Introduction
SCLC, accounting for 15–20% of newly diagnosed lung
cancer, is an extremely aggressive malignancy with early metastasis
and poor prognosis. Despite a prompt response to chemotherapy,
relapses occur in the majority of patients with SCLC. Therefore the
development of an alternative therapy against SCLC becomes
imperative (1).
ATO has been proven to be an effective therapeutic
agent in acute promyelocytic leukemia (APL) with high complete
remission rate and prolonged survival (2,3). ATO
can induce apoptosis through PML-RARα-independent pathways in APL
or other cancer cells via p53 activation (4,5),
Bcl-2 downregulation (6,7), mitochondrial membrane depolarization
and cytochrome c release (8–10),
depletion of intracellular reduced glutathione (GSH) content and
elevation of reactive oxygen species (11,12).
More recently, the application of ATO in lung cancer treatment has
been explored in preclinical models, mainly in non-small cell lung
cancer (NSCLC). ATO induces growth inhibition and apoptosis in
NSCLC cells through G2/M cell cycle arrest (13,14),
Bcl-2 downregulation and GSH depletion (15). Recently, downregulation of
thymidylate synthase and E2F1 were observed in lung adenocarcinoma
(16,17) and mesothelioma (18). Synergistic effects have been
observed when combining ATO with cisplatin (19), buthionine sulfoximine (BSO)
(15), suberoylanilide hydroxamic
acid (20), and sulindac (21). The effect of ATO in SCLC is less
reported. In a panel of lung cancer cell lines, ATO is highly
cytotoxic to SCLC preferentially mediated through necrosis rather
than apoptosis (22).
Biologically, ATO is known to bind to proteins with
sulfhydryl (-SH) groups (23). It
targets GSH which is a thiol molecule involved in detoxification of
arsenicals (11,24). Evidently, loss of GSH and its
cytotoxic effect is not directly caused by damage of GSH-related
enzymes (glutathione peroxidase, glutathione reductase and
glutathione transferase) (25).
Nonetheless, thioredoxin (Trx) system serves as a key antioxidant
target of ATO in breast cancer cells (26). Trx system, composed of thioredoxin
reductase (TrxR), Trx and NADPH, plays an important role in
maintaining somatic redox homeostasis. Moreover, TrxR and Trx are
overexpressed in many cancers (27–29),
which take part in redox regulation of transcription factors
(30). Therefore, Trx system has
become a new target in cancer therapy (31).
In the present study, we aimed to elucidate the
mechanisms of ATO in SCLC using a cell line model, which may
provide a scientific base for future application of ATO in
treatment of SCLC.
Materials and methods
Chemicals and reagents
ATO, N-acetyl-L-cystine (NAC), buthionine
sulphoximine (BSO) and butylated hydroxyanisole (BHA) were obtained
from Sigma-Aldrich (St. Louis, MO, USA). Apoptosis inducing factor
(AIF), Bcl-2, cleaved caspase-3, cytochrome c, PARP, SMAC,
thioredoxin and XIAP antibodies were purchased from Cell Signaling
Technology (Danvers, MA, USA).
Cell lines and culture
Five SCLC cell lines (H187, DMS79, H526, H69 and
H841) were obtained from the American Type Culture Collection
(Manassas, VA, USA). H187, DMS79, H526 and H69 cells were cultured
in RPMI-1640 medium (Gibco®, Life Technologies,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS,
Gibco), while H841 cells were grown in HETIS medium supplemented
with 5% FBS. Cells were incubated in 37°C with a humidified
atmosphere of 5% CO2.
Cell proliferation assay
The cytotoxicity of ATO in SCLC cell lines was
measured using
3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl-tetrazolium bromide
(MTT) assay (ATCC) as previously performed (32).
Annexin V-phycoerythrin
(PE)/7-aminoactinomycin D (AAD) staining
Apoptosis was determined by PE-conjugated Annexin
V/7-AAD kit (BD Biosciences, CA, USA). Briefly, treated cells were
collected, washed and re-suspended in binding buffer. Cells were
stained for 15 min at room temperature with PE-Annexin V (Ex/Em =
488/578 nm)/7-AAD (Ex/Em = 546/647 nm), then read-out by Cytomics
FC 500 analyzer with FL2/FL4 channels (Beckman Coulter, CA, USA).
The cell populations in PE+/7-AAD− and
PE+/7-AAD+ quadrants were calculated.
Measurement of mitochondrial membrane
depolarization (MMD)
Briefly, cells after treatment were collected,
washed, and stained with JC-1 (5 μg/ml) at 37°C for 15 min before
reading with FL1/FL2 channels of Cytomics FC 500 analyzer.
Detection of glutathione (GSH) and
reactive oxygen species (ROS)
Cellular GSH level was measured by using
5-chloro-methylfluorescein diacetate (CMFDA, Ex/Em = 522/595 nm,
Invitrogen, CA, USA) fluorescence as described previously (33). After treatment, cells were washed
and incubated for 30 min at 37°C with 5 μM CMFDA in FBS-free
medium, followed by incubation for 40 min at 37°C with complete
medium. CMF fluorescence intensity was determined using a Cytomics
FC500 flow cytometer. Hydrogen peroxide
(H2O2) and superoxide were measured by
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA,
Ex/Em = 500/520 nm) and dihydroethidium (DHE, Ex/Em = 518/605 nm)
(Invitrogen) staining, respectively. Treated cells were washed and
incubated for 20 min at 37°C with 1 μM of H2DCFDA or 1
μM DHE in FBS-free medium following flow cytometry.
Immunofluorescence
Oxidative DNA damage was assessed using
8-hydroxy-2-deoxyguanosine (8-OHdG) immunofluorescence. Cells were
seeded on slides before treatment. Slides were washed and blocked
for 60 min with 10% normal goat serum in PBS, followed by
incubation at 4°C overnight with monoclonal anti-8-OHdG antibody
(Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). The slides
were then incubated for 2 h with FITC-conjugated goat anti-mouse
IgG (Santa Cruz Biotechnology, Ex/Em = 500/520 nm), cover-slipped
with UltraCruz™ Mounting Medium (Santa Cruz Biotechnology) and
observed with Eclipse E-800 (Nikon, Tokyo, Japan). DAPI (Santa Cruz
Biotechnology) staining was used to visualize the nucleus.
Western blotting
Total protein was extracted with RIPA buffer (PBS,
1% Nonidet-P-40, 0.1% deoxycholate, 0.1% sodium dodecyl sulfate)
containing protease inhibitors. Nuclear protein and mitochondrial
protein were collected with NE-PER Nuclear and Cytoplasmic
Extraction kit and Mitochondria Isolation kit respectively (Pierce
Biotech, Rockford, IL, USA) as described by the manufacturer.
Proteins (40 μg) were separated in sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred
to nitrocellulose membrane (GE Healthcare, Buckinghamshire, UK),
immunoblotted with corresponding primary and secondary antibodies,
and detected using ECL detection kit (GE Healthcare). Image
analysis was carried out with ImageJ (Research Services Branch,
National Institute of Mental Health, Bethesda, MD, USA).
DNA fragmentation assay
Cellular DNA was extracted with standard DNA
phenol/chloroform extraction method (34). DNA (0.1 μg/sample) was separated in
1.5% agarose gel and bands were visualized on a UV transilluminator
at 302 nm.
Redox western blot analysis
Redox forms of Trx1 were separated as previously
described (26,35). Trx1 was carboxymethylated in
guanidine-Tris solution (6 M guanidine-HCl, 50 mM Tris, 1 mM EDTA,
30 mM iodoacetic acid) at pH 7.5. After incubation at 37°C for 30
min, proteins were desalted and concentrated using centrifugal
filter device (Millipore, Billerica, MA, USA). Proteins were
separated with native gel and western blotting was performed. Redox
potential was calculated with Nernst equation: Eh, Trx1 = 240 mV +
30 × ln (ratio of Trx1Ox and Trx1Red)
(36).
Statistical analysis
Data were presented as mean ± standard deviation
(SD), with between-group differences analyzed by two-tailed
Student’s t-test. A p-value <0.05 was considered statistically
significant. Linear regression was used to calculate
IC50 values. All statistical analyses were performed
using Prism 5 (GraphPad Software, La Jolla, CA, USA).
Results
Cell growth inhibition and apoptosis
induced by ATO
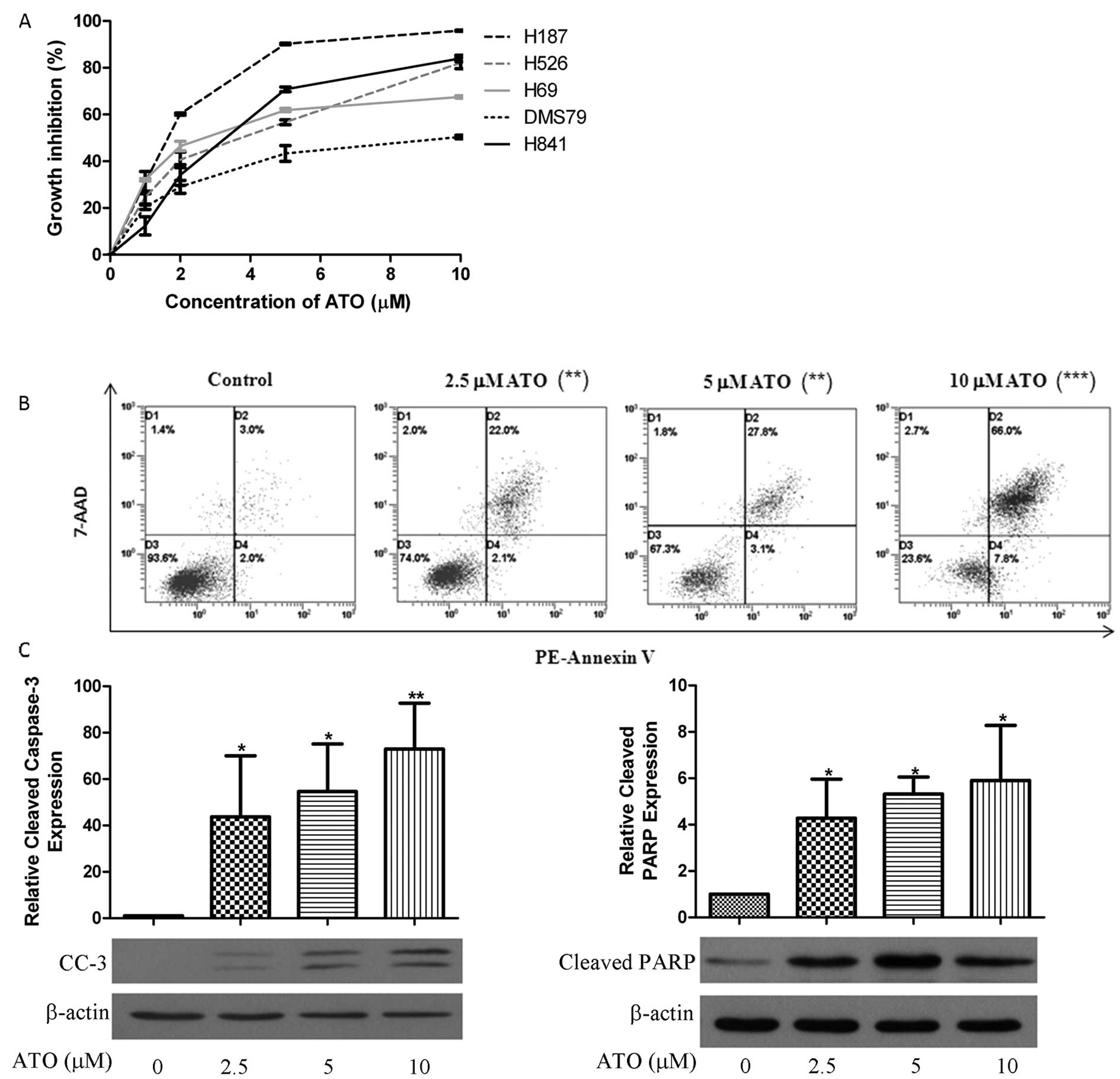
Five SCLC cell lines were incubated for 48 h with
different concentrations of ATO. Cell proliferation was suppressed
in a dose-dependent manner with IC50 values in
clinically reachable levels (Fig.
1A). H841 cell line was chosen due to its adherent property for
apoptotic assay. ATO induced dose-dependent apoptosis (Fig. 1B) in H841 cells accompanied by
increased cleaved caspase-3 and PARP (Fig. 1C).
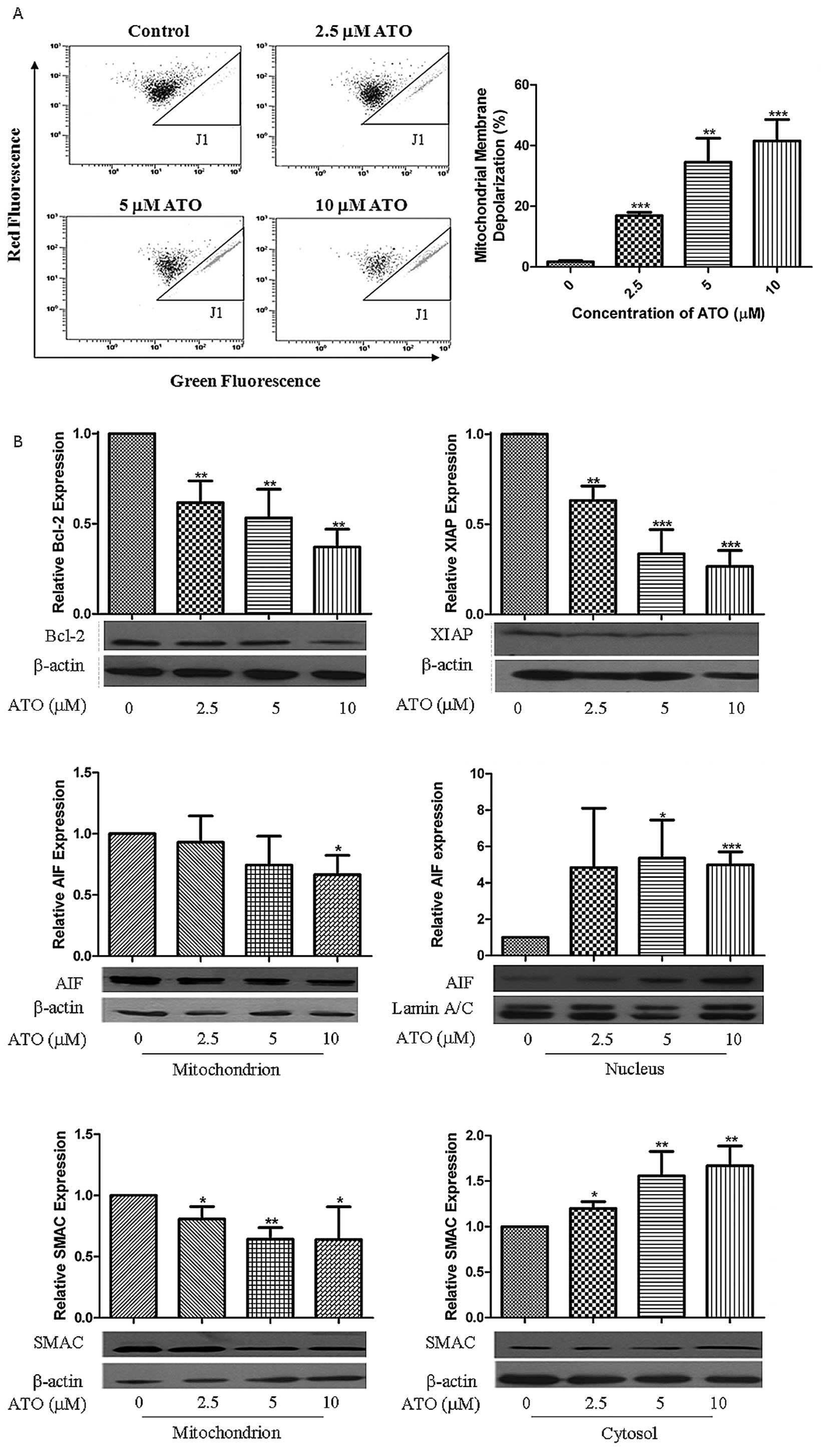
ATO-induced MMD, GSH depletion and ROS
elevation
ATO prompted a dose-dependent shift from red to
green fluorescence that indicated MMD in H841 cells (Fig. 2A). This was associated with
downregulation of Bcl-2 and XIAP as well as release of AIF and SMAC
to cytosol (Fig. 2B). Low
concentration of ATO (2.5 μM) reduced 60% of GSH content (Fig. 2C) and increased
H2O2 by >1.5-fold compared with control
H841 cells (30500±6150 vs 13522±3146) (Fig. 2D). Nonetheless, superoxide content
remained unchanged except at the highest concentration (10 μM) of
ATO treatment (Fig. 2D).
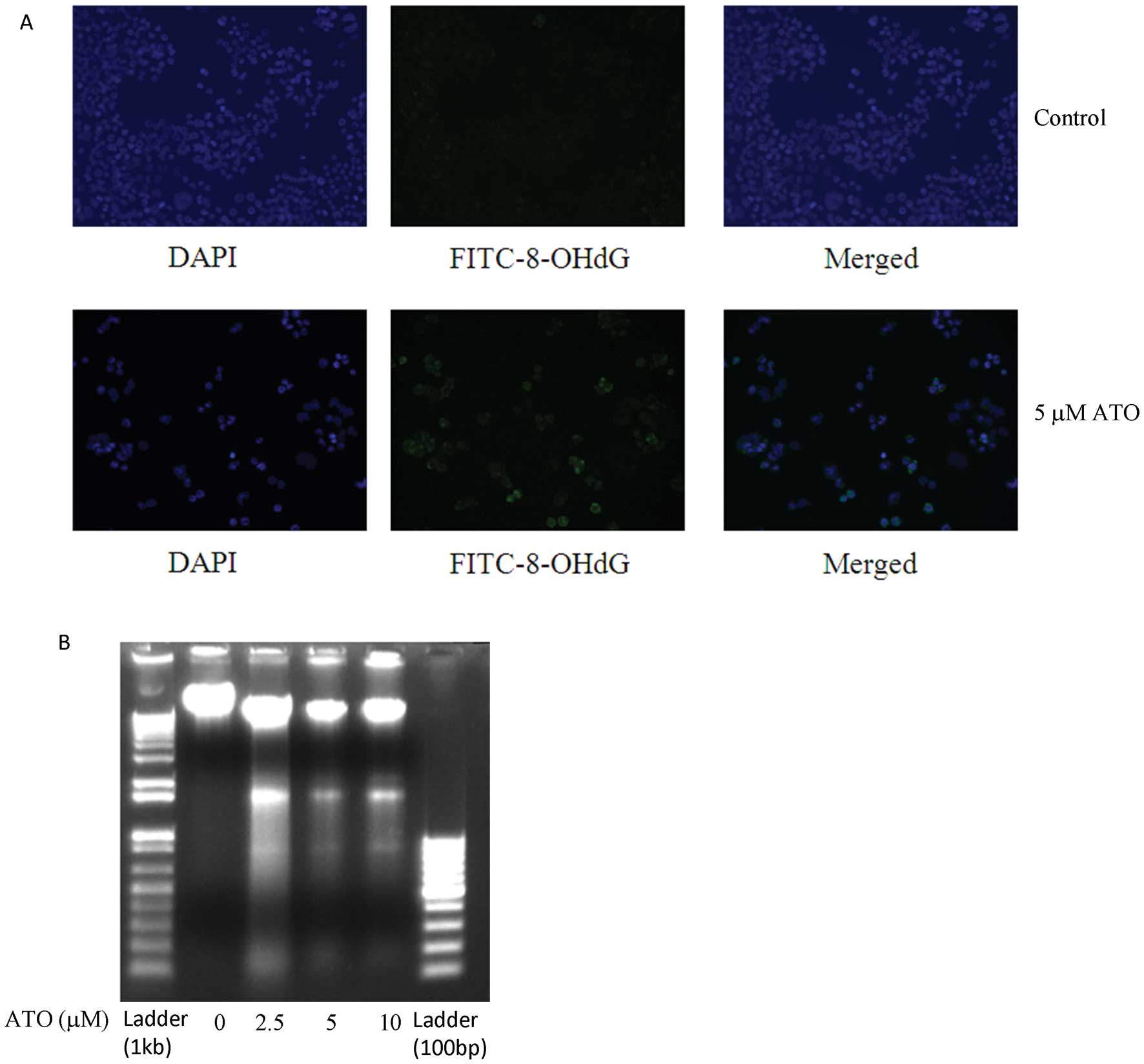
ATO induced oxidative DNA damage and
fragmentation
Treatment with ATO (5 μM) resulted in stronger FITC
fluorescence (green) than in the control group, in keeping with
increased 8-OHdG content in H841 cells upon exposure to ATO
(Fig. 3A). This is associated with
appearance of lower molecular weight DNA fragments with ATO
treatment (Fig. 3B).
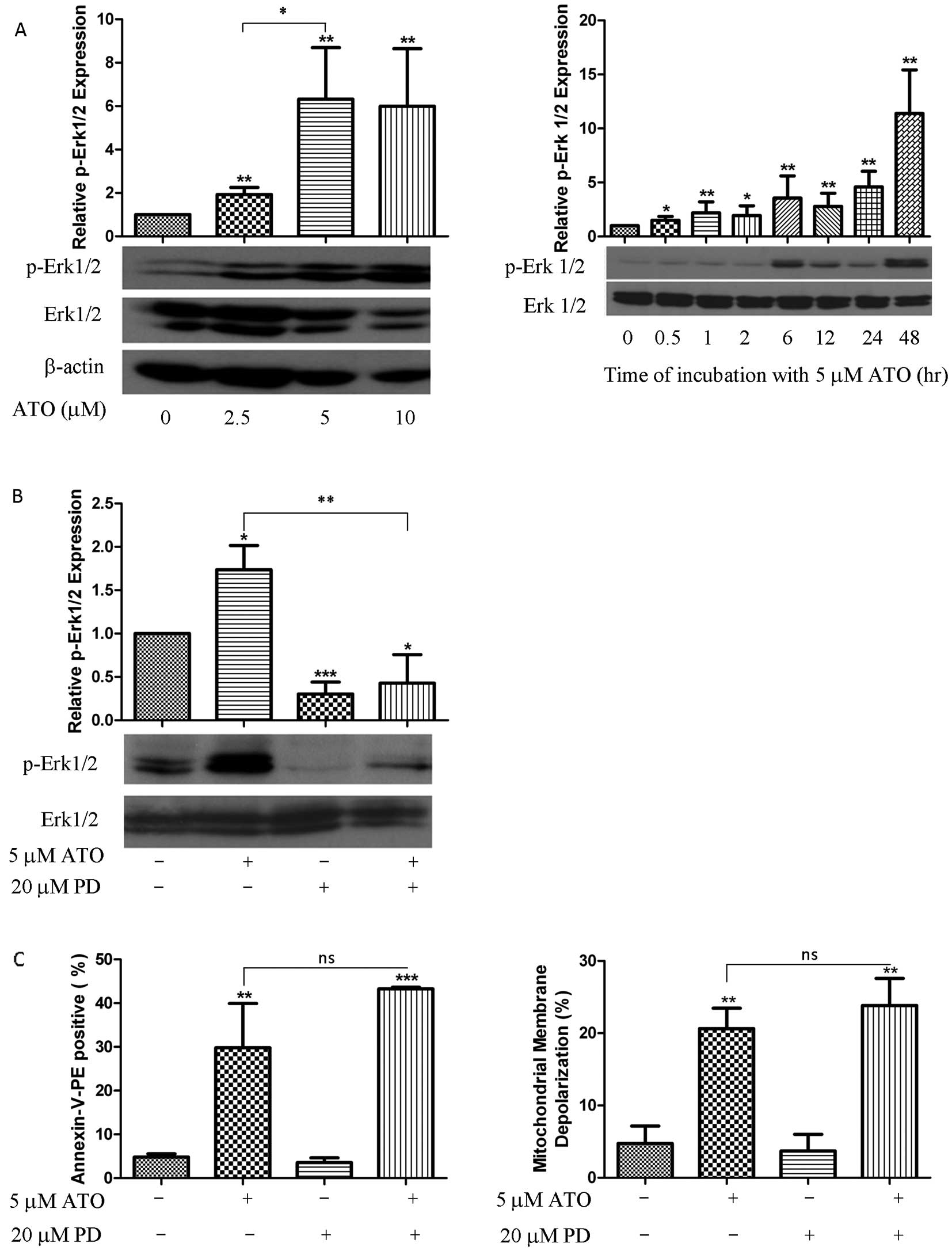
Changes in MAPKs expression during ATO
treatment
Elevation of p-Erk1/2 was detected after 30 min of
ATO treatment and it was sustained for ≥48 h (Fig. 4A) while p-P38 and p-JNK were
unaltered (data not shown). Nonetheless, application of specific
p-Erk1/2 inhibitor (PD98059) (20 μM) decreased p-Erk1/2 expression
induced by ATO (Fig. 4B) but did
not reverse ATO-induced cell death and MMD (Fig. 4C).
Effects of antioxidants on cell death
induced by ATO
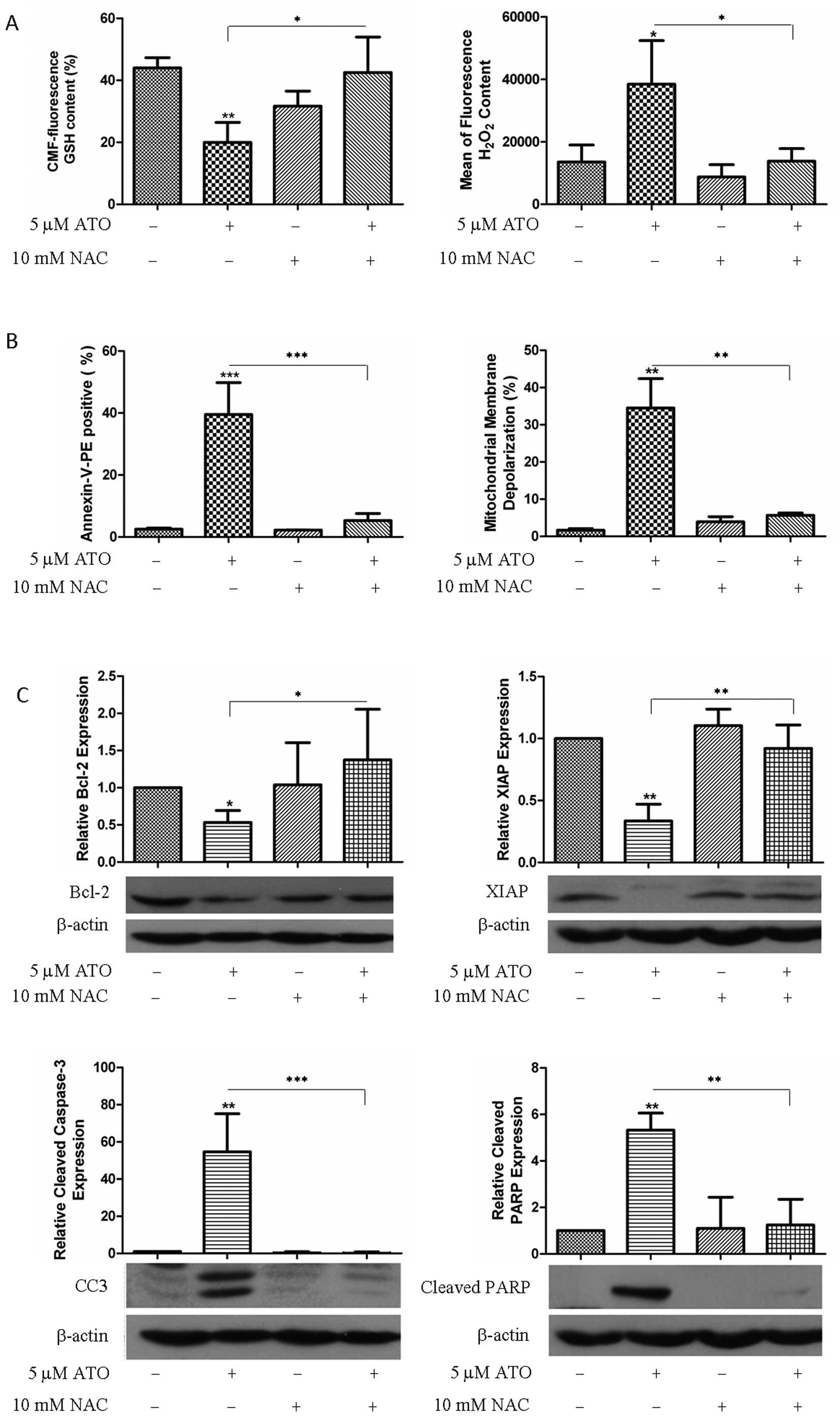
NAC, a cysteine donor in GSH synthesis, was used to
determine the role of GSH and ROS in ATO-induced cell death in H841
cells. NAC (10 mM) was shown to effectively restore GSH content and
reduce H2O2 after ATO exposure (Fig. 5A). This was associated with
reversal of apoptotic cell death (39.6±5.3 vs. 5.3±1.2% for ATO vs
ATO + NAC) and MMD (34.5±4.5 vs. 5.6±0.4% for ATO vs. ATO + NAC)
(Fig. 5B). In addition, NAC
reversed the alterations in apoptosis-related proteins induced by
ATO (Bcl-2, XIAP, cleaved caspase-3 and cleaved PARP) (Fig. 5C). On the other hand, a different
antioxidant BHA suppressed H2O2 induced by
ATO but failed to restore GSH content. Consequently, BHA only
partially reversed apoptosis and MMD induced by ATO (Fig. 5D).
