Introduction
Non-small cell lung cancer (NSCLC) is one of the
common causes of death in the world (1). Although cisplatin treatment after
surgery is known as a standard and effective chemotherapy,
metastatic spread in NSCLC patients often occurs (2–4). In
addition, it is still controversial whether an adjuvant
chemotherapy with cisplatin gives a significant survival advantage
in stage I NSCLC patients compared with surgery alone (5–7).
This may lead to the possibility that metastatic NSCLCs already
acquire resistance to chemotherapy during tumor progression.
Cancer metastasis is one of the major causes of the
high mortality rate in cancer patients and it consists of multiple
biological steps, such as dissemination from primary tumor,
intravasation, attachment to vessel of target tissue,
extravasation, angiogenesis, and subsequent growth at the
metastasis site (8). Some of these
biological steps have been shown to be related to
epithelial-to-mesenchymal transition (EMT) (9–12).
Although EMT also has been shown to limit the sensitivities of
cancer cells to chemotherapeutic drugs (13,14),
the potential mechanism by which cancer cells acquire resistance to
anticancer drugs associated with the EMT process is not well
defined.
In this study, we demonstrated that EMT in human
NSCLC cell line A549 induced by TGF-β treatment limits the
sensitivities to various anticancer drugs, and further identified
MCL-1 as a critical molecule of such EMT-associated
chemo-resistance of A549 cells. Importantly, we showed that
targeting MCL-1 by siRNA delivery or the pan-BCL2 inhibitor
treatment could overcome the EMT-associated chemo-resistance in
A549 cells.
Materials and methods
Reagents and plasmids
The reagents used were recombinant human TGF-β from
Peprotech (London, UK), obatoclax from Selleck Chemicals (Houston,
TX, USA), ABT-737 from AdooQ BioScience (Irvine, CA, USA),
cisplatin, vinorelbine, gemcitabine and paclitaxel from Wako Pure
Chemical Industries (Osaka, Japan). siRNAs against MCL-1
(L-004501-00, J-004501-16, and J-004501-17), Bcl2A1 (L-003306-00),
and control (D-00181-02) siRNA were purchased from Thermo Fisher
Scientific (Rockford, IL, USA). The human MCL-1 cDNA was amplified
from normal human cDNA and subcloned into pcDNA3.1-HA (from David
E. Fisher, MGH, MA, USA).
Cell cultures
Human lung adenocarcinoma A549 cells were cultured
in RPMI-1640 medium (Life Technologies Corp., Carlsbad, CA, USA)
with 10% fetal bovine serum (FBS; ICN Biomedicals, Aurora, OH,
USA), 2 mM L-glutamine (Life Technologies Corp.), 100 U/ml
penicillin and 100 μg/ml streptomycin in 5% CO2 at 37°C.
A549 cells stably expressing MCL-1 or vector control were
established by transfecting pcDNA3.1-HA/MCL-1 or pcDNA3.1 under
G418 (1 mg/ml). For siRNA transfection, each 25 nM of siRNAs was
reverse-transfected using Lipofectamine™ RNAiMAX (Life Technologies
Corp.) following the manufacturer’s instructions, and the
transfected cells were used for each experiment.
Cell viability assay
Cell viability was quantified using the cell
proliferation reagent WST-1 (Dojindo, Japan) or CellTiter-Glo
(Promega, Madison, WI, USA). A549 cells, siRNA-reverse transfected
A549 cells, or stable MCL-1 expressing A549 cells were incubated
for 24 h. The antitumor drugs were then added after pre-treatment
with TGF-β for 48 h. After additional incubation for the indicated
time, WST-1 solution or CellTiter-Glo reagent was added. Absorbance
was measured at 450 nm using a Microplate reader for WST-1 assay
and luminescence was measured using a GloMax Multi-detection system
(Promega) for CellTiter-Glo assay. The cell viability was
determined as percent viability compared with the vehicle
control.
Western blot analysis
Whole cell lysates were prepared as described
previously (15). The primary
antibodies used were E-cadherin, N-cadherin, Snail, MCL-1,
BCL2A1/Bfl-1, BCL-xL, PARP and caspase-3 (Cell Signaling
Technology, Beverly, MA, USA), hemagglutinin (HA) (Roche,
Indianapolis, IN, USA) and β-actin (Santa Cruz Biotechmology, Santa
Cruz, CA, USA). All antibodies were used by ×2000 dilution.
Real-time RT-PCR
Expression of MCL-1 and BCL2A1 mRNA
was quantitatively determined by real-time PCR on an ABI PRISM 7300
Real Time PCR System (Life Technologies Corp.). Total RNAs were
prepared using the RNeasy Plus Mini kit (Qiagen, Hilden, Germany).
Expression level of the targeted mRNAs was normalized to
β-actin mRNA. The primers used were: 5′-TCG TAA GGA CAA AAC
GGG AC-3′ (sense) and 5′-CAT TCC TGA TGC CAC CTT CT-3′ (antisense)
for MCL-1 mRNA, 5′-CCC GGA TGT GGA TAC CTA TAA GGA GA-3′
(sense) and 5′-GTC ATC CAG CCA GAT TTA GGT TCA-3′ (antisense) for
BCL2A1 mRNA, and 5′-GCA CAG AGC CTC GCC TT-3′ (sense) and
5′-GTT GTC GAC GAC GAG CG-3′ (antisense) for β-actin mRNA.
Apoptosis assay
Apoptotic cell number was determined using the MUSE
Annexin V and Dead Cell kit (Merck KGaA, Darmstadt, Germany)
according to the manufacturer’s instructions. Briefly, the stable
cells were harvested after being treated with cisplatin and diluted
with PBS containing 1% bovine serum albumin (BSA) as a dilution
buffer to a concentration of 5×105 cells/ml. Cell
suspension (100 μl) was then added to 100 μl MUSE Annexin V and
Dead Cell reagent (2× dilution), incubated for 20 min at room
temperature, and analyzed using the MUSE Cell Analyzer. Total
Annexin V-positive cells were determined as apoptotic cells.
Results
Acquired chemo-resistance in A549 cells
associates with TGF-β-induced EMT
Although the relationship between
epithelial-to-mesenchymal transition (EMT) and chemo-resistance has
been implicated (14,16), the detailed molecular mechanism of
such EMT-accompanying chemo-resistance has not been determined yet.
Therefore, we first tested tthe various antitumor reagents, such as
cisplatin, paclitaxel, gemcitabine, and vinorelbine, in A549 lung
adenocarcinoma cell lines with or without inducing EMT by
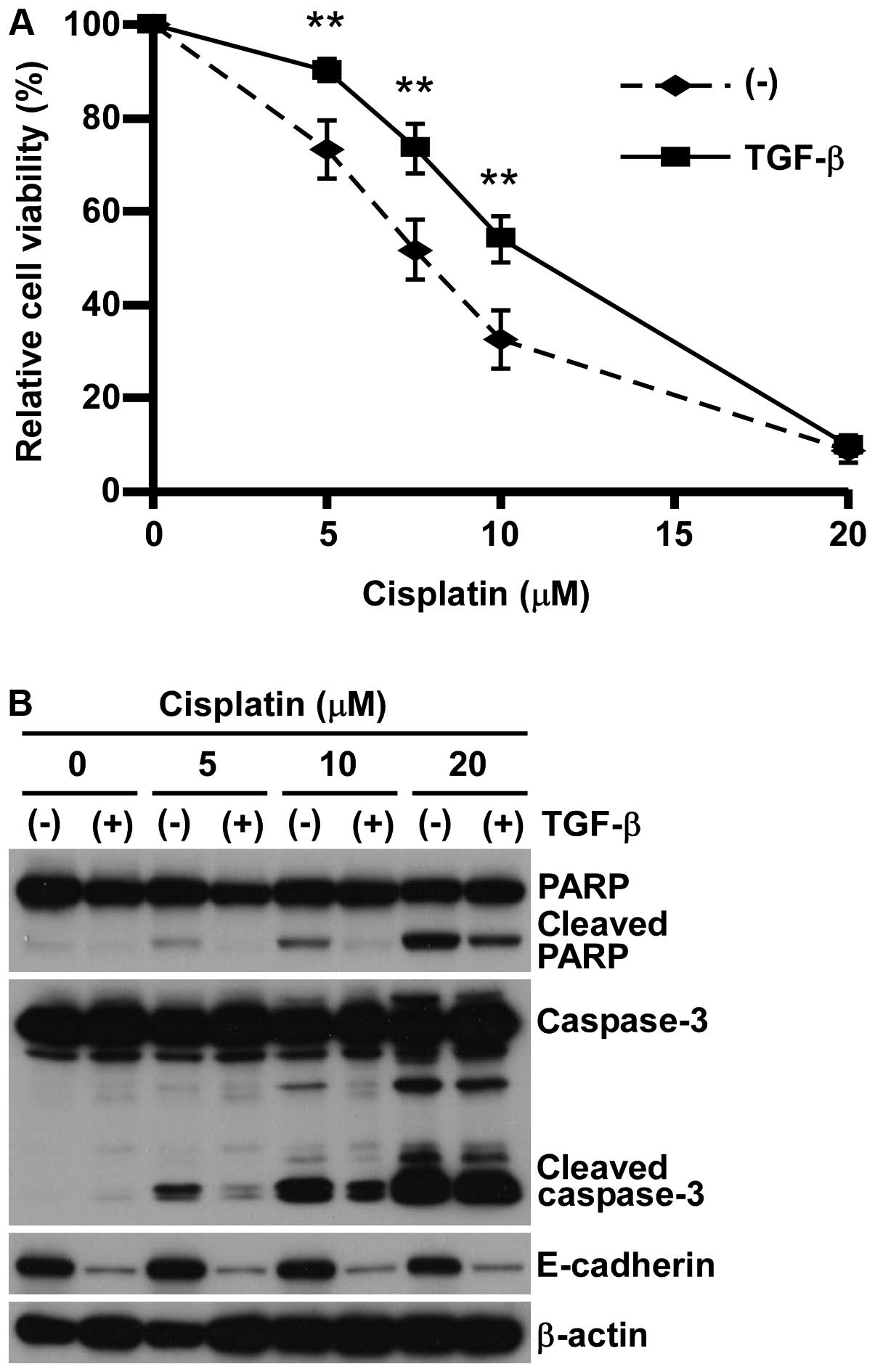
pre-treating with 5 ng/ml TGF-β (11,17).
Consistent with our previous studies (17,18),
A549 pre-treated with TGF-β showed EMT phenotype in both cell
morphology and expression of protein markers such as E-cadherin
reduction and N-cadherin induction (data not shown). In concert
with EMT-induction, A549 cells pre-treated with TGF-β showed
significant resistance against all anticancer reagents tested
(Fig. 1A and Table I), which was associated with the
reduction of apoptosis marker expression (Fig. 1B). These findings indicate that
A549 cells acquired a wide spectrum of chemo-resistance, possibly
through apoptosis inhibition associated with EMT induced by
TGF-β.
 | Table IIC50 of various anticancer
drugs in TGF-β-induced chemo-resistance. |
Table I
IC50 of various anticancer
drugs in TGF-β-induced chemo-resistance.
|
IC50a (mean ± SD) |
|---|
|
|
|---|
| A549 | A549/TGF-β |
|---|
| Cisplatin (μM) | 7.6±0.8 | 10.6±0.8b |
