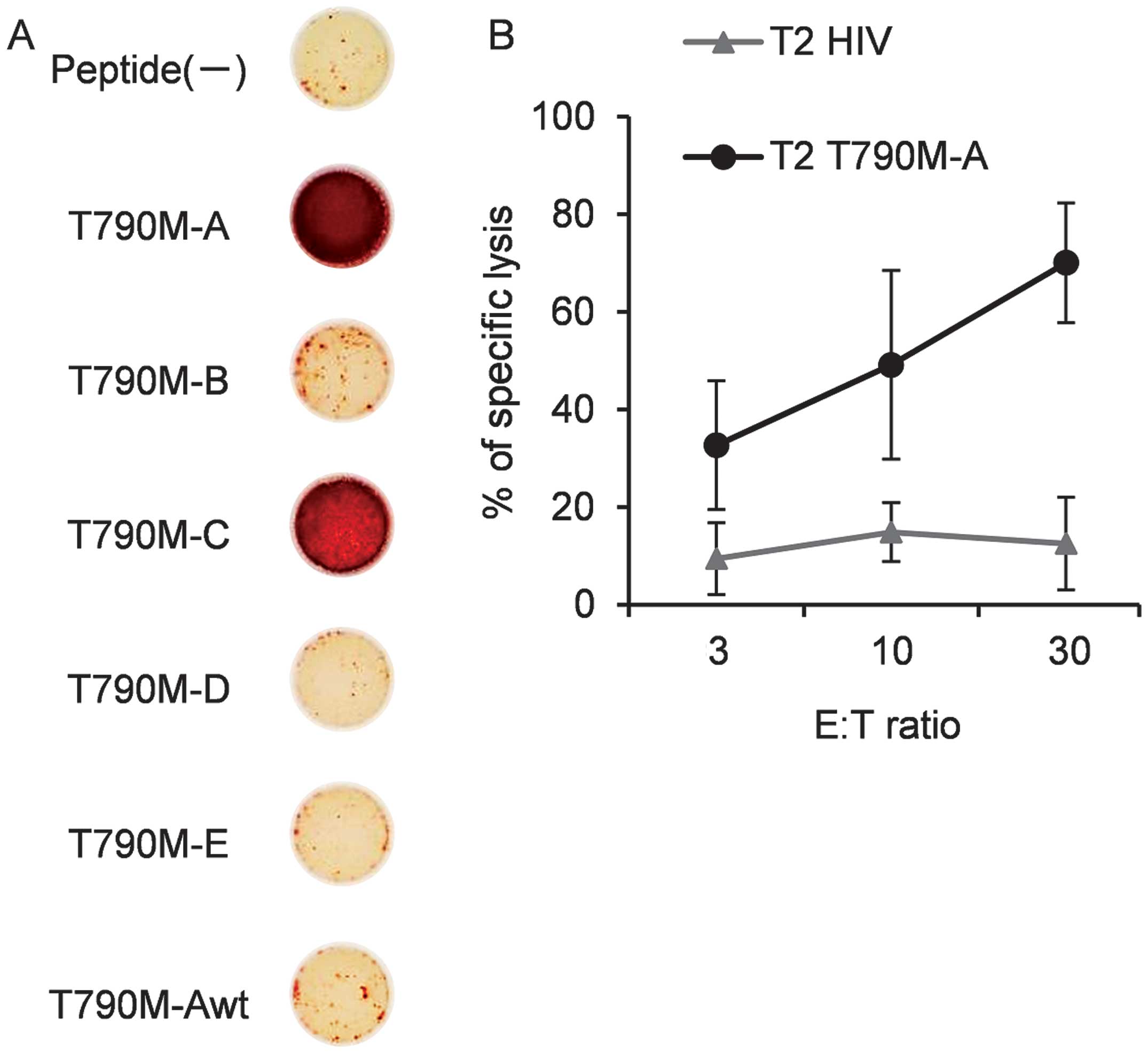
T790M-derived peptides, the T790M-A-specific CTL line was cultured
with T2 cells pulsed with each peptide, and IFN-γ production was
measured by ELISPOT assay. The T790M-A-specific CTL line
specifically recognized T2 cells pulsed with T790M-A (789–797) but
not non-peptide-pulsed T2 cells. The T790M-A-specific CTL line did
not recognize T2 cells pulsed with the T790M-A (789–797) wild-type
(ITQLMPFGC) peptide. Also, T2 cells pulsed with T790M-B, -D, and -E
were not recognized by the T790M-A-specific CTL line (Fig. 3A). However, the T790M-A-specific
CTL line showed cross-reactivity with T2 cells pulsed with
T790M-C.