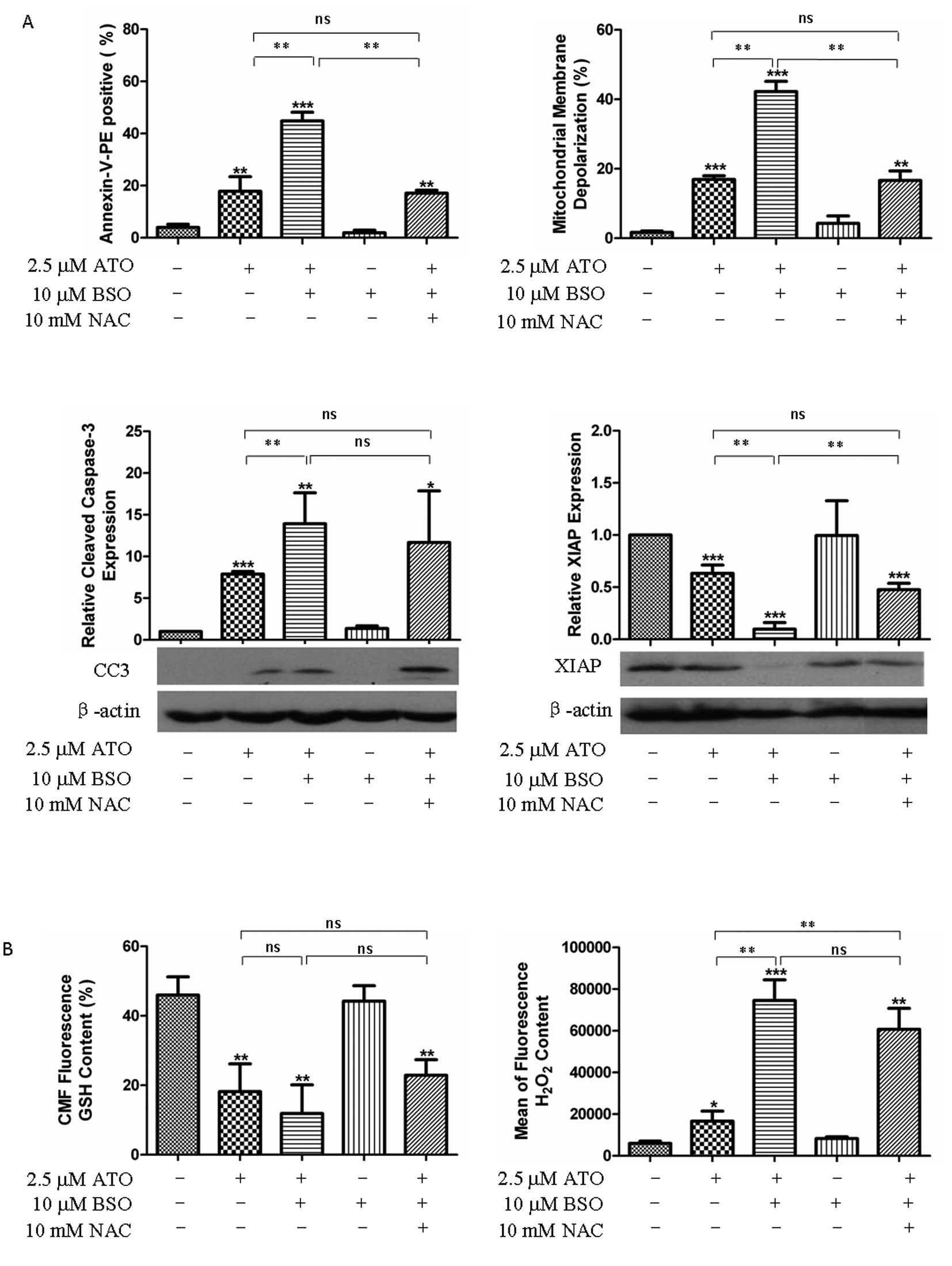
BSO potentiated cytotoxic effect of ATO
by further disturbing the redox status
BSO, an inhibitor of γ-glutamylcysteine synthetase,
was used as a GSH inhibitor. BSO potentiated the cytotoxic effect
of ATO in H841 cells, as shown by increased apoptotic death
(PE+ by flow cytometry in 17 vs. 44%: ATO vs. ATO +
BSO), enhanced MMD (17 vs. 42%: ATO vs. ATO + BSO), increased
cleavage of caspase-3 and downregulated XIAP (Fig. 6A). Nonetheless, administration of
10 mM NAC only partially rescued the H841 cells from apoptosis and
MMD (Fig. 6A). Interestingly, BSO
caused more accumulation of H2O2 than further
depletion of GSH when combined with ATO (Fig. 6B), resulting in increased oxidative
DNA damage (8-OHdG-FITC signal) in BSO/ATO group compared with
either ATO or BSO alone (Fig.
6C).
Trx1 is one of the important components of Trx redox
system responsible for maintaining redox equilibrium in mammalian
cells. ATO reduced total Trx1 protein expression in H841 cells,
which could be further downregulated by BSO but was reversed by NAC
(Fig. 6D). Based on Trx redox
western blot analysis, both oxidized and reduced forms of Trx1 were
downregulated by ATO, nevertheless, redox potential (Eh) remained
the same in different treatment groups (Fig. 6E).
Discussion
In the past decade, ATO has been used as a highly
effective anticancer therapy in leukemia (particularly APL) with or
without all-trans retinoic acid and achieving high rate of complete
remission (2,3). Nonetheless, it has been increasingly
reported to exert cytotoxic effect in other forms of haematological
malignancies and solid cancers such as breast (37), ovarian (38), cervical (39), and lung (20) cancers. The mechanisms and pathways
involved are highly diverse and cell type-specific, though
knowledge is scarce in SCLC.
In this study, a panel of five SCLC cell lines was
tested for the effect of ATO on cellular proliferation, with
IC50 values in clinical achievable concentrations
(3).
Caspase-dependent apoptosis is classified into
intrinsic (mitochondria-mediated) and extrinsic (death
receptor-mediated) pathways (40).
Both mechanisms converge to activate the apoptotic executioner
caspase-3 and -7, which consequently cleave polyADP-ribose
polymerase (PARP) that impairs DNA repair. Classically, cleaved
caspase-3 and cleaved PARP are widely adopted apoptotic markers. In
this study, both markers were elevated after ATO treatment in H841
cells, indicating cell death via the caspase-dependent pathway
(41).
AIF is one of the main mediators in cell death,
which is liberated from mitochondria and translocated to nucleus
resulting in DNA damage. It has been suggested that nuclear
translocation of AIF accompanied by activation of receptor
interacting protein 1 (RIP1) are markers of necroptosis (42). Nonetheless, the binding of AIF with
DNA provokes caspase-independent chromatin condensation and DNA
fragmentation (43–46). In our H841 cell line model, apart
from triggering MMD accompanied by AIF release and nuclear
translocation, ATO treatment also increased RIP1 expression (data
not shown). Taken together, cell death initiated by ATO in H841
cells could be mediated through both caspase-dependent apoptosis
and AIF-induced apoptosis or necroptosis.
