Introduction
Leukaemia is the most common type of cancer in
children. Chemoresistance represents a great obstacle to leukaemia
therapy that might be responsible for refractory and relapsing
cases. Various mechanisms underlying drug resistance have been
recognised, such as those involving drug exporters, altered drug
target sites, enhanced effectiveness of DNA repair mechanisms,
altered pharmacokinetics, the downregulation of apoptosis and
resistant tumour stem cells (1).
In recent years, autophagy, which plays an intricate role in cell
death and survival, has also been considered as a potential
mechanism underlying resistance to chemotherapy, radiation therapy
and immunotherapy in cancer cells (2–5).
During the autophagic process, energy production is
maintained via the degradation of cellular components and the
recycling of organelles. The mammalian autophagic process begins
with autophagosome formation, which can be separated into three
steps: initiation and nucleation, elongation, maturation. During
the initiation stage, the Ulk1-Atg13-FIP200 complex is activated by
an autophagic signal and participates in autophagosome formation
(6,7). Then, the class III PI3K (PI3KC3)
complex, which is composed of PI3KC3, Beclin1, p150 and ATG14,
plays an essential role in isolation membrane nucleation (8). Subsequently, the membrane is
elongated by a series of autophagy-related proteins (ATG), with the
transition from LC3-I to LC3-II. At last, the autophagosome is
maturated with the action of LC3-II and Beclin1 (9,11).
In the absence of autophagy stimulation, Beclin1
binds to Bcl-2 and remains stable. Upon sensing cell stress, high
mobility group box 1 (HMGB1), which is primarily localised to the
nucleus, translocates to the cytoplasm and promotes the
phosphorylation of Bcl-2, thereby promoting the escape of Beclin1
from Bcl-2 and subsequently facilitating PI3KC3-Beclin1 complex
assembly (12,13).
As critical effector of autophagosome formation, the
Ulk13-FIP200 complex, the PI3KC3 complex and the HMGB1 complex are
not independent modules. In addition, the translocation of HMGB1 is
a crucial molecular event during autophagy. However, the
association between the activities of these complexes and the
function of HMGB1 translocation, specifically in leukaemia, have
yet to be clearly elucidated.
In the present study, we demonstrated that both the
Ulk1-Atg13-FIP200 complex and the HMGB1-Beclin1 complex acted as
critical regulators of autophagy. Their core components, FIP200 and
HMGB1, were essential for autophagy-related drug resistance in
leukaemia. The Ulk1-Atg13-FIP200 complex acted upstream of the
HMGB1-Beclin1 and PI3KC3-Beclin1 complexes. Additionally, we first
discovered that the translocation of HMGB1 represented a decisive
step in the autophagic process and was involved in a regulatory
relationship between Ulk1-Atg13-FIP200 and HMGB1-Beclin1 complex.
These findings revealed that the inhibition of autophagy may
augment chemotherapy and may help to overcome drug resistance.
Materials and methods
Subjects and cell isolation
This study was approved by the Ethics Committee of
the Sun Yat-Sen Memorial Hospital of Sun Yat-Sen University.
Written informed consent was obtained from all the participants or
their parents.
Bone marrow samples were collected from patients
aged from 0 to 14 years who suffered from childhood acute
lymphoblastic leukaemia (ALL) or a non-malignant blood disease. In
total, 30 subjects with B-cell ALL (12 patients in the primary
phase, 12 in the complete remission phase who were still undergoing
treatment, 3 patients in the complete remission phase who had
completed their treatment course and 3 in the relapse or
non-remission phase) and 8 subjects with non-malignant blood
disease were enrolled. The diagnoses of ALL were based on
morphologic and flow cytometric analyses of the immunophenotype,
and the stratification of ALL was based on the IC-BFM 2002 criteria
for ALL. Bone marrow mononuclear cells (BMMCs) were isolated via
Ficoll (MP Biomedicals, Santa Ana, CA, USA) density gradient
centrifugation (14).
Quantitative real-time PCR (RT-qPCR)
Total RNA was isolated from BMMCs using TRIzol
reagent (Takara Bio Inc., Otsu, Shiga, Japan) and then
reverse-transcribed into cDNA. The sequences of primers used were
as follows: GAPDH: forward, 5′-GGTCGGAGTCAACGGATTTGGTCG-3′ and
reverse, 5′-CCTCCGACGCCTGCTTCACCAC-3′; for HMGB1: forward,
5′-TTTCAAACAAAGATGCCACA-3′ and reverse,
5′-GTTCCCTAAACTCCTAAGCAGATA-3′; for Beclin1: forward,
5′-ATCCTGGACCGTGTCACCATCCAGG-3′ and reverse,
5′-GTTGAGCTGAGTGTCCAGCTGG-3′; for FIP200: forward,
5′-TCAGTAAGTTGCGCAGTAGCA-3′ and reverse,
5′-TTCAGGTGCACAAGCTCCAT-3′. Reactions were carried out in a Roche
Light Cycler 480 system (Roche Diagnostics GmbH, Mannheim, Germany)
with a SYBR Premix ExTaq kit (Takara Bio Inc., Otsu, Shiga, Japan).
With the 2−ΔΔCt method, Data were normalized to GAPDH
expression. The control group was set as 1.
Cell culture, antibodies and
reagents
The human leukaemia cell line, Jurkat and Reh, were
purchased from the Type Culture Collection of the Chinese Academy
of Sciences, Shanghai, China and cultured in RPMI-1640 medium
(Gibco, Grand Island, NY, USA) with 10% FBS (Hyclone, Logan, UT,
USA) in a humidified incubator with 5% CO2 and 95% air
(15).
The antibodies to FIP200, ULK1, mAtg13, PI3KC3,
Phospho-ULK1 (Ser555) and Rabbit (DA1E) mAb IgG XPTM Isotype
Control were obtained from Cell Signaling Technology (Danvers, MA,
USA). The antibodies to LC3 were obtained from Sigma-Aldrich (St.
Louis, MO, USA). The antibodies to HMGB1, Beclin1, Bcl-2 and P62
were obtained from Abcam (Cambridge, UK). The antibodies to actin,
GAPDH and tubulin were obtained from EarthOx Life Science
(Millbrae, CA, USA). All secondary antibodies were purchased from
Bioss (Beijing Biosynthesis Biotechnology Co., Ltd.). Vincristine
(VCR), daunorubicin (DNR), 3-methyladenine (3-MA), chloroquine (CQ)
and ethyl pyruvate (EP) were purchased from Sigma-Aldrich.
Lentivirus infection and cell
sorting
Lentivirus with shRNA was purchased from GenePharma
(Shanghai, China). Transfection was conducted with lentivirus using
5 μg/ml polybrene and selected with 1 μg/ml l puromycin under the
instruction of GenePharma Recombinant Lentivirus Operation Manual
(16). The interference efficiency
was evaluated by RT-qPCR and western blotting. We selected the best
sequence which processed the highest interference efficiency out
from 3 different sequences. The optimal target sequence for FIP200
was 5′-GCCGTCAAACTATTGCCAAAC-3′, for HMGB1 was
5′-CCCGTTATGAAAGAGAAATGA-3′ and for control was
5′-TTCTCCGAACGTGTCACGTTTC-3′.
Cell viability analysis
Cells were plated in 96-well plate at a density of
~6×104 cells/well. Cell viability was measured after
medication treatments by CCK-8 test (Dojindo Molecular Technologies
Inc., Kumamoto, Kyushu, Japan) according to the manufacturer’s
instructions.
Western blot analysis
Cells with different treatments were collected and
lysed. The cell lysate was separated by 8% (10, 12 and 15%) sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
electrophoretically transferred onto polyvinylidene fluoride
membrane (Millpore, Billerica, MA, USA). The membrane was blocked
with 5% non-fat milk, and then incubated with the primary
antibodies at a dilution of 1:1,000 overnight at 4°C. After
rinsing, the membranes were incubated for 1 h at room temperature
with secondary antibodies at a dilution of 1:1,000 and then washed
with TBST (15,17,18).
The membranes were detected with enhanced chemiluminescence reagent
(Millpore) by G: BOX XT4 (Syngene, Cambridge, UK). The expressions
of the target proteins were quantified by Quantity One version
4.6.2.
Immunoprecipitation analysis
Immunoprecipitation (IP) analysis was performed by
Pierce Classic IP kit (Pierce Biotechnology, Rockford, IL, USA)
according to the manufacturer’s protocol. In brief, cells were
collected and lysed by ice cold IP lysis buffer. Then the protein
was pre-cleared with the control agarose resin at 4°C for 1 h. The
pre-cleared cell lysate was incubated with the Rabbit (DA1E) mAb
IgG XP™ Isotype Control or specific antibodies overnight at 4°C to
form the immune complex and then with protein A/G plus agarose for
1 h at 4°C to capture the immune complex. After washed the resin
with wash buffer, the immune complex was eluted at 100°C for 10
min. The eluate was collected and subjected to SDS-PAGE for
immunoblot analysis (15,18). The Clean-Blot IP Detection reagent
(Pierce Biotechnology) was used as secondary antibody at a dilution
of 1:1,000 for detecting the target proteins without interference
from denatured IgG in western blot analysis.
Immunofluorescence analysis
Cells were collected and fixed in 4% formaldehyde
for 20 min, permeabilized in 0.3% Triton X-100 for 5 min and
incubated in blocking buffer for 1 h. Then cells were incubated
overnight at 4°C with primary antibodies. After rinsing, cells were
incubated with FITC- or Cy3-conjugated secondary antibodies for 1 h
at room temperature in the dark and then with DAPI for 3 min. After
rinsing, cells were resuspended. Several drops of cell suspension
were added in a laser confocal Petri dish (12,19).
The images were captured by a Carl Zeiss LSM 710 (Carl Zeiss Inc.,
Oberkochen, Germany).
Transmission electron microscopic
analysis
Cells were fixed with 2% paraformaldehyde and 2.5%
glutaraldehyde in 0.1 mol/l phosphate buffer (pH 7.4), followed by
1% OsO4. After dehydration, thin sections were stained
with uranyl acetate and lead citrate for observation by Tecnai G2
Spirit Twin electron microscope (FEI, Hillsboro, OR, USA).
Transmission electron microscopic (TEM) assessment of
autophagosome-like structures was carried out as previously
described (19,20).
Statistical analysis
Data are shown as mean ± standard deviation (SD).
The difference among groups was examined using the Student’s t-test
or ANOVA in measurement data and Fisher’s exact probabilities in
small size enumeration data. The Pearson product-moment correlation
method was used to analyze the linear correlation between two
groups. A value of p<0.05 was regarded as statistically
significant. All statistical analyses were conducted by SPSS
version 18.0.
Results
HMGB1 expression correlates with the
childhood acute lymphoblastic leukaemia stage
In total, 30 children with B-cell ALL and 8 control
subjects with a non-malignant blood disease were included in the
present study.
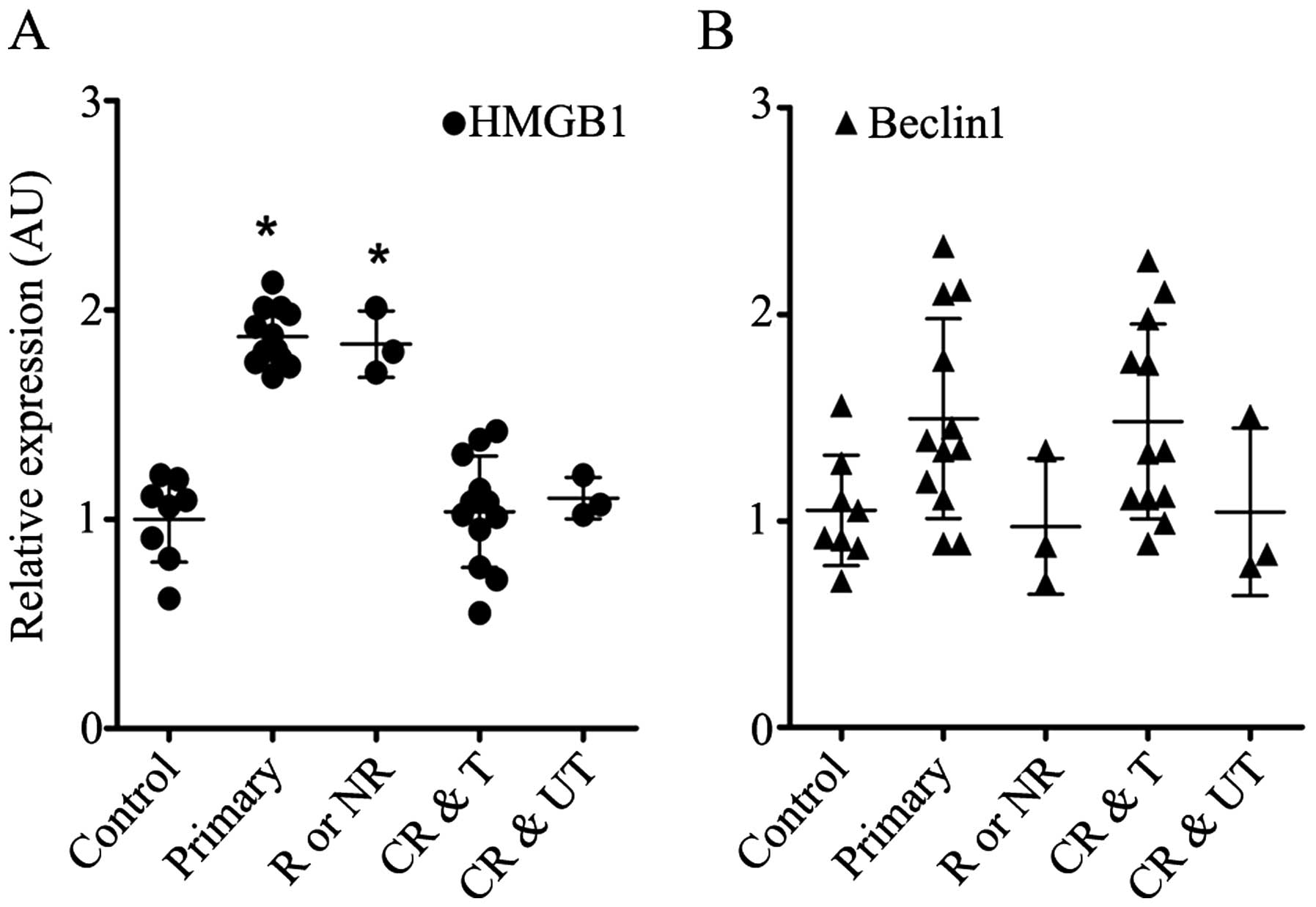
As shown in Fig.
1A, HMGB1 was upregulated in the BMMCs obtained from the
patients with primary, relapsed or non-remission leukaemia, whereas
its expression level in the BMMCs from the ALL patients who
exhibited complete remission was as low as that from those with a
non-malignant blood disease. The expression level of Beclin1 was
nearly identical between the groups (Fig. 1B). The expression levels of HMGB1
and Beclin1 appeared to be irrelevant to risk stratification
(p>0.05, data not shown).
The inhibition of autophagy and FIP200
depletion increases the sensitivity of leukaemia cells to
chemotherapy
DNR and VCR are basic drugs used for leukaemia
therapy. As shown in Fig. 2A, both
drugs caused striking damage to Jurkat and Reh cells in a
dose-dependent manner, and this trend was particularly apparent in
the Reh cells. Interestingly, in the presence of 3-methyladenine
(3-MA), an inhibitor of early-phase autophagy, the cytotoxic effect
was much stronger and cell viability was more sharply reduced
(Fig. 2B). This result suggested
that the inhibition of autophagy may augment the lethality of
anticancer agents.
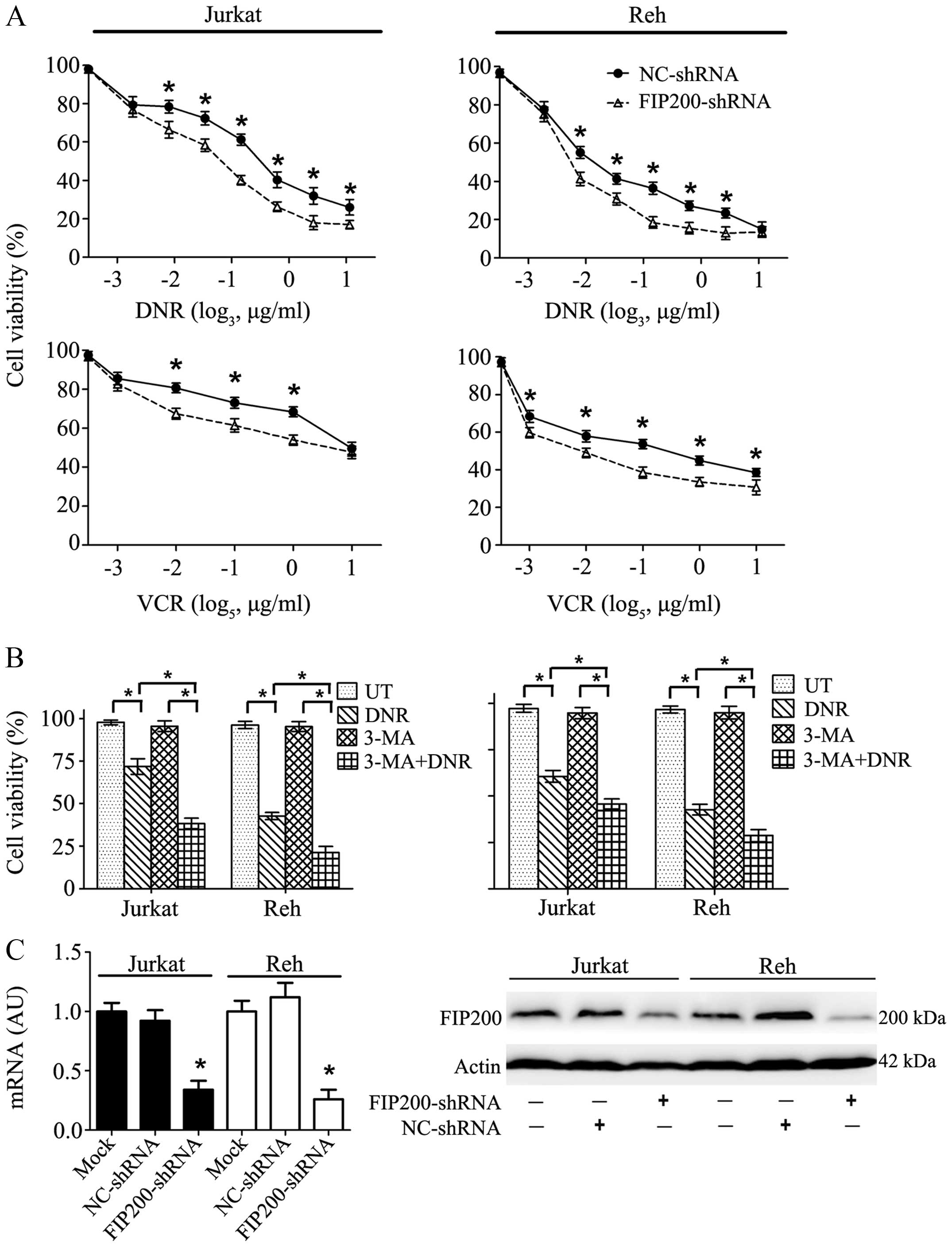 | Figure 2Autophagy inhibitor treatment and
FIP200 depletion increased the sensitivity of leukaemia cells to
chemotherapy. (A) Chemotherapeutic drugs exerted damaging effects
on the leukaemia cells in a dose-dependent manner, and this effect
was exacerbated by FIP200 depletion. Jurkat and Reh cells were
treated with DNR (0, 0.05, 0.1, 0.2, 0.4, 0.8, 1.6 μg/ml, or 3.2
μg/ml) or VCR (0, 0.008, 0.04, 0.2, 1, or 5 μg/ml) for 24 h. Cell
viability was analysed using a CCK-8 kit. (B) The inhibition of
autophagy ameliorated drug resistance. Jurkat and Reh cells were
treated with DNR (0.4 μM) or VCR (1 μM) for 24 h with or without
3-MA (5 mM). Cell viability was analysed using a CCK-8 kit (n=3,
*p<0.001). (C) Jurkat and Reh cells were transfected
with control or FIP200 shRNA. Then, the mRNA and protein levels of
FIP200 were assessed via RT-qPCR and western blot analysis,
respectively (n=3, *p<0.05). UT, untreated group. The
viability of the control cells was normalised to 100%. |
To explore the potential role of FIP200, a key
protein involved in autophagy, a target-specific shRNA against
FIP200 was transfected into Jurkat and Reh cells. This treatment
resulted in a significant decrease in FIP200 at both the mRNA and
protein levels (Fig. 2C). Cell
viability analysis showed that the silencing of FIP200 mimicked the
inhibitory effect of 3-MA and rendered the leukaemia cells more
sensitive to DNR and VCR (Fig.
2A).
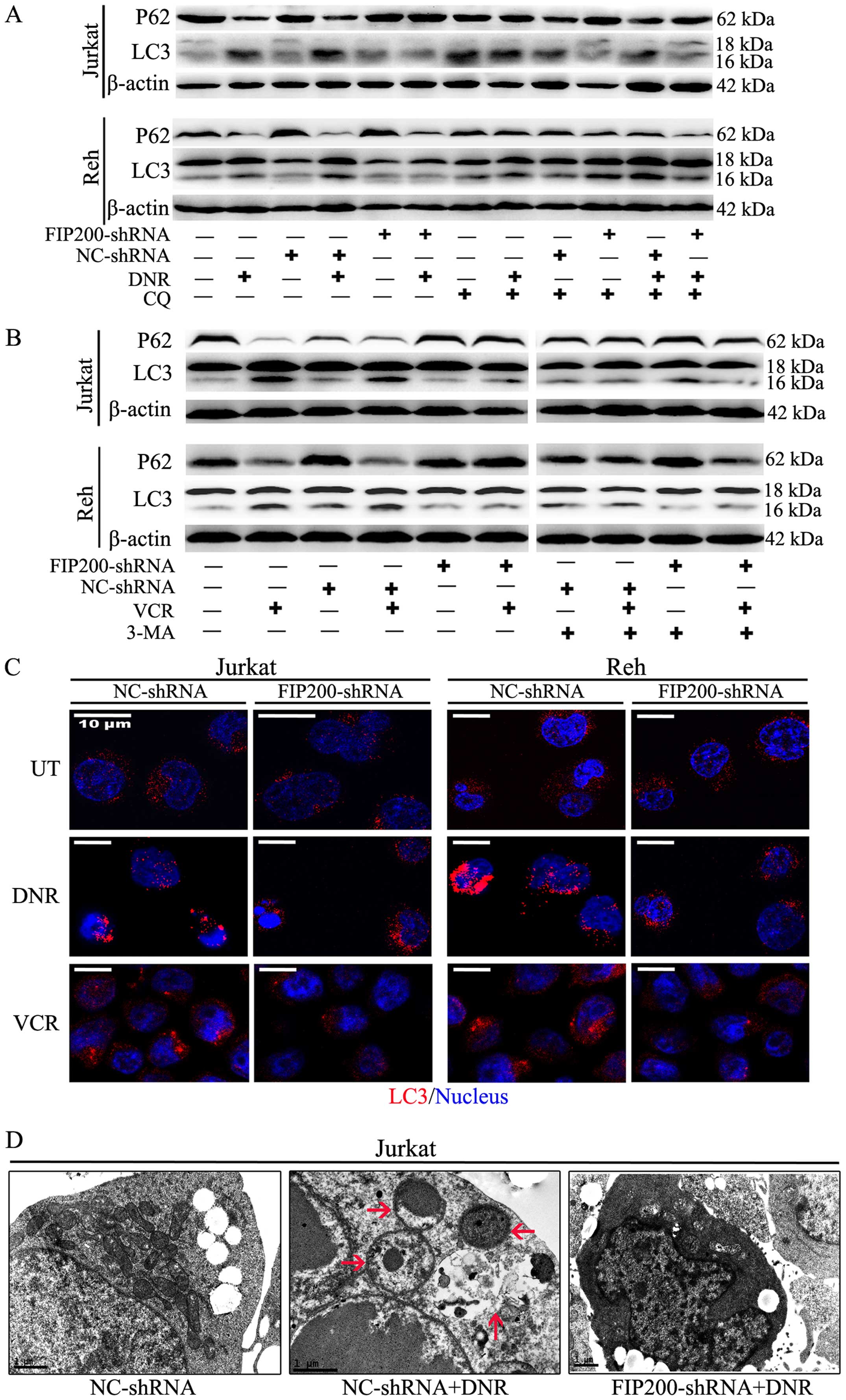
Silencing of FIP200 suppresses
chemotherapy-induced autophagy in leukaemia cells
Autophagy is a highly dynamic, multi-step process.
To assess the entire autophagic process, we measured autophagosomes
containing LC3 puncta using immunofluorescence staining and
analysed the LC3-II/I ratio via western blot analysis.
Additionally, we observed ultrastructural changes via electron
microscopy and assessed autophagic flux based on lysosomal turnover
and the P62 level via western blot analysis (19,21,22).
Based on western blot analysis, we found that
silencing FIP200 suppressed the chemotherapy-induced high
expression of LC3-II and the corresponding degradation of P62
(Fig. 3A and B). The accumulation
of LC3-II in the mock-treated and control cells was enhanced in the
presence of the lysosomal inhibitor chloroquine (CQ) in the
presence or absence of stimuli, but this effect was not observed in
the FIP200 shRNA-treated cells (Fig.
3A). These changes demonstrated that the accumulation of LC3-II
was not due to its decreased degradation but rather increased
autophagic flux. However, the changes of LC3-II and P62 were not
observed in the presence of 3-MA (Fig.
3B). Based on immunofluorescence analysis, the change in
endogenous LC3 puncta was consistent with the results of western
blot analysis in both cell lines (Fig.
3C). Moreover, ultrastructural analysis revealed that the
FIP200 shRNA-treated cells exhibited fewer autophagosomes during
chemotherapy compared with the control shRNA-treated cells
(Fig. 3D).
The data shown in Figs.
2 and 3 demonstrate that in
leukaemia, the anticancer agent-induced cytotoxic effects were due
to the autophagy-related drug resistance, and these effects were
ameliorated by the depletion of FIP200 or the application of an
autophagy inhibitor.
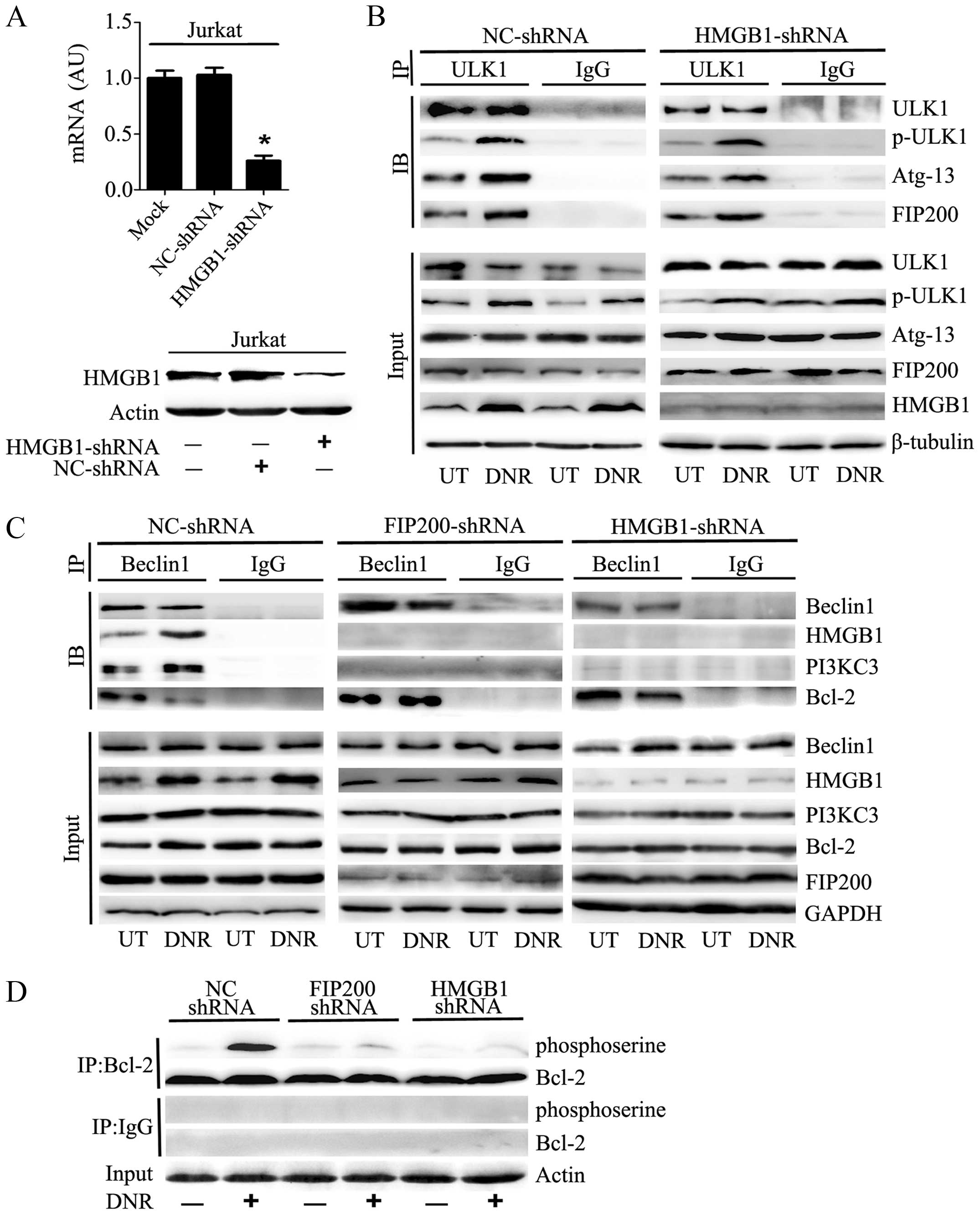
The activation of the ULK1-mAtg13-FIP200
complex served as an upstream signal to HMGB1-Beclin1 formation
during autophagy
We transfected Jurkat cells with HMGB1 shRNA, which
resulted in a prominent decrease in the mRNA and protein expression
of HMGB1 (Fig. 4A). As shown in
Fig. 4B, the phosphorylation of
ULK1 at Ser555, which plays a critical role in responding to
autophagic stimuli, was significantly increased after anticancer
agent treatment. Immunoprecipitation analysis indicated that
silencing HMGB1 did not affect ULK1-mAtg13-FIP200 complex assembly
or ULK1 phosphorylation following DNR treatment.
Conversely, the absence of FIP200 did inhibit the
assembly of the HMGB1-Beclin1 complex or the subsequent formation
of the PI3KC3-Beclin1 complex after chemotherapy stimulation
(Fig. 4C). In the control
shRNA-treated Jurkat cells, Beclin1 separated from Bcl-2 after DNR
treatment and subsequently complexed with HMGB1 and PI3KC3. In
contrast, Beclin1-Bcl-2 was not separated after DNR treatment in
the absence of FIP200 or HMGB1. Thus, the HMGB1-Beclin1 and
PI3KC3-Beclin1 complexes failed to coalesce. Furthermore, we
observed that the depletion of HMGB1 or FIP200 reduced the
phosphorylation of Bcl2 (Fig. 4D),
which may result in the stable binding of Bcl2 to Beclin1.
The translocation of HMGB1 is required
for autophagy-mediated chemoresistance in leukaemia cells
Although primarily localised to the nucleus, HMGB1
can translocate to the cytoplasm and the extracellular space during
cell activation and cell death. To investigate the role of HMGB1 in
autophagy, we focused on its localization. By detecting the
expression of HMGB1 in the nucleus and the cytosol separately, we
found that the chemotherapy promoted the translocation of HMGB1
from the nucleus to the cytosol in the cultured Jurkat and Reh
cells (Fig. 5A). This
translocation was also detected using immunofluorescence techniques
(Fig. 5B). Remarkably, HMGB1 did
not translocate in the absence of FIP200 (Fig. 5A and B). When combining the data of
Figs. 4B and C and 5A and B, we concluded that the activation
of the ULK1-mAtg13-FIP200 complex induced the translocation of
HMGB1 and the subsequent dissociation of Beclin1-Bcl-2 and
formation of the HMGB1-Beclin1 complex, ultimately influencing
autophagy-related chemoresistance.
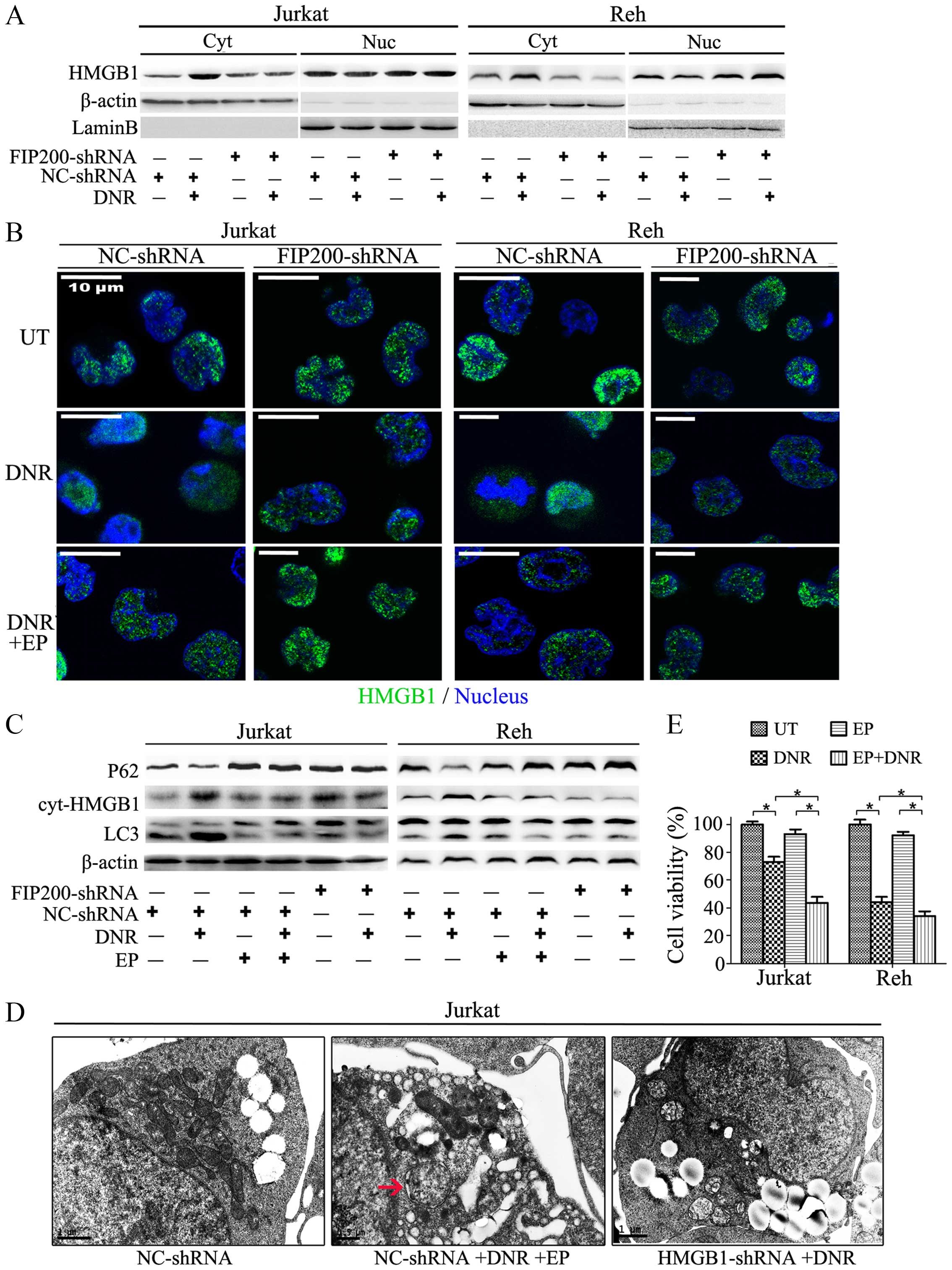 | Figure 5The translocation of HMGB1 is
essential for autophagy-mediated chemoresistance in leukaemia
cells. (A) Jurkat and Reh cells were transfected with control or
FIP200 shRNA and then treated with or without DNR (0.4 μM) for 24
h. The cell lysates were separated into the cytosolic and nuclear
fractions, which were subjected to western blot analysis of HMGB1
expression. Actin and lamin-B were used as loading controls for
cytosolic and nuclear proteins, respectively. (B) Jurkat and Reh
cells were transfected with control or FIP200 shRNA followed by DNR
(0.4 μM) treatment for 24 h. The cells were subjected to confocal
microscopic analysis to detect the localisation of HMGB1 (green,
HMGB1; blue, nucleus). (C) Jurkat and Reh cells were transfected
with control shRNA or FIP200 shRNA followed by DNR (0.4 μM)
treatment in the presence or absence of EP (5 mM) for 24 h. Then,
the cells were subjected to western blot analysis of LC3-II/I, p62
and cytosolic HMGB1 expression. Actin was used as a loading
control. (D) Jurkat cells were transfected with control or HMGB1
shRNA followed by treatment with DNR (0.4 μM) in the presence or
absence of EP (5 mM) for 24 h. The cells were subjected to
transmission electron microscopy to observe autophagosome-like
structures (indicated by the red arrows). (E) Jurkat and Reh cells
were treated with DNR (0.4 μM) for 24 h in the presence or absence
of EP (5 mM). Then, cell viability was analysed using a CCK-8 kit
(n=3, *p<0.05). The viability of the control cells
was normalised to 100%. Cyt, cytoplasm. Nuc, nucleus. UT, untreated
group. |
As shown in Fig. 5B and
C, the Jurkat and Reh cells were treated with DNR in the
presence or absence of EP, an inhibitor of HMGB1 translocation. We
found that blocking HMGB1 translocation mimicked the knockdown of
FIP200 and suppressed autophagy. At the ultrastructural level
(Fig. 5D), EP reduced the
formation of autophagosomes and autophagolysosomes to a great
extent, and this effect was similar to that of HMGB1 or FIP200
depletion (Figs. 3D and 5D). Moreover, in the absence of HMGB1
translocation, autophagy was suppressed, and the leukaemia cells
became more sensitive to the chemotherapy (Fig. 5E).
The above results suggested that HMGB1 is involved
in the positive regulation and maintenance of autophagy in stressed
leukaemia cells and that HMGB1 translocation is a necessary
component of the autophagic process. Blocking HMGB1 translocation
by interrupting the upstream signal or using a translocation
inhibitor may represent a useful strategy for reducing
autophagy-induced drug resistance.
Discussion
Resistance to chemotherapy is an intractable but
common barrier to the clinical treatment of leukaemia. The
adaptation of leukaemia cells to drug-induced stress in the tumour
microenvironment has been proposed to be a key mechanism underlying
chemotherapy resistance. Autophagy, a lysosomal degradation process
that plays a paradoxical role in anticancer treatment, typically
exerts a protective effect by facilitating the acquisition of drug
resistance in leukaemia (9,12,23).
Consistent with previous reports, our results showed that the
detrimental effects of chemotherapy were exacerbated by the
inhibition of autophagy (Fig. 2),
which indicated that the antitumour agents induced
autophagy-related drug resistance. In this study, we further
elucidated the crucial function and regulatory relationships of the
ULK1-Atg13-FIP200 and HMGB1-Beclin1 complexes. HMGB1 translocation
played a crucial role in dissociation of these complexes and in the
subsequent complexation.
Vast amount of data have demonstrated that the
ULK1-Atg13-FIP200 complex integrates incoming autophagic signals
into autophagosome formation, an essential event for autophagy
induction (24–26). FIP200, a key signalling node that
regulates various cellular processes, is a central components of
the ULK1-Atg13-FIP200 complex and is important for the appropriate
phosphorylation of ULK1 (27,28).
Wei et al observed that the ablation of FIP200 led to
autophagic defects, significantly reduced cell proliferation and
inhibited mammary tumourigenesis (29). Liu et al found that the
deletion of FIP200 suppressed autophagy and caused osteopenia in
mice (30). Bae and Guan reported
that the inhibition of autophagy via the inactivation of FIP200
inhibited the repair of damaged DNA and enhanced the efficacy of
cancer treatments (31).
Consistently, our study showed that silencing FIP200 increased cell
death, suppressed autophagy and subsequently sensitised the
leukaemia cells to anticancer therapy. FIP200 knockdown achieved a
similar effect to that of other pharmacological autophagy
inhibitors, such as 3-MA and CQ (Figs.
2 and 3).
HMGB1-Beclin1 is another important complex that is
assembled after HMGB1 translocation (13). We examined BMMCs from 38 children
and found that HMGB1 was significantly upregulated in the ALL
patients who exhibited primary, relapsed or recurrent disease
(Fig. 1A) and was equivalent to
baseline in the ALL patients who exhibited complete remission. This
result was consistent with those of previous studies (23,32)
and indicated that HMGB1 contributes to the pathogenesis of
leukaemia and even drug resistance. In vitro, we detected
HMGB1 translocation from the nucleus to the cytosol in the presence
of autophagic stimuli (Fig. 5).
Moreover, either the absence of HMGB1 or the inhibition of HMGB1
translocation distinctly attenuated autophagy and ameliorated
chemotherapy resistance, and these effects mimicked those of the
autophagy inhibitors (Fig. 5).
Another autophagic complex component, Beclin1, has been considered
as a tumour suppressor (33–35).
Reportedly, overexpressing Beclin1 induces significant autophagic
cell death in leukaemia (36).
However, several investigators measured the Beclin1 expression
level in PBMCs or BMMCs from Chinese leukaemia patients and
obtained inconsistent results (37,38).
More recently, others have disputed the original hypothesis and
concluded that Beclin1 mutation might not be significant in human
cancer and that Beclin1 is likely not a tumour suppressor gene
(39). In our study, no
significant difference in Beclin1 expression was detected between
the groups (Fig. 1B). This result
could be because the decreased Beclin1 expression in leukaemia
patient may have been offset by an increase in Beclin1 expression
due to the activation of autophagy. However, the small sample size
may reduce the reliability of these results. A long-term study
using a greater number of patients is required in the future.
Substantial research has been dedicated to the
examination of the functions of the ULK1-mAtg13-FIP200 and
HMGB1-Beclin1 complexes, but little attention has been paid to
their association. Pan et al recently reported that HMGB1
facilitates the formation of the Beclin-1-PI3K-III complex through
activating the MEK/ERK1/2 signalling pathway and then promotes
docetaxel resistance in human lung adenocarcinoma (40). Their results support future
investigation of HMGB1 as a strategic target. However, they failed
to mention the ULK1-mAtg13-FIP200 complex. Huang et al
proposed that HMGB1 serves as a downstream signal from
ULK1-mAtg13-FIP200 complex formation and facilitates autophagy by
interacting with Beclin1 (41).
However, their study only used osteosarcoma cells and did not
elucidate the potential mechanism. In the present study, we showed
that in leukaemia cells, the ULK1-mAtg13-FIP200 complex functions
upstream of HMGB1 and is required for the assembly of the
HMGB1-Beclin1 and PI3KC3-Beclin1 complexes (Fig. 4). Remarkably, we demonstrated that
the ULK1-mAtg13-FIP200 complex partially depended on HMGB1
translocation to facilitate the formation of these downstream
complexes (Figs. 4 and 5). This finding is consistent with a
recent report on murine pseudomonas infection (42). Moreover, previous reports have
suggested that Beclin1 is bound to Bcl-2 under normal conditions
but that during autophagy Beclin1 is bound to cytosolic HMGB1
following Bcl-2 phosphorylation (43–46).
Our results strongly corroborated this concept (Fig. 4C and D), as these changes were no
longer observed after FIP200 or HMGB1 depletion, revealing a
potential approach to prevent autophagy-related drug
resistance.
To date, CQ and its derivative hydroxychloroquine
(HCQ) are the only autophagy inhibitors that have been used in
dozens of clinical trials of human tumours (47). Due to their effectiveness in
autophagy inhibition and their anti-angiogenic properties, effects
of chemotherapy drugs are clearly enhanced when CQ or HCQ is
administered as an adjuvant therapy. However, the clinical
application of CQ and HCQ has been limited because of their
prolonged half-life, low potency and ocular toxicity. Therefore,
the identification of more specific targets and the establishment
of safer and more effective therapeutic strategies are highly
desirable. Our data provide insight into that which may meet the
demand.
