Introduction
According to cancer statistics 2014, breast cancer
is the top leading cancer in incidence (232,340 cases in USA) with
the second highest mortality rate (39,620 death in USA) in women in
the United States (1).
Triple-negative breast cancer (TNBC), comprising 10–20% of all
breast cancers, is a subgroup of breast cancer showing diverse and
heterogeneous features with lack of estrogen receptor (ER) and
progesterone receptor (PR) expression as well as human epidermal
growth factor receptor 2 (HER2) amplification (2,3) and
is inadequate to established hormonal therapy and/or HER2 targeted
therapy due to the lack of these proteins (4). The TNBC shows poor prognosis due to
aggressive biological behavior of tumors as well as earlier
involvement of distant metastasis (5). No proven optimistic therapies against
TNBCs are established yet and the development of new method on the
basis of the weak points of TNBCs is needed (6).
Epidermal growth factor receptor (EGFR) is a member
of membrane anchored receptor tyrosine kinase ERBB/HER family
comprising of EGFR, HER2, HER3 and HER4 (7,8). The
EGFR in normal cells is essential for cell proliferation and
survival. Aberrant activation of EGFR by copy number amplification,
protein overexpression or point mutation is closely related with
unregulated proliferation, malignant transformation, invasion,
metastasis and resistance to apoptosis of cancer cells (7,8). Up
to 70–80% of metastatic breast cancers shows overexpression of
EGFR, but without significant association of HER2 overexpression
(9,10). EGFR was found to be expressed at a
high level in ~50% of TNBCs and in ~70% of basal-like breast
cancers (11,12). Among the groups of TNBC classified
by Lehmann et al, basal-like 2 (BL2) and mesenchymal
stem-like (MSL) subtypes show active EGFR signaling (2). More than 50% of MSL type TNBC is
comprised of basal-like features according to intrinsic subtype
(13). The germline mutations of
BRCA1 and early onset of TNBC is also associated with EGFR
activation in breast cancers (14,15).
Along with cytokeratin 5/6 as a marker of basal-like breast
cancers, the EGFR expression is a marker of poor prognosis
regardless of the expression of ER or PR (12,14,16–18).
Nevertheless, the results from clinical trials with anti-EGFR
combined with platinum or other neoadjuvant agents revealed
disappointing results (19–21).
Gefitinib (Iressa) is an orally administrable
anticancer agent against EGFR kinase and shows efficacies against
various cancers with EGFR activation including breast, lung, colon
and other cancers (22–24). Although gefitinib has effects on
EGFR activated cancer cells, apparently most TNBC cells with
elevated level of EGFR exhibit resistance to EGFR inhibitor
treatment. Previously, we found that combination of gefitinib and
PI3K/AKT pathway inhibitors synergistically inhibit subsets of TNBC
cells in vitro (25). On
the contrary, regardless of high level expression of EGFR, TNBC
cells in MSL subtype including HS578T, MDA-MB-231, and MDA-MB-436
are relatively resistant to these combinations (25). Receptor tyrosine kinase crosstalk,
providing surrogate or redundant pathways of cell survival against
kinase targeted therapy, is one of the mechanisms of drug
resistance (26–31). As an attempt to identify potential
receptor tyrosine kinase inhibitors (RTKIs) which induce synthetic
lethality in the presence of gefitinib, we performed an MTT
screening in MDA-MB-231 cells. We further characterized a MET
(mesenchymal-epithelial transition factor) inhibitor SU11274 as a
synthetic lethal agent with gefitinib in MSL subtype TNBC
cells.
Materials and methods
Cell culture and reagents
Reagents for cell culture were purchased from
Invitrogen (Carlsbad, CA, USA), Lonza (Basel, Switzerland), or
Cellgro (Manassas, VA, USA). HS578T, MDA-MB-231, and MDA-MB-436
were obtained from the Tissue Culture Shared Resource of Georgetown
University Medical Center and maintained in the Dulbecco’s modified
Eagle’s medium (DMEM) (Lonza) containing 10% heat inactivated fetal
bovine serum (Omega Scientific, Inc., Tarzana, CA, USA) and 100
U/ml penicillin/streptomycin (Lonza). SUM149PT was maintained
according to the manufacturer’s recommendation (Asterand, Detroit,
MI, USA). The viability of cultured cells was monitored by the
trypan blue dye exclusion method using the Luna Automated Cell
Counter (Logos Biosystems, Gyunggi-Do, Korea). Receptor tyrosine
kinase inhibitors were purchased from the following sources: AEW541
from Cayman Chemical (Ann Arbor, MI, USA); AG1024 from Enzo Life
Sciences (Farmingdale, NY, USA); BMS-754807 and OSI-906 from MedKoo
Biosciences (Chapel Hill, NC, USA); ABT-869, AV-951, BAY 73-4506,
BMS-536924, BMS-599626, brivaninb, cediranib, CYC116, E-7080,
ENMD-2076, GSK1838705A, GSK1904529A, JNJ-38877605, LDN193189,
MGCD265, motesanib, MP-470, NVP-TAE684, OSI-930, PF-2341066
(crizotinib), PHA-665752, SB431542, SB525334, SU11274, Tie2 kinase
inhibitor, XL184, and XL880 from Selleck Chemicals (Houston, TX,
USA); axitinib, dovitinib, gefitinib, GW-2580, lapatinib,
lestaurtinib, masitinib, pazopanib, sorafenib, sunitinib,
tandutinib, vandetanib, and vatalanib from LC Labs (Woburn, MA,
USA). Genistein and MG132 was purchase from Sigma (St. Louis, MO,
USA). Stock solutions of compounds were made in dimethyl sulfoxide
(DMSO) and stored at −20°C in small aliquots.
Synthetic lethal screening
MDA-MB-231 cells (2,500 cells/ well) in 96-well
plates were treated with increasing amount of gefitinib and
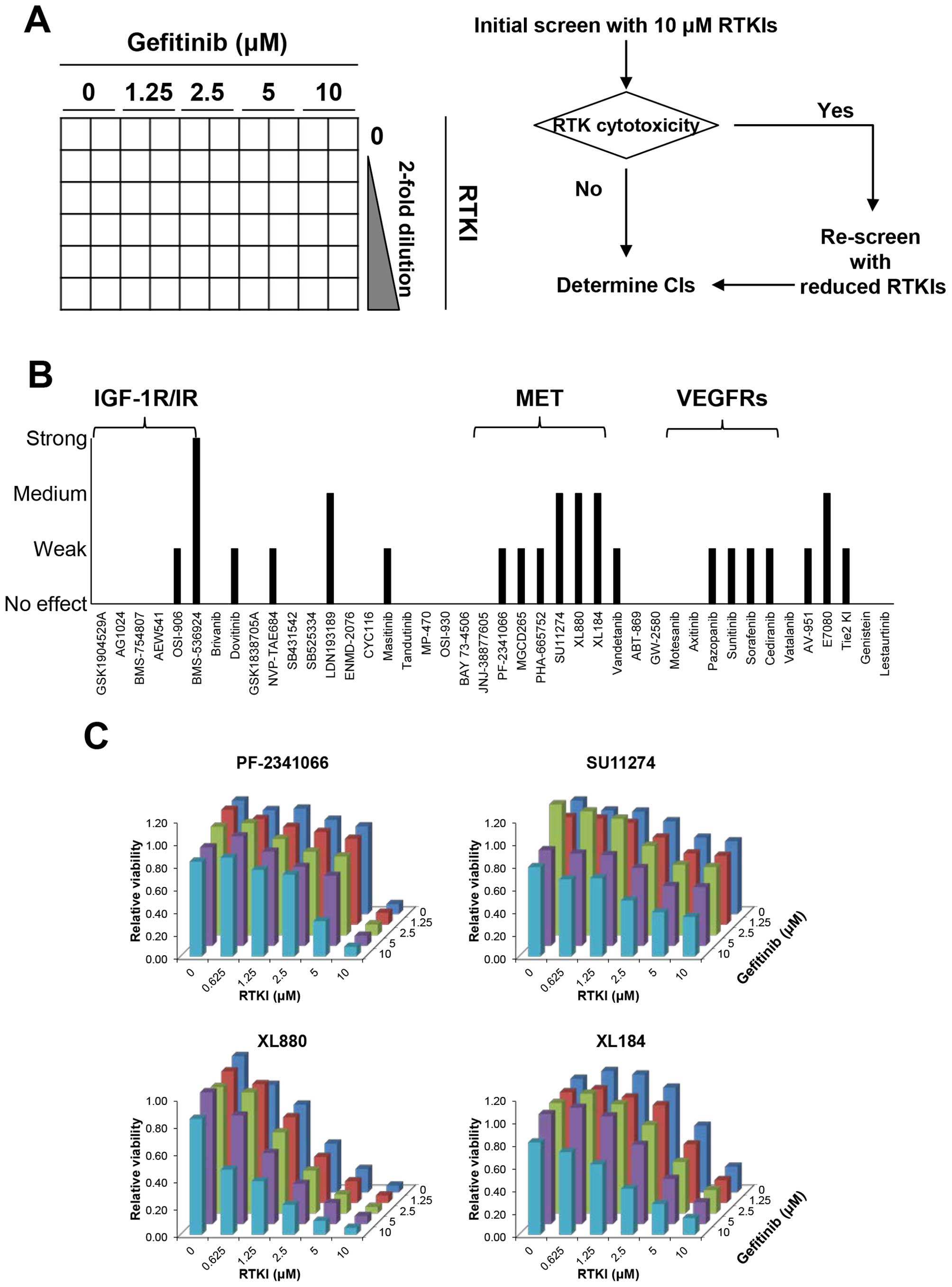
increasing amount of RTKIs in duplicates in a 6×5 matrix (Fig. 1A). In an initial screening, the
highest concentration of RTKIs was 10 μM. The highest
concentrations of RTKIs were reduced when significant reduction of
cell viability was observed in single agent treatments. The
synergism was determined by calculating classification index (CI)
with equation of A × B / AB, where A and
B are the cell viability with individual agent and AB
is the cell viability with the combination (32). We further indexed as follows:
strong synergism as index 3 when the CI>1.3 at >5 combination
points; medium synergism as index 2 when the CI>1.3 at 3 or 4
combination points; weak synergism as index 1 when the CI>1.3 at
1 or 2 combination points. Cell viability was determined at ~72 h
after treatment of compounds by MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)
assay as described previously except for using 4 mg/ml of MTT
solution (25,33).