On the other hand, MMD is associated with
mitochondrial release of another apoptosis mediator SMAC, in which
cytosolic SMAC can inhibit apoptosis inhibitor protein (XIAP)
leading to subsequent release of caspases that initiate apoptosis
(40). Our finding of SMAC release
and XIAP downregulation during treatment with ATO in H841 cells
suggests the possibility of caspase-3 activation via SMAC
release/XIAP inhibition rather than cytochrome c
release/caspase-9 activation. This is in keeping with previously
reported mechanism of caspase-dependent apoptosis via
SMAC/XIAP/caspase activation in other cancer cell line models with
ATO treatment (47,48).
GSH depletion and H2O2
accumulation with ATO treatment in H841 cells detected in this
study are in keeping with previous reports (11,49–52).
Nonetheless, the type and amount of ROS needed to initiate cell
death by ATO are still controversial, which can vary with different
cell lines and dose of ATO (53).
Indeed, the role of ROS in causing cell death induced by ATO was
uncertain in other studies (54,55).
Based on our findings in H841 SCLC model, oxidative stress with ATO
treatment was due to both H2O2 production and
GSH depletion, in which NAC (a GSH precursor) (56) could reverse these alterations
resulting in restoration of mitochondrial membrane potential. In
leukemia cell line U973, intracellular GSH depletion could trigger
Bax translocation to mitochondria and provoke membrane
permeabilization resulting in apoptosis (57). In H841 cell line, we demonstrated
downregulation of Bcl-2 upon ATO treatment, abrogating the
protection on mitochondria. The mechanism of ATO-induced Bcl-2
downregulation has not been clearly elucidated. Nonetheless Bcl-2
has manifested an antioxidant-like effect in response to either
oxidative stress or GSH depletion (58,59).
Reversal of ATO-induced Bcl-2 downregulation with NAC in our
experiments has suggested the mechanistic role of oxidative stress
and GSH depletion.
BHA is an effective scavenger of free radicals
widely used in food industry. It has been shown to be effective in
scavenging H2O2 to rescue cells from the DNA
damage (60–63). In our study, adding BHA with ATO in
H841 cells was capable of reducing H2O2
without effective restoration of GSH content compared with ATO
alone, resulting in partial reversal of cell inhibition and
apoptosis induced by ATO. It is possible that depletion of GSH with
ATO treatment is not only due to consumption by
H2O2 mediated via glutathione peroxidase
(GPx) (64) but also direct
consumption by ATO (65). In
addition, oxidative DNA damage was more pronounced in ATO treatment
with BSO compared with ATO alone, associated with significant GSH
depletion due to increased H2O2 as previously
reported (66,67). Taken together, GSH depletion plays
a more important role in ATO-induced cell death in SCLC, similar to
previous findings in NSCLC (66).
Thioredoxin system was reported to be an important
target of ATO in some cancers (26,68).
In H841 cell line model, ATO downregulated both reduced and
oxidative form of Trx1 without affecting redox status. This is
discrepant from previous report, which indicated that ATO could
inhibit thioredoxin reductase and consequently decrease the reduced
form of Trx1 (68). Although the
activity or expression of thioredoxin reductase has not been
measured, our findings suggest that ATO targets the total Trx1
rather than just the reduced form in H841 cells.
MAPKs are potential ROS response factors,
nonetheless, p-JNK and p-p38 were not involved in ATO-induced cell
death in our H841 cell line model (data not shown), which is
different from other cancer models (69–71).
Although upregulated p-Erk1/2 was detected after ATO treatment in
H841 cells, specific Erk inhibitor PD98059 failed to revert the
effects induced by ATO on MMD and cell death. It suggests that
p-Erk1/2 is not an effective mediator leading to cell death induced
by ATO in SCLC, which may serve in other biological actions
(72,73).
In conclusion, the mechanisms of cell death induced
by ATO in SCLC are mainly dependent on altered redox homeostasis
that comprises GSH depletion, H2O2
accumulation and mitochondrial depolarization. Both
caspase-dependent pathway mediated via Bcl-2/SMAC/XIAP/Caspase-3
and caspase-independent pathway mediated via AIF would take part in
causing apoptosis and necroptosis in SCLC.
Abbreviations:
|
ATO
|
arsenic trioxide
|
|
BHA
|
butylated hydroxyanisole
|
|
GSH
|
glutathione
|
|
NAC
|
N-acetyl-L-cysteine
|
|
SMAC/DIABLO
|
second mitochondria-derived activator
of caspases
|
|
Trx1
|
thioredoxin 1
|
|
XIAP
|
X-linked inhibitor of apoptosis
protein
|
References
|
1
|
Lally BE, Urbanic JJ, Blackstock AW,
Miller AA and Perry MC: Small cell lung cancer: have we made any
progress over the last 25 years? Oncologist. 12:1096–1104. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shen ZX, Chen GQ, Ni JH, et al: Use of
arsenic trioxide (As2O3) in the treatment of
acute promyelocytic leukemia (APL): II. Clinical efficacy and
pharmacokinetics in relapsed patients. Blood. 89:3354–3360.
1997.PubMed/NCBI
|
|
3
|
Chen GQ, Shi XG, Tang W, et al: Use of
arsenic trioxide (As2O3) in the treatment of
acute promyelocytic leukemia (APL): I. As2O3
exerts dose-dependent dual effects on APL cells. Blood.
89:3345–3353. 1997.PubMed/NCBI
|
|
4
|
Yoda A, Toyoshima K, Watanabe Y, et al:
Arsenic trioxide augments Chk2/p53-mediated apoptosis by inhibiting
oncogenic Wip1 phosphatase. J Biol Chem. 283:18969–18979. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li Y, Qu X, Qu J, et al: Arsenic trioxide
induces apoptosis and G2/M phase arrest by inducing Cbl to inhibit
PI3K/Akt signaling and thereby regulate p53 activation. Cancer
Lett. 284:208–215. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Scholz C, Richter A, Lehmann M,
Schulze-Osthoff K, Dorken B and Daniel PT: Arsenic trioxide induces
regulated, death receptor-independent cell death through a
Bcl-2-controlled pathway. Oncogene. 24:7031–7042. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen GQ, Zhu J, Shi XG, et al: In vitro
studies on cellular and molecular mechanisms of arsenic trioxide
(As2O3) in the treatment of acute
promyelocytic leukemia: As2O3 induces NB4
cell apoptosis with downregulation of Bcl-2 expression and
modulation of PML-RAR alpha/PML proteins. Blood. 88:1052–1061.
1996.PubMed/NCBI
|
|
8
|
Kang YH, Yi MJ, Kim MJ, et al:
Caspase-independent cell death by arsenic trioxide in human
cervical cancer cells: reactive oxygen species-mediated
poly(ADP-ribose) polymerase-1 activation signals apoptosis-inducing
factor release from mitochondria. Cancer Res. 64:8960–8967. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shen ZY, Shen J, Cai WJ, Hong C and Zheng
MH: The alteration of mitochondria is an early event of arsenic
trioxide induced apoptosis in esophageal carcinoma cells. Int J Mol
Med. 5:155–158. 2000.PubMed/NCBI
|
|
10
|
Dalton WS: Targeting the mitochondria: an
exciting new approach to myeloma therapy. Commentary re: NJ Bahlis
et al: Feasibility and correlates of arsenic trioxide combined with
ascorbic acid-mediated depletion of intracellular glutathione for
the treatment of relapsed/refractory multiple myeloma. Clin Cancer
Res. 8:3658–3668. 2002.
Clin Cancer Res. 8:3643–3645. 2002.
|
|
11
|
Bhalla S, Gordon LI, David K, et al:
Glutathione depletion enhances arsenic trioxide-induced apoptosis
in lymphoma cells through mitochondrial-independent mechanisms. Br
J Haematol. 150:365–369. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Davison K, Cote S, Mader S and Miller WH:
Glutathione depletion overcomes resistance to arsenic trioxide in
arsenic-resistant cell lines. Leukemia. 17:931–940. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Walker AM, Stevens JJ, Ndebele K and
Tchounwou PB: Arsenic trioxide modulates DNA synthesis and
apoptosis in lung carcinoma cells. Int J Environ Res Public Health.
7:1996–2007. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qu GP, Xiu QY, Li B, Liu YA and Zhang LZ:
Arsenic trioxide inhibits the growth of human lung cancer cell
lines via cell cycle arrest and induction of apoptosis at both
normoxia and hypoxia. Toxicol Ind Health. 25:505–515. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Han YH, Kim SZ, Kim SH and Park WH:
Induction of apoptosis in arsenic trioxide-treated lung cancer A549
cells by buthionine sulfoximine. Mol Cells. 26:158–164.
2008.PubMed/NCBI
|
|
16
|
Lam SK, Li YY, Zheng CY, Leung LL and Ho
JC: E2F1 downregulation by arsenic trioxide in lung adenocarcinoma.
Int J Oncol. 45:2033–2043. 2014.PubMed/NCBI
|
|
17
|
Lam SK, Mak JC, Zheng CY, Li YY, Kwong YL
and Ho JC: Downregulation of thymidylate synthase with arsenic
trioxide in lung adenocarcinoma. Int J Oncol. 44:2093–2102.
2014.PubMed/NCBI
|
|
18
|
Lam SK, Li YY, Zheng CY and Ho JC:
Downregulation of thymidylate synthase and E2F1 by arsenic trioxide
in mesothelioma. Int J Oncol. 46:113–122. 2015.
|
|
19
|
Li H, Zhu X, Zhang Y, Xiang J and Chen H:
Arsenic trioxide exerts synergistic effects with cisplatin on
non-small cell lung cancer cells via apoptosis induction. J Exp
Clin Cancer Res. 28:1102009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chien CW, Yao JH, Chang SY, Lee PC and Lee
TC: Enhanced suppression of tumor growth by concomitant treatment
of human lung cancer cells with suberoylanilide hydroxamic acid and
arsenic trioxide. Toxicol Appl Pharmacol. 257:59–66. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Park JH, Kim EJ, Jang HY, et al:
Combination treatment with arsenic trioxide and sulindac enhances
apoptotic cell death in lung cancer cells via activation of
oxidative stress and mitogen-activated protein kinases. Oncol Rep.
20:379–384. 2008.PubMed/NCBI
|
|
22
|
Pettersson HM, Pietras A, Munksgaard
Persson M, et al: Arsenic trioxide is highly cytotoxic to small
cell lung carcinoma cells. Mol Cancer Ther. 8:160–170. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Miller WH Jr, Schipper HM, Lee JS, Singer
J and Waxman S: Mechanisms of action of arsenic trioxide. Cancer
Res. 62:3893–3903. 2002.PubMed/NCBI
|
|
24
|
Carney DA: Arsenic trioxide mechanisms of
action - looking beyond acute promyelocytic leukemia. Leuk
Lymphoma. 49:1846–1851. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chouchane S and Snow ET: In vitro effect
of arsenical compounds on glutathione-related enzymes. Chem Res
Toxicol. 14:517–522. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lu J, Chew EH and Holmgren A: Targeting
thioredoxin reductase is a basis for cancer therapy by arsenic
trioxide. Proc Natl Acad Sci USA. 104:12288–12293. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim HJ, Chae HZ, Kim YJ, et al:
Preferential elevation of Prx I and Trx expression in lung cancer
cells following hypoxia and in human lung cancer tissues. Cell Biol
Toxicol. 19:285–298. 2003. View Article : Google Scholar
|
|
28
|
Hedley D, Pintilie M, Woo J, et al:
Up-regulation of the redox mediators thioredoxin and
apurinic/apyrimidinic excision (APE)/Ref-1 in hypoxic microregions
of invasive cervical carcinomas, mapped using multispectral,
wide-field fluorescence image analysis. Am J Pathol. 164:557–565.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Choi JH, Kim TN, Kim S, et al:
Overexpression of mitochondrial thioredoxin reductase and
peroxiredoxin III in hepatocellular carcinomas. Anticancer Res.
22:3331–3335. 2002.
|
|
30
|
Powis G, Mustacich D and Coon A: The role
of the redox protein thioredoxin in cell growth and cancer. Free
Radic Biol Med. 29:312–322. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tonissen KF and Di Trapani G: Thioredoxin
system inhibitors as mediators of apoptosis for cancer therapy. Mol
Nutr Food Res. 53:87–103. 2009. View Article : Google Scholar
|
|
32
|
Zheng CY, Lam SK, Li YY, Fong BM, Mak JC
and Ho JC: Combination of arsenic trioxide and chemotherapy in
small cell lung cancer. Lung Cancer. 82:222–230. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tauskela JS, Hewitt K, Kang LP, et al:
Evaluation of glutathione-sensitive fluorescent dyes in cortical
culture. Glia. 30:329–341. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sordet O, Liao Z, Liu H, et al:
Topoisomerase I-DNA complexes contribute to arsenic
trioxide-induced apoptosis. J Biol Chem. 279:33968–33975. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Watson WH, Pohl J, Montfort WR, et al:
Redox potential of human thioredoxin 1 and identification of a
second dithiol/disulfide motif. J Biol Chem. 278:33408–33415. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Go YM and Jones DP: Thioredoxin redox
western analysis. Curr Protoc Toxicol. Chapter 17(Unit 17.12)
View Article : Google Scholar : 2009.PubMed/NCBI
|
|
37
|
Sun RC, Board PG and Blackburn AC:
Targeting metabolism with arsenic trioxide and dichloroacetate in
breast cancer cells. Mol Cancer. 10:1422011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Askar N, Cirpan T, Toprak E, et al:
Arsenic trioxide exposure to ovarian carcinoma cells leads to
decreased level of topoisomerase II and cytotoxicity. Int J Gynecol
Cancer. 16:1552–1556. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wen X, Li D, Zhang Y, Liu S, Ghali L and
Iles RK: Arsenic trioxide induces cervical cancer apoptosis, but
specifically targets human papillomavirus-infected cell
populations. Anticancer Drugs. 23:280–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lam HK, Li K, Chik KW, et al: Arsenic
trioxide mediates intrinsic and extrinsic pathways of apoptosis and
cell cycle arrest in acute megakaryocytic leukemia. Int J Oncol.
27:537–545. 2005.PubMed/NCBI
|
|
42
|
Delavallee L, Cabon L, Galan-Malo P,
Lorenzo HK and Susin SA: AIF-mediated caspase-independent
necroptosis: a new chance for targeted therapeutics. IUBMB Life.