Next, we evaluated the cytolytic activity of the
T790M-A-specific CTL line against cognate peptide-pulsed T2 cells.
The T790M-A-specific CTL line specifically lysed T790M-A
peptide-pulsed T2 cells but not HIV-peptide-pulsed T2 cells
(Fig. 3B). These results suggest
that the T790M-A-specific CTL line showed cross-reactivity against
some EGFR T790M-derived peptides, but not the corresponding
wild-type EGFR-derived peptide. This cross-reactivity seems to be
favorable for efficacy against EGFR T790M+ cancer
cells.
The T790M-A-specific CTL line recognizes
and lyses HLA-A2+ T790M+ NCSLC cells
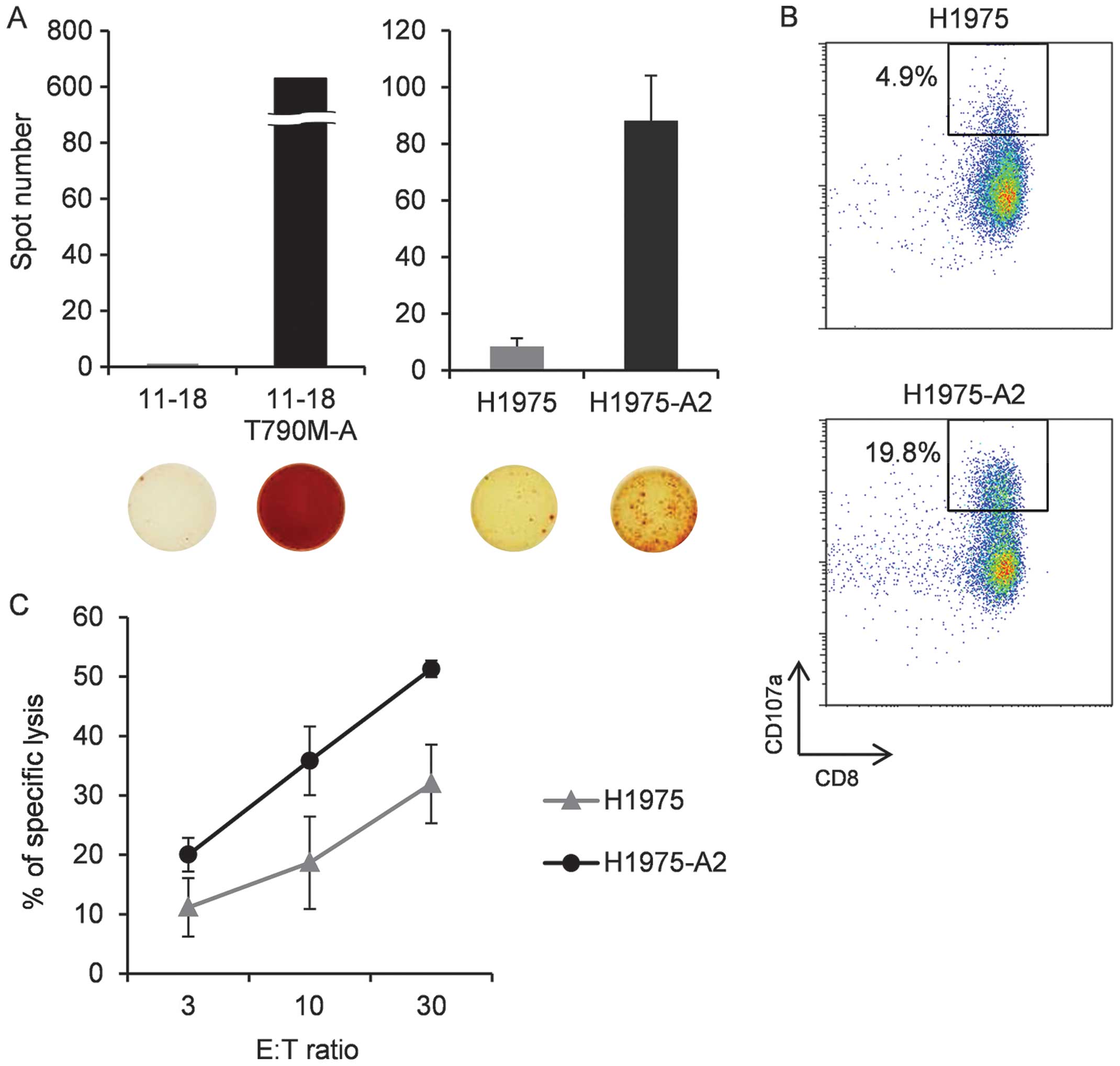
Next, we assessed the ability of the
T790M-A-specific CTL line to recognize the HLA-A2+
T790M+ NCSLC cell line. This CTL line was incubated with
11–18 (T790M−, HLA-A2+), T790M-A-pulsed
11–18, H-1975 (T790M+ HLA-A2−), or H-1975-A2
(T790M+ HLA-A2+), and IFN-γ production was
evaluated. We confirmed that the T790M-A-specific CTL line
recognized peptide-pulsed 11–18 and H-1975-A2, but not 11–18 and
H-1975, cells by IFN-γ ELISPOT assay (Fig. 4A). Similar data were obtained using
CTLs from healthy donor 1 stimulated with T790M-A peptide-pulsed DC
and aAPC-A2 in vitro, which were not purified by the CD107a
assay (data not shown).
To evaluate the function of the T790M-A-specific CTL
line against H1975-A2, a CD107a assay was performed.
CD107a+ cells were detected more frequently in culture
with H-1975-A2 than with H-1975 cells (Fig. 4B).
Finally, we investigated the cytotoxic activity of
the T790M-A-specific CTL line against H-1975-A2. Target cells were
labeled with calcein-AM and co-cultured with the effector cells for
4–6 h. The T790M-A-specific CTL line showed cytotoxic activity
against H1975-A2 cells, but not H1975 cells (Fig. 4C). These results suggest that the
T790M-A-specific CTL line can recognize NSCLC cells harboring the
EGFR T790M mutation in an HLA-A2-restricted manner.
Discussion
Mutated antigens associated with tumor cell
progression and survival or drug resistance represent novel targets
for cancer vaccine therapy. Warren et al evaluated
computationally the antigenic potential of somatic mutations that
occur in human cancers (26). They
showed that several gene mutation-derived epitopes have immunogenic
potential, at least computationally. Moreover, point mutations
within the ABL kinase domain of the BCR-ABL gene are the
most common causes of resistance to imatinib in chronic myeloid
leukemia (CML) patients (27). Cai
et al reported that the mutated BCR-ABL gene was
associated with a TKI-resistance-generated CTL epitope in CML
patients (28). These results
suggest new immunotherapeutic approaches based on a TKI-resistant
mutation-derived neoantigen. That is, mutations associated with
acquired resistance to TKI therapy can be targeted by immune-based
treatment strategies. This strategy may be an option to treat the
gene mutation-mediated drug-resistant cancer cells. In the present
study, we demonstrated the immunogenicity of antigens from mutated
EGFR that are involved in TKI resistance in NCSLC.
TAAs can be classified into several categories, such
as cancer-testis (CT) antigens, overexpressed antigens,
differentiation antigens, and mutated antigens. Of these, only
mutated antigens are unique, because they are not expressed in
normal tissues. Previous reports have shown that peptide vaccine
therapy can occasionally induce ineffective CTL responses, contrary
to expectations (29–31). One possibility is that the induced
antigen-specific CTLs have a low affinity, and thus recognize only
target cells pulsed with high concentrations of the peptide and not
naturally presented epitopes on tumor cells. Several EGFR-derived
CTL epitopes have been identified (32,33);
however, the frequency of high-avidity EGFR-specific CTLs seems to
be low in patients with EGFR-expressing cancers, because EGFR is a
self-antigen that induces tolerance. The ability of low-avidity
CTLs to recognize antigen-expressing tumor cells is considered to
be weak. However, mutation-derived antigens are not self-antigens;
thus, they would not be expected to induce immunotolerance, and so
may have high immunogenicity. Indeed, in melanoma patients who
experienced dramatic therapeutic effects after adoptive cell
therapy with tumor-infiltrating lymphocytes (TILs), the mutated
antigen-derived epitope was immunodominant and was recognized by
tumor-reactive T cells (34,35).
In the present study, BIMAS was used to select EGFR
T790M-derived candidate peptides that bind to
HLA-A*02:01 according to computer algorithms, and
T790M-A-specific CTLs could be induced from PBMCs of all four
healthy donors by stimulation with peptide-pulsed DCs and aAPCs.
Amino acid substitution of anchor residues (at position 2 and the
C-terminus for HLA-A2) can alter the binding affinity (36–38).
Leucine and methionine are the preferred anchor residues at
position 2 of HLA-A2 (36,37). T790M-A.(IMQLMPFGC) harbors a
substitution of threonine to methionine at the anchor site, which
confers immunogenicity. Also, valine and leucine are the preferred
anchor residues at the C-terminus (36,37).
Then, we designed the modified peptides, T790M-D
(IMQLMPFGV, substitution of cysteine to valine at the C-terminus)
and T790M-E.(IMQLMPFGL, substitution of cysteine to leucine at the
C-terminus). These peptides bound to the HLA-A*02:01
molecule strongly (Fig. 1), but
could not induce specific CTLs. T790M-D and -E are not
self-antigens, being similar in this respect to T790M-A; this
difference may be due in part to the difference in the frequency of
peptide-specific CTL precursors. To confirm that the predicted
candidate peptides are naturally presented peptides on tumor cells,
peptide-specific CTL clones or lines induced by the peptides must
recognize the tumor cells. A mass spectrometry (MS)-based method
facilitates identification of peptide presentation by tumor cells
(39). In this study, we confirmed
the peptide-specific recognition of tumor cells by a
peptide-specific CTL line, but not a CTL clone. However, CTL lines
may contain distinct CTL clones that recognize irrelevant peptides,
leading to apparent tumor reactivity (40). To avoid misleading tumor
recognition and to evaluate the antigen-specific response of a CTL
line, we used a peptide-specific CTL line established by CD107a
sorting. An IFN-γ ELISPOT assay suggested that the specific CTL
line recognized NSCLC cells harboring the EGFR T790M mutation in an
HLA-A*02:01-restricted manner.