Clonogenic cell survival assay
Cells were subcultured into 6-well plates with
appropriate densities: 500–1,000 cells/well for HS578T and 3,000
cells/well for MDA-MB-231. The day after subculture, the cells were
treated with indicated concentrations of compounds for 24 h, and
then the cells were supplemented with fresh normal growth media
without compounds. The cells were further cultured for 10–14 days
after treatment with replacement of fresh normal growth media twice
per week. The survived colonies were stained as described
previously (34). After intensive
washing, the images of colonies were captured by scanner. The
relative number of colonies was determined as follows: crystal
violet stain of colonies was solubilized by solubilization buffer
[1:1 mixture (v/v) of 0.1 M sodium phosphate
(NaH2PO4, pH 4.5) and ethanol] and the
observance of solubilized crystal violet was measured by ELx808
microplate reader (BioTek, Winooski, VT, USA).
Western blot analyses and antibodies
Western blot analyses were performed as described
previously (25). Antibodies used
in this study were as follows: MET (sc-161), ERK1 (sc-94), and PARP
(sc-7150) from Santa Cruz (Santa Cruz, CA, USA); p-EGFR (Y1068)
(#2237), EGFR (#4405), p-MET (Y1234/Y1235) (#3123), phospho-AKT
(Ser473) (#9271), AKT (#9272), p-ERK1/2 (T202/Y204) (#4370), p-p70
S6K (T389) (#9205), p70 S6K (#9202), p-S6 (S235/S236) (#4856), S6
(#2217) and XIAP (#2045) from Cell Signaling (Danvers, MA, USA);
α-tubulin, β-actin, and horseradish peroxidase-conjugated secondary
antibodies from Sigma.
Transfection of siRNA and cell
proliferation assay
Transfection of siRNA was performed with
Lipofectamine 2000 (from Invitrogen) as described previously
(35). In brief, HS578T
(0.4–0.6×105 cells/well) or MDA-MB-231
(1.0×105 cells/well) cells in 6-well plates were
transfected with 100 pmoles of siRNA mixed with 2.5 μl of
Lipofectamine 2000 in serum-free DMEM. After 4-h incubation, cells
were supplemented with equal volume of DMEM containing 20% FBS and
200 U/ml penicillin/streptomycin to maintain normal growth
condition and further incubated for 3 days. After 3-day incubation,
cells were further supplemented with equal volume of DMEM
containing 20% FBS and 200 U/ml penicillin/streptomycin and
incubated for ≤2 more days. The proliferation of cells was
determined by counting viable cells which were stained by acridine
orange (AO)/propidium iodide (PI) with the Luna-FLDual Fluorescence
Cell Counter (Logos Biosystems). The siRNAs were purchased from
Bioneer (Seoul, Korea) with following sequences: control-siRNA,
5′-GAC GAG CGG CAC GUG CAC AUU-3′; and RPS6-siRNA, 5′-GAA GCA GCG
UAC CAA GAA A(dTdT)-3′.
Cell cycle analysis
Cells were treated as indicated and the cells, both
attached and floating, were harvested to analyze the cell cycle at
the Flow Cytometry and Cell Sorting Shared Resource of Georgetown
University Medical Centers described previously (33).
Statistical analysis
The two-tailed Student’s t-test was applied for
statistical analysis. *P<0.05;
**P<0.01; ***P<0.001.
Results
Synthetic lethal screening of RTKIs in
MDA-MB-231 cells
Since our previous study identified synergistic
effects of EGFR and PI3K/AKT inhibition in a subset of TNBC cells
(25), we reasoned that
combination of kinase inhibitors with EGFR inhibition might induce
synthetic lethality in TNBC cells. We noted that TNBC cells of MSL
subtype showed innate resistance to EGFR inhibition. Because
overcoming resistance is an unmet need to treat human cancer
(36,37), we performed an MTT screening for
synthetic lethality with a small library of various RTKIs in
MDA-MB-231 cells in the presence of an EGFR inhibitor gefitinib
(Fig. 1A). We identified various
MET inhibitors (Table I) as
potential agents that induced synthetic lethal effects with
gefitinib in MDA-MB-231 cells (Fig.
1B). Gefitinib and MET inhibitor combinations synergistically
reduced the viable cells in MDA-MB-231 cells in a range of various
molar ratios of these drugs (Fig.
1C).
 | Table IMET inhibitors identified in this
study. |
Table I
MET inhibitors identified in this
study.
| Inhibitor | Other name | Known targets
(IC50 in nM) | (Refs.) |
|---|
| PF-2341066 | Crizotinib | MET (4), ALK
(24) | (72) |
| MGCD265 | | MET (1), VEGFR1/2
(3) VEGFR3 (4), RON (2), TIE2 (7) | (73) |
| PHA-665752 | | MET (9) | (41) |
| SU11274 | | MET (10), RON
(4000) | (39) |
| XL880 | Foretinib
EXEL-2880
GSK1369089 | MET (0.4), RON (3),
VEGFR2 (0.86), FLT1 (6.8), FLT4 (2.8),
KIT (6.7), FLT3 (3.6), PDGFRα (3.6), PDGFRβ (9.6),
TIE2 (1.1), FGFRI1 (660) | (74) |
| XL184 | Cabozantinib | Met (1.3), VEGFR2
(0.035), KIT (4.6), FLT1 (12), FLT3 (11.3), FLT4 (6), Tie2 (14.3),
Ret (4) | (75) |
Cytotoxic effect of EGFR and MET
inhibitors in human TNBC cell lines
One distinct feature of TNBC is overexpression of
EGFR (6,11,12).
In addition, a recent study reported that high level expression of
MET is an adverse prognostic factor in TNBC patients (38). We determined the level of these
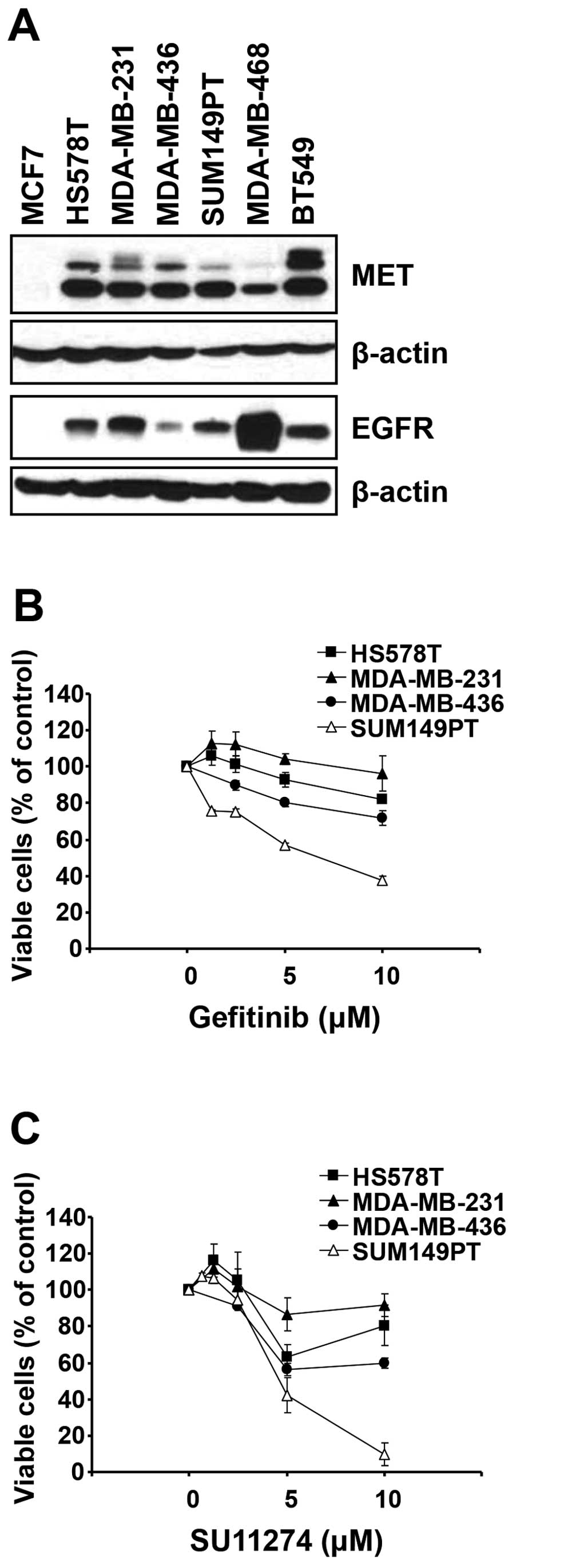
proteins in a set of TNBC cell lines by western blot analysis. As
reported previously (25), the
level of EGFR was high in all TNBC cell lines tested compared to
the luminal breast cancer cell line MCF7 (Fig. 2A). The level of MET was also higher
in TNBC cells than in MCF7 (Fig.
2A).
Since the levels of both EGFR and MET are elevated
in human TNBC cell lines, we further determined the cytotoxic
effect of EGFR and MET inhibitors as a single agent toward four
different human TNBC cell lines. The cells were treated with the
EGFR inhibitor gefitinib or the MET inhibitor SU11274 (39) for ~72 h and the viable cells were
determined by MTT cell viability assay. Consistent with a previous
report (25), three cell lines
(HS578T, MDA-MB-231 and MDA-MB-436) of MSL subtype were relatively
resistant to gefitinib compared to a BL2 subtype cell line SUM149PT
(Fig. 2B). Gefitinib reduced the
viable SUM149PT cells in a dose-dependent manner. On the contrary,
the effect of gefitinib was limited on three cell lines of MSL
subtype. Additionally, three MSL subtype cell lines were more
resistant to SU11274 than SUM149PT cell line (Fig. 2C). Notably, near complete loss of
viable cells was observed in SUM149PT cells treated with 10 μM of
SU11274, while the effect of SU11274 was less potent toward HS578T,
MDA-MB-231 and MDA-MB-436 cells.
Synergistic cytotoxic effect of EGFR/MET
inhibitor combination in human TNBC cell lines
Since MET inhibitors were identified as potent
synthetic lethal agents in combination of gefitinib, we further
determined whether the combination of EGFR/MET inhibitors has any
beneficial effect in treatment of human TNBC cell lines of MSL
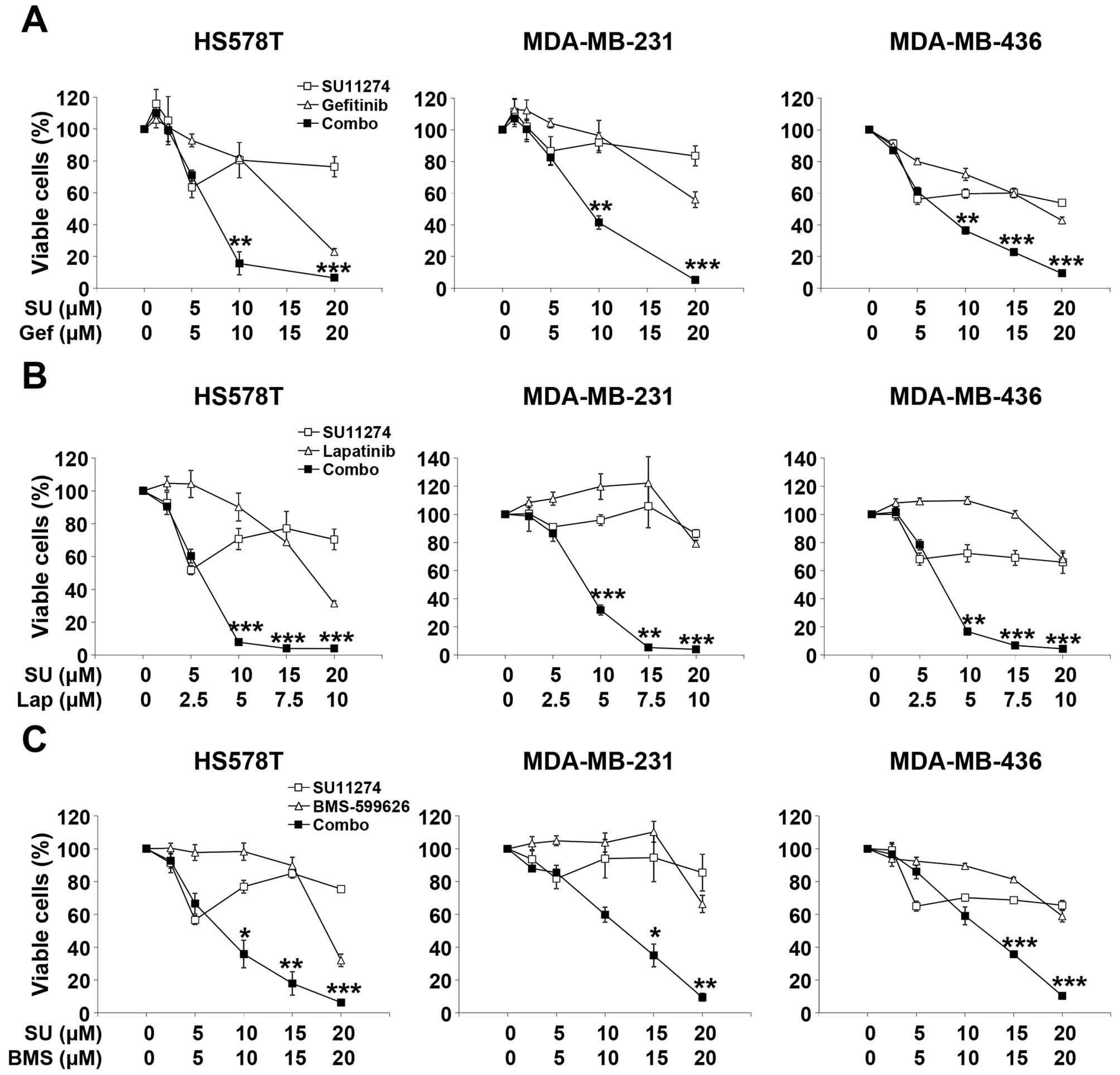
subtype. Three types of TNBC cells were treated with increasing
concentrations of both EGFR (gefitinib) and MET (SU11274)
inhibitors for ~72 h and the viable cells were determined by MTT
assay. Combination of gefitinib and SU11274 with fixed molar ratio
of 1:1 markedly reduced the viable cells in HS578T, MDA-MB-231, and
MDA-MB-436 cells (Fig. 3A). In
addition to gefitinib, lapatinib and BMS-599626 (EGFR/HER2 dual
inhibitors) also showed marked synergism with SU11274 (Fig. 3B and C). These results suggest that
combination of EGFR/MET inhibitors synergistically reduces the cell
viability of MSL subtype cell lines of human TNBC.
Gefitinib/SU11274 combination reduces the
survival of human TNBC cell lines
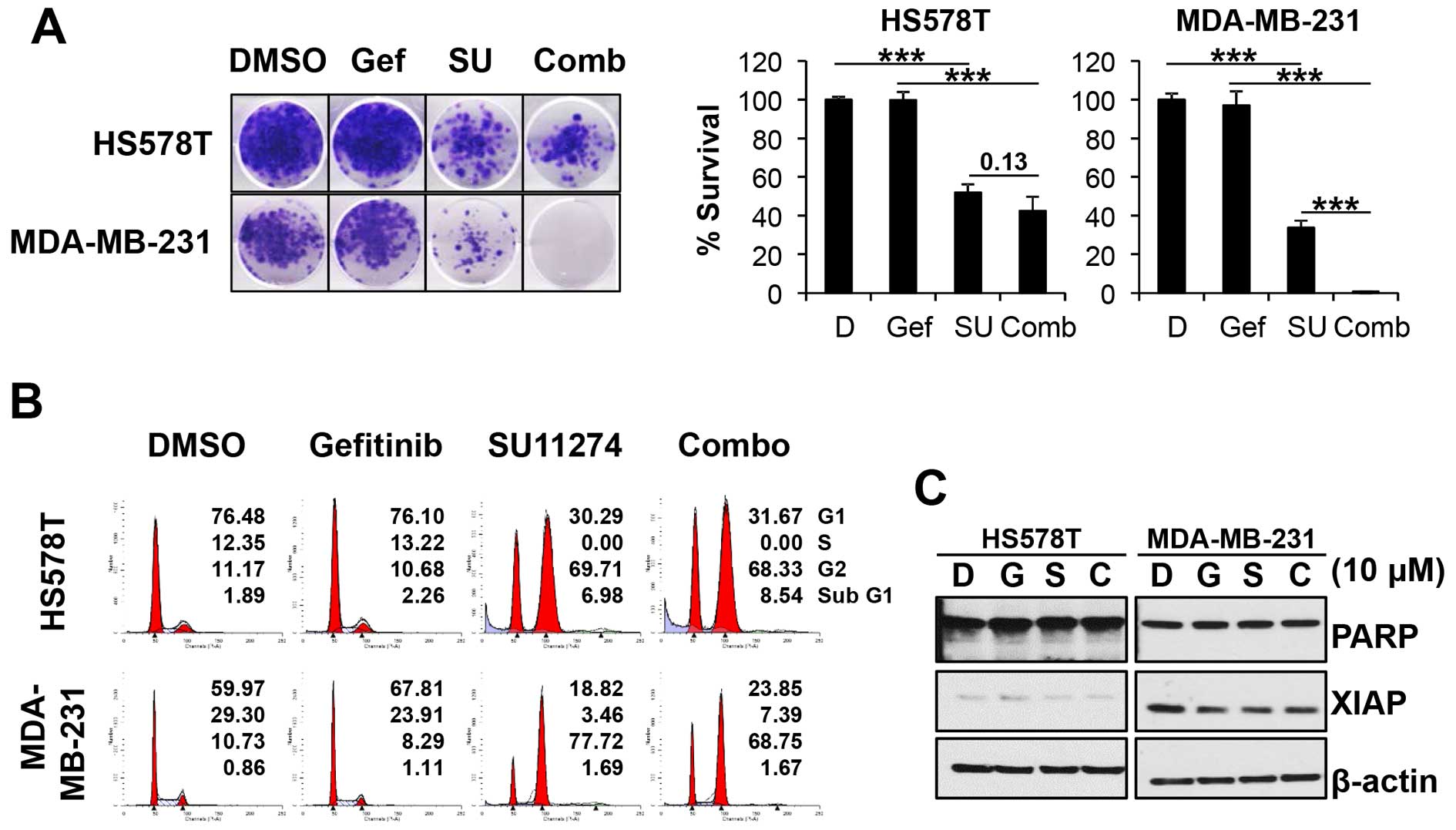
The effect of gefitinib/SU11274 combination was
further evaluated by clonogenic cell survival assay. TNBC cells
were subcultured in 6-well plates in an appropriate density and
treated with drug combinations for 24 h. After wash out of drugs,
cells were further cultivated in normal growth media. As shown in
Fig. 4A, gefitinib alone could not
suppress the number of survived colonies in either HS578T or
MDA-MB-231 cells. On the contrary, SU11274, as a single agent,
significantly reduced the number of surviving colonies. Consistent
with MTT assay, gefitinib/SU11274 combination reduced the colony
formation in both cell lines.
