Introduction
Chronic myeloid leukemia (CML) is a clonal
myeloproliferative neoplasm characterized by Philadelphia
chromosome, the t(9;22) (q34;q11) reciprocal translocation,
resulting in the expression of BCR-ABL fusion protein (1). BCR-ABL is a tyrosine kinase
responsible for malignant transformation by activating multiple
signal pathways, including the PI3K/Akt, MAPK/ERK and STATs
(2–4), leading to aberrant cell survival
(5,6). The successful introduction of the
tyrosine kinase inhibitors (TKIs), constitutes an effective
strategy in CML. The US Food and Drug Administration (FDA) has
approved imatinib as the first-line treatment for Philadelphia
chromosome-positive CML. Unfortunately, a portion of patients may
relapse upon drug discontinuation, or drug resistance (7). Studies reported that different point
mutations were associated with resistance to imatinib, and T315I
mutation in BCR-ABL, was particularly frequent (8). To overcome imatinib resistance,
second generation TKIs have been developed, such as dasatinib,
nilotinib, bosutinib and bafetinib. However, the T315I mutation
confers resistance to all these TKIs (9,10).
Hence, novel compounds or strategies to override this challenging
problem are urgently required for CML treatment.
Aurora kinase A (Aur-A), an oncogenic protein of
serine/threonine kinases family, is central for mitotic progression
(11). Aur-A was aberrantly
overexpressed in solid tumors such as prostate, colon, pancreas,
and breast cancer, as well as leukemia (12). Recent study suggested that Aur-A
kinase activity was responsible for chemoresistance and tumorigenic
ability (13). Small molecule
kinase inhibitors of Aur-A have attracted great interest, such as
MLN8237, ZM447439 and VX-680 (MK0457). VX-680 effectively inhibited
multiple myeloma growth, especially in RHAMM overexpressing
patients, and CML with BCR-ABL mutations (14–16).
Recently, we synthesized and identified a novel compound against
Aur-A termed AKI603 with IC50=12.3±1.5 nM in
vitro by kinase activity assay. AKI603 inhibited breast cancer
cell line proliferation, especially abolished enrichment of tumor
initiating cells (TICs) induced by epirubicin, suggesting its
potent use in solid cancer treatment (17).
In this study, we investigated the effects of the
novel Aur-A small molecule inhibitor AKI603 in CML cells including
imatinib-resistant cells. The results showed that AKI603 inhibited
cell proliferation with G2/M phase cell cycle arrest.
Interestingly, imatinib-resistant cells displayed high Aur-A kinase
activity, while targeting Aur-A kinase activity by AKI603 potently
suppressed colony formation ability and promoted cell
differentiation. Thus, inhibition of Aur-A kinase overcame imatinib
resistance, providing a novel therapeutic strategy against CML.
Materials and methods
Chemicals and cell culture
AKI603, designed and synthesized by our lab
(17,18), was dissolved in dimethyl sulfoxide
(DMSO) to a stock concentration of 100 mM and stored at −20°C.
Imatinib (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in DMSO
at 10 mM stock solution and stored at −20°C. KBM5 and KBM5-T315I
cell lines were gifts from Dr Peng Huang (MD Anderson Cancer
Center, Houston, TX, USA). All the cells were cultured in RPMI-1640
medium supplemented with 10% (v/v) fetal bovine serum (Gibco,
Invitrogen, Carlsbad, CA, USA). Cord blood mononuclear cells
(CBMCs) were obtained from 3 healthy donors. All donors provided
written informed consent, and the study was approved by the
Institute Research Ethics Committee at Sun Yat-sen University.
Cell viability assay
Cell viability was evaluated by WST-8 assay (Dojindo
Laboratories, Kumamoto, Japan) according to the manufacturer’s
instructions. Briefly, 1×104 cells were seeded in each
well of the 96-well flat-bottomed plate. After treatment with
imatinib or AKI603 for 48 h, 10 μl WST-8 solution was added to each
well and cells were incubated at 37°C for 4 h. The absorbance was
finally determined at 450 nm using an Eon Microplate
Spectrophotometer (BioTek, Winooski, VT, USA).
Cell cycle analysis
Cells were collected, fixed, and resuspended in PBS
containing 0.2% Triton X-100, 100 μg/ml RNaseA, and 50 μg/ml PI.
Cell cycle analysis was carried out using a FACSCalibur flow
cytometer (BD Biosciences, San Jose, CA, USA).
Cell differentiation assessment
To measure CD11b expression, cells were treated with
indicated concentration of AKI603. After culture, cells were
collected and incubated with PE-CD11b antibody (BD Biosciences).
Expression of CD11b on cell surface was measured by flow
cytometry.
Colony formation assay
Cells were cultured in RPMI-1640 medium supplemented
with 0.9% methylcellulose and 10% FBS. The colonies (containing ≥40
cells) were counted by light microscopy after 10 days.
Western blot analysis
Total cellular proteins were isolated with lysis
buffer. Equal amounts of protein were subjected to SDS-PAGE and
transferred to nitrocellulose membranes. The membranes were blocked
and then incubated with phospho-Aur-A, phospho-c-Abl, pRb, Rb (Cell
Signaling Technology Corp., Beverly, MA, USA), Aur-A
(Sigma-Aldrich), c-Abl and GAPDH antibodies (Santa Cruz
Biotechnology, Santa Cruz, CA, USA). The protein bands were
visualized using an enhanced chemiluminescence reagent
(Sigma-Aldrich), according to the manufacturer’s instructions.
Annexin V/PI analysis
Cells were treated with the indicated concentration
of AKI603, collected and resuspended in the binding buffer.
Annexin-V-FITC and PI was added to the cells according to the
protocol by the Annexin V-FITC apoptosis detection kit (BD
Biosciences). The cells were then incubated for 15 min in the dark
and subjected to flow cytometry analysis.
Cell morphology analysis
Cells were incubated with AKI603 for the indicated
dose and collected for analysis. Cytospin slides were prepared, and
the cells were counterstained with Wright-Giemsa. The morphology of
cells was observed by microscopy (IX81, Olympus, Japan).
Nitrotetrazolium blue chloride (NBT)
reduction assay
The same amount of cells untreated or treated with
AKI603 were incubated at 37°C in RPMI-1640 medium containing 10%
FBS and 0.1% NBT (Sigma-Aldrich). The cells were then centrifuged
and dissolved in DMSO. The optical density was read at 570 nm.