The T790M-A-specific CTL line did not show activity
against the corresponding wild-type peptide. This suggests that
EGFR T790M-targeted immunotherapy has no effect on NSCLC prior to
EGFR-TKI treatment, with the exception of any pre-existing
population of T790M-harboring cells, at least theoretically. Thus,
consideration of combination therapy, EGFR-TKI and EGFR
T790M-targeted immunotherapy, seems reasonable. Several studies
have suggested that combination therapy could improve the efficacy
of cancer immunotherapy. For instance, some chemotherapeutic agents
can lead to upregulation of TAA expression or improvement of tumor
cell resistance to specific CTLs (41). Use of an EGFR-TKI or anti-EGFR
antibody augments the IFN-γ-induced expression of MHC classes I and
II by A431 malignant human keratinocytes (42). Moreover, gefitinib improved the
cytotoxic activity of natural killer cells against H1975 by
modulating the interaction between NK cells and cancer cells, and
by inhibiting STAT3 expression (43). These results indicate that the
combination of EGFR-TKI and immunotherapy may have synergistic
activity against NSCLC cells. The concept of combination therapy is
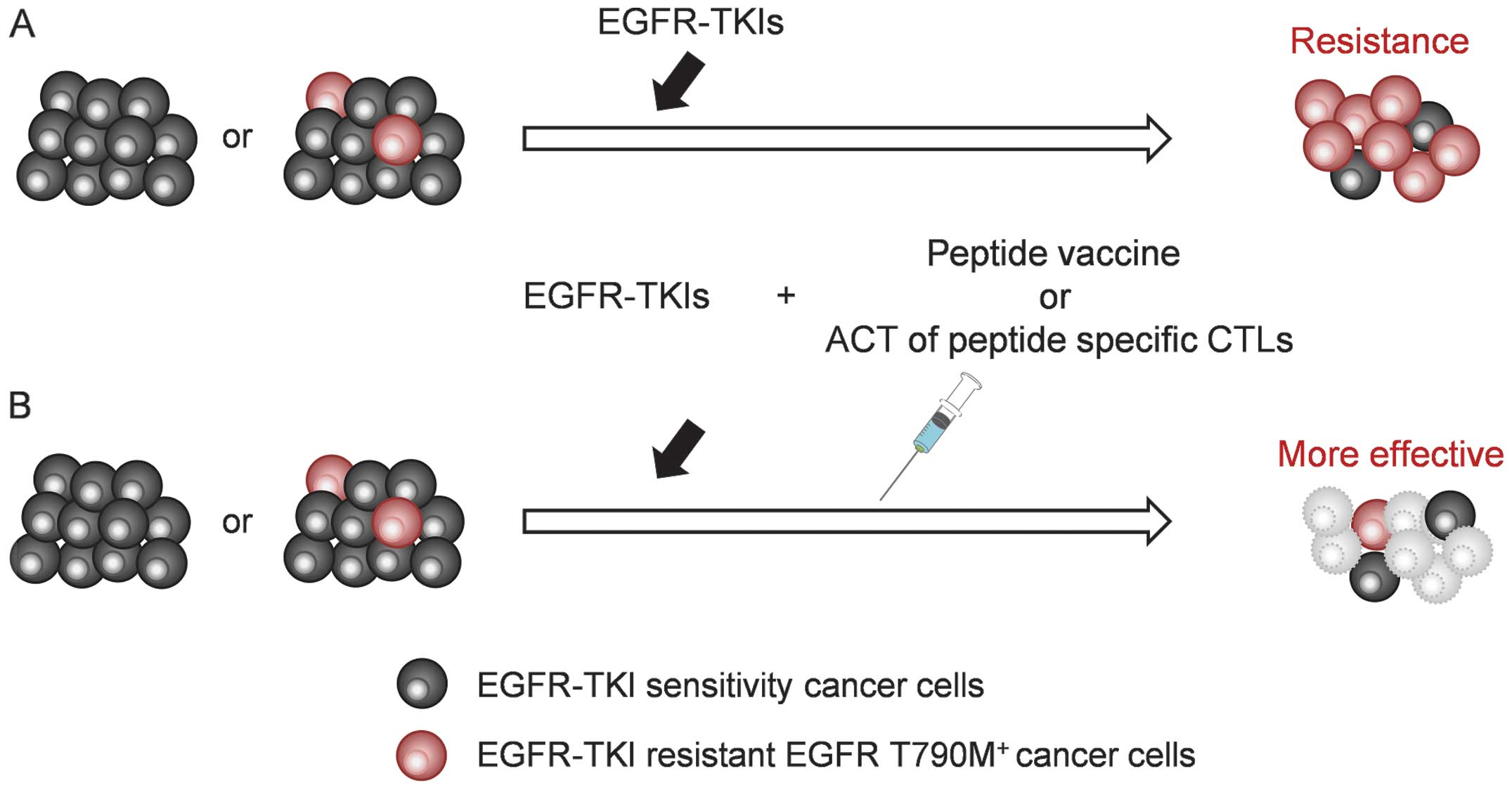
shown in Fig. 5. Adding EGFR
T790M-targeted immunotherapy to EGFR-TKI treatment could control
the progression of cancer cells harboring T790M.