| Paclitaxel (nM) | 11.1±1.1 | 22.4±7.4b |
| Gemcitabine (μM) | 0.75±0.2 | >40b |
| Vinorelbine (nM) | 15.8±3.0 | 29.1±6.9b |
Critical role of MCL-1 in A549
chemo-resistance associated with TGF-β-induced EMT
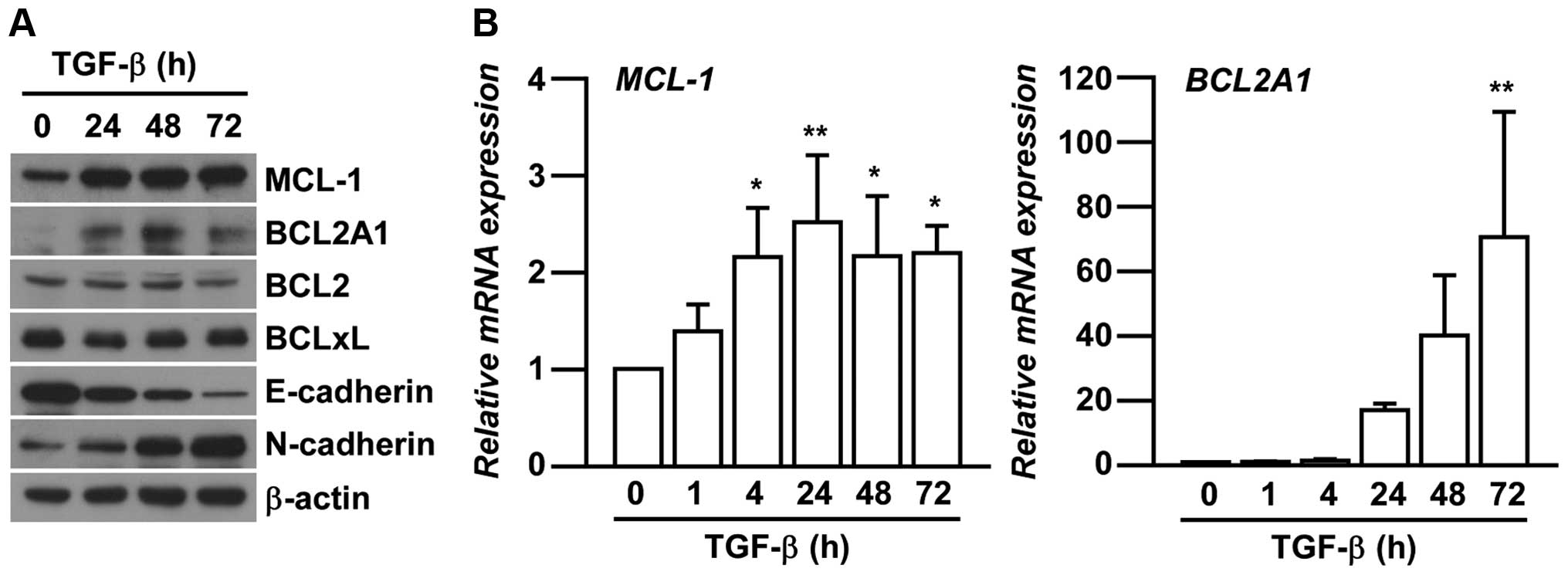
We next investigated the expression levels of BCL2
family members in EMT-induced A549 cells. The members are related
to chemo-resistance in various cancers by inhibiting apoptosis
(15,19,20).
Amongst many of BCL2 family members, the expression of MCL-1 and
BCL2A1 were specifically increased associated with TGF-β-induced
EMT in A549 cells in a time-dependent manner (Fig. 2A). We further confirmed the
increased mRNA expression of MCL-1 and BCL2A1 after
TGF-β treatments (Fig. 2B)
consistent with their protein expression.
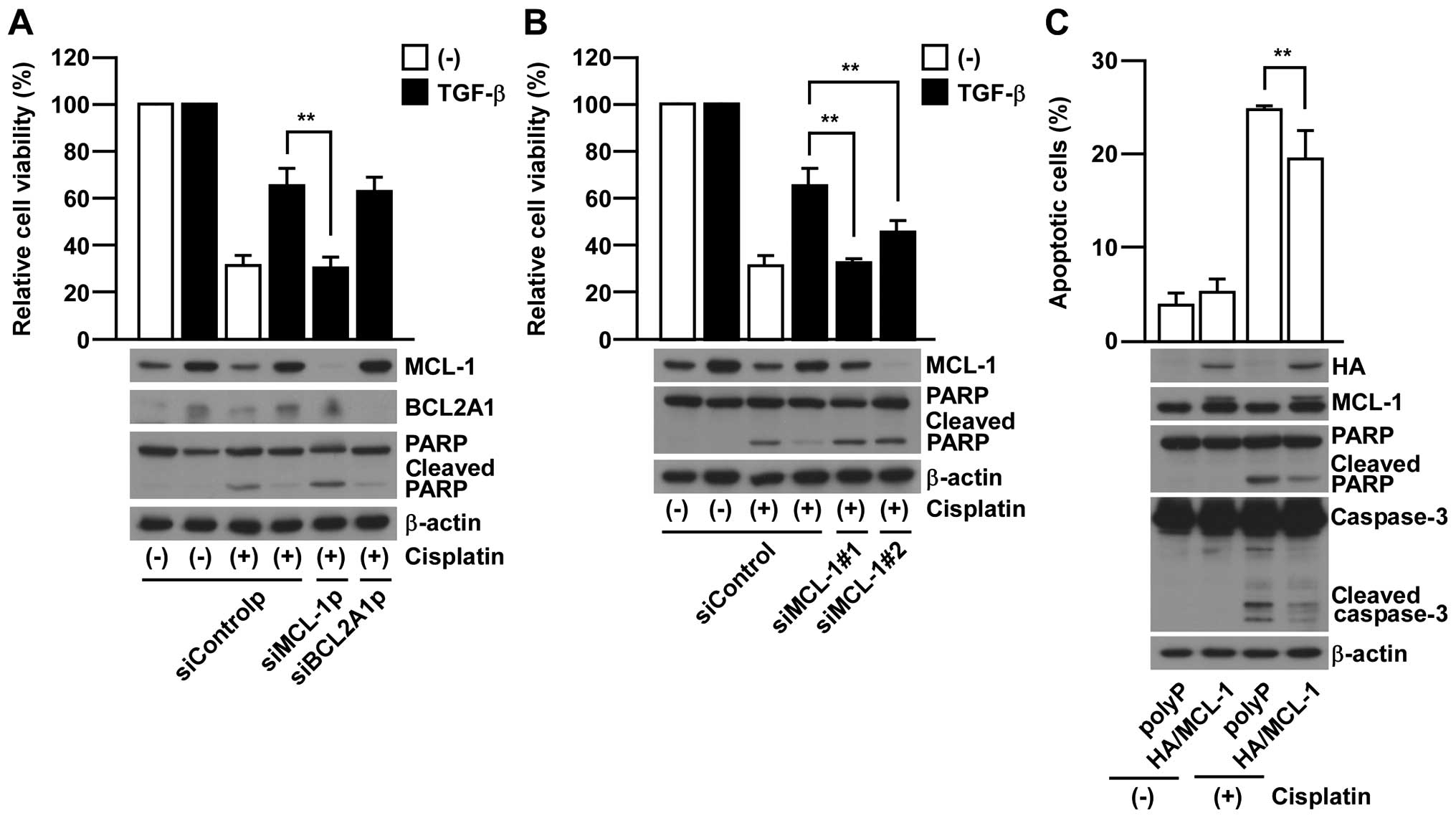
In order to further investigate whether either MCL-1
or BCL2A1 is required for EMT-associated chemo-resistance, we
employed gene knock-down of MCL-1 or BCL2A1 by using siRNA pools in
which four different siRNAs are contained. MCL-1 knock-down rescued
the sensitivity to cisplatin treatment in EMT-induced A549 cells
contrary to BCL2A1 knock-down which did not show any significant
effect (Fig. 3A). Similar results
were also confirmed in additional experiments using two siRNAs
against MCL-1 with different target sequences (Fig. 3B). Furthermore,
MCL-1-overexpression was able to significantly suppress the
cisplatin-induced apoptosis in non-EMT-induced parental A549 cells
(Fig. 3C). Collectively, these
results strongly support the critical contribution of MCL-1 in
acquiring EMT-associated chemo-resistance in A549 cells.
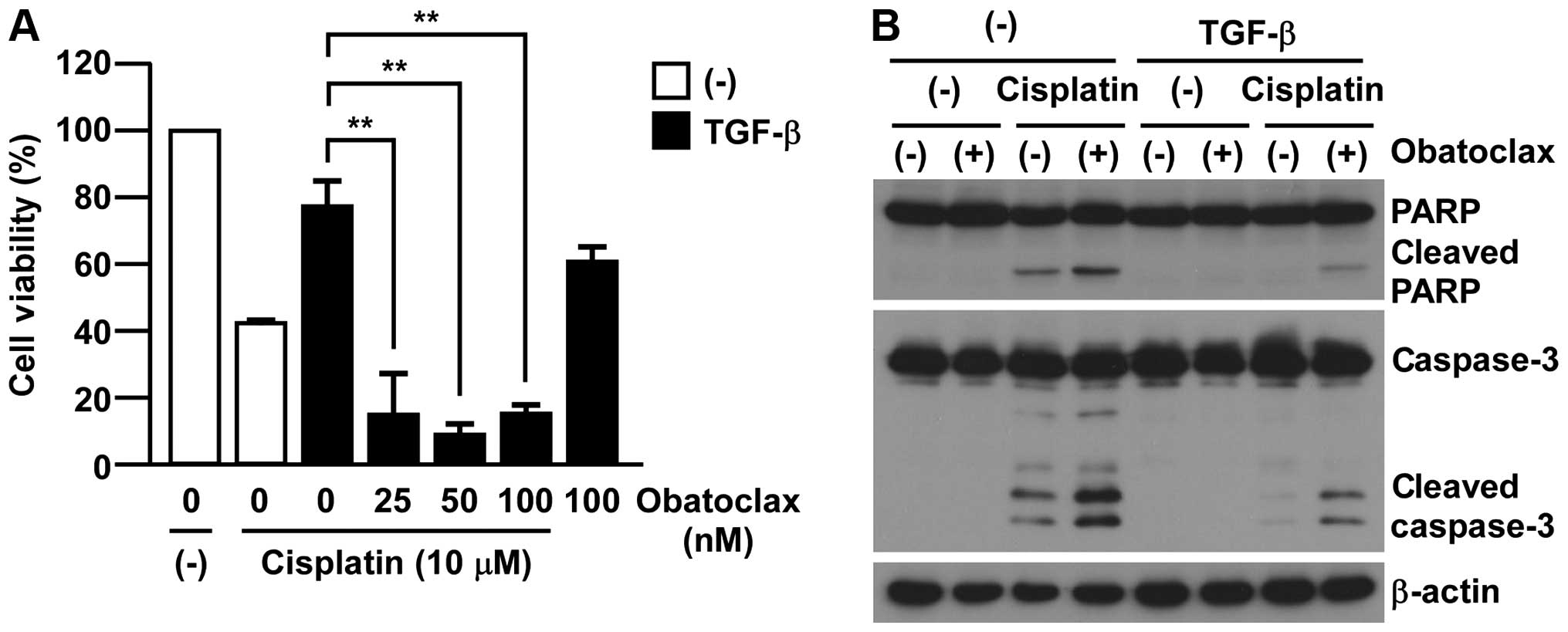
MCL-1 inhibition by pan-BCL2 inhibitor
treatment overcomes TGF-β-induced chemo-resistance
To test the clinical application of our findings, we
examined the effects of pan-BCL2 inhibitors in EMT-associated
chemo-resistance in A549 cells. As shown in Fig. 4A, obatoclax, which has a broad
range of inhibition in BCL2 family members including MCL-1,
re-sensitized EMT-induced A549 cells to cisplatin toxicity. Of
note, the treatment with obatoclax (100 nM) showed only minor
effect on TGF-β-treated A549 cell viability (Fig. 4A). In accordance with
re-sensitizing to cisplatin toxicity, we also detected the cleavage
of both caspase-3 and PARP in the combination of obatoclax with
cisplatin in EMT-induced A549 cells by TGF-β (Fig. 4B). Collectively, these results
implicate a clinical advantage for targeting MCL-1 in
EMT-associated cisplatin-resistance in A549 cells.
Discussion
In this study, we demonstrated that human NSCLC A549
acquired chemo-resistance upon TGF-β-induced EMT and such
EMT-associated chemo-resistance was mediated through
MCL-1-dependent anti-apoptotic pathway. By treating with pan-BCL2
inhibitor, obatoclax, EMT-associated chemo-resistance in A549 cells
can be reversed, therefore we propose that pharmacological
inhibition of MCL-1 could be an attractive target to overcome
EMT-associated chemo-resistance and further inhibit metastasis
spread in NSCLC patients.
Although MCL-1 was a key molecule in EMT-associated
chemo-resistance in this study (Fig.
3), other BCL-2 family members are known to contribute for
chemo-resistance in general. For example, enhanced BCL-2 expression
is involved in nicotine- or matrilysin-induced cisplatin-resistance
in lung cancer cells (21,22) and BCL2A1 confers resistance to BRAF
inhibitors in melanoma (15).
Considering other EMT inducers in tumor microenvironment, EGF or
HGF, have been reported to induce both EMT and MCL-1 expression
(20,23–26),
MCL-1 induction might be a common mechanism for EMT-associated
chemo-resistance. Although we do not show any direct connection
between TGF-β and MCL-1, there are several reports that the
EMT-related transcription factors, ZEB1 or Twist1, can regulate
MCL-1 expression (27,28). In this context, we observed the
induction of ZEB1 expression in A549 cells after TGF-β treatment
(data not shown). Collectively, these observations suggest that
ZEB1-mediated transcriptional control can be involved in
EMT-associated chemo-resistance by regulating MCL-1 expression.