Statistical analysis
Data are presented as mean ± SD. Statistical
analysis was performed using SPSS version 16.0 (SPSS, Inc.) and
Prism 6 (GraphPad Software, Inc.). The Kruskal-Wallis test,
followed by Dunn’s multiple comparison test, was used to perform
statistical comparison for colony size distribution. The unpaired
Student’s t-test was used to perform statistical comparison between
two groups. The ANOVA test, followed by least significant
difference test, was used for multiple comparisons. A value of
p<0.05 was considered statistically significant.
Results
AKI603 inhibits Aur-A activity and cell
viability in CML cells
Chemical structure of AKI603 is shown in Fig. 1A. In this study, we applied two CML
cells lines, KBM5 and imatinib-resistant KBM5-T315I cells to study
the effects of AKI603. WST-8 assay showed that KBM-T315I cells
exhibited significant resistance towards imatinib compared with the
wild-type KBM5 cells (Fig. 1B). To
evaluate CML cell proliferation by AKI603 treatment, cell viability
was also assessed by WST-8 assay. The CML cell lines tested were
sensitive to AKI603 ranging from 0.3125 to 5.0 μM (Fig. 1C). In 0.3125 μM treatment, the cell
viability was decreased to 68.8±1.6% in KBM5 cells and 78.2±3.0% in
KBM5-T315I cells. In 5.0 μM treatment, the cell viability was
decreased to 25.0±1.4% in KBM5 cells while 40.7±0.9% in KBM5-T315I
cells. Moreover, mononuclear cells from cord blood were exposed to
various concentrations of AKI603. Results showed that cell
viability was slightly changed after 48-h incubation (Fig. 1D). To assess whether AKI603
inhibited Aur-A kinase activity, western blot analysis was
performed. As shown in Fig. 1E,
KBM-T315I cells displayed high level of Aur-A Thr288
phosphorylation. Treatment with AKI603 for 48 h significantly
inhibited Aur-A phosphorylation (labeled pAur-A) at 0.3125 μM or
higher concentrations in KBM5 and KBM5-T315I cells, while total
Aur-A protein was not changed. Moreover, we detected the change of
BCR-ABL phosphorylation (labeled pBCR-ABL) after AKI603 treatment.
We found that either phosphorylation or total protein of BCR-ABL
was stable in different treatments of AKI603, ruling out the
possibility that the effect of AKI603 was through inhibition of
BCR-ABL activity. Our data implied that AKI603 might have a
potential therapeutic application in leukemia treatment.
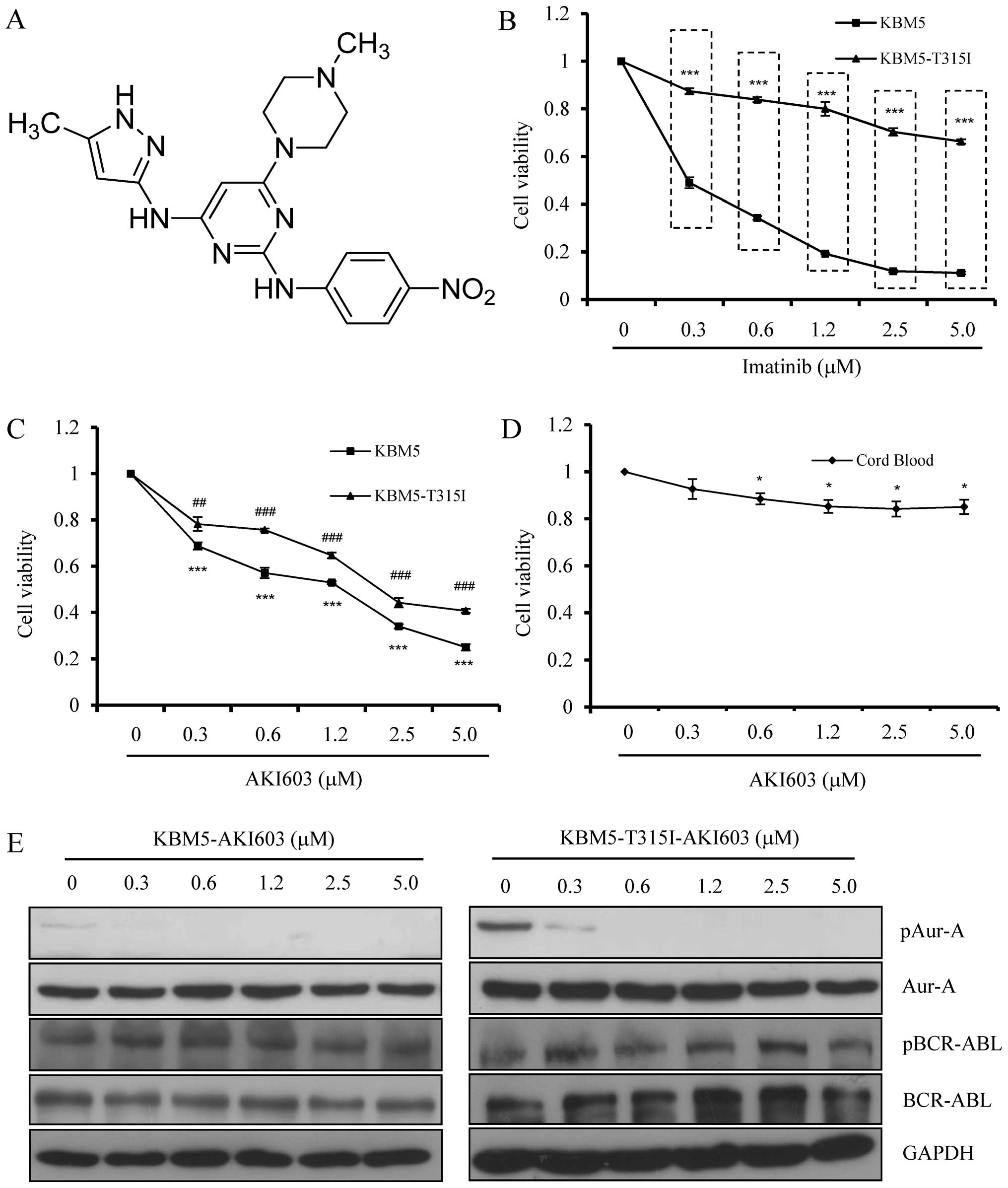 | Figure 1AKI603 inhibits Aur-A activity and
cell viability in CML cells. (A) Chemical structure of AKI603. (B)
KBM5 and KBM5-T315I cells were treated with different doses of
imatinib for 48 h and subjected to WST-8 assay. Data are the mean
of three independent experiments (***p<0.001,
Student’s t-test). (C) Cells treated with various concentrations of
AKI603 for 48 h were subjected to WST-8 assay. Data are the mean of
three independent experiments (** or
##p<0.01, *** or ###p<0.001.
The ANOVA test, followed by least significant difference test, was
used for statistical comparison). (D) CBMCs of three healthy donors
were exposed to different concentrations of AKI603 for 48 h. WST-8
assay was performed. (E) Cells were treated with various
concentrations of AKI603 (0, 0.3125, 0.625, 1.25, 2.5 and 5.0 μM)
for 48 h. The lysates were subjected for western blot analysis of
pAur-A (active form), Aur-A, pBCR-ABL (active form) and BCR-ABL
expression. |
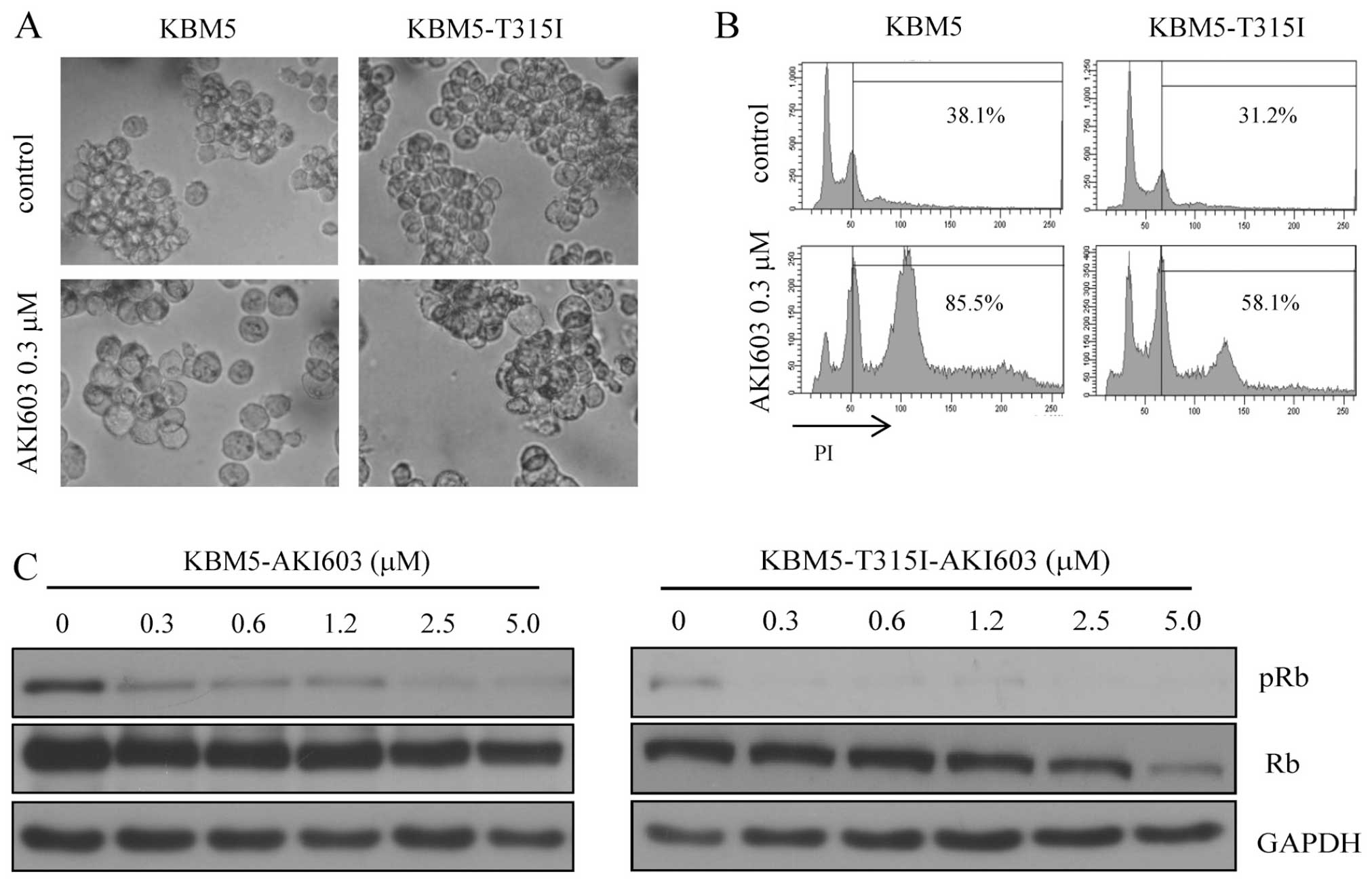
AKI603 induces cell cycle arrest and
decreases Rb phosphorylation in CML cells
Due to the crucial role of Aur-A in mitosis, the
cell cycle blocking effect on KBM5 and KBM5-T315I was examined.
AKI603 significantly induced polyploidization in CML cells as
assessed by microscopy observation (Fig. 2A). These morphological changes were
confirmed by flow cytometry. DNA content analysis by PI staining
showed that treatment of AKI603 induced polyploidy of both KBM5 and
KBM-T315I cells (Fig. 2B). These
results suggested that the proliferative inhibition induced by
AKI603 could be associated with cell cycle blockage. In addition,
western blot analysis showed that phosphorylation of Rb was reduced
in AKI603 treated cells, even at the concentration of 0.3125 μM
(Fig. 2C). Thus, our data
suggested that AKI603 might inhibit cell proliferation by inducing
G2/M cell cycle arrest, and reducing Rb phosphorylation.