63:221–232. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cande C, Cecconi F, Dessen P and Kroemer
G: Apoptosis-inducing factor (AIF): key to the conserved
caspase-independent pathways of cell death? J Cell Sci.
115:4727–4734. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Susin SA, Lorenzo HK, Zamzami N, et al:
Molecular characterization of mitochondrial apoptosis-inducing
factor. Nature. 397:441–446. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lorenzo HK and Susin SA: Therapeutic
potential of AIF-mediated caspase-independent programmed cell
death. Drug Resist Updat. 10:235–255. 2007. View Article : Google Scholar
|
|
46
|
Ye H, Cande C, Stephanou NC, et al: DNA
binding is required for the apoptogenic action of apoptosis
inducing factor. Nat Struct Biol. 9:680–684. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Choi YJ, Park JW, Suh SI, et al: Arsenic
trioxide-induced apoptosis in U937 cells involve generation of
reactive oxygen species and inhibition of Akt. Int J Oncol.
21:603–610. 2002.PubMed/NCBI
|
|
48
|
Calvino E, Estan MC, Simon GP, et al:
Increased apoptotic efficacy of lonidamine plus arsenic trioxide
combination in human leukemia cells. Reactive oxygen species
generation and defensive protein kinase (MEK/ERK, Akt/mTOR)
modulation. Biochem Pharmacol. 82:1619–1629. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xiao G, Tang X, Yao C and Wang C:
Potentiation of arsenic trioxide-induced apoptosis by
8-bromo-7-methoxychrysin in human leukemia cells involves depletion
of intracellular reduced glutathione. Acta Biochim Biophys Sin.
43:712–721. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Konig H, Hartel N, Schultheis B, et al:
Enhanced Bcr-Abl-specific antileukemic activity of arsenic trioxide
(Trisenox) through glutathione-depletion in imatinib-resistant
cells. Haematologica. 92:838–841. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ramos AM, Fernandez C, Amran D, Sancho P,
de Blas E and Aller P: Pharmacologic inhibitors of PI3K/Akt
potentiate the apoptotic action of the antileukemic drug arsenic
trioxide via glutathione depletion and increased peroxide
accumulation in myeloid leukemia cells. Blood. 105:4013–4020. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hu XM, Hirano T and Oka K: Arsenic
trioxide induces apoptosis in cells of MOLT-4 and its
daunorubicin-resistant cell line via depletion of intracellular
glutathione, disruption of mitochondrial membrane potential and
activation of caspase-3. Cancer Chemother Pharmacol. 52:47–58.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: Effects of arsenic trioxide on cell death, reactive
oxygen species and glutathione levels in different cell types. Int
J Mol Med. 25:121–128. 2010.
|
|
54
|
Yi J, Yang J, He R, et al: Emodin enhances
arsenic trioxide-induced apoptosis via generation of reactive
oxygen species and inhibition of survival signaling. Cancer Res.
64:108–116. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Morales AA, Gutman D, Cejas PJ, Lee KP and
Boise LH: Reactive oxygen species are not required for an arsenic
trioxide-induced antioxidant response or apoptosis. J Biol Chem.
284:12886–12895. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sun SY: N-acetylcysteine, reactive oxygen
species and beyond. Cancer Biol Ther. 9:109–110. 2010. View Article : Google Scholar :
|
|
57
|
Guha P, Dey A, Sen R, Chatterjee M,
Chattopadhyay S and Bandyopadhyay SK: Intracellular GSH depletion
triggered mitochondrial Bax translocation to accomplish
resveratrol-induced apoptosis in the U937 cell line. J Pharmacol
Exp Ther. 336:206–214. 2011. View Article : Google Scholar
|
|
58
|
Voehringer DW and Meyn RE: Redox aspects
of Bcl-2 function. Antioxid Redox Signal. 2:537–550. 2000.
View Article : Google Scholar
|
|
59
|
Hochman A, Sternin H, Gorodin S, et al:
Enhanced oxidative stress and altered antioxidants in brains of
Bcl-2-deficient mice. J Neurochem. 71:741–748. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Rajesh K, Vedamurthy J, Prakash D,
Thammanna Gowda SS, Satish BP and Dinesha R: Antioxidant activity
of spathodea campanulata in prevention of TBOOH and
H2O2 induced DNA damage. Int J Curr
Pharmaceut Res. 3:32011.
|
|
61
|
Keser S, Celik S, Turkoglu S, Yilmaz Ö and
Turkoglu I: Hydrogen peroxide radical scavenging and total
antioxidant activity of Hawthorn. Chem J. 2:42012.
|
|
62
|
Cherouny PH, Ghodgaonkar RB, Gurtner GH
and Dubin NH: The effect of the antioxidant, butylated hydroxy
anisole, on peroxide-induced and spontaneous activity of the uterus
from the pregnant rat. Biol Reprod. 41:98–103. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Gulcin I, Alici HA and Cesur M:
Determination of in vitro antioxidant and radical scavenging
activities of propofol. Chem Pharm Bull. 53:281–285. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lushchak VI: Glutathione homeostasis and
functions: potential targets for medical interventions. J Amino
Acids. 7368372012.PubMed/NCBI
|
|
65
|
Patrick L: Toxic metals and antioxidants:
Part II. The role of antioxidants in arsenic and cadmium toxicity.
Altern Med Rev. 8:106–128. 2003.PubMed/NCBI
|
|
66
|
Han YH, Kim SH, Kim SZ and Park WH:
Apoptosis in arsenic trioxide-treated Calu-6 lung cells is
correlated with the depletion of GSH levels rather than the changes
of ROS levels. J Cell Biochem. 104:862–878. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Reliene R and Schiestl RH: Glutathione
depletion by buthionine sulfoximine induces DNA deletions in mice.
Carcinogenesis. 27:240–244. 2006. View Article : Google Scholar
|
|
68
|
Tian C, Gao P, Zheng Y, et al: Redox
status of thioredoxin-1 (TRX1) determines the sensitivity of human
liver carcinoma cells (HepG2) to arsenic trioxide-induced cell
death. Cell Res. 18:458–471. 2008. View Article : Google Scholar
|
|
69
|
Kang YH and Lee SJ: The role of p38 MAPK
and JNK in arsenic trioxide-induced mitochondrial cell death in
human cervical cancer cells. J Cell Physiol. 217:23–33. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: The effect of MAPK inhibitors on arsenic
trioxide-treated Calu-6 lung cells in relation to cell death, ROS
and GSH levels. Anticancer Res. 29:3837–3844. 2009.PubMed/NCBI
|
|
71
|
Zhang S, Guo W, Ren TT, Lu XC, Tang GQ and
Zhao FL: Arsenic trioxide inhibits Ewing’s sarcoma cell
invasiveness by targeting p38(MAPK) and c-Jun N-terminal kinase.
Anticancer Drugs. 23:108–118. 2012. View Article : Google Scholar
|
|
72
|
Chiu HW, Ho SY, Guo HR and Wang YJ:
Combination treatment with arsenic trioxide and irradiation
enhances autophagic effects in U118-MG cells through increased
mitotic arrest and regulation of PI3K/Akt and ERK1/2 signaling
pathways. Autophagy. 5:472–483. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Ellington AA, Berhow MA and Singletary KW:
Inhibition of Akt signaling and enhanced ERK1/2 activity are
involved in induction of macroautophagy by triterpenoid B-group
soyasaponins in colon cancer cells. Carcinogenesis. 27:298–306.
2006. View Article : Google Scholar
|