The effect of gefitinib/SU11274 combination on the
cell cycle distribution was further analyzed. Cells were treated
with drugs for 24 h and the cells, both attached and floating, were
collected to determine the cell cycle. As shown in Fig. 4B, gefitinib alone could not
significantly affect the cell cycle distribution of HS578T and
MDA-MB-231 cells. However, treatment of SU11274 markedly induced
the accumulation of G2 accompanying by reduction of both G1 and S
phase in both cell lines. The cell cycle distribution induced by
SU11274 was sustained in the cells which were treated with
gefitinib/ SU11274 combination.
To detect apoptotic cell death, we further analyzed
the Poly (ADP-ribose) polymerase (PARP) cleavage and the level of
X-linked inhibitor of apoptosis protein (XIAP) by western blot
analysis. HS578T and MDA-MB-231 cells were treated with compounds
for 24 h and the lysates were subjected to western blot analysis.
As shown in Fig. 4C, no apparent
induction of PARP cleavage was observed. The level of XIAP protein
was also marginally reduced by gefitinib/SU11274 combination.
Combination of gefitinib/SU11274
synergistically reduces the level of RPS6 in MSL subtype TNBC
cells
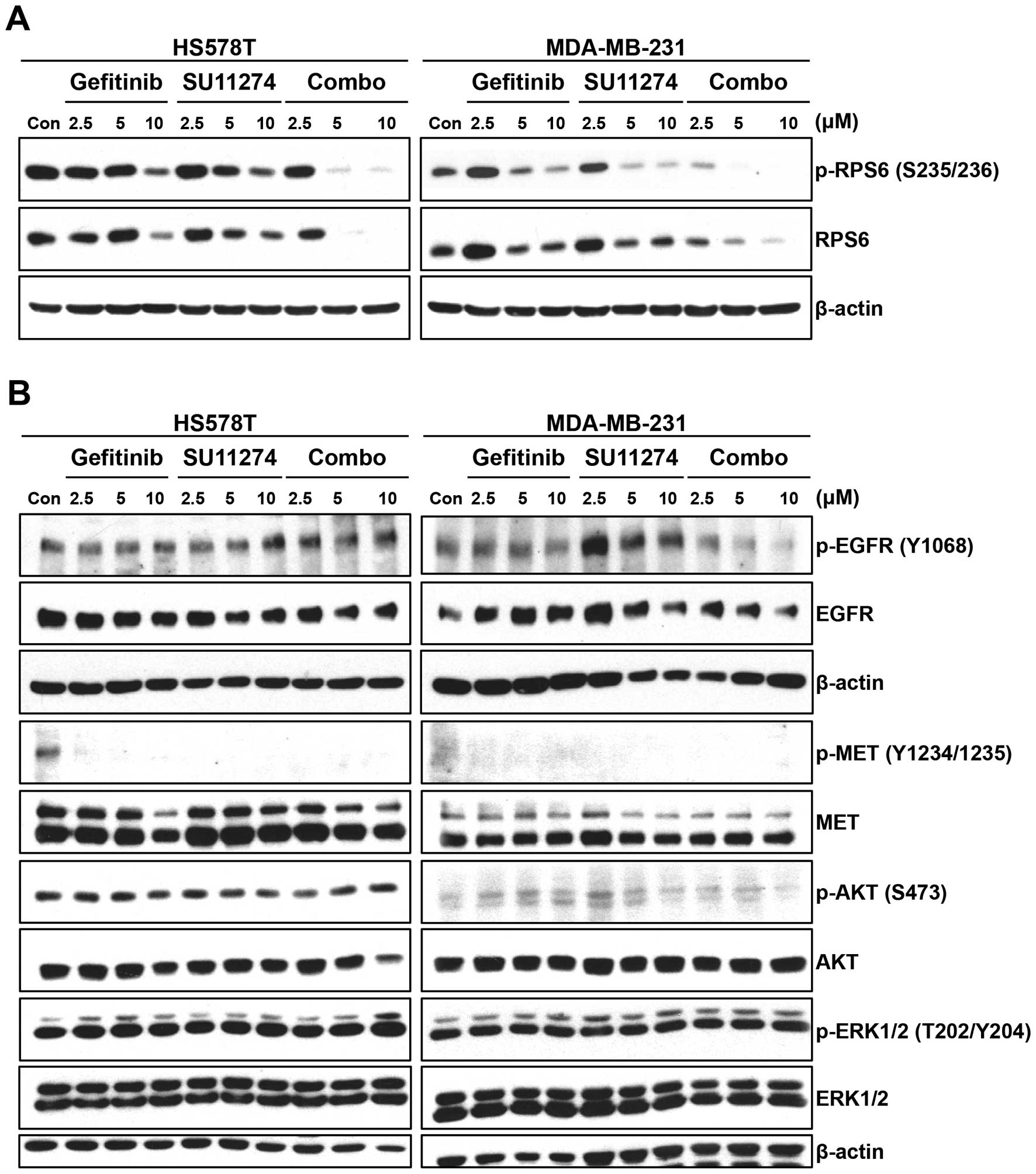
To determine signaling pathways mediating the
gefitinib/SU11274 effect, we performed a series of western blot
analyses. HS578T and MDA-MB-231 cells were treated with increasing
concentrations of drugs for 24 h, either single agents or
combination, then the lysates from these cells were subjected to
western blot analysis. Interestingly, single agent treatment,
either gefitinib or SU11274 for 24 h, reduced the level of
phospho-ribosomal protein S6 (RPS6) (S235/S236) in HS578T and
MDA-MB-231 cells in a dose-dependent manner (Fig. 5A). In addition, gefitinib/SU11274
combination synergistically reduced the level of phospho-RPS6 in
these cells. Surprisingly, the level of RPS6 protein itself was
reduced by these drugs as single agents and further reduced by
combination treatment.
Unexpectedly, 24-h treatment of gefitinib did not
reduce the level of phospho-EGFR (Y1068) in these cells, while
gefitinib/ SU11274 combination reduced the level of phospho-EGFR
(Y1068) only in MDA-MB-231 cells (Fig.
5B). As expected, SU11274 reduced the level of phospho-MET
(Y1234/Y1235) in these cells (Fig.
5B). In addition, the level of phospho-MET was also reduced by
gefitinib in both cell types. However, neither gefitinib nor
SU11274 could reduce the levels of phospho-AKT (S473) and
phospho-ERK1/2 (T202/Y204). The gefitinib/SU11274 combination could
not reduce either phospho-AKT or phospho-ERK1/2 (Fig. 5B). These results suggest that 24-h
treatment of gefitinib or SU11274 could not inhibit the AKT and ERK
pathways in these cell lines.