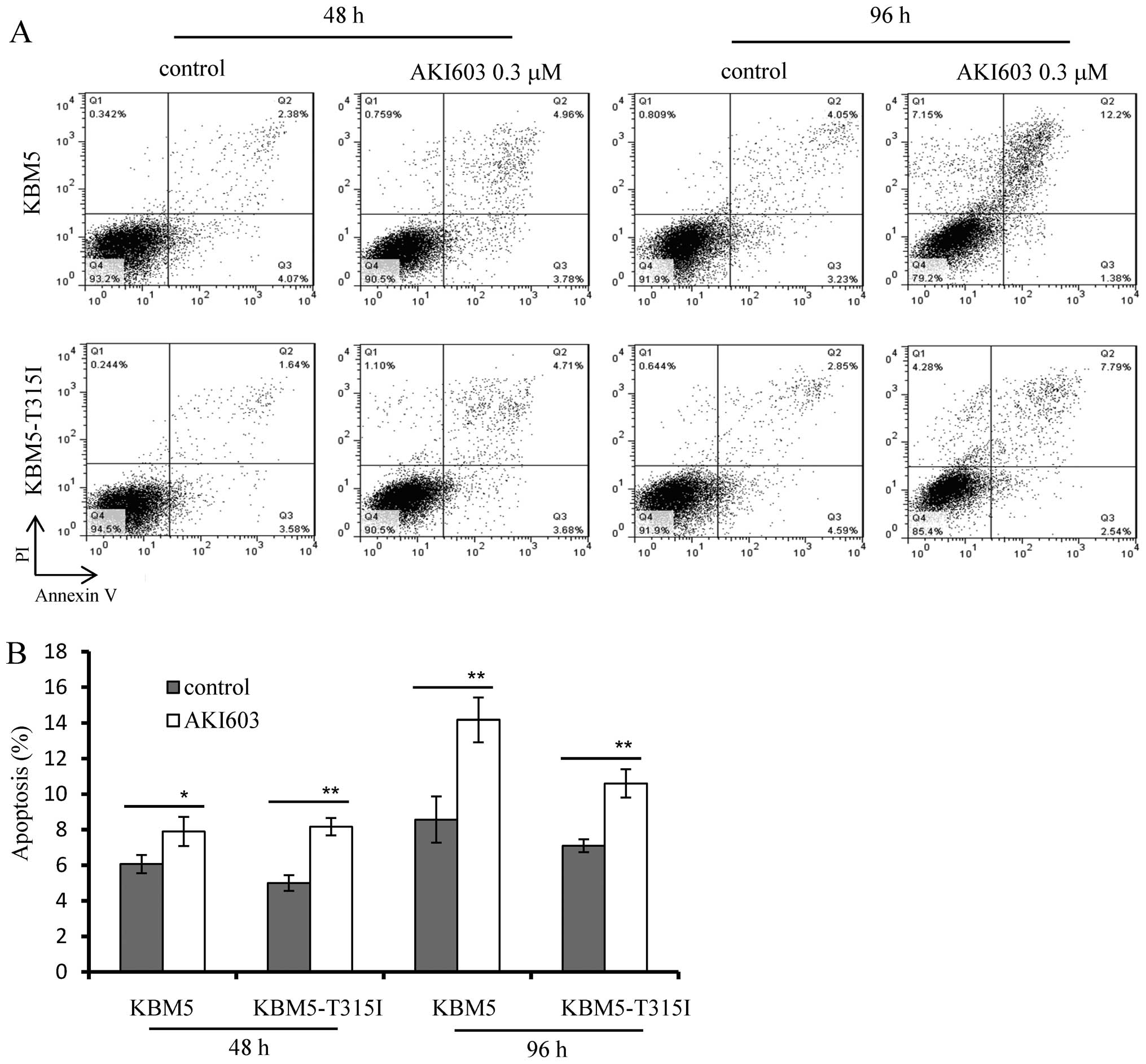
AKI603 does not induce obvious apoptosis
in CML cells
To evaluate whether AKI603 induced apoptosis, cells
were collected for flow cytometry analysis of Annexin V/PI
staining. As shown in Fig. 1,
0.3125 μM AKI603 significantly suppressed cell viability, thus we
chose this concentration for apoptosis detection. The results
showed that both KBM5 and KBM5-T315I cells treated with indicated
concentration (0.3125 μM) of AKI603 did not undergo obvious
apoptosis either in 48 or 96 h (Fig.
3A). In 96-h treatment, AKI603 increased the percentage of
apoptotic cells from 8.6±1.3 to 14.2±1.3% in KBM5 cells and from
7.1±0.4 to 10.6±0.8% in KBM5-T315I cells (Fig. 3B), indicating that cell viability
inhibited by AKI603 was not largely due to apoptosis.
AKI603 suppresses colony formation
ability in CML cells
We next examined the long-term effect (10 days) of
AKI603 against cell colony formation capacity. To this end, the two
cell types were maintained in methylcellulose culture medium with
or without different treatment, and the diameter and number of the
colonies were measured. First, we investigated the long-term effect
of the cells under imatinib treatment. As shown in Fig. 4A and B, 0.3125 μM imatinib
decreased the size of colony formation (56.8±26.0 μm, p<0.001)
compared with the control group (206.7±69.2 μm) in KBM5 cells.
However, 0.3125 μM imatinib did not obviously decrease the size of
colony formation in KBM5-T315I cells. The results further suggested
that imatinib could not inhibit the cell colony formation capacity
in KBM5-T315I cells. We next examined the long-term effect of
AKI603. The colonies in AKI603 treatment groups were fewer than the
control group. Under the concentration of 0.3125 μM, the cells
could not form colonies in either cell type (Fig. 4C). Thus, we reduced the treatment
concentration of AKI603 and found that 0.1563 μM treatment
completely suppressed the colony formation (Fig. 4D). Under low concentrations, the
colony size was smaller when cultured with AKI603 compared with
control (Fig. 4E and F). For
example, 0.0781 μM AKI603 decreased the size of colony formation
(112.8±44.7 μm, p<0.001) compared with the control group
(218.2±64.2 μm) in KBM5 cells. In KBM5-T315I cells, AKI603
decreased the size of colony formation (169.8±70.7 μm, p<0.001)
compared with the control group (243.7±84.6 μm).