Yamada et al reported two HLA-A2-restricted
EGFR T790M-derived CTL epitopes (790–799 MQLMPFGCLL and 788–798 LI
MQLMPFGCL) (44). In addition to
these epitopes, we identified the HLA-A*02:01-restricted
CTL epitope T790M-A.(789–797 IMQLMPFGC). We found that a
T790M-A-specific CTL line established from human PBMCs had the
ability to recognize and lyse the
HLA-A*02:01+ T790M+ NCSLC cell
line, and importantly, did not show cross-reactivity with the
corresponding wild-type EGFR peptide. These results suggest that
the EGFR T790M-A-specific CTL line recognizes single amino acid
substitutions, leading to a low level of auto-immune reaction. The
combination of an EGFR-TKI and T790M-targeted immunotherapy may be
useful for treatment of NSCLC with the T790M mutation.
Acknowledgements
We thank Professor Seiji Yano for providing the
NSCLC cell line H1975, Dr Tetsuro Sasada for providing the NSCLC
cell line H1975-A2, and Professor Naoto Hirano for providing
aAPC-A2. This study was supported in part by the National Cancer
Center Research and Development Fund (25-A-7), as well as Research
for the Promotion of Cancer Control Programmes, Research on
Applying Health Technology, and Third Term Comprehensive Control
Research for Cancer from the Ministry of Health, Labour, and
Welfare, Tokyo, Japan.
Abbreviations:
|
aAPC
|
artificial antigen-presenting cell
|
|
ELISPOT
|
enzyme-linked immuno spot
|
|
HLA
|
human leukocyte antigen
|
|
IFN-γ
|
interferon-γ
|
|
MAPK
|
mitogen-activated protein kinase
|
|
PBMC
|
peripheral blood mononuclear cell
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
PFS
|
progression-free survival
|
|
STAT
|
signal transducer and activator of
transcription
|
References
|
1
|
Jemal A, Siegel R, Ward E, Murray T, Xu J
and Thun MJ: Cancer statistics, 2007. CA Cancer J Clin. 57:43–66.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Youlden DR, Cramb SM and Baade PD: The
international epidemiology of lung cancer: geographical
distribution and secular trends. J Thorac Oncol. 3:819–831. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Normanno N, Maiello MR and De Luca A:
Epidermal growth factor receptor tyrosine kinase inhibitors
(EGFR-TKIs): simple drugs with a complex mechanism of action? J
Cell Physiol. 194:13–19. 2003. View Article : Google Scholar
|
|
4
|
Hynes NE and Lane HA: ERBB receptors and
cancer: the complexity of targeted inhibitors. Nat Rev Cancer.
5:341–354. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mok TS, Wu YL, Thongprasert S, et al:
Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N
Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mitsudomi T, Morita S, Yatabe Y, et al:
Gefitinib versus cisplatin plus docetaxel in patients with
non-small-cell lung cancer harbouring mutations of the epidermal
growth factor receptor (WJTOG3405): an open label, randomised phase
3 trial. Lancet Oncol. 11:121–128. 2010. View Article : Google Scholar
|
|
7
|
Maemondo M, Inoue A, Kobayashi K, et al:
Gefitinib or chemotherapy for non-small-cell lung cancer with
mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou C, Wu YL, Chen G, et al: Erlotinib
versus chemotherapy as first-line treatment for patients with
advanced EGFR mutation-positive non-small-cell lung cancer
(OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase
3 study. Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rosell R, Carcereny E, Gervais R, et al:
Erlotinib versus standard chemotherapy as first-line treatment for
European patients with advanced EGFR mutation-positive
non-small-cell lung cancer (EURTAC): a multicentre, open-label,
randomised phase 3 trial. Lancet Oncol. 13:239–246. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pao W, Miller VA, Politi KA, et al:
Acquired resistance of lung adenocarcinomas to gefitinib or
erlotinib is associated with a second mutation in the EGFR kinase
domain. PLoS Med. 2:e732005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kobayashi S, Boggon TJ, Dayaram T, et al:
EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fujita Y, Suda K, Kimura H, et al: Highly
sensitive detection of EGFR T790M mutation using colony
hybridization predicts favorable prognosis of patients with lung
cancer harboring activating EGFR mutation. J Thorac Oncol.