Gefitinib/SU11274 combination reduces the
level of RPS6 in a proteasome-independent manner
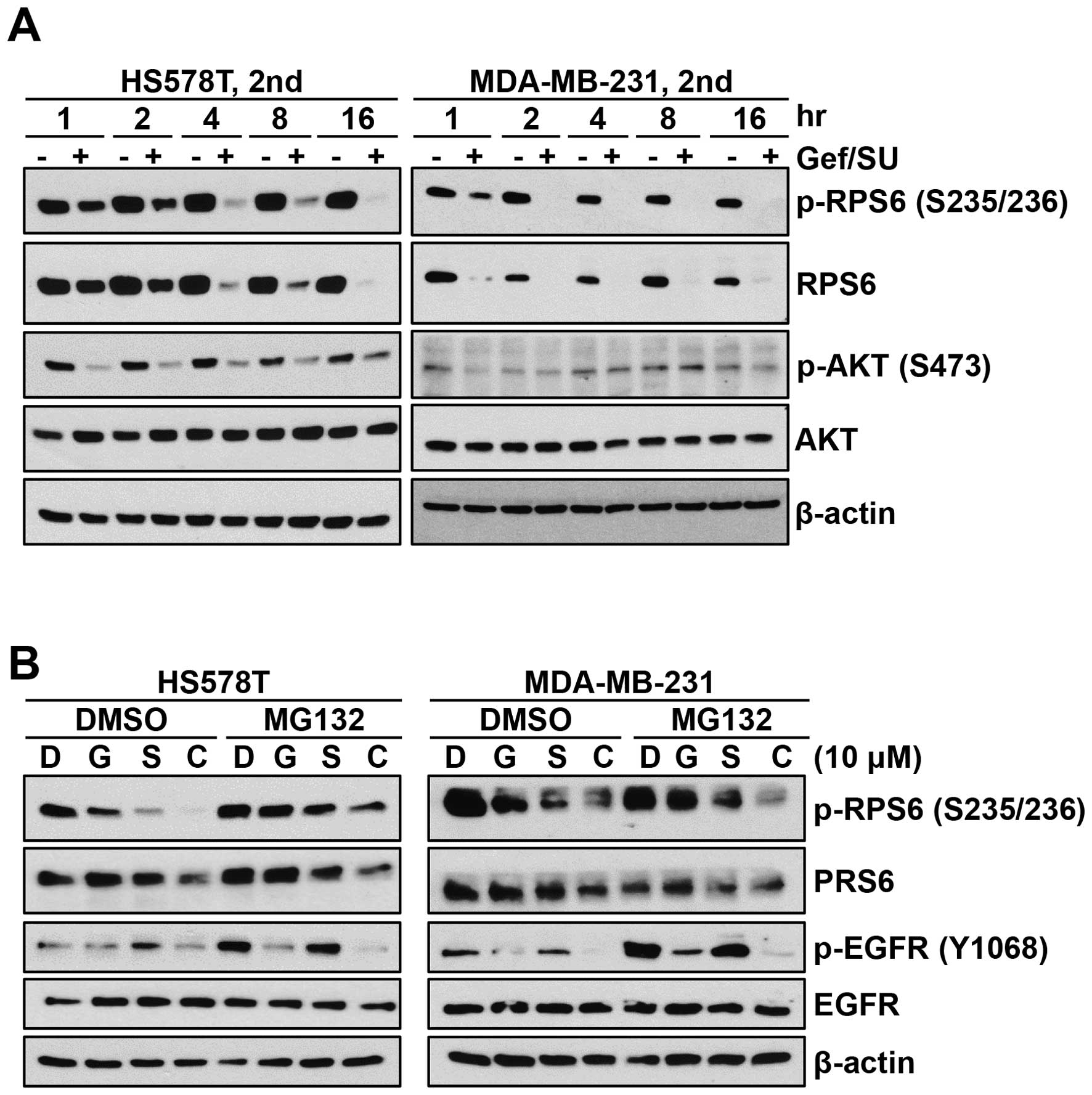
To determine the level of RPS6 over time, cells were
treated with gefitinib/SU11274 combination for several time
intervals and the level of RPS6 proteins was detected by western
blot analysis (Fig. 6A).
Interestingly, the decrease of both phospho-RPS6 (S235/236) and
RPS6 itself was evident as early as 1 h after treatment (Fig. 6A). In addtion, the decrease of RPS6
protein level was sustained for up to 16 h. The level of
phospho-AKT (S473) was decreased at 1 h after combination
treatment. However, the decrease of phospho-AKT (S473) was reversed
over time in both cell lines (Fig.
6A).
The effect of proteasome inhibition on the level of
RPS6 was also determined by western blot analysis (Fig. 6B). Cells were treated with 10 μM of
either gefitinib or SU11274 and combination of both drugs for 4 h
in the presence of the proteasome inhibitor MG132. Consistently,
gefitinib/SU11274 combination markedly reduced the level of RPS6 in
both cell lines. However, the treatment of MG132 did not affect
gefitinib/SU11274-mediated reduction of RPS6 (Fig. 6B). Contrary to 24-h treatment, 4-h
treatment of 10 μM gefitinib reduced the level of phospho-EGFR
(Y1068) in these cells. In addition, gefitinib/SU11274 combination
further reduced the level of phospho-EGFR. These results suggest
that gefitinib/ SU11274 combination induces irreversible reduction
of RPS6 in a proteasome-independent manner.
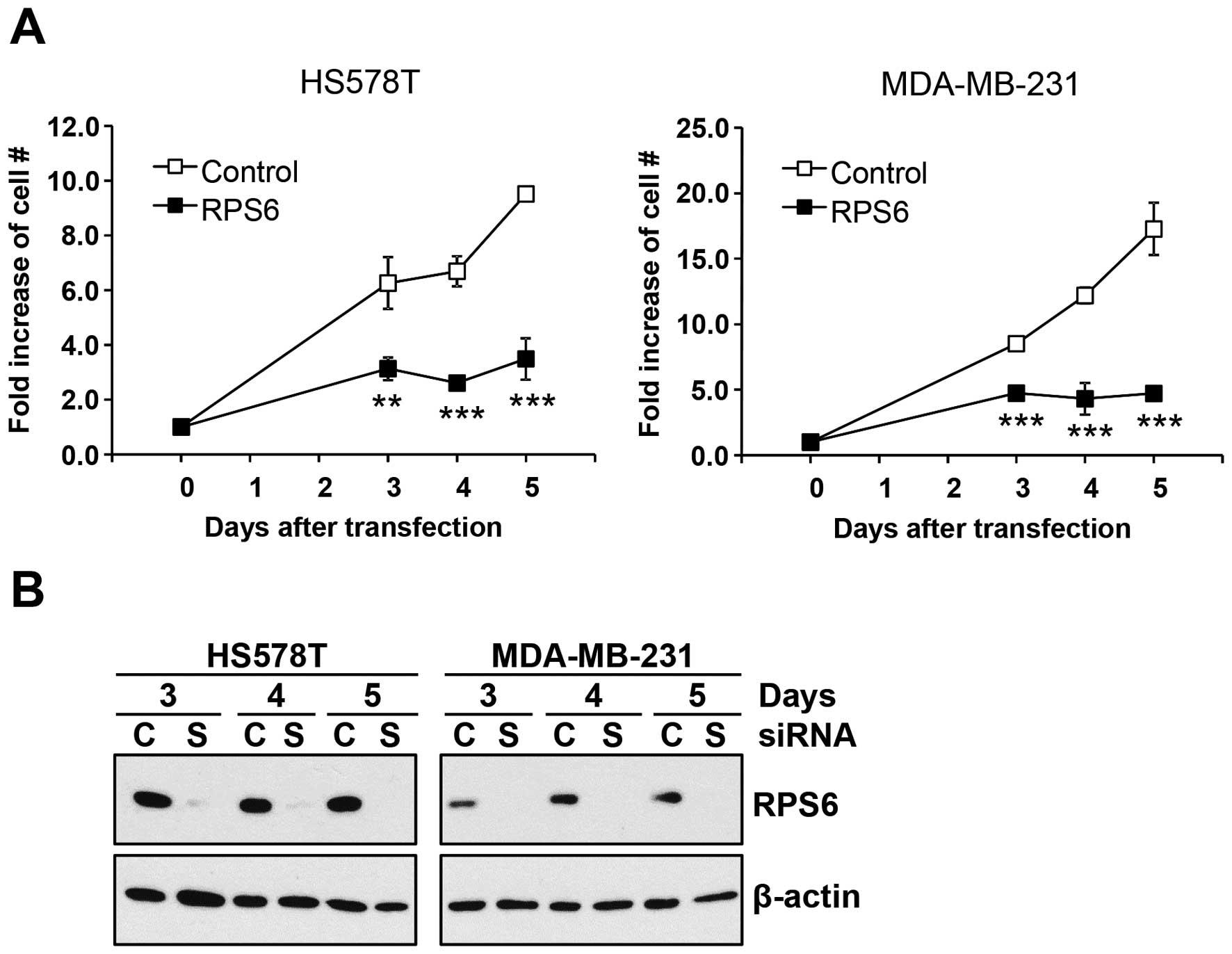
Knockdown of RPS6 reduces the
proliferation of TNBC cells
Since gefitinib/SU11274 combination synergistically
reduced the level of RPS6 in MSL subtype TNBC cells, we questioned
whether RPS6 is important to the proliferation of these cells. To
address this, we knocked down the RPS6 protein by specific siRNA.
HS578T cells and MDA-MB-231 cells were transfected with either
control- or RPS6-siRNA and cultivated for up to 5 days. The
difference of cell proliferation was traced by viable cell counting
at indicated days. As shown in Fig.
7A, knockdown of RPS6 profoundly reduced the proliferation of
both HS578T and MDA-MB-231 cells as early as 3 days after siRNA
transfection. Western blot analysis confirmed the knockdown of RPS6
under these conditions (Fig. 7B).
Taken together, our data suggest that gefitinib/SU11274 combination
reduced the proliferation of a subset of TNBC cells through
downregulation of RPS6 proteins.
Discussion
In the present study, we demonstrated that the MET
inhibitor SU11274 is a synthetic lethal agent in the combination
with EGFR inhibitors for the MSL subtype of TNBC cells. The levels
of EGFR and MET are highly elevated in TNBC cells tested.
Nevertheless, EGFR inhibitors (gefitinib, lapatinib, and
BMS-599626) and SU11274 has limited potency in TNBC cells of MSL
subtype such as HS578T, MDA-MB-231, and MDA-MB-436 in MTT assay.
However, the combination of these drugs markedly reduced the viable
cells in MTT assays and survival of these cells in clonogenic
assays. One notable feature of these combinations is the reduction
of RPS6 protein levels. Treatment of gefitinib/SU11274 combination
for 24 h did not affect various signaling pathways including AKT
and ERK. However, the level of phospho-RPS6 (S235/236) was
synergistically reduced by this combination. The reduction of
phospho-RPS6 (S235/236) was due to the reduction of RPS6 protein
itself as early as 1 h after combination treatment. Although the
level of phospho-AKT (S473) was reduced by this combination in
early time points, it was reversed over time and near completely
recovered at 16 h after treatment. On the contrary, the initial
reduction of RPS6 protein level was maintained over time.
Proteasome inhibition did not reverse the reduction of RPS6 by
gefitinib/ SU11274 combination. Interestingly, siRNA-based
knockdown of RPS6 itself was enough to reduce the proliferation of
HS578T and MDA-MB-231 cells. Taken together our data suggest that
dual inhibition of EGFR and MET induces synthetic lethality in a
subtype of TNBC cells through downregulation of RPS6 protein.
MET, a member of receptor tyrosine kinase, is
activated by hepatocyte growth factor/scatter factor (HGF/SF)
(40). The binding of HGF to MET
activates various signal pathways including RAS/MAPK, PI3K/AKT, SRC
and STAT3/5 and these signal pathways mediate normal cell
proliferation, cell scattering, invasion, migration, embryogenesis,
evading apoptosis, angiogenesis, and tissue regeneration (41). MET and HGF are highly expressed in
a wide variety of cancers including lung, ovary, renal, gastric,
pancreas, head and neck and colon cancers and are also considered
to contribute to unregulated cell proliferation, reduced apoptosis,
altered cytoskeletal function, tumor cell scattering, migration,
dissemination, and invasion during cancer cell metastasis (41–43).
However, the roles of MET in the proliferation and/or survival of
TNBC cells is largely unappreciated. A recent study demonstrated
that paracrine activation of MET by fibroblast-secreted HGF induces
gefitinib resistance in two TNBC cell lines, SUM102 and SUM149PT
(44). It has also been reported
that MET is colocalized with AXL receptor kinase complex which
includes EGFR, HER2/3, MET and platelet-derived growth factor
receptor β (PDGFRβ) in TNBC cells (45). Inhibition of MET was also reported
as a potential opportunity of Notch targeting for TNBC patients
with MET overexpression and Notch hyper-activation (46). More recently, the MET inhibitor
PHA-665752 with the EGFR inhibitor erlotinib was demonstrated to
reduce the viability of the BL1 subtype TNBC MDA-MB-468 cells
(47). In the present study, we
found that 10 μM treatment of the MET inhibitor SU11274 exhibited a
limited potency toward MSL subtype TNBC cells in MTT assay, while
24-h treatment of 10 μM SU11274 showed significant reduction of
TNBC cell survival in clonogenic assay. Interestingly, SU11274
alone induced significant increase of cells in G2 phase of cell
cycle. These results suggest that MET itself might have potential
role in the regulation of the cell cycle and/or long-term survival
of MSL subtype TNBC cells. Further study will be needed to decipher
the role of MET in the proliferation and/or survival of TNBC
cells.
Our present data suggest that co-targeting EGFR and
MET trigger an irreversible reduction of RPS6 protein: while the
inhibition of upstream signaling pathway such as EGFR and AKT was
reversed with time, the initial reduction of RPS6 protein level was
sustained. In addition, knockdown of RPS6 itself significantly
reduced the proliferation of MSL subtype TNBC cells in the present
study. These results suggest that reduction of RPS6 by
gefitinib/SU11274 combination is sufficient to inhibit the
proliferation of MSL subtype TNBC cells. RPS6 protein is
evolutionarily conserved from yeast to vertebrate and indispensable
for protein synthesis (48).
Despite the fact that increased phosphorylation and mRNA
upregulation of RPS6 has been reported in several human cancers
(49–61), the role of RPS6 in cancer
initiation and/or progression has not been well appreciated.
Recently, the potential implication of RPS6 in human cancer was
revealed by knockdown experiments. Knockdown of RPS6 by siRNA
reduced the survival of Ewing family tumor cell lines with near
complete cell death in a siRNA library screening (62). Knockdown of RPS6 by shRNA was also
reported to reduce the proliferation of diffuse large B-cell
lymphoma (DLBCL) cell lines (57).
Phospho-RPS6 has been reported to attenuate KRAS-induced DNA damage
in acinar cells and in acinar-to-ductal metaplasia (ADM) and
p53-mediated tumor suppression during initiation of pancreatic
cancer (63). Collectively,
targeting RPS6 may provide alternative therapeutic regimen to treat
human cancers with high level of RPS6.
RPS6 protein is phosphorylated at multiple sites by
various upstream kinases such as RPS6 kinase α1 (RPS6KA1), RPS6KA3,
death-associated protein kinase 1 (DAPK1), and PAS domain
containing serine/threonine kinase (PASK) (64–69).
The phosphorylation of RPS6 is involved in the regulation of global
protein synthesis that determines the size of cells, cell
proliferation, and glucose homeostasis (48). RPS6 is also known to be important
to cap-dependent protein translation (67). Unfortunately, the regulation of
RPS6 protein stability has not yet been explored. Heat shock
protein 90 (HSP90), a molecular chaperone, binds to RPS6 (70) and regulates its degradation through
ubiquitin-dependent proteolysis (71). In addition, the HSP90 inhibitor
geldanamycin reduces the level of RPS6 (71).
In the present study, the reduction of RPS6 by
gefitinib/ SU11274 combination was not reversed by the proteasome
inhibitor MG132. Since the reduction of RPS6 itself was as rapid as
1 h after treatment, it is plausible that active proteolysis
regulates the gefitinib/SU11274-mediated reduction of RPS6 level.
Further study is needed to address how RPS6 stability is regulated
by this combination in TNBC cells.
Acknowledgements
This study was supported by the Susan G. Komen for
the Cure (FAS0703858) and by the Lombardi Comprehensive Cancer
Center, Georgetown University Medical Center (P30-CA051008) awarded
to the late Professor Insoo Bae.
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lehmann BD, Bauer JA, Chen X, Sanders ME,
Chakravarthy AB, Shyr Y and Pietenpol JA: Identification of human
triple-negative breast cancer subtypes and preclinical models for
selection of targeted therapies. J Clin Invest. 121:2750–2767.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morris GJ, Naidu S, Topham AK, Guiles F,
Xu Y, McCue P, Schwartz GF, Park PK, Rosenberg AL, Brill K, et al:
Differences in breast carcinoma characteristics in newly diagnosed
African-American and Caucasian patients: A single-institution
compilation compared with the National Cancer Institute’s
Surveillance, Epidemiology, and End Results database. Cancer.
110:876–884. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Irvin WJ Jr and Carey LA: What is
triple-negative breast cancer? Eur J Cancer. 44:2799–2805. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liedtke C, Mazouni C, Hess KR, André F,
Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B,
Green M, et al: Response to neoadjuvant therapy and long-term
survival in patients with triple-negative breast cancer. J Clin
Oncol. 26:1275–1281. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Podo F, Buydens LM, Degani H, Hilhorst R,
Klipp E, Gribbestad IS, Van Huffel S, van Laarhoven HW, Luts J,
Monleon D, et al: FEMME Consortium: Triple-negative breast cancer:
Present challenges and new perspectives. Mol Oncol. 4:209–229.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eccles SA: The epidermal growth factor
receptor/Erb-B/HER family in normal and malignant breast biology.
Int J Dev Biol. 55:685–696. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yarden Y and Pines G: The ERBB network: At
last, cancer therapy meets systems biology. Nat Rev Cancer.
12:553–563. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reis-Filho JS, Milanezi F, Carvalho S,
Simpson PT, Steele D, Savage K, Lambros MB, Pereira EM, Nesland JM,
Lakhani SR, et al: Metaplastic breast carcinomas exhibit EGFR, but
not HER2, gene amplification and overexpression:
Immunohistochemical and chromogenic in situ hybridization analysis.
Breast Cancer Res. 7:R1028–R1035. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reis-Filho JS, Pinheiro C, Lambros MB,
Milanezi F, Carvalho S, Savage K, Simpson PT, Jones C, Swift S,
Mackay A, et al: EGFR amplification and lack of activating
mutations in metaplastic breast carcinomas. J Pathol. 209:445–453.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Livasy CA, Karaca G, Nanda R, Tretiakova
MS, Olopade OI, Moore DT and Perou CM: Phenotypic evaluation of the
basal-like subtype of invasive breast carcinoma. Mod Pathol.
19:264–271. 2006. View Article : Google Scholar
|
|
12
|
Nielsen TO, Hsu FD, Jensen K, Cheang M,
Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler
L, et al: Immunohistochemical and clinical characterization of the
basal-like subtype of invasive breast carcinoma. Clin Cancer Res.
10:5367–5374. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lehmann BD and Pietenpol JA:
Identification and use of biomarkers in treatment strategies for
triple-negative breast cancer subtypes. J Pathol. 232:142–150.
2014. View Article : Google Scholar :
|
|
14
|
Dawson SJ, Provenzano E and Caldas C:
Triple negative breast cancers: Clinical and prognostic
implications. Eur J Cancer. 45(Suppl 1): 27–40. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lakhani SR, Reis-Filho JS, Fulford L,
Penault-Llorca F, van der Vijver M, Parry S, Bishop T, Benitez J,
Rivas C, Bignon YJ, et al: Prediction of BRCA1 status in patients
with breast cancer using estrogen receptor and basal phenotype.
Clin Cancer Res. 11:5175–5180. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cheang MC, Voduc D, Bajdik C, Leung S,
McKinney S, Chia SK, Perou CM and Nielsen TO: Basal-like breast
cancer defined by five biomarkers has superior prognostic value
than triple-negative phenotype. Clin Cancer Res. 14:1368–1376.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rakha EA, El-Sayed ME, Green AR, Lee AH,
Robertson JF and Ellis IO: Prognostic markers in triple-negative
breast cancer. Cancer. 109:25–32. 2007. View Article : Google Scholar
|
|
18
|
Tischkowitz M, Brunet JS, Bégin LR,
Huntsman DG, Cheang MC, Akslen LA, Nielsen TO and Foulkes WD: Use
of immunohistochemical markers can refine prognosis in triple
negative breast cancer. BMC Cancer. 7:1342007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baselga J, Gómez P, Greil R, Braga S,
Climent MA, Wardley AM, Kaufman B, Stemmer SM, Pêgo A, Chan A, et
al: Randomized phase II study of the anti-epidermal growth factor
receptor monoclonal antibody cetuximab with cisplatin versus
cisplatin alone in patients with metastatic triple-negative breast
cancer. J Clin Oncol. 31:2586–2592. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bernsdorf M, Ingvar C, Jörgensen L, Tuxen
MK, Jakobsen EH, Saetersdal A, Kimper-Karl ML, Kroman N, Balslev E
and Ejlertsen B: Effect of adding gefitinib to neoadjuvant
chemotherapy in estrogen receptor negative early breast cancer in a
randomized phase II trial. Breast Cancer Res Treat. 126:463–470.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Carey LA, Rugo HS, Marcom PK, Mayer EL,
Esteva FJ, Ma CX, Liu MC, Storniolo AM, Rimawi MF, Forero-Torres A,
et al: TBCRC 001: Randomized phase II study of cetuximab in
combination with carboplatin in stage IV triple-negative breast
cancer. J Clin Oncol. 30:2615–2623. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Anderson NG, Ahmad T, Chan K, Dobson R and
Bundred NJ: ZD1839 (Iressa), a novel epidermal growth factor
receptor (EGFR) tyrosine kinase inhibitor, potently inhibits the
growth of EGFR-positive cancer cell lines with or without erbB2
overexpression. Int J Cancer. 94:774–782. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ciardiello F, Caputo R, Bianco R, Damiano
V, Pomatico G, De Placido S, Bianco AR and Tortora G: Antitumor
effect and potentiation of cytotoxic drugs activity in human cancer
cells by ZD-1839 (Iressa), an epidermal growth factor
receptor-selective tyrosine kinase inhibitor. Clin Cancer Res.
6:2053–2063. 2000.PubMed/NCBI
|
|
24
|
Wakeling AE, Guy SP, Woodburn JR, Ashton
SE, Curry BJ, Barker AJ and Gibson KH: ZD1839 (Iressa): An orally
active inhibitor of epidermal growth factor signaling with
potential for cancer therapy. Cancer Res. 62:5749–5754.
2002.PubMed/NCBI
|
|
25
|
Yi YW, Hong W, Kang HJ, Kim HJ, Zhao W,
Wang A, Seong YS and Bae I: Inhibition of the PI3K/AKT pathway
potentiates cytotoxicity of EGFR kinase inhibitors in
triple-negative breast cancer cells. J Cell Mol Med. 17:648–656.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jin Q and Esteva FJ: Cross-talk between
the ErbB/HER family and the type I insulin-like growth factor
receptor signaling pathway in breast cancer. J Mammary Gland Biol
Neoplasia. 13:485–498. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Karamouzis MV, Konstantinopoulos PA and
Papavassiliou AG: Targeting MET as a strategy to overcome
crosstalk-related resistance to EGFR inhibitors. Lancet Oncol.
10:709–717. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu P, Cheng H, Roberts TM and Zhao JJ:
Targeting the phos-phoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nahta R, Yu D, Hung MC, Hortobagyi GN and
Esteva FJ: Mechanisms of disease: Understanding resistance to
HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol.
3:269–280. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yamaguchi H, Chang SS, Hsu JL and Hung MC:
Signaling crosstalk in the resistance to HER family receptor
targeted therapy. Oncogene. 33:1073–1081. 2014. View Article : Google Scholar
|
|
31
|
Baselga J: Targeting tyrosine kinases in
cancer: The second wave. Science. 312:1175–1178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Morgillo F, Kim WY, Kim ES, Ciardiello F,
Hong WK and Lee HY: Implication of the insulin-like growth
factor-IR pathway in the resistance of non-small cell lung cancer
cells to treatment with gefitinib. Clin Cancer Res. 13:2795–2803.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yi YW, Kang HJ, Kim HJ, Kong Y, Brown ML
and Bae I: Targeting mutant p53 by a SIRT1 activator YK-3-237
inhibits the proliferation of triple-negative breast cancer cells.
Oncotarget. 4:984–994. 2013.PubMed/NCBI
|
|
34
|
Duong HQ, Yi YW, Kang HJ, Hong YB, Tang W,
Wang A, Seong YS and Bae I: Inhibition of NRF2 by PIK-75 augments
sensitivity of pancreatic cancer cells to gemcitabine. Int J Oncol.
44:959–969. 2014.
|
|
35
|
Hou S, Yi YW, Kang HJ, Zhang L, Kim HJ,
Kong Y, Liu Y, Wang K, Kong HS, Grindrod S, et al: Novel carbazole
inhibits phospho-STAT3 through induction of protein-tyrosine
phosphatase PTPN6. J Med Chem. 57:6342–6353. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lovly CM and Shaw AT: Molecular pathways:
resistance to kinase inhibitors and implications for therapeutic
strategies. Clin Cancer Res. 20:2249–2256. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Szakács G, Paterson JK, Ludwig JA,
Booth-Genthe C and Gottesman MM: Targeting multidrug resistance in
cancer. Nat Rev Drug Discov. 5:219–234. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zagouri F, Bago-Horvath Z, Rössler F,
Brandstetter A, Bartsch R, Papadimitriou CA, Dimitrakakis C,
Tsigginou A, Papaspyrou I, Giannos A, et al: High MET expression is
an adverse prognostic factor in patients with triple-negative
breast cancer. Br J Cancer. 108:1100–1105. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang X, Le P, Liang C, Chan J, Kiewlich D,
Miller T, Harris D, Sun L, Rice A, Vasile S, et al: Potent and
selective inhibitors of the Met [hepatocyte growth factor/scatter
factor (HGF/SF) receptor] tyrosine kinase block HGF/SF-induced
tumor cell growth and invasion. Mol Cancer Ther. 2:1085–1092.
2003.PubMed/NCBI
|
|
40
|
Ma PC, Maulik G, Christensen J and Salgia
R: c-Met: Structure, functions and potential for therapeutic
inhibition. Cancer Metastasis Rev. 22:309–325. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Christensen JG, Schreck R, Burrows J,
Kuruganti P, Chan E, Le P, Chen J, Wang X, Ruslim L, Blake R, et
al: A selective small molecule inhibitor of c-Met kinase inhibits
c-Met-dependent phenotypes in vitro and exhibits cytoreductive
antitumor activity in vivo. Cancer Res. 63:7345–7355.
2003.PubMed/NCBI
|
|
42
|
Sierra JR and Tsao MS: c-MET as a
potential therapeutic target and biomarker in cancer. Ther Adv Med
Oncol. 3(Suppl): S21–S35. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Trusolino L and Comoglio PM:
Scatter-factor and semaphorin receptors: Cell signalling for
invasive growth. Nat Rev Cancer. 2:289–300. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mueller KL, Madden JM, Zoratti GL,
Kuperwasser C, List K and Boerner JL: Fibroblast-secreted
hepatocyte growth factor mediates epidermal growth factor receptor
tyrosine kinase inhibitor resistance in triple-negative breast
cancers through paracrine activation of Met. Breast Cancer Res.
14:R1042012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Meyer AS, Miller MA, Gertler FB and
Lauffenburger DA: The receptor AXL diversifies EGFR signaling and
limits the response to EGFR-targeted inhibitors in triple-negative
breast cancer cells. Sci Signal. 6:ra662013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang S, Chung WC, Miele L and Xu K:
Targeting Met and Notch in the Lfng-deficient, Met-amplified
triple-negative breast cancer. Cancer Biol Ther. 15:633–642. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kim YJ, Choi JS, Seo J, Song JY, Lee SE,
Kwon MJ, Kwon MJ, Kundu J, Jung K, Oh E, et al: MET is a potential
target for use in combination therapy with EGFR inhibition in
triple-negative/ basal-like breast cancer. Int J Cancer.
134:2424–2436. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Meyuhas O: Physiological roles of
ribosomal protein S6: One of its kind. Int Rev Cell Mol Biol.
268:1–37. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Barrows BD, Rutkowski MJ, Gültekın SH,
Parsa AT and Tıhan T: Evidence of ambiguous differentiation and
mTOR pathway dysregulation in subependymal giant cell astrocytoma.
Turk Patoloji Derg. 28:95–103. 2012.PubMed/NCBI
|
|
50
|
Bellizzi AM, Bloomston M, Zhou XP, Iwenofu
OH and Frankel WL: The mTOR pathway is frequently activated in
pancreatic ductal adenocarcinoma and chronic pancreatitis. Appl
Immunohistochem Mol Morphol. 18:442–447. 2010.PubMed/NCBI
|
|
51
|
Chaisuparat R, Rojanawatsirivej S and
Yodsanga S: Ribosomal protein S6 phosphorylation is associated with
epithelial dysplasia and squamous cell carcinoma of the oral
cavity. Pathol Oncol Res. 19:189–193. 2013. View Article : Google Scholar
|
|
52
|
Chaisuparat R, Yodsanga S, Montaner S and
Jham BC: Activation of the Akt/mTOR pathway in dentigerous cysts,
odontogenic keratocysts, and ameloblastomas. Oral Surg Oral Med
Oral Pathol Oral Radiol. 116:336–342. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chakraborty S, Mohiyuddin SM, Gopinath KS
and Kumar A: Involvement of TSC genes and differential expression
of other members of the mTOR signaling pathway in oral squamous
cell carcinoma. BMC Cancer. 8:1632008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chen W, Drakos E, Grammatikakis I,
Schlette EJ, Li J, Leventaki V, Staikou-Drakopoulou E, Patsouris E,
Panayiotidis P, Medeiros LJ, et al: mTOR signaling is activated by
FLT3 kinase and promotes survival of FLT3-mutated acute myeloid
leukemia cells. Mol Cancer. 9:2922010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chiang DY, Villanueva A, Hoshida Y, Peix
J, Newell P, Minguez B, LeBlanc AC, Donovan DJ, Thung SN, Solé M,
et al: Focal gains of VEGFA and molecular classification of
hepatocellular carcinoma. Cancer Res. 68:6779–6788. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Golfinopoulos V, Pentheroudakis G, Goussia
A, Siozopoulou V, Bobos M, Krikelis D, Cervantes A, Ciuleanu T,
Marselos M, Fountzilas G, et al: Intracellular signalling via the
AKT axis and downstream effectors is active and prognostically
significant in cancer of unknown primary (CUP): a study of 100 CUP
cases. Ann Oncol. 23:2725–2730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hagner PR, Mazan-Mamczarz K, Dai B, Balzer
EM, Corl S, Martin SS, Zhao XF and Gartenhaus RB: Ribosomal protein
S6 is highly expressed in non-Hodgkin lymphoma and associates with
mRNA containing a 5′ terminal oligopyrimidine tract. Oncogene.
30:1531–1541. 2011. View Article : Google Scholar :
|
|
58
|
Iwenofu OH, Lackman RD, Staddon AP,
Goodwin DG, Haupt HM and Brooks JS: Phospho-S6 ribosomal protein: a
potential new predictive sarcoma marker for targeted mTOR therapy.
Mod Pathol. 21:231–237. 2008. View Article : Google Scholar
|
|
59
|
Kouvaraki MA, Liakou C, Paraschi A, Dimas
K, Patsouris E, Tseleni-Balafouta S, Rassidakis GZ and Moraitis D:
Activation of mTOR signaling in medullary and aggressive papillary
thyroid carcinomas. Surgery. 150:1258–1265. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Villanueva A, Chiang DY, Newell P, Peix J,
Thung S, Alsinet C, Tovar V, Roayaie S, Minguez B, Sole M, et al:
Pivotal role of mTOR signaling in hepatocellular carcinoma.
Gastroenterology. 135:1972–1983. 1983 e1971–1911. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Qian ZR, Ter-Minassian M, Chan JA, Imamura
Y, Hooshmand SM, Kuchiba A, Morikawa T, Brais LK, Daskalova A,
Heafield R, et al: Prognostic significance of MTOR pathway
component expression in neuroendocrine tumors. J Clin Oncol.
31:3418–3425. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Potratz JC, Saunders DN, Wai DH, Ng TL,
McKinney SE, Carboni JM, Gottardis MM, Triche TJ, Jürgens H, Pollak
MN, et al: Synthetic lethality screens reveal RPS6 and MST1R as
modifiers of insulin-like growth factor-1 receptor inhibitor
activity in childhood sarcomas. Cancer Res. 70:8770–8781. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Khalaileh A, Dreazen A, Khatib A, Apel R,
Swisa A, Kidess-Bassir N, Maitra A, Meyuhas O, Dor Y and Zamir G:
Phosphorylation of ribosomal protein S6 attenuates DNA damage and
tumor suppression during development of pancreatic cancer. Cancer
Res. 73:1811–1820. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Dephoure N, Zhou C, Villén J, Beausoleil
SA, Bakalarski CE, Elledge SJ and Gygi SP: A quantitative atlas of
mitotic phosphorylation. Proc Natl Acad Sci USA. 105:10762–10767.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Olsen JV, Vermeulen M, Santamaria A, Kumar
C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, et al:
Quantitative phosphoproteomics reveals widespread full
phosphorylation site occupancy during mitosis. Sci Signal.
3:ra32010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Rigbolt KT, Prokhorova TA, Akimov V,
Henningsen J, Johansen PT, Kratchmarova I, Kassem M, Mann M, Olsen
JV and Blagoev B: System-wide temporal characterization of the
proteome and phosphoproteome of human embryonic stem cell
differentiation. Sci Signal. 4:rs32011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Roux PP, Shahbazian D, Vu H, Holz MK,
Cohen MS, Taunton J, Sonenberg N and Blenis J: RAS/ERK signaling
promotes site-specific ribosomal protein S6 phosphorylation via RSK
and stimulates cap-dependent translation. J Biol Chem.
282:14056–14064. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Schläfli P, Tröger J, Eckhardt K, Borter
E, Spielmann P and Wenger RH: Substrate preference and
phosphatidylinositol monophosphate inhibition of the catalytic
domain of the Per-Arnt-Sim domain kinase PASKIN. FEBS J.
278:1757–1768. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Stevens C, Lin Y, Harrison B, Burch L,
Ridgway RA, Sansom O and Hupp T: Peptide combinatorial libraries
identify TSC2 as a death-associated protein kinase (DAPK) death
domain-binding protein and reveal a stimulatory role for DAPK in
mTORC1 signaling. J Biol Chem. 284:334–344. 2009. View Article : Google Scholar
|
|
70
|
Falsone SF, Gesslbauer B, Tirk F,
Piccinini AM and Kungl AJ: A proteomic snapshot of the human heat
shock protein 90 interactome. FEBS Lett. 579:6350–6354. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kim TS, Jang CY, Kim HD, Lee JY, Ahn BY
and Kim J: Interaction of Hsp90 with ribosomal proteins protects
from ubiquitination and proteasome-dependent degradation. Mol Biol
Cell. 17:824–833. 2006. View Article : Google Scholar :
|
|
72
|
Zou HY, Li Q, Lee JH, Arango ME, McDonnell
SR, Yamazaki S, Koudriakova TB, Alton G, Cui JJ, Kung PP, et al: An
orally available small-molecule inhibitor of c-Met, PF-2341066,
exhibits cytoreductive antitumor efficacy through antiproliferative
and antiangiogenic mechanisms. Cancer Res. 67:4408–4417. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Bonfils C, Beaulieu N, Fournel M,
Ste-Croix H, Besterman JM and Maroun CR: The combination of
MGCD265, a Met/VEGFR inhibitor in clinical development, and
erlotinib potently inhibits tumor growth by altering multiple
pathways including glycolysis. Cancer Res. 72(Suppl 8): S17902012.
View Article : Google Scholar
|
|
74
|
Qian F, Engst S, Yamaguchi K, Yu P, Won
KA, Mock L, Lou T, Tan J, Li C, Tam D, et al: Inhibition of tumor
cell growth, invasion, and metastasis by EXEL-2880 (XL880,
GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine
kinases. Cancer Res. 69:8009–8016. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
You WK, Sennino B, Williamson CW, Falcón
B, Hashizume H, Yao LC, Aftab DT and McDonald DM: VEGF and c-Met
blockade amplify angiogenesis inhibition in pancreatic islet
cancer. Cancer Res. 71:4758–4768. 2011. View Article : Google Scholar : PubMed/NCBI
|