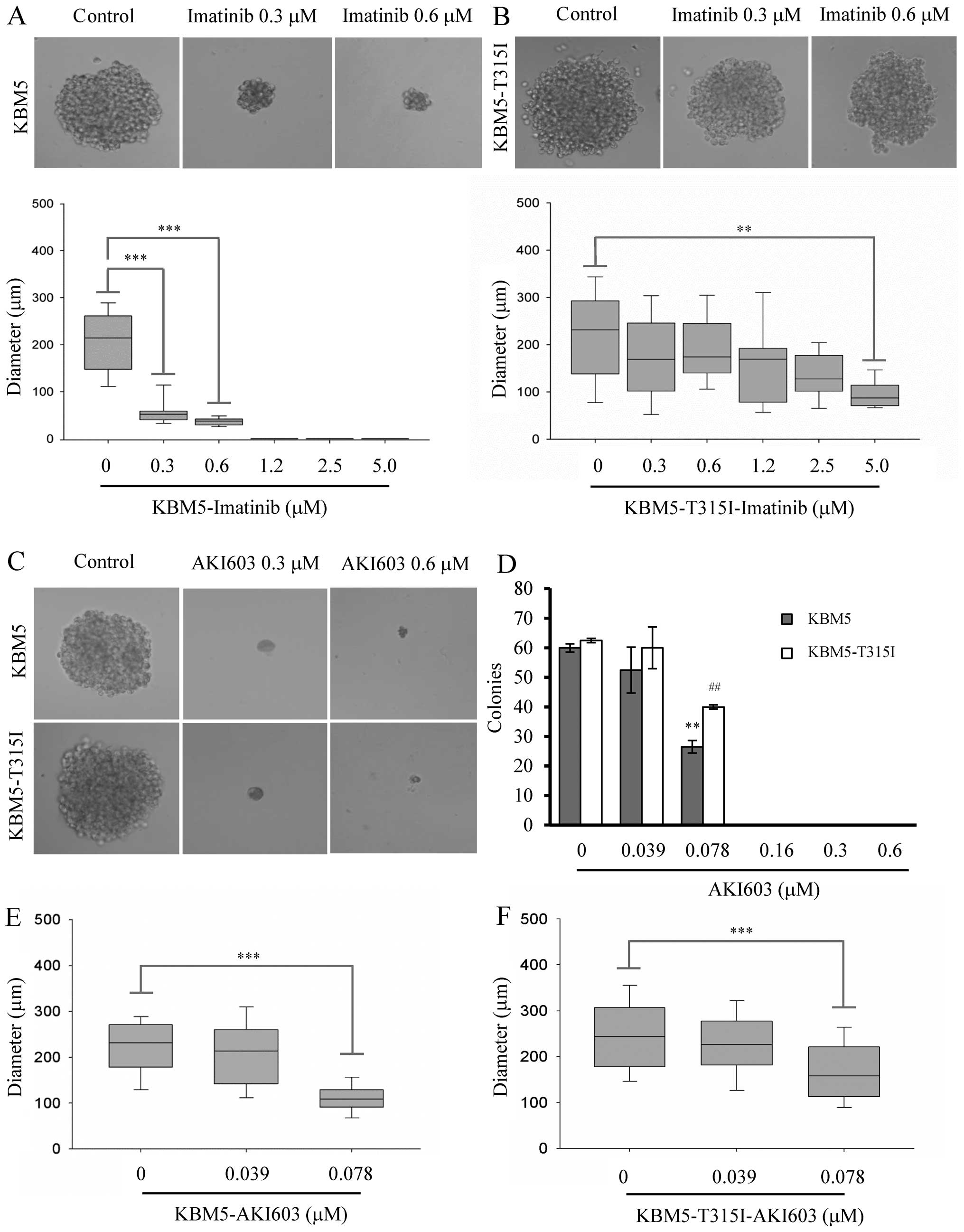 | Figure 4AKI603 suppresses colony formation
ability in CML cells. The colony formation of KBM5 and KBM5-T315I
cells treated with imatinib for 10 days were analyzed. Ten-day-old
KBM5 (A) and KBM5-T315I (B) colonies cultured in medium containing
indicated concentrations of imatinib were photographed.
Representative images and the diameters of colonies are shown. The
values from three independent experiments are presented in a box
plot graph and the size distribution of the colonies is shown. The
horizontal line within each box represent the median value. The
Kruskal-Wallis test, followed by Dunn’s multiple comparison test,
was used to make statistical comparisons (**p<0.01,
***p<0.001). (C) The colony formation of KBM5 and
KBM5-T315I treated with 0, 0.0391, 0.0781, 0.1563, 0.3125, 0.625,
1.25, 2.5 and 5.0 μM of AKI603 for 10 days were analyzed.
Representative images are shown. (D) The number of colonies is
presented as a bar graph. The bar represents means ± SD of three
independent experiments. The diameters of 10-day-old KBM5 (E) and
KBM5-T315I (F) colonies cultured in medium containing indicated
concentrations of AKI603 were measured as described above
(***p<0.001). |
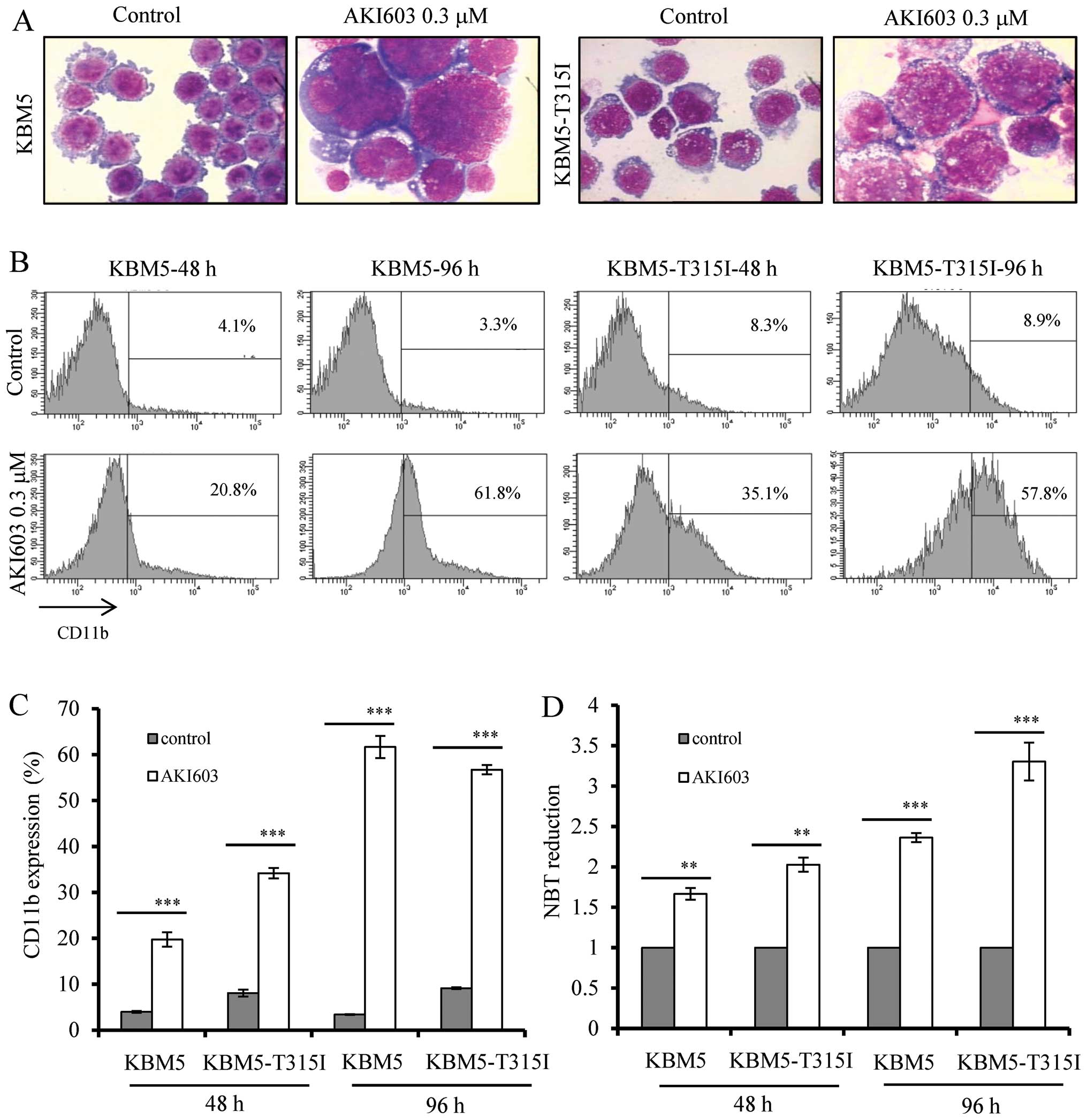
AKI603 promotes cell differentiation in
CML cells
After 96-h incubation with AKI603, we found that
both cell types presented polyploidization and obvious phenotype
changes by Wright-Giemsa staining (Fig. 5A). To investigate the mechanism of
AKI603 inhibition of colony formation without inducing apoptosis,
the KBM5 and KBM5-T315I cells were treated with AKI603 and cell
differentiation was evaluated by quantifying CD11b expression, a
marker of myeloid differentiation. After exposure of KBM5 and
KBM-T315I cells to AKI603 (0.3125 μM) for 48 h, the percentage of
CD11b-positive cells induced by AKI603 was significantly increased
from 4.0±0.2 to 19.7±1.6% in KBM5 cells and from 8.1±0.8 to
34.2±1.1% in KBM5-T315I cells. Moreover, 96-h treatment increased
CD11b expression from 3.4±0.1 to 61.7±2.4% in KBM5 cells and from
9.1±0.2 to 56.7±1.0% in KBM5-T315I cells (Fig. 5B and C). The differentiation state
was also confirmed by NBT reduction assay (Fig. 5D). Results showed that both cells
displayed increased NBT reduction after AKI603 treatment,
indicating the sensitivity of both KBM5 and KBM5-T315I cells to
AKI603 might be due to induced cell differentiation.
Discussion
To date, imatinib represents a major success in the
era of targeted cancer therapy. However, point mutation in BCR-ABL,
which prevents imatinib binding, is responsible for most of the
cases of acquired clinical resistance to imatinib. Different point
mutations in the BCR-ABL kinase domain have been isolated from
patients expressing BCR-ABL (19,20).
In addition to imatinib, other novel TKIs are also ineffective
against the T315I mutation. Patients with T315I have a poor
prognosis, with a short survival from the initiation of imatinib
therapy (21,22). Therefore, we aimed to identify
effective targeted therapy against CML cells carrying the wild-type
BCR-ABL or BCR-ABL-T315I mutation. In the present study, we showed
that novel small molecular inhibitor AKI603 effectively inhibited
Aur-A kinase activity. Surprisingly, AKI603 potently inhibited cell
proliferation, suppressed colony formation ability and promoted
cell differentiation in both wild-type BCR-ABL and BCR-ABL-T315I
mutant CML cells, suggesting a new approach to overcome imatinib
resistance.
Aurora kinase regulated multiple critical mitotic
processes. Disruption of Aurora kinase activity induces abnormal
spindle pole organization, centrosome separation and chromosome
congression (23). Ultimately,
cells treated with Aurora kinase inhibitor undergo cell growth
inhibition through the development of deleterious aneuploidy
(24,25). In this report, we found that AKI603
inhibited Aur-A kinase and presented anti-leukemia effects in KBM5
cells, as well as KBM5-T315I cells, suggesting a possible novel and
potent target in treating imatinib-resistant CML. We clearly showed
that AKI603 inhibited viability of CML cells (Fig. 1). At the dose range used, AKI603
inhibited Aur-A phosphorylation at Thr288 (Fig. 1). In addition, AKI603 treatment
initially resulted in G2/M cell cycle arrest and significant degree
of aneuploidy (Fig. 2), a typical
phenotype of Aurora suppression (24). These data were consistent with our
finding in AML and solid tumor cells that inhibition of Aurora
induced cell cycle arrest, promoted polyploidy formation and
inhibited cell proliferation (24–26).
Drug resistance is a major hindrance for effective
leukemic treatment. The mechanism of resistance is complicated,
such as accelerated drug efflux, the activation of oncogene or
inactivation of tumor suppressor gene, metabolic disturbance and
leukemia stem cells (LSCs) enrichment. Previous reports showed that
CD34+/CD38− leukemic precursors exhibited
resistance to daunorubicin in comparison with the
CD34+/CD38+ blasts with highly expressed
multidrug resistance genes (27).
Our study reported that CD34+ leukemic progenitor cells
displayed drug resistance resulting from high levels of Bcl-2
expression (28). These data
indicated that leukemic progenitor cells largely contributed to
clinical drug resistance. It has been reported that Aur-A was
overexpressed in both primary AML cells and LSCs compared to normal
hematopoietic stem cells (29–31).
Our recent data showed that inhibition of Aur-A activity by AKI603
reduced CD24Low/CD44High TICs, and
mammosphere formation (17),
indicating that Aur-A kinase activity was important for maintaining
TICs population. In this study, we found that Aur-A was active in
imatinib-resistant cells (Fig. 1),
suggesting that imatinib resistance might result from Aur-A
activation and be associated with LSCs enrichment.
Consistent with our recent finding that Aur-A
inhibition increased the sensitivity of conventional
chemotherapeutic drugs (17),
previous studies indicated that Aur-A was critical for overriding
cell cycle checkpoint in cancer, and therefore responsible for the
chemoresistance (32,33). Aurora kinase inhibitor CCT129202
increased the sensitivity of ABCB1/ABCG2 overexpressing cells and
cancer stem-like cells to chemotherapeutics (34). Inhibition of Aur-A plus cytarabine
treatment in LSCs resulted in increased cytotoxicity compared to
cytarabine treatment alone (30).
MLN8237, under investigation in multiple phase I and II studies,
was active in resistant CML and significantly increased the
efficacy of nilotinib treatment (35). Moreover, it reduced the formation
of spheroids, attenuated the self-renewal of spheroid forming
cells, and promoted cell differentiation (36). Notably, the Aurora inhibitor MK0457
and histone deacetylase inhibitor vorinostat combination was highly
active against primary CD34+ CML cells and Ba/F3 cells
with BCR-ABL mutations, such as T315I, E255K, and M351T (37). All these studies indicated that
Aur-A activity might be a potent target to abolish LSCs, thus
overcoming drug resistance. In the present study, we showed that
cells treated with AKI603 did not undergo obvious apoptosis
(Fig. 3). Importantly, AKI603
inhibited cell colony formation (Fig.
4) and promoted cells differentiation (Fig. 5) under a long time culture in
imatinib-resistant CML cells, indicating AKI603 might shift
leukemic progenitor cells to differentiated population, rather than
induce cell death.
In conclusion, the present study unveiled a
potential anti-leukemia function of the potential Aurora kinase
inhibitor AKI603. We also presented a novel mechanism that
inhibition of Aur-A kinase by AKI603 overcame drug resistance
through promoting cell differentiation in CML cells, suggesting a
novel strategy in leukemia treatment.
Acknowledgements
We thank Mr. Wen-Xing Lai, the members of Quentin
Liu lab and Medical Research Center, The Third Affiliated Hospital,
Sun Yat-sen University for their critical comments and technical
support. This study was supported by the National High Technology
Research and Development Program of China (863 Program; no.
2015AA020926 to Z.-J. Long), the Science and Technology Project of
Guangzhou (no. 2012J2200077 to Z.-J. Long), the Fundamental
Research Funds for the Central Universities (no. 11ykpy37 to Z.-J.
Long), the National Natural Science Foundation of China (no.
81130040 to Q. Liu) and the National Basic Research Program of
China (973 Program; no. 2012CB967000 to Q. Liu).
Abbreviations:
|
CML
|
chronic myeloid leukemia
|
|
AML
|
acute myeloid leukemia
|
|
TKIs
|
tyrosine kinase inhibitors
|
|
LSCs
|
leukemia stem cells
|
|
TICs
|
tumor initiating cells
|
|
CBMCs
|
cord blood mono-nuclear cells
|
References
|
1
|
Savona M and Talpaz M: Getting to the stem
of chronic myeloid leukaemia. Nat Rev Cancer. 8:341–350. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gesbert F, Sellers WR, Signoretti S, Loda
M and Griffin JD: BCR/ABL regulates expression of the
cyclin-dependent kinase inhibitor p27Kip1 through the
phosphatidylinositol 3-kinase/AKT pathway. J Biol Chem.
275:39223–39230. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lugo TG, Pendergast AM, Muller AJ and
Witte ON: Tyrosine kinase activity and transformation potency of
bcr-abl oncogene products. Science. 247:1079–1082. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Danial NN and Rothman P: JAK-STAT
signaling activated by Abl oncogenes. Oncogene. 19:2523–2531. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Airiau K, Mahon FX, Josselin M, Jeanneteau
M, Turcq B and Belloc F: ABT-737 increases tyrosine kinase
inhibitor-induced apoptosis in chronic myeloid leukemia cells
through XIAP downregulation and sensitizes CD34(+)CD38(−)population
to imatinib. Exp Hematol. 40:367–378. 2012. View Article : Google Scholar
|
|
6
|
Soliera AR, Mariani SA, Audia A, Lidonnici
MR, Addya S, Ferrari-Amorotti G, Cattelani S, Manzotti G,
Fragliasso V, Peterson L, et al: Gfi-1 inhibits proliferation and
colony formation of p210BCR/ABL- expressing cells via
transcriptional repression of STAT5 and Mcl-1. Leukemia.
26:1555–1563. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kantarjian HM, Talpaz M, Giles F, O’Brien
S and Cortes J: New insights into the pathophysiology of chronic
myeloid leukemia and imatinib resistance. Ann Intern Med.
145:913–923. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shah NP: Loss of response to imatinib:
mechanisms and management. Hematology Am Soc Hematol Educ Program.
183–187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bradeen HA, Eide CA, O’Hare T, Johnson KJ,
Willis SG, Lee FY, Druker BJ and Deininger MW: Comparison of
imatinib mesylate, dasatinib (BMS-354825), and nilotinib (AMN107)
in an N-ethyl-N-nitrosourea (ENU)-based mutagenesis screen: high
efficacy of drug combinations. Blood. 108:2332–2338. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Deguchi Y, Kimura S, Ashihara E, Niwa T,
Hodohara K, Fujiyama Y and Maekawa T: Comparison of imatinib,
dasatinib, nilotinib and INNO-406 in imatinib-resistant cell lines.
Leuk Res. 32:980–983. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Meraldi P, Honda R and Nigg EA: Aurora
kinases link chromosome segregation and cell division to cancer
susceptibility. Curr Opin Genet Dev. 14:29–36. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marumoto T, Zhang D and Saya H: Aurora-A -
a guardian of poles. Nat Rev Cancer. 5:42–50. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cammareri P, Scopelliti A, Todaro M,
Eterno V, Francescangeli F, Moyer MP, Agrusa A, Dieli F, Zeuner A
and Stassi G: Aurora-A is essential for the tumorigenic capacity
and chemoresistance of colorectal cancer stem cells. Cancer Res.
70:4655–4665. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi Y, Reiman T, Li W, Maxwell CA, Sen S,
Pilarski L, Daniels TR, Penichet ML, Feldman R and Lichtenstein A:
Targeting aurora kinases as therapy in multiple myeloma. Blood.
109:3915–3921. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Giles FJ, Cortes J, Jones D, Bergstrom D,
Kantarjian H and Freedman SJ: MK-0457, a novel kinase inhibitor, is
active in patients with chronic myeloid leukemia or acute
lymphocytic leukemia with the T315I BCR-ABL mutation. Blood.
109:500–502. 2007. View Article : Google Scholar
|
|
16
|
Shah NP, Skaggs BJ, Branford S, Hughes TP,
Nicoll JM, Paquette RL and Sawyers CL: Sequential ABL kinase
inhibitor therapy selects for compound drug-resistant BCR-ABL
mutations with altered oncogenic potency. J Clin Invest.
117:2562–2569. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zheng FM, Long ZJ, Hou ZJ, Luo Y, Xu LZ,
Xia JL, Lai XJ, Liu JW, Wang X, Kamran M, et al: A novel small
molecule aurora kinase inhibitor attenuates breast tumor-initiating
cells and overcomes drug resistance. Mol Cancer Ther. 13:1991–2003.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Luo Y, Deng YQ, Wang J, Long ZJ, Tu ZC,
Peng W, Zhang JQ, Liu Q and Lu G: Design, synthesis and
bioevaluation of N-trisubstituted pyrimidine derivatives as potent
aurora A kinase inhibitors. Eur J Med Chem. 78:65–71. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Melo JV and Chuah C: Resistance to
imatinib mesylate in chronic myeloid leukaemia. Cancer Lett.
249:121–132. 2007. View Article : Google Scholar
|
|
20
|
Branford S: Chronic myeloid leukemia:
molecular monitoring in clinical practice. Hematology Am Soc
Hematol Educ Program. 376–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nicolini FE, Hayette S, Corm S, Bachy E,
Bories D, Tulliez M, Guilhot F, Legros L, Maloisel F, Kiladjian JJ,
et al: Clinical outcome of 27 imatinib mesylate-resistant chronic
myelogenous leukemia patients harboring a T315I BCR-ABL mutation.
Haematologica. 92:1238–1241. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Soverini S, Colarossi S, Gnani A, Rosti G,
Castagnetti F, Poerio A, Iacobucci I, Amabile M, Abruzzese E,
Orlandi E, et al: Contribution of ABL kinase domain mutations to
imatinib resistance in different subsets of Philadelphia-positive
patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia.
Clin Cancer Res. 12:7374–7379. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Carmena M and Earnshaw WC: The cellular
geography of aurora kinases. Nat Rev Mol Cell Biol. 4:842–854.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu LL, Long ZJ, Wang LX, Zheng FM, Fang
ZG, Yan M, Xu DF, Chen JJ, Wang SW, Lin DJ, et al: Inhibition of
mTOR pathway sensitizes acute myeloid leukemia cells to aurora
inhibitors by suppression of glycolytic metabolism. Mol Cancer Res.
11:1326–1336. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Long ZJ, Xu J, Yan M, Zhang JG, Guan Z, Xu
DZ, Wang XR, Yao J, Zheng FM, Chu GL, et al: ZM 447439 inhibition
of aurora kinase induces Hep2 cancer cell apoptosis in
three-dimensional culture. Cell Cycle. 7:1473–1479. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu DR, Huang S, Long ZJ, Chen JJ, Zou ZZ,
Li J, Lin DJ and Liu Q: Inhibition of mitotic kinase Aurora
suppresses Akt-1 activation and induces apoptotic cell death in
all-trans retinoid acid-resistant acute promyelocytic leukemia
cells. J Transl Med. 9:742011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Costello RT, Mallet F, Gaugler B, Sainty
D, Arnoulet C, Gastaut JA and Olive D: Human acute myeloid leukemia
CD34+/CD38− progenitor cells have decreased
sensitivity to chemotherapy and Fas-induced apoptosis, reduced
immunogenicity, and impaired dendritic cell transformation
capacities. Cancer Res. 60:4403–4411. 2000.PubMed/NCBI
|
|
28
|
Rao J, Xu DR, Zheng FM, Long ZJ, Huang SS,
Wu X, Zhou WH, Huang RW and Liu Q: Curcumin reduces expression of
Bcl-2, leading to apoptosis in daunorubicin-insensitive
CD34+ acute myeloid leukemia cell lines and primary
sorted CD34+ acute myeloid leukemia cells. J Transl Med.
9:712011. View Article : Google Scholar
|
|
29
|
Yang J, Ikezoe T, Nishioka C, Nobumoto A,
Udaka K and Yokoyama A: CD34+/CD38− acute
myelogenous leukemia cells aberrantly express Aurora kinase A. Int
J Cancer. 133:2706–2719. 2013.PubMed/NCBI
|
|
30
|
Kim SJ, Jang JE, Cheong JW, Eom JI, Jeung
HK, Kim Y, Hwang DY and Min YH: Aurora A kinase expression is
increased in leukemia stem cells, and a selective Aurora A kinase
inhibitor enhances Ara-C-induced apoptosis in acute myeloid
leukemia stem cells. Korean J Hematol. 47:178–185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ye D, Garcia-Manero G, Kantarjian HM, Xiao
L, Vadhan-Raj S, Fernandez MH, Nguyen MH, Medeiros LJ and
Bueso-Ramos CE: Analysis of Aurora kinase A expression in CD34(+)
blast cells isolated from patients with myelodysplastic syndromes
and acute myeloid leukemia. J Hematop. 2:2–8. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ouchi M, Fujiuchi N, Sasai K, Katayama H,
Minamishima YA, Ongusaha PP, Deng C, Sen S, Lee SW and Ouchi T:
BRCA1 phosphorylation by Aurora-A in the regulation of G2 to M
transition. J Biol Chem. 279:19643–19648. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Katayama H, Wang J, Treekitkarnmongkol W,
Kawai H, Sasai K, Zhang H, Wang H, Adams HP, Jiang S, Chakraborty
SN, et al: Aurora kinase-A inactivates DNA damage-induced apoptosis
and spindle assembly checkpoint response functions of p73. Cancer
Cell. 21:196–211. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng C, Liu ZG, Zhang H, Xie JD, Chen XG,
Zhao XQ, Wang F, Liang YJ, Chen LK, Singh S, et al: Enhancing
chemosensitivity in ABCB1- and ABCG2-overexpressing cells and
cancer stem-like cells by an Aurora kinase inhibitor CCT129202. Mol
Pharm. 9:1971–1982. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kelly KR, Ecsedy J, Medina E, et al: The
novel Aurora A kinase inhibitor MLN8237 is active in resistant
chronic myeloid leukaemia and significantly increases the efficacy
of nilotinib. J Cell Mol Med. 15:2057–2070. 2011. View Article : Google Scholar
|
|
36
|
Mohan P, Castellsague J, Jiang J, Allen K,
Chen H, Nemirovsky O, Spyra M, Hu K, Kluwe L, Pujana MA, et al:
Genomic imbalance of HMMR/RHAMM regulates the sensitivity and
response of malignant peripheral nerve sheath tumour cells to
aurora kinase inhibition. Oncotarget. 4:80–93. 2013.PubMed/NCBI
|
|
37
|
Dai Y, Chen S, Venditti CA, Pei XY, Nguyen
TK, Dent P and Grant S: Vorinostat synergistically potentiates
MK-0457 lethality in chronic myelogenous leukemia cells sensitive
and resistant to imatinib mesylate. Blood. 112:793–804. 2008.
View Article : Google Scholar : PubMed/NCBI
|