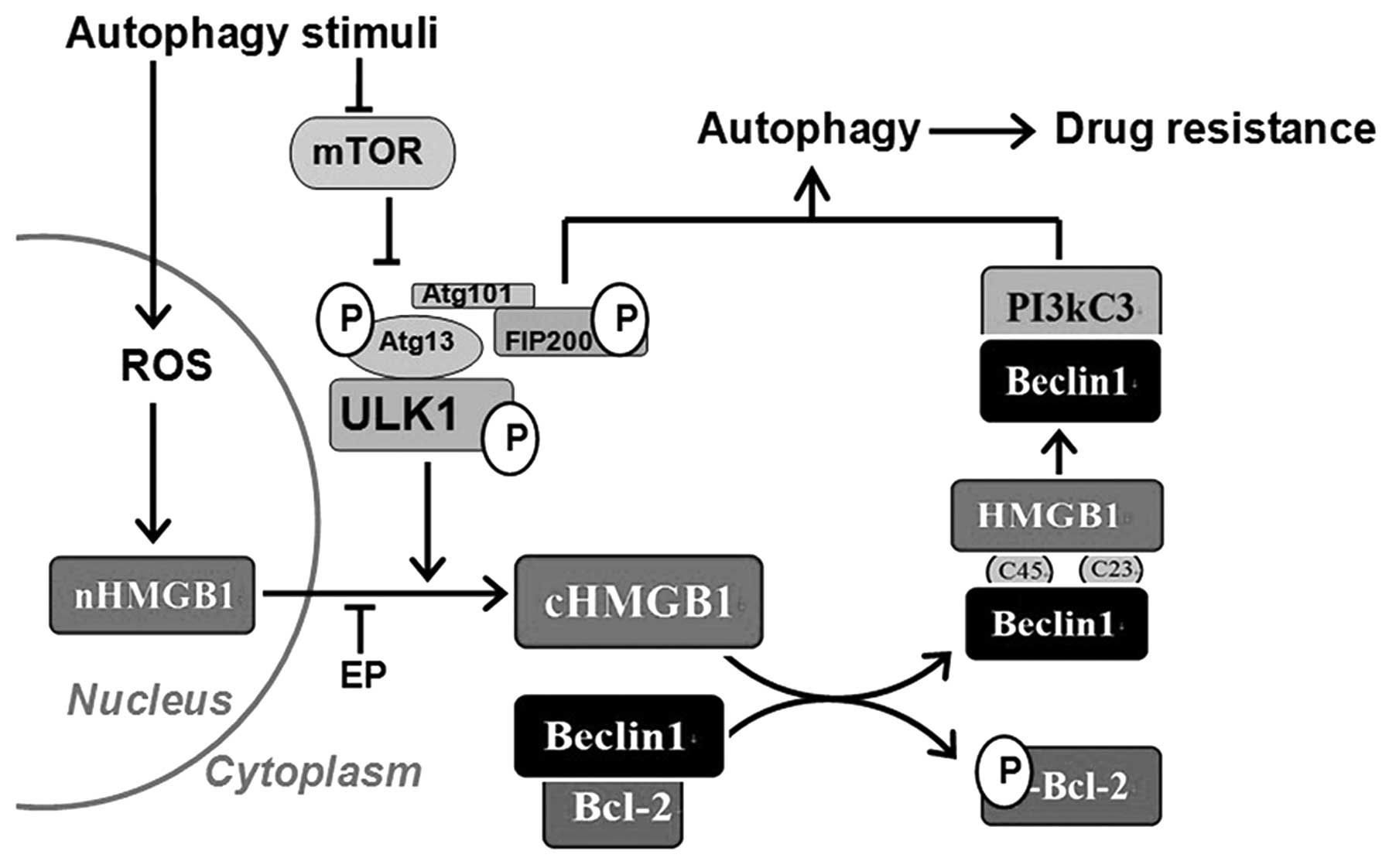
In conclusion, by combining previous data with our
present results, we have elucidated the process underlying
autophagy-related drug resistance in leukaemia (Fig. 6). The results of the present study
suggest that the Ulk1-Atg13-FIP200 complex functions upstream of
the HMGB1-Beclin1 and PI3KC3-Beclin1 complexes and HMGB1
translocation is involved in their regulatory relationships.
However, this unidirectional regulation model appears to be
oversimplified, and additional crosstalk should be considered in
future research.
The dissociation and recoupling of autophagic
complexes are essential events involved in the autophagy-related
chemoresistance. By inhibiting the transformation of these
complexes or HMGB1 trafficking, we are likely to render tumour
cells more susceptible to conventional therapies and overcome drug
resistance in leukaemia.
Acknowledgements
This study was supported by National Natural Science
Foundation of China (grant no. 81100370) and Sun Yat-Sen University
Clinical Research 5010 Program (grant no. 2007016).
References
|
1
|
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu
Y, Xie M, Yin X, Livesey KM, Loze MT, et al: DAMP-mediated
autophagy contributes to drug resistance. Autophagy. 7:112–114.
2011. View Article : Google Scholar :
|
|
2
|
Livesey KM, Tang D, Zeh HJ and Lotze MT:
Autophagy inhibition in combination cancer treatment. Curr Opin
Investig Drugs. 10:1269–1279. 2009.PubMed/NCBI
|
|
3
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sui X, Chen R, Wang Z, Huang Z, Kong N,
Zhang M, Han W, Lou F, Yang J, Zhang Q, et al: Autophagy and
chemotherapy resistance: A promising therapeutic target for cancer
treatment. Cell Death Dis. 4:e8382013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen S, Rehman SK, Zhang W, Wen A, Yao L
and Zhang J: Autophagy is a therapeutic target in anticancer drug
resistance. Biochim Biophys Acta. 1806:220–229. 2010.PubMed/NCBI
|
|
6
|
Alers S, Löffler AS, Wesselborg S and
Stork B: Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy:
Cross talk, shortcuts, and feedbacks. Mol Cell Biol. 32:2–11. 2012.
View Article : Google Scholar :
|
|
7
|
Wong PM, Puente C, Ganley IG and Jiang X:
The ULK1 complex: Sensing nutrient signals for autophagy
activation. Autophagy. 9:124–137. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wirth M, Joachim J and Tooze SA:
Autophagosome formation - the role of ULK1 and Beclin1-PI3KC3
complexes in setting the stage. Semin Cancer Biol. 23:301–309.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nencioni A, Cea M, Montecucco F, Longo VD,
Patrone F, Carella AM, Holyoake TL and Helgason GV: Autophagy in
blood cancers: Biological role and therapeutic implications.
Haematologica. 98:1335–1343. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pyo JO, Nah J and Jung YK: Molecules and
their functions in autophagy. Exp Mol Med. 44:73–80. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Viry E, Paggetti J, Baginska J,
Mgrditchian T, Berchem G, Moussay E and Janji B: Autophagy: An
adaptive metabolic response to stress shaping the antitumor
immunity. Biochem Pharmacol. 92:31–42. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu
Y, Xie M, Yin X, Livesey KM, Lotze MT, et al: HMGB1-induced
autophagy promotes chemotherapy resistance in leukemia cells.
Leukemia. 25:23–31. 2011. View Article : Google Scholar
|
|
13
|
Tang D, Kang R, Livesey KM, Cheh CW,
Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ III,
et al: Endogenous HMGB1 regulates autophagy. J Cell Biol.
190:881–892. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yeo C, Saunders N, Locca D, Flett A,
Preston M, Brookman P, Davy B, Mathur A and Agrawal S: Ficoll-Paque
versus Lymphoprep: A comparative study of two density gradient
media for therapeutic bone marrow mononuclear cell preparations.
Regen Med. 4:689–696. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kang R, Tang D, Yu Y, Wang Z, Hu T, Wang H
and Cao L: WAVE1 regulates Bcl-2 localization and phosphorylation
in leukemia cells. Leukemia. 24:177–186. 2010. View Article : Google Scholar
|
|
16
|
Valizadeh A, Ahmadzadeh A, Teimoori A,
Khodadadi A and Saki G: Effects of TNF secreting HEK cells on B
lymphocytes’ apoptosis in human chronic lymphocytic leukemias.
Asian Pac J Cancer Prev. 15:9885–9889. 2014. View Article : Google Scholar
|
|
17
|
Mizushima N and Yoshimori T: How to
interpret LC3 immuno-blotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tang D, Kang R, Xiao W, Jiang L, Liu M,
Shi Y, Wang K, Wang H and Xiao X: Nuclear heat shock protein 72 as
a negative regulator of oxidative stress (hydrogen
peroxide)-induced HMGB1 cytoplasmic translocation and release. J
Immunol. 178:7376–7384. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ylä-Anttila P, Vihinen H, Jokitalo E and
Eskelinen EL: Monitoring autophagy by electron microscopy in
mammalian cells. Methods Enzymol. 452:143–164. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang XJ, Chen S, Huang KX and Le WD: Why
should autophagic flux be assessed? Acta Pharmacol Sin. 34:595–599.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang L, Yu Y, Kang R, Yang M, Xie M, Wang
Z, Tang D, Zhao M, Liu L, Zhang H, et al: Up-regulated autophagy by
endogenous high mobility group box-1 promotes chemoresistance in
leukemia cells. Leuk Lymphoma. 53:315–322. 2012. View Article : Google Scholar
|
|
24
|
Alers S, Löffler AS, Paasch F, Dieterle
AM, Keppeler H, Lauber K, Campbell DG, Fehrenbacher B, Schaller M,
Wesselborg S, et al: Atg13 and FIP200 act independently of Ulk1 and
Ulk2 in autophagy induction. Autophagy. 7:1423–1433. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM,
Cao J, Kundu M and Kim DH: ULK-Atg13-FIP200 complexes mediate mTOR
signaling to the autophagy machinery. Mol Biol Cell. 20:1992–2003.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ganley IG, Lam H, Wang J, Ding X, Chen S
and Jiang X: ULK1.ATG13FIP200 complex mediates mTOR signaling and
is essential for autophagy. J Biol Chem. 284:12297–12305. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gan B and Guan JL: FIP200, a key signaling
node to coordinately regulate various cellular processes. Cell
Signal. 20:787–794. 2008. View Article : Google Scholar :
|
|
28
|
Hara T, Takamura A, Kishi C, Iemura S,
Natsume T, Guan JL and Mizushima N: FIP200, a ULK-interacting
protein, is required for autophagosome formation in mammalian
cells. J Cell Biol. 181:497–510. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wei H, Wei S, Gan B, Peng X, Zou W and
Guan JL: Suppression of autophagy by FIP200 deletion inhibits
mammary tumorigenesis. Genes Dev. 25:1510–1527. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu F, Fang F, Yuan H, Yang D, Chen Y,
Williams L, Goldstein SA, Krebsbach PH and Guan JL: Suppression of
autophagy by FIP200 deletion leads to osteopenia in mice through
the inhibition of osteoblast terminal differentiation. J Bone Miner
Res. 28:2414–2430. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bae H and Guan JL: Suppression of
autophagy by FIP200 deletion impairs DNA damage repair and
increases cell death upon treatments with anticancer agents. Mol
Cancer Res. 9:1232–1241. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kang R, Tang DL, Cao LZ, Yu Y, Zhang GY
and Xiao XZ: High mobility group box 1 is increased in children
with acute lymphocytic leukemia and stimulates the release of tumor
necrosis factor-alpha in leukemic cell. Zhonghua Er Ke Za Zhi.
45:329–333. 2007.(In Chinese). PubMed/NCBI
|
|
33
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Aita VM, Liang XH, Murty VV, Pincus DL, Yu
W, Cayanis E, Kalachikov S, Gilliam TC and Levine B: Cloning and
genomic organization of beclin 1, a candidate tumor suppressor gene
on chromosome 17q21. Genomics. 59:59–65. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yue Z, Jin S, Yang C, Levine AJ and Heintz
N: Beclin 1, an autophagy gene essential for early embryonic
development, is a haploinsufficient tumor suppressor. Proc Natl
Acad Sci USA. 100:15077–15082. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tong Y, You L, Liu H, Li L, Meng H, Qian Q
and Qian W: Potent antitumor activity of oncolytic adenovirus
expressing Beclin-1 via induction of autophagic cell death in
leukemia. Oncotarget. 4:860–874. 2013.PubMed/NCBI
|
|
37
|
Hu XY, Bai H, Pan YZ, Wnag CB, Wu B, Zhao
Q, Ai H, Chen Z and Han X: Expression of autophagy related gene
Beclin1 and MAPLC3 in bone marrow mononuclear cells isolated from
acute leukemia patients and its significance. Zhongguo Shi Yan Xue
Ye Xue Za Zhi. 19:598–601. 2011.(In Chinese). PubMed/NCBI
|
|
38
|
Wan SY, Zhang R, Wang YY, Cen JN, Zhou J,
Yang Y, Jiang F and Chen ZX: Expression of autophagy related gene
Beclin1 in myelodysplastic syndrome patients and its significance.
Zhongguo Shi Yan Xue Ye Xue Za Zhi. 21:936–939. 2013.(In Chinese).
PubMed/NCBI
|
|
39
|
Laddha SV, Ganesan S, Chan CS and White E:
Mutational landscape of the essential autophagy gene BECN1 in human
cancers. Mol Cancer Res. 12:485–490. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pan B, Chen D, Huang J, Wang R, Feng B,
Song H and Chen L: HMGB1-mediated autophagy promotes docetaxel
resistance in human lung adenocarcinoma. Mol Cancer. 13:1652014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Huang J, Ni J, Liu K, Yu Y, Xie M, Kang R,
Vernon P, Cao L and Tang D: HMGB1 promotes drug resistance in
osteosarcoma. Cancer Res. 72:230–238. 2012. View Article : Google Scholar
|
|
42
|
Li Y, Gan CP, Zhang S, Zhou XK, Li XF, Wei
YQ, Yang JL and Wu M: FIP200 is involved in murine pseudomonas
infection by regulating HMGB1 intracellular translocation. Cell
Physiol Biochem. 33:1733–1744. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Marquez RT and Xu L: Bcl-2:Beclin 1
complex: multiple, mechanisms regulating autophagy/apoptosis toggle
switch. Am J Cancer Res. 2:214–221. 2012.PubMed/NCBI
|
|
44
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Saeki K, Yuo A, Okuma E, Yazaki Y, Susin
SA, Kroemer G and Takaku F: Bcl-2 down-regulation causes autophagy
in a caspase-independent manner in human leukemic HL60 cells. Cell
Death Differ. 7:1263–1269. 2000. View Article : Google Scholar
|
|
46
|
Wei Y, Pattingre S, Sinha S, Bassik M and
Levine B: JNK1-mediated phosphorylation of Bcl-2 regulates
starvation-induced autophagy. Mol Cell. 30:678–688. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Amaravadi RK, Lippincott-Schwartz J, Yin
XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT and White E:
Principles and current strategies for targeting autophagy for
cancer treatment. Clin Cancer Res. 17:654–666. 2011. View Article : Google Scholar : PubMed/NCBI
|