7:1640–1644. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Su KY, Chen HY, Li KC, et al: Pretreatment
epidermal growth factor receptor (EGFR) T790M mutation predicts
shorter EGFR tyrosine kinase inhibitor response duration in
patients with non-small-cell lung cancer. J Clin Oncol. 30:433–440.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rosell R, Molina MA, Costa C, et al:
Pretreatment EGFR T790M mutation and BRCA1 mRNA expression in
erlotinib-treated advanced non-small-cell lung cancer patients with
EGFR mutations. Clin Cancer Res. 17:1160–1168. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shepherd FA, Douillard JY and Blumenschein
GR Jr: Immunotherapy for non-small cell lung cancer: novel
approaches to improve patient outcome. J Thorac Oncol. 6:1763–1773.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rosenberg SA, Yang JC and Restifo NP:
Cancer immunotherapy: moving beyond current vaccines. Nat Med.
10:909–915. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kawakami Y, Wang X, Shofuda T, et al:
Isolation of a new melanoma antigen, MART-2, containing a mutated
epitope recognized by autologous tumor-infiltrating T lymphocytes.
J Immunol. 166:2871–2877. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lennerz V, Fatho M, Gentilini C, et al:
The response of autologous T cells to a human melanoma is dominated
by mutated neoantigens. Proc Natl Acad Sci USA. 102:16013–16018.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yoshikawa T, Nakatsugawa M, Suzuki S, et
al: HLA-A2-restricted glypican-3 peptide-specific CTL clones
induced by peptide vaccine show high avidity and antigen-specific
killing activity against tumor cells. Cancer Sci. 102:918–925.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Valmori D, Fonteneau JF, Lizana CM, et al:
Enhanced generation of specific tumor-reactive CTL in vitro by
selected Melan-A/MART-1 immunodominant peptide analogues. J
Immunol. 160:1750–1758. 1998.PubMed/NCBI
|
|
21
|
Parkhurst MR, Salgaller ML, Southwood S,
et al: Improved induction of melanoma-reactive CTL with peptides
from the melanoma antigen gp100 modified at
HLA-A*0201-binding residues. J Immunol. 157:2539–2548.
1996.PubMed/NCBI
|
|
22
|
Fong L, Hou Y, Rivas A, et al: Altered
peptide ligand vaccination with Flt3 ligand expanded dendritic
cells for tumor immunotherapy. Proc Natl Acad Sci USA.
98:8809–8814. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hirano N, Butler MO, Xia Z, et al:
Engagement of CD83 ligand induces prolonged expansion of
CD8+ T cells and preferential enrichment for antigen
specificity. Blood. 107:1528–1536. 2006. View Article : Google Scholar
|
|
24
|
Yoshimura M, Tada Y, Ofuzi K, Yamamoto M
and Nakatsura T: Identification of a novel
HLA-A*02:01-restricted cytotoxic T lymphocyte epitope
derived from the EML4-ALK fusion gene. Oncol Rep. 32:33–39.
2014.PubMed/NCBI
|
|
25
|
Rubio V, Stuge TB, Singh N, et al: Ex vivo
identification, isolation and analysis of tumor-cytolytic T cells.
Nat Med. 9:1377–1382. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Warren RL and Holt RA: A census of
predicted mutational epitopes suitable for immunologic cancer
control. Hum Immunol. 71:245–254. 2010. View Article : Google Scholar
|
|
27
|
Soverini S, Hochhaus A, Nicolini FE, et
al: BCR-ABL kinase domain mutation analysis in chronic myeloid
leukemia patients treated with tyrosine kinase inhibitors:
recommendations from an expert panel on behalf of European
LeukemiaNet. Blood. 118:1208–1215. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cai A, Keskin DB, DeLuca DS, et al:
Mutated BCR-ABL generates immunogenic T-cell epitopes in CML
patients. Clin Cancer Res. 18:5761–5772. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yamshchikov GV, Barnd DL, Eastham S, et
al: Evaluation of peptide vaccine immunogenicity in draining lymph
nodes and peripheral blood of melanoma patients. Int J Cancer.
92:703–711. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen W, Yewdell JW, Levine RL and Bennink
JR: Modification of cysteine residues in vitro and in vivo affects
the immunogenicity and antigenicity of major histocompatibility
complex class I-restricted viral determinants. J Exp Med.
189:1757–1764. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Meadows L, Wang W, den Haan JM, et al: The
HLA-A*0201-restricted H-Y antigen contains a
posttranslationally modified cysteine that significantly affects T
cell recognition. Immunity. 6:273–281. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shomura H, Shichijo S, Matsueda S, et al:
Identification of epidermal growth factor receptor-derived peptides
immunogenic for HLA-A2(+) cancer patients. Br J Cancer.
90:1563–1571. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shomura H, Shichijo S, Komatsu N, et al:
Identification of epidermal growth factor receptor-derived peptides
recognised by both cellular and humoral immune responses in
HLA-A24+ non-small cell lung cancer patients. Eur J
Cancer. 40:1776–1786. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lu YC, Yao X, Li YF, et al: Mutated
PPP1R3B is recognized by T cells used to treat a melanoma patient
who experienced a durable complete tumor regression. J Immunol.
190:6034–6042. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Robbins PF, Lu YC, El-Gamil M, et al:
Mining exomic sequencing data to identify mutated antigens
recognized by adoptively transferred tumor-reactive T cells. Nat
Med. 19:747–752. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Falk K, Rötzschke O, Stevanović S, Jung G
and Rammensee HG: Allele-specific motifs revealed by sequencing of
self-peptides eluted from MHC molecules. Nature. 351:290–296. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sidney J, Southwood S, Mann DL,
Fernandez-Vina MA, Newman MJ and Sette A: Majority of peptides
binding HLA-A*0201 with high affinity crossreact with
other A2-supertype molecules. Hum Immunol. 62:1200–1216. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Matsushita H, Vesely MD, Koboldt DC, et
al: Cancer exome analysis reveals a T-cell-dependent mechanism of
cancer immunoediting. Nature. 482:400–404. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schirle M, Keilholz W, Weber B, et al:
Identification of tumor-associated MHC class I ligands by a novel T
cell-independent approach. Eur J Immunol. 30:2216–2225. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Parkhurst MR, Riley JP, Igarashi T, Li Y,
Robbins PF and Rosenberg SA: Immunization of patients with the
hTERT:540–548 peptide induces peptide-reactive T lymphocytes that
do not recognize tumors endogenously expressing telomerase. Clin
Cancer Res. 10:4688–4698. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Matar P, Alaniz L, Rozados V, et al:
Immunotherapy for liver tumors: present status and future
prospects. J Biomed Sci. 16:302009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pollack BP, Sapkota B and Cartee TV:
Epidermal growth factor receptor inhibition augments the expression
of MHC class I and II genes. Clin Cancer Res. 17:4400–4413. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
He S, Yin T, Li D, et al: Enhanced
interaction between natural killer cells and lung cancer cells:
involvement in gefitinib-mediated immunoregulation. J Transl Med.
11:1862013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yamada T, Azuma K, Muta E, et al: EGFR
T790M mutation as a possible target for immunotherapy;
identification of HLA-A*0201-restricted T cell epitopes
derived from the EGFR T790M mutation. PLoS One. 8:e783892013.
View Article : Google Scholar
|