Nevertheless, our current results implicate that
mesenchymal-transitioned NSCLC could acquire the chemo-resistance
through the induction of MCL-1. Consistent with our findings, it is
reported that EMT can be observed in the tumor specimens resected
from NSCLC patients after chemo-radiotherapy (14) to acquire chemo-resistance (14,16);
therefore, these lines of evidence support a clinical relevance of
our presented findings. Importantly, we have demonstrated the
importance of pharmacological targeting of MCL-1 to re-sensitize
cisplatin treatment in A549 cells. In addition to its importance in
EMT-associated chemo-resistance shown in this study, MCL-1 is
involved in anoikis-resistance in NSCLCs, which can be critical for
the survival of tumor cells during the metastatic process (29).
In conclusion, we newly identified MCL-1 as a key
molecule for acquiring EMT-associated chemo-resistance in human
NSCLC. Considering EMT-associated MCL-1 induction might play
critical roles not only in chemo-resistance, but also metastatic
spread and survival in distant tissue, pharmacological targeting of
MCL-1 provides a new therapeutic opportunity in NSCLC particularly
for combining with postoperative chemotherapies.
Acknowledgements
The authors would like to thank David E. Fisher
(MGH, Boston, MA, USA) for kindly giving the plasmids and all
members of the Saiki laboratory for discussions and suggestions.
This study was supported in part by Grant-in-Aid for Young
Scientists (B) 24701023 (M.T.), and 24700971 (S.Y.), by
Grants-in-aid for Challenging Exploratory Research 24659348 (I.S.)
from the Ministry of Education, Culture, Sports, Science, and
Technology (Japan), and by Grant for young scientists from
Hokuriku-Bank (S.Y.).
Abbreviations:
|
NSCLC
|
non-small cell lung cancer
|
|
EMT
|
epithelial-to-mesenchymal
transition
|
References
|
1
|
Ohe Y, Ohashi Y, Kubota K, et al:
Randomized phase III study of cisplatin plus irinotecan versus
carboplatin plus paclitaxel, cisplatin plus gemcitabine, and
cisplatin plus vinorelbine for advanced non-small-cell lung cancer:
Four-Arm Cooperative Study in Japan. Ann Oncol. 18:317–323. 2007.
View Article : Google Scholar
|
|
2
|
Groome PA, Bolejack V, Crowley JJ, et al:
The IASLC Lung Cancer Staging Project: validation of the proposals
for revision of the T, N, and M descriptors and consequent stage
groupings in the forthcoming (seventh) edition of the TNM
classification of malignant tumours. J Thorac Oncol. 2:694–705.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ginsberg RJ and Rubinstein LV: Randomized
trial of lobectomy versus limited resection for T1 N0 non-small
cell lung cancer. Lung Cancer Study Group. Ann Thorac Surg.
60:613–622. 1995. View Article : Google Scholar
|
|
4
|
Carr SR, Schuchert MJ, Pennathur A, et al:
Impact of tumor size on outcomes after anatomic lung resection for
stage 1A non-small cell lung cancer based on the current staging
system. J Thorac Cardiovasc Surg. 143:390–397. 2012. View Article : Google Scholar
|
|
5
|
Douillard JY, Rosell R, De Lena M, et al:
Adjuvant vinorelbine plus cisplatin versus observation in patients
with completely resected stage IB-IIIA non-small-cell lung cancer
(Adjuvant Navelbine International Trialist Association [ANITA]): a
randomised controlled trial. Lancet Oncol. 7:719–727. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arriagada R, Bergman B, Dunant A, Le
Chevalier T, Pignon JP and Vansteenkiste J: Cisplatin-based
adjuvant chemotherapy in patients with completely resected
non-small-cell lung cancer. N Engl J Med. 350:351–360. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Winton T, Livingston R, Johnson D, et al:
Vinorelbine plus cisplatin vs. observation in resected
non-small-cell lung cancer. N Engl J Med. 352:2589–2597. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fidler IJ: The pathogenesis of cancer
metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev
Cancer. 3:453–458. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chaffer CL and Weinberg RA: A perspective
on cancer cell metastasis. Science. 331:1559–1564. 2012. View Article : Google Scholar
|
|
10
|
Ledford H: Cancer theory faces doubts.
Nature. 472:2732011. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsai JH, Donaher JL, Murphy DA, Chau S and
Yang J: Spatiotemporal regulation of epithelial-mesenchymal
transition is essential for squamous cell carcinoma metastasis.
Cancer Cell. 22:725–736. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rho JK, Choi YJ, Lee JK, et al: Epithelial
to mesenchymal transition derived from repeated exposure to
gefitinib determines the sensitivity to EGFR inhibitors in A549, a
non-small cell lung cancer cell line. Lung Cancer. 63:219–226.
2009. View Article : Google Scholar
|
|
14
|
Shintani Y, Okimura A, Sato K, et al:
Epithelial to mesenchymal transition is a determinant of
sensitivity to chemoradiotherapy in non-small cell lung cancer. Ann
Thorac Surg. 92:1794–1804. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Haq R, Yokoyama S, Hawryluk EB, et al:
BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that
confers resistance to BRAF inhibition. Proc Natl Acad Sci USA.
110:4321–4326. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thomson S, Buck E, Petti F, et al:
Epithelial to mesenchymal transition is a determinant of
sensitivity of non-small-cell lung carcinoma cell lines and
xenografts to epidermal growth factor receptor inhibition. Cancer
Res. 65:9455–9462. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kato S, Hayakawa Y, Sakurai H, Saiki I and
Yokoyama S: Mesenchymal-transitioned cancer cells instigate the
invasion of epithelial cancer cells through secretion of WNT3 and
WNT5B. Cancer Sci. 105:281–289. 2014. View Article : Google Scholar
|
|
18
|
Kin R, Kato S, Kaneto N, et al:
Procyanidin C1 from Cinnamomi Cortex inhibits TGF-β-induced
epithelial-to-mesenchymal transition in the A549 lung cancer cell
line. Int J Oncol. 43:1901–1906. 2013.PubMed/NCBI
|
|
19
|
Li J, Viallet J and Haura EB: A small
molecule pan-Bcl-2 family inhibitor, GX15-070, induces apoptosis
and enhances cisplatin-induced apoptosis in non-small cell lung
cancer cells. Cancer Chemother Pharmacol. 61:525–534. 2008.
View Article : Google Scholar
|
|
20
|
Henson ES, Gibson EM, Villanueva J,
Bristow NA, Haney N and Gibson SB: Increased expression of Mcl-1 is
responsible for the blockage of TRAIL-induced apoptosis mediated by
EGF/ErbB1 signaling pathway. J Cell Biochem. 89:1177–1192. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu H, Zhang T, Wu B, Huang J, Zhou Y and
Zhu J: Chronic exposure to exogenous matrilysin induces
chemoresistance and enhances Bcl-2 expression in A549 lung
adenocarcinoma cells. Mol Biol Rep. 36:2099–2109. 2009. View Article : Google Scholar
|
|
22
|
Nishioka T, Luo LY, Shen L, et al:
Nicotine increases the resistance of lung cancer cells to cisplatin
through enhancing Bcl-2 stability. Br J Cancer. 110:1785–1792.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Leu CM, Chang C and Hu C: Epidermal growth
factor (EGF) suppresses staurosporine-induced apoptosis by inducing
mcl-1 via the mitogen-activated protein kinase pathway. Oncogene.
19:1665–1675. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lu Z, Ghosh S, Wang Z and Hunter T:
Downregulation of caveolin-1 function by EGF leads to the loss of
E-cadherin, increased transcriptional activity of beta-catenin, and
enhanced tumor cell invasion. Cancer Cell. 4:499–515. 2003.
View Article : Google Scholar
|
|
25
|
Hu P, Chu GC, Zhu G, et al: Multiplexed
quantum dot labeling of activated c-Met signaling in
castration-resistant human prostate cancer. PLoS One. 6:e286702011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schulze-Bergkamen H, Brenner D, Krueger A,
et al: Hepatocyte growth factor induces Mcl-1 in primary human
hepatocytes and inhibits CD95-mediated apoptosis via Akt.
Hepatology. 39:645–654. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jin HO, Hong SE, Woo SH, et al: Silencing
of Twist1 sensitizes NSCLC cells to cisplatin via AMPK-activated
mTOR inhibition. Cell Death Dis. 3:e3192012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sanchez-Tillo E, Fanlo L, Siles L, et al:
The EMT activator ZEB1 promotes tumor growth and determines
differential response to chemotherapy in mantle cell lymphoma. Cell
Death Differ. 21:247–257. 2014. View Article : Google Scholar :
|
|
29
|
Li Z, Zhao J, Du Y, et al: Downregulation
of 14-3-3zeta suppresses anchorage-independent growth of lung
cancer cells through anoikis activation. Proc Natl Acad Sci USA.
105:162–167. 2008. View Article : Google Scholar
|