1. Introduction
The results from a breast cancer prevention clinical
trial in the past have shown that 4-hydroxyphenylretinamide (4-HPR,
fenretinide), a synthetic retinoid, given for more than 5 years to
women with removed primary breast cancer suppressed by 30% the
development of second cancer in the contra-lateral breast (1). Most importantly, 4-HPR decreased the
incidence of both, ER+ and ER− tumors that is
not the case with tamoxifen and aromatase inhibitors. 4-HPR was
particularly efficacious in premenopausal women, suggesting
potential involvement of ER/PR signaling in mediating the antitumor
potential of retinoids (2).
However, because of some side-effects of 4-HPR, these early
clinical studies were not extended and over the last 25 years no
further large scale breast cancer prevention trials with retinoids
have been performed (3). In
addition to 4-HPR, all-trans retinoic acid (atRA,
tretinoin), 9-cis retinoic acid (9-cis RA,
alitretinoin), 13-cis retinoic acid (13-cis RA,
isotretinoin) and rexinoid, LGD1069 (targretin, bexarotene) have
been also used for treatment of breast and other types of cancer,
but in most cases disappointing clinical results have been reported
(4). Surprisingly, the combination
of retinoids with temoxifen (5,6) or
with chemotherapy agents (taxol, cisplatin and histone deacethylase
inhibitors) did not significantly improve the clinical outcome in
patients with metastatic breast cancer (7). Most studies suggest that retinoids
suppress cell and tumor growth by receptor dependent and
independent mechanisms (3,4). Retinoids are ligands of retinoic acid
receptors alpha, beta, gamma (RARs, α, β and γ), whereas rexinoids
are ligands of retinoid X receptors alpha, beta, gamma (RXRs, α, β
and γ). Both, retinoids and rexinoids affect normal and tumor cells
by modulating transcriptional activity of the above receptors, as
well as by exploring receptor independent mechanisms (8,9).
Retinoids and rexinoids are cell differentiation agents, which
induce differentiation of both, epithelial and non-epithelial cells
that consequentially leads to inhibition of proliferation (10). Previously, we have shown in
vivo that retinoids (atRA, 9cRA and 4-HPR), rexinoids
(LGD1069), tamoxifen, aromatase inhibitors (vorazole) and DHEA, in
addition to inhibition of cell proliferation can also induce CS in
premalignant lesions and tumors of MNU-model of mammary
carcinogenesis which develops ER+ tumors in rats
(11,12). For both, retinoids and rexinoids,
lower doses preferentially suppressed cell proliferation and
induced CS, whereas higher doses induced apoptosis (13). Recently, we found that rexinoids
(bexarotene, LGD1069, targretin) are also efficacious inhibitors of
mammary carcinogenesis in MMTV-Neu mice, which spontaneously
develop ER− mammary tumors similar to those of triple
negative Her2/Neu positive breast cancers (14). The antitumor potential of rexinoids
in this model was associated with decreased cell proliferation and
increased CS. Cytotoxic agents, which cause DNA damage and gene
instability can also induce CS by activating p53-p21 signaling
(15,16). Each of the above cellular
mechanisms is consequence of multiple and well orchestrated gene
alterations recently summarized in several excellent reviews
(17–19). Over the last several years,
intensive research has been done on the role of oncogenes in the
development and maintenance of senescence phenotype in normal and
tumor cells. Among various oncogenes, the level of MYC and RAS
expression appears to play critical role. It was found that they
may promote or suppress tumor progression and in the latter CS
plays a significant role (20,21).
Increasing evidence indicates that SC are metabolically active and
may secrete various cytokines, which may not only inhibit, but also
promote cell proliferation and eventually tumor progression
(18,22,23).
2. Retinoids and rexinoids differentially
modulate senescence associated genes in ER+ and
ER− breast cancer cells
Studies from our and other laboratories have shown
that in ER+ breast cancer cell line retinoids (atRA,
9cRA and 4-HPR) are more efficacious than rexinoids (LGD1069,
bexarotene, targretin) in inhibiting cell growth and in inducing
CS, whereas rexinoids have very similar effect in both,
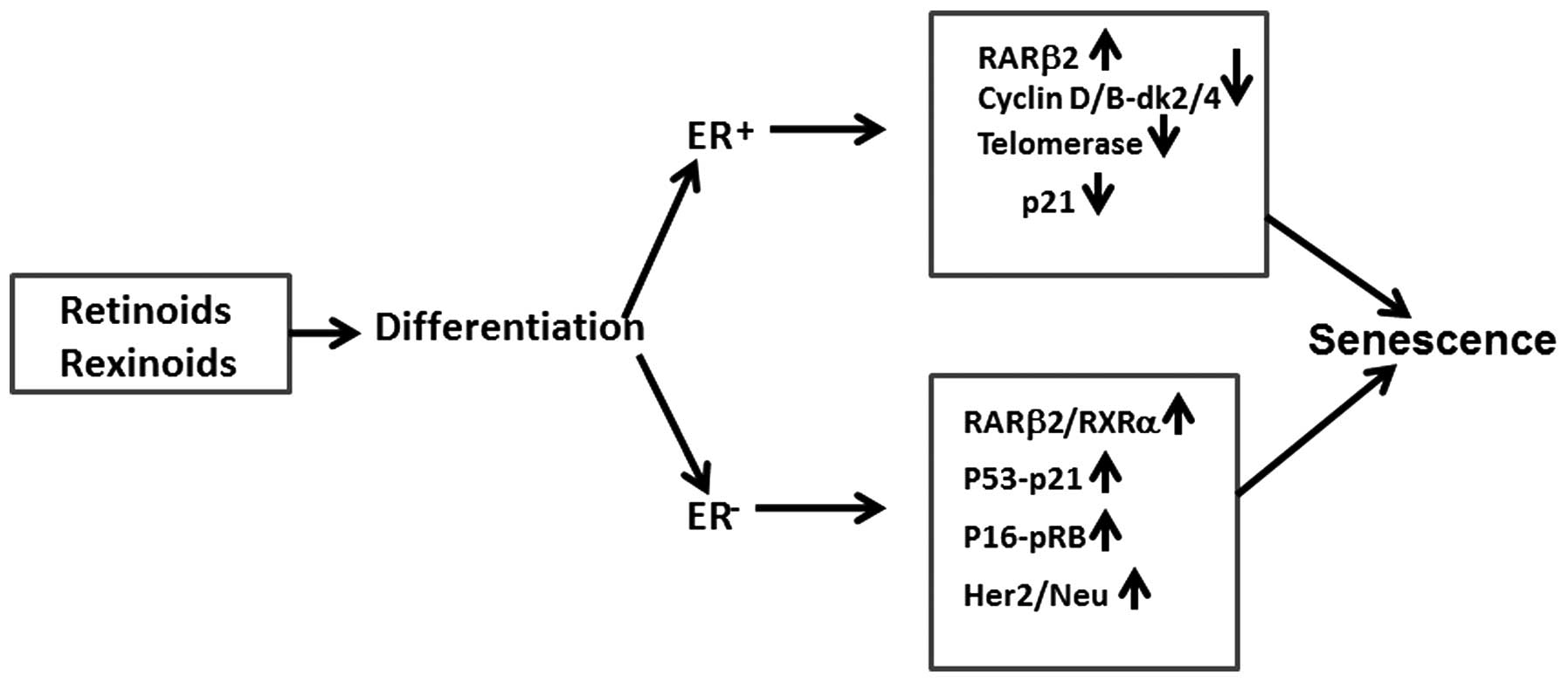
ER+ and ER− cell lines (4,10,14,17).
ER+ breast cancer cells when cultured for a long time,
for instance in colony formation assay, are prone spontaneously to
senesce contrary to ER− cells, which rarely senesce, but
rather develop stem cell phenotype (24). Further analysis of breast cancer
cell types revealed that, luminal A and normal-like luminal cells
are those that senesce, contrary to luminal B and basal-like cells,
which rarely senesce and behave as stem cells. These data are
important because human breast carcinomas could be divided into the
above subtypes and, thus, their cellular mechanisms of response to
treatment could be predicted. In addition to ER status, p21
expression appears also to modulate the retinoid/rexinoid induced
CS in normal human mammary epithelial cells (HMECs) and in most
breast cancer cell lines (Table
I). p21 induction is usually result of DNA damage that leads to
p53 activation and consequently to cell cycle arrest, CS and/or
apoptosis (16,19). This is well documented for MCF-7
cells treated with doxorubicin, but little is known whether
retinoids and rexinoids may also affect p53 and p21 expression.
Gene analysis of MCF-7 cells treated with atRA or doxorubicin
revealed overlapping of gene alterations, suggesting that in
inducing CS retinoids may explore, at least in part, the signaling
pathways of genotoxic agents (25). This was also confirmed in our
studies on MDA-MB-231 cells treated for 24 h with bexarotene and
doxorubicin, where p21 was upregulated (14). The extension of treatment with
bexarotene from 1 to 3 days increased not only p21, but p53
expression as well and this correlated with increased gH2A.X level,
an indicator of DNA damage. Since, in MCF-7 cells, bexarotene
decreased p21 expression, it appears that ER status may
differentially modulate the molecular mechanisms of response of
breast cancer cells to rexinoids and retinoids (Fig. 1). In addition to p53-p21 axe,
retinoids and rexinoids may explore other signaling pathways in
cell growth inhibition and CS. For instance, decreased cyclin
D1/E-cdk4/6 expression by ubiquitination and protein degradation
may suppress pRb phosphorylation and E2F expression, leading to
temporary (quiescence) or permanent (senescence) cell proliferation
arrest (26). Similar data were
recently reported for cyclin D1KE/KE deficient mouse
MECs that express ErbB2 (27).
MECs developed autophagy, but failed to implement ErbB2-induced
senescence in vivo. Downregulation of autophagy or cdk4/6
activity in MECs led to decreased autophagy and increased CS. We
also found that bexarotene decreased ATG4B (autophagy related 4B
cystein peptidase) in ER+, T47D cells but had opposite
effect in resistant to senescence ER−, MDA-MB-231 cells
(Table II). It was also found
that p95HER2 expression, a constitutively active fragment of the
tyrosine kinase receptor HER2 may result in either increased cell
proliferation or senescence (28).
In SC, p95HER2 elicits a secretome enriched in proteases, cytokines
and growth factors which eventually may increase cell growth and
metastatic capacity of breast tumor cells (22). The data support previous studies
showing that co-culturing of SC with fibroblasts stimulate
proliferation of the latter (18).
Thus, depending on the HER2 signaling, CS may play a double role,
to suppress or increase tumor growth. This information is important
because ~20% of human breast cancers express HerB2/Neu, suggesting
potential involvement of CS as biomarker of response in clinical
trials with Herceptine and other antitumor agents (Fig. 1). By cDNA microarray hybridization
and RT-PCR analysis it was shown that the retinoid-induced (atRA
and 4-HPR) growth arrest in MCF-7 cells is associated with strong
induction of 13 genes (25). Four
of these genes (IGF-binding protein 3, EPLIN, β IG-H3 and FAT10)
have anti-proliferative activity that may lead to CS. The function
of the induced genes may also account for other cellular effects of
retinoids, including proteosome-mediated protein degradation,
increased cell adhesion, and retinoic acid synthesis all of them
promoting CS (23,26). In normal HMEC, rexinoids
(bexarotene) modulate the activity of more than 100 genes
(upregulated and downregulated) (29). Sixteen of these genes have been
validated by using quantitative RT-PCR and western blotting, among
them: RARβ, growth regulatory genes, transcription factors and
differentiation markers. Some of these genes are associated with
cell cycle arrest, inflammation and CS. It is still an open
question whether retinoids/rexinoid, first induce cell
differentiation and consequently cell cycle arrest, which when
continues for a long time may lead to senescence, or senescence is
independent cellular mechanisms not necessarily associated with
differentiation, as has been reported for lymphocytic leukemia with
remarkable clinical benefits (30). In another study, Wainwright at
al (31) developed two
sub-clones from SK-N-SH neroblastoma cell line: in SH-N sub-clone
atRA induced neuronal differentiation with characteristic
neurofilaments, whereas in SH-F sub-clone cells senesce with
concomitant p16Ink4a and p18Ink4b
upregulation and surprisingly with decreased p21 expression. This
did not happen with differentiated SH-N sub-clone where atRA
induced p21. Thus, it appears that p21 may play distinctive role in
mediating cell differentiation and senescence induced by retinoids
in various tumor cell types. Cooperation between RARs/RXRs,
EGF/TGF/IGF and TGFα, TGFβ1/TGFβ2 signaling has been reported
recently (32). In addition to
RARs, retinoids may modulate WNT/NOTCH, PI3K/AKT, MAPKs and PKA/PKC
signaling and thus reduce cell proliferation and eventually induce
CS (33). In a recent study from
our laboratory, T47D and MDA-MB-231 cells were treated for 24 h
with bexarotene, and gene alterations, some of them associated with
CS were identified (Table II).
Since, it takes 5–7 days for both retinoids and rexinoids to induce
CS in vitro, the selected in Table II genes do not directly represent
those expressed in SC. However, they do indicate that even at very
early time-points, rexinoids may modulate the activity of certain
genes that contribute to CS. For instance, bexarotene induced DHRS3
and RARRES3 genes, which are associated with retinoid metabolism
and storage and thus by collateral mechanisms may affect RARs and
RXRs, including RARβ and consequently CS (34,35).
It appears that bexarotene is more efficacious inducer of
differentiation in ER+, T47D cells than in
ER−, MDA-MB-231 cells, as demonstrated by upregulation
of GDF15, KRT13 and CEND1, genes associated with cell
differentiation. In both cell lines bexarotene suppressed cell
cycle progression (telomerase reversed transcriptase-TERT, CEND1
and CDK11B), intercellular matrix protein stromolysin 3 (MMP11) and
basal membrane (laminin alpha 3-LAMA3) proteins, which indirectly
or by paracrine mechanisms potentiate CS (36). Modulation of RAS oncogene (RAB26)
and IGFBP6 may also contribute to the LGD1069 induced CS in breast
cancer cells (37,38).
 | Table IEffects of atRA and LGD1069 on
cellular senescence in breast cancer cell lines. |
Table I
Effects of atRA and LGD1069 on
cellular senescence in breast cancer cell lines.
| Cell line | Type | ER/PR | p21 | atRA-SC-%, 1.0
μM | LGD1069-SC-%, 1.0
μM |
|---|
| HMEC | Normal | − | + | 38 | 23 |
| MCF10 | Benign | − | + | 22 | 20 |
| MCF10AT | AH | − | + | 30 | 15 |
| MCFCA1a | Tumor | − | − | 15 | 6 |
| | | | 26.2±9.9 | 16±7.4 |
| MCF-7 | Tumor | + | + | 65 | 12 |
| T47D | Tumor | + | + | 52 | 24 |
| BT474 | Tumor | + | + | 28 | 15 |
| ZR-75-1 | Tumor | + | + | 33 | 18 |
| | | | 44.5±17.1a–c | 17.2±5.1c |
| MDA-MB-468 | Tumor | − | − | 17 | 8 |
| MDA-MB-231 | Tumor | − | + | 10 | 13 |
| MDA-MB-453 | Tumor | − | + | 15 | 16 |
| BT-20 | Tumor | − | + | 27 | 20 |
| SK-BR-3 | Tumor | − | + | 21 | 11 |
| | | | 18±6.4b | 13.6±4.6 |
| BCA-1 | Tumor | − | + | 22 | 12 |
| BCA-2 | Tumor | − | − | 3 | 4 |
| BCA-3 | Tumor | − | + | 26 | 15 |
| BCA-7 | Tumor | − | + | 37 | 20 |
| | | | 22.0±14.1b | 12.7±6.7 |
| Table IIRexinoids differentially affect
senescence associated genes in T47D and MDA-MB-231 cells. |
Table II
Rexinoids differentially affect
senescence associated genes in T47D and MDA-MB-231 cells.
| Gene symbol | T47D Bex/Con | MB231 Bex/Con | Gene name |
|---|
| DHRS3 | 9.84 | 6.48 |
Dehydrogenase/reductase (SDR family)
member 3 |
| RARRES3 | 5.61 | 2.08 | Retinoic acid
receptor responder 3 |
| GDF15 | 3.08 | 2.41 | Growth
differentiation factor 15 |
| TERT | 0.49 | 0.26 | Telomerase reverse
transcriptase |
| CDK11B | 0.47 | 0.24 | Cyclin-dependent
kinase 11B |
| MMP11 | 0.44 | 0.16 | Matrix
metallopeptidase 11 (stromelysin 3) |
| LAMA3 | 0.32 | 0.48 | Laminin, α 3 |
| KRT13 | 2.78 | 0.41 | Keratin 13 |
| UBE2E2 | 0.49 | 2.03 |
Ubiquitin-conjugating enzyme E2E2 |
| ATG4B | 0.48 | 2.47 | Autophagy related
4B, cysteine peptidase |
| APOD | 3.04 | 0.46 | Apolipoprotein
D |
| CEND1 | 5.05 | 0.45 | Cell cycle exit and
neuronal differentiation 1 |
| RAB26 | 3.49 | 0.98 | RAB26, member RAS
oncogene family |
| IGFBP6 | 1.99 | 2.00 | Insulin-like growth
factor binding protein 6 |
| RAB40AL | 1.03 | 0.41 | RAB40A, member RAS
oncogene family-like |
| p53 | 1.08 | 0.91 | Tumor protein
p53 |
3. RARβ isoforms and cellular senescence in
breast cancer cells
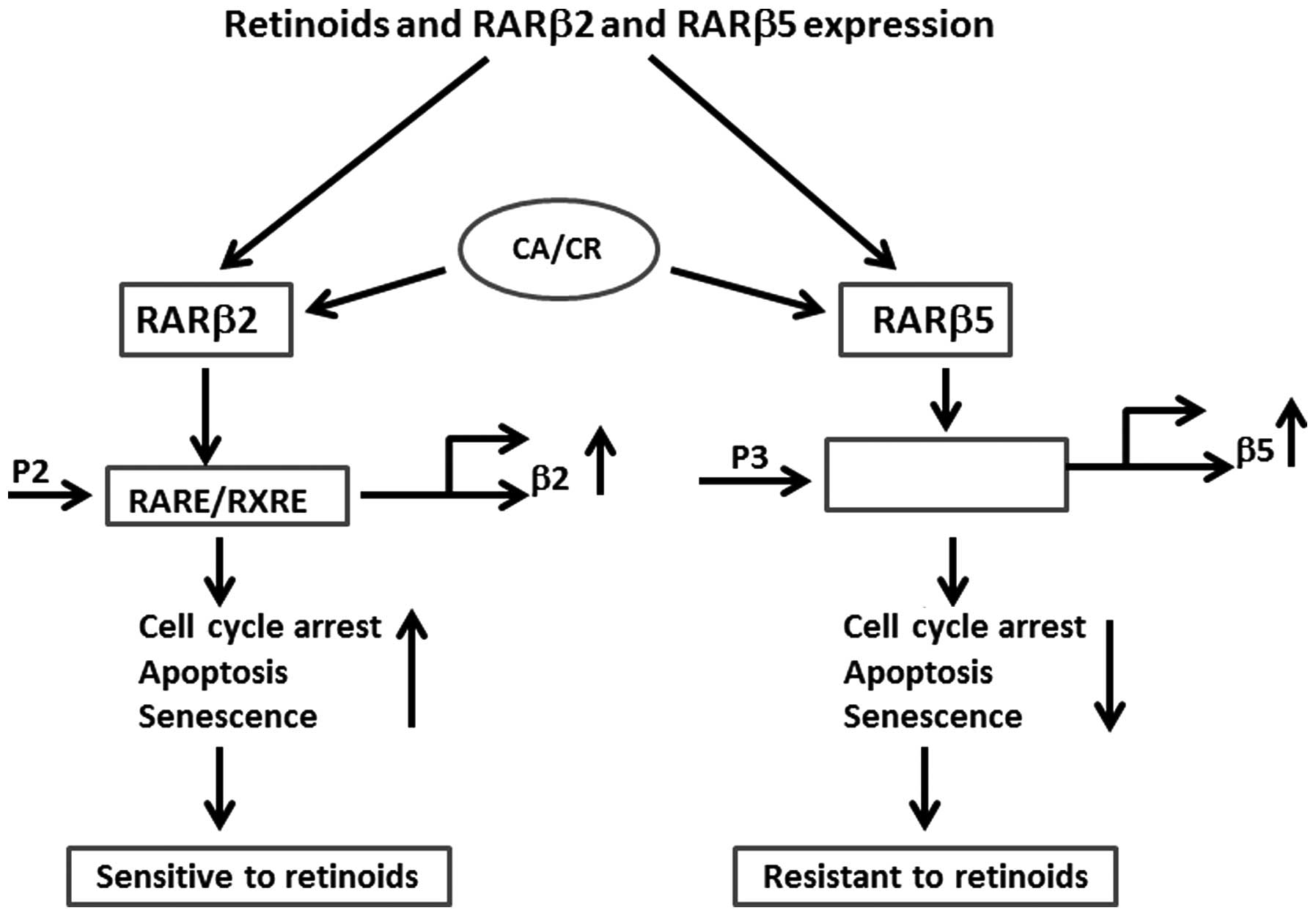
RARβ has five isoforms: β1, β2, β3, β4 and β5
(8,9). RARβ2 and RARβ4 isoforms are mostly
examined in breast normal and tumor cells, but they may also
mediate the effect of retinoids in other epithelial cell types
(8,10). RARβ2 is expressed in normal MECs,
but is lost in most breast cancer cells and in most premalignant
lesions and tumors, suggesting its tumor suppressor role (39). Activation of RARβ2 by retinoids or
by epigenetic approaches, as well as by gene transduction to cells
lacking the receptor may lead to decreased proliferation and
increased senescence (40,41). Previously, we have identified a
novel RARβ (β5) isoform (GenBank: AC133141.2 and AC098477.2) which
has an independent P3 promoter and appears to play a dominant
negative role in RARβ signaling (42). Breast cancer cells that express
RARβ5 were resistant to retinoids and when treated with atRA did
not senesce. RARβ5 inhibition by siRNA in MDA-MB-231 and BCA2 cells
increased their sensitivity to retinoids, as determined by cell
growth inhibition and CS (43). As
shown in the Fig. 2,
retinoids/rexinoids induce cell cycle arrest and CS by activating
P2 promoter and RARβ2 transcription, whereas upregulation of P3
promoter and RARβ5 expression have opposite effect and suppresses
CS. At mRNA level, the high RARβ2/RARβ5 ratio was associated with
increased cell sensitivity to retinoids (atRA, 9cRA), further
supporting the role of RARβ5 as potential dominant negative
regulator of RARβ2 (Fig. 2).
However, there are breast cancer cell lines, which do not express
RARβ5, but are also resistant to retinoids, suggesting involvement
of other transcription factors in mediating the cellular effect of
retinoids (19,36,38).
Recently, it was shown that breast carcinomas with high RARα/RARγ
ratio are more sensitive to atRA and have better prognosis than
those with inversed ratio of the above receptors (44). By microarray analysis it was found
that both, RARs agonists and antagonists produced similar effects
on gene expression, suggesting that the RARE-dependent RARβ2 gene
transcription is only a partial component of the retinoid-induced
cell growth inhibition and CS (45). The ability of retinoids and
rexinoids to induce CS depends also on the cell type and genetic
background. Thus, in a recent study it was shown that antisense
oligonucleotides against RARβ2 reduced proliferation and caused
apoptosis in 3 lung cancer cell lines, but had no effect in 2 other
cell lines lacking RARβ2, suggesting that RARβ2 may not only
suppress, but also promote proliferative activity of tumor cells
and thus plays a role of proto-oncogene (41). RARβ isoforms may directly or
indirectly cooperate with other RARs and RXRs, ER, and other
nuclear receptors (PPARβ/γ, vitamin D, thyroid) and thus affect
cellular responses to retinoids and rexinoids (9,41).
In most breast carcinomas RARβ is downregulated by hypermethylation
of its promoter and/or by alterations of chromatin structure
(39,46). Therefore, a combination of
retinoids with dimethylating agents, methyltransferase inhibitors
or histone deacethylase inhibitors have shown promising efficacy in
cell and tumor growth inhibition.
4. Retinoids and rexinoids induce CS in
mammary premalignant lesions and tumors
To identify SC in vitro and in vivo
β-galactosidase (SA-β-Gal) reaction was employed (47). The protocol for conducting this
reaction in tissues and tumors and potential alternative methods
for identification of SC are described in our previous studies
(11–14). We showed that retinoids (9cRA and
4-HPR) at doses that suppress MNU-induced mammary carcinogenesis in
rats in addition to inhibition of cell proliferation can also
induce CS (11,12). Surprisingly, 4-HPR given for 4
weeks also suppressed telomerase activity that correlated with
decreased cell proliferation and increased CS, suggesting the
potential involvement of telomerase in the retinoid-induced CS
(48). It has been shown that
shortening of telomere and decreased telomerase activity lead to
gene instability, activate p53 expression, and thus promote CS in
p53-dependent manner (49). By
employing MMTV-Neu mice, which spontaneously develop ER−
mammary tumors, bexarotene given for 4 weeks at 80 or 40 mg/kg body
weight, suppressed tumor frequency and growth and this was
associated with inhibition of cell proliferation and induction of
CS (14). Bexarotene was more
efficacious in inducing CS in normal MEC and premalignant lesions
than in tumors. SC were predominantly identified in differentiated
tumor areas, suggesting that differentiated breast carcinomas are
more prone to develop CS than non-differentiated ones (Fig. 3). By double labeling, first with
SA-β-Gal to identify SC and then by antibodies that recognize
biomarkers expressed in SC, we found that LGD1069 induced RARβ2,
p21, p16 and pRB, but not p53 expression in MMTV-Neu mammary
tumors, suggesting a p53 independent mechanisms of CS (Fig. 3). To further understand the role of
RARβ expression on the retinoid-induced CS in vivo, RARβ
wild-type (+/+) and RARβ deficient (−/−) mice were employed. We
obtained these mice from the laboratory of Pierre Chambon in
Strasburg, France. Mice were cross-bread in our laboratory and RARβ
expression was determined by DNA analysis. Mammary gland
architecture of RARβ homozygous (−/−) mice did not differ from that
of wild-type RARβ mice (+/+). Mice of both genotypes were followed
up for more than 12 months and no mammary tumors were identified,
suggesting that RARβ deficiency alone does not promote mammary
carcinogenesis. Mice of both genotypes were treated with 9cRA at 80
mg/kg for 4 weeks and cell proliferation and apoptosis were
determined. No difference in the values of BrdU-labeled and SC was
found in mammary terminal end buds (TEBs) and lobules of both
genotypes, suggesting that RARβ deficiency alone in normal MECs is
not the critical target of retinoids in inhibiting cell
proliferation and in inducing CS. Thus, it appears that other
transcription factors may contribute to the retinoid-induced CS in
normal and tumor MECs.
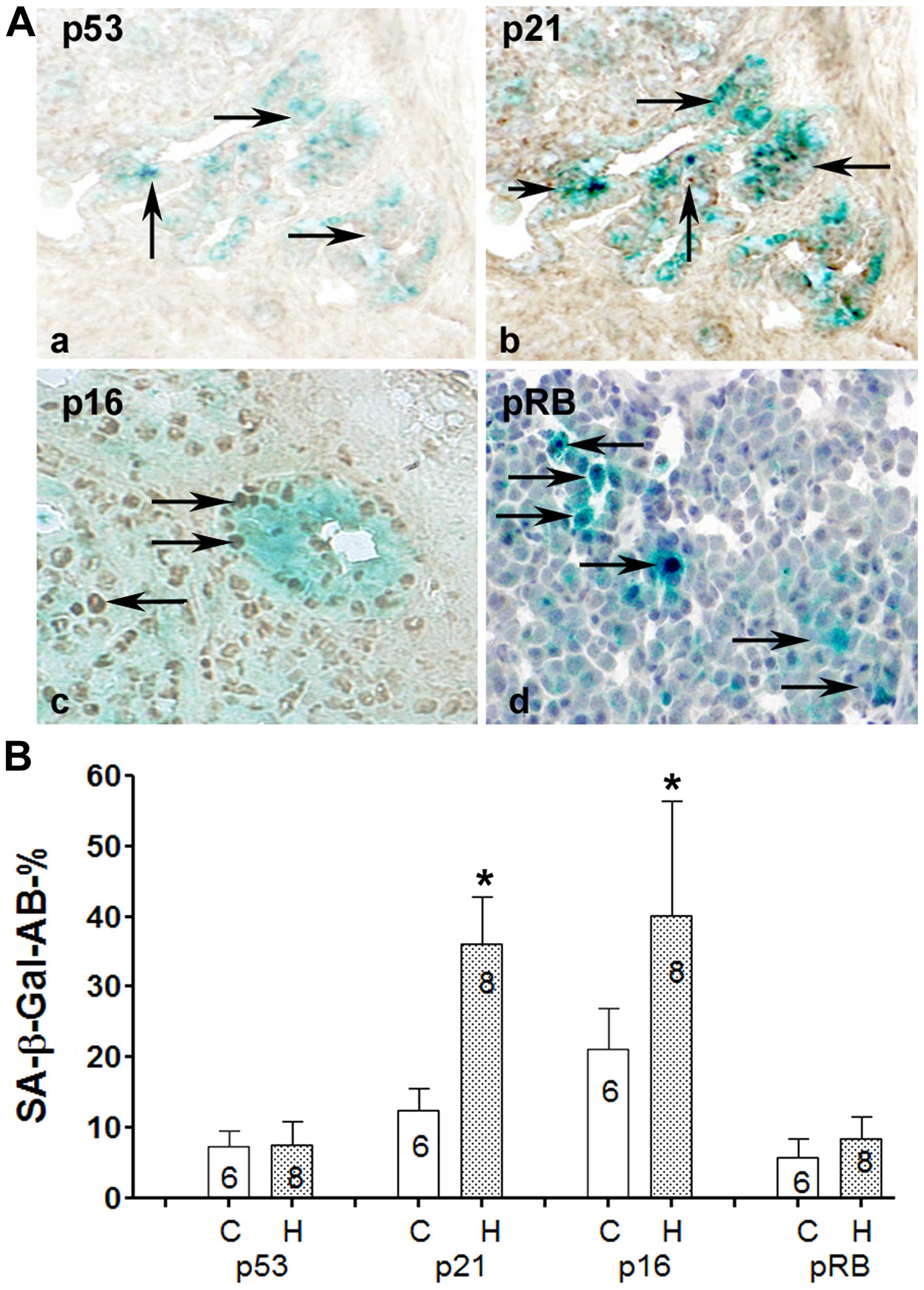 | Figure 3(A) A double-labeling method was
developed for identification of genes overexpressed in SC (14). (B) Values of SA-β-Gal cells and the
expression of p53, p21, p16 and pRB proteins in senescent and
non-senescent cells. C, control values, H, values in treated with
bexarotene tumors. In the columns is given the number of animals
with tumors examined. (A-a) Frozen sections from mammary tumor of
MMTV-Neu mice treated for 4 weeks with bexarotene. Slides were
first stained by SA-β-Gal kit and then overnight with antibody
against p53. SC (blue stained) are detected among differentiated
(alveolar) tumor area. No positive p53 staining in SC as compared
to control animals is presented (B, first columns; magnification,
×200). (A–b) Parallel sections from the same tumor stained by
SA-β-Gal and p21 antibody, note overexpression of p21
(brown-stained nuclei) among SC also as shown in B (second columns)
as well, asterisk indicates significant difference with control
values (P<0.05). (A–c) Frozen tumor section from a mammary tumor
of animal treated with bexarotene for 4 weeks and double stained by
SA-β-Gal and p16 antibody. p16 appears overexpressed (brown-stained
nuclei) in SC, as confirmed also in B (third columns;
*P<0.05). (A–d) Double staining of frozen tumor
section with SA-β-Gal kit and pRB antibody (magnification, ×400).
Although, there is increase in pRB in some tumor cells, no
significant difference with pRB values in non-senescent cells was
found (B, last column). |
5. Potential clinical implications
Cellular senescence as biomarker of
prognosis
Most studies suggest that malignant transformation
is associated with lost potential of cells to senesce (18,19).
We confirmed this in vivo, in mammary premalignant lesions
(AH, MIN and CIS) and tumors of rats and mice (12–14).
Studies by Collado et al (50) on a mouse model of lung
carcinogenesis that expresses K-ras12 V12 oncogene showed SC in
benign lesions (adenomas), but not in carcinomas, thus supporting
our in vivo data on mammary carcinogenesis. Clinical data
from patients with benign skin lesions (nevi) (51) and human breast cancer (52), further support the role of CS as
suppressor mechanism in tumor development and progression. In
addition to CS, low proliferating activity and high apoptosis have
been also considered biomarkers of good prognosis (13,53).
Thus, based on the values of proliferating cells, CS and apoptosis
mammary premalignant lesions and tumors could be divided into two
categories; one with high percentage of proliferating cells and low
percentage of SC and apoptosis, suggesting progression and poor
prognosis and another one, with small number of proliferating cell
and high number of CS and apoptosis, suggesting regression,
disintegration and favorable prognosis. Based on the percentage of
SC in premalignant lesions and tumors, Collado and Serrano
(54) suggested the implementation
of senescence index (SI), as biomarker of prognosis and treatment
efficacy, similarly to cell proliferation and apoptosis indexes.
Since, ARF-p53-p21 and/or p16-pRB are involved in mediating the
senescent program of antitumor agents including retinoids, the lack
or decreased expression of the above genes in breast premalignant
lesions and tumors may also have a negative effect on spontaneous
CS and thus promote carcinogenesis and tumor progression (55). Recent in vitro studies on
cell lines transfected with HER2/New support our in vivo
studies on MMTV-Neu mice indicating that activation of this
oncogene can promote CS (56).
Since, SC can produce cytokines that not only suppress, but also
promote cell proliferation, it has been speculated that CS can play
a double role, to suppress or promote tumor development and
progression (18,22).
Cellular senescence as biomarker of
efficacy
Efficacy studies with established and novel cancer
prevention and therapy agents are another avenue for potential
clinical implication of CS. By employing MNU-, and MMTV-Neu models
of mammary carcinogenesis, we found that retinoids (9cRA and
4-HPR), rexinoids (bexarotene), tamoxifen and aromatase inhibitors
(vorazole), in addition to inhibition of cell proliferation also
induced CS and this correlated with their efficacy to suppress
tumor growth (12,14). The role of CS as a biomarker of
response was also confirmed in a clinical trial with antitumor
agents. Patients with p53 wild-type and mutated-types of breast
carcinomas have been treated with neo-adjuvant chemotherapy, a
combination of cyclophosphamide, adriamycin and 5-fluorouracil
(CAF). In tumors removed 12–87 days after chemotherapy, SC have
been identified in p53 wild-type tumors only, suggesting p53
involvement in mediating CS. In addition to p53, p21 and p16
expression appears also involved in mediating CS induced by
cytotoxic agents (52). After
termination of treatment with antitumor agents, SC remain
detectable in tissues and tumors for a long time (weeks, months),
as compared to cell proliferation and apoptosis, which are
short-term cellular events, therefore, CS may have advantage as
biomarkers of response in long-term cancer prevention and therapy
studies. However, this need to be confirmed in future large scale
cancer prevention and therapy studies.
Development of novel agents that
preferentially induce CS
Development of novel agents that selectively induce
CS is another avenue that could be explored in cancer prevention
and treatment. These agents apparently need to modulate the
activity of genes involved in initiation and maintenance of
senescent phenotype. As was shown above, modulation of ER,
Her2/New, RAS, RARβ2 signaling may affect the decision of cells to
stop proliferating, senesce or die by apoptosis. Since, SC produce
in vitro cytokines that suppress cell growth by inducing CS
it has been suggested also to be employed for selective cytokines
in vivo experiments (55,56).
Some cytokines can also stimulate cell growth, therefore, selection
need to be strongly monitored.
Acknowledgements
The present study was supported by Susan G. Komen
Breast Cancer Research Foundation, KG100509 and NIH-R03CA137739
grants to K.C.
Abbreviations:
|
CS
|
cellular senescence
|
|
SC
|
senescent cells
|
|
MNU
|
N-methylnitrosourea
|
|
4HPR
|
4-hydroxyphenylretinamide
|
|
RARs
|
retinoid acid receptors α, β, γ
|
|
RXRs
|
retinoid X receptors α, β, γ
|
References
|
1
|
Veronesi U, De Palo G, Marubini E, Costa
A, Formelli F, Mariani L, Decensi A, Camerini T, Del Turco MR, Di
Mauro MG, et al: Randomized trial of fenretinide to prevent second
breast malignancy in women with early breast cancer. J Natl Cancer
Inst. 91:1847–1856. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Veronesi U, Mariani L, Decensi A, Formelli
F, Camerini T, Miceli R, Di Mauro MG, Costa A, Marubini E, Sporn
MB, et al: Fifteen-year results of a randomized phase III trial of
fenretinide to prevent second breast cancer. Ann Oncol.
17:1065–1071. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lazzeroni M and DeCensi A: Breast cancer
prevention by anti-hormones and other drugs: Where do we stand?
Hematol Oncol Clin North Am. 27:657–672. vii2013. View Article : Google Scholar
|
|
4
|
Garattini E, Bolis M, Garattini SK,
Fratelli M, Centritto F, Paroni G, Gianni’ M, Zanetti A, Pagani A,
Fisher JN, et al: Retinoids and breast cancer: From basic studies
to the clinic and back again. Cancer Treat Rev. 40:739–749. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chiesa MD, Passalacqua R, Michiara M,
Franciosi V, Di Costanzo F, Bisagni G, Camisa R, Buti S, Tomasello
G and Cocconi G; Italian Oncology Group for Clinical Research.
Tamoxifen vs Tamoxifen plus 13-cis-retinoic acid vs Tamoxifen plus
Interferon alpha-2a as first-line endocrine treatments in advanced
breast cancer: Updated results of a phase II, prospective,
randomised multicentre trial. Acta Biomed. 78:204–209. 2007.
|
|
6
|
Decensi A, Robertson C, Guerrieri-Gonzaga
A, Serrano D, Cazzaniga M, Mora S, Gulisano M, Johansson H,
Galimberti V, Cassano E, et al: Randomized double-blind 2 × 2 trial
of low-dose tamoxifen and fenretinide for breast cancer prevention
in high-risk premenopausal women. J Clin Oncol. 27:3749–3756. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bryan M, Pulte ED, Toomey KC, Pliner L,
Pavlick AC, Saunders T and Wieder R: A pilot phase II trial of
all-trans retinoic acid (Vesanoid) and paclitaxel (Taxol) in
patients with recurrent or metastatic breast cancer. Invest New
Drugs. 29:1482–1487. 2011. View Article : Google Scholar
|
|
8
|
Chambon P: A decade of molecular biology
of retinoic acid receptors. FASEB J. 10:940–954. 1996.PubMed/NCBI
|
|
9
|
Tang XH and Gudas LJ: Retinoids, retinoic
acid receptors, and cancer. Annu Rev Pathol. 6:345–364. 2011.
View Article : Google Scholar
|
|
10
|
Wang Q, Lee D, Sysounthone V, Chandraratna
RAS, Christakos S, Korah R and Wieder R: 1,25-dihydroxyvitamin D3
and retonic acid analogues induce differentiation in breast cancer
cells with function- and cell-specific additive effects. Breast
Cancer Res Treat. 67:157–168. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Christov KT, Shilkaitis AL, Kim ES, Steele
VE and Lubet RA: Chemopreventive agents induce a senescence-like
phenotype in rat mammary tumours. Eur J Cancer. 39:230–239. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shilkaitis A, Green A, Christov K,
Shilkaitis A, Punj V and Steele VE: DHEA inhibits the progression
phase of mammary carcinogenesis by inducing cellular senescence via
a p16 dependent but p53 independent mechanism. Breast Cancer Res.
7:132–140. 2005. View
Article : Google Scholar
|
|
13
|
Christov K, Grubbs CJ, Shilkaitis A,
Juliana MM and Lubet RA: Short-term modulation of cell
proliferation and apoptosis and preventive/therapeutic efficacy of
various agents in a mammary cancer model. Clin Cancer Res.
13:5488–5496. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shilkaitis A, Bratescu L, Green A, Yamada
T and Christov K: Bexarotene induces cellular senescence in
MMTV-Neu mouse model of mammary carcinogenesis. Cancer Prev Res
(Phila). 6:299–308. 2013. View Article : Google Scholar
|
|
15
|
Sugrue MM, Shin DY, Lee SW and Aaronson
SA: Wild-type p53 triggers a rapid senescence program in human
tumor cells lacking functional p53. Proc Natl Acad Sci USA.
94:9648–9653. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chang BD, Xuan Y, Broude EV, Zhu H, Schott
B, Fang J and Roninson IB: Role of p53 and p21waf1/cip1
in senescence-like terminal proliferation arrest induced in human
tumor cells by chemotherapeutic drugs. Oncogene. 18:4808–4818.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chang BD, Broude EV, Dokmanovic M, Zhu H,
Ruth A, Xuan Y, Kandel ES, Lausch E, Christov K and Roninson IB: A
senescence-like phenotype distinguishes tumor cells that undergo
terminal proliferation arrest after exposure to anticancer agents.
Cancer Res. 59:3761–3767. 1999.PubMed/NCBI
|
|
18
|
Campisi J: Senescent cells, tumor
suppression, and organismal aging: Good citizens, bad neighbors.
Cell. 120:513–522. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bartkova J, Rezaei N, Liontos M,
Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E,
Niforou K, Zoumpourlis VC, et al: Oncogene-induced senescence is
part of the tumorigenesis barrier imposed by DNA damage
checkpoints. Nature. 444:633–637. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Collado M and Serrano M: The power and the
promise of oncogene-induced senescence markers. Nat Rev Cancer.
6:472–476. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sarkisian CJ, Keister BA, Stairs DB, Boxer
RB, Moody SE and Chodosh LA: Dose-dependent oncogene-induced
senescence in vivo and its evasion during mammary tumorigenesis.
Nat Cell Biol. 9:493–505. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kuilman T and Peeper DS:
Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev
Cancer. 9:81–94. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Capparelli C, Chiavarina B,
Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, Andò S, Howell
A, Martinez-Outschoorn UE, Sotgia F, et al: CDK inhibitors
(p16/p19/p21) induce senescence and autophagy in cancer-associated
fibroblasts, ‘fueling’ tumor growth via paracrine interactions,
without an increase in neoangiogenesis. Cell Cycle. 11:3599–3610.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mumcuoglu M, Bagislar S, Yuzugullu H,
Alotaibi H, Senturk S, Telkoparan P, Gur-Dedeoglu B, Cingoz B,
Bozkurt B, Tazebay UH, et al: The ability to generate senescent
progeny as a mechanism underlying breast cancer cell heterogeneity.
PLoS One. 5:e112882010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dokmanovic M, Chang BD, Fang J and
Roninson IB: Retinoid-induced growth arrest of breast carcinoma
cells involves co-activation of multiple growth-inhibitory genes.
Cancer Biol Ther. 1:24–27. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tanaka T, Rodríguez de la Concepción ML
and De Luca LM: Involvement of all-trans-retinoic acid in the
breakdown of retinoic acid receptors alpha and gamma through
proteasomes in MCF-7 human breast cancer cells. Biochem Pharmacol.
61:1347–1355. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brown NE, Jeselsohn R, Bihani T, Hu MG,
Foltopoulou P, Kuperwasser C and Hinds PW: Cyclin D1 activity
regulates autophagy and senescence in the mammary epithelium.
Cancer Res. 72:6477–6489. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Angelini PD, Zacarias Fluck MF, Pedersen
K, Parra-Palau JL, Guiu M, Bernadó Morales C, Vicario R,
Luque-García A, Navalpotro NP, Giralt J, et al: Constitutive HER2
signaling promotes breast cancer metastasis through cellular
senescence. Cancer Res. 73:450–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim HT, Kong G, Denardo D, Li Y, Uray I,
Pal S, Mohsin S, Hilsenbeck SG, Bissonnette R, Lamph WW, et al:
Identification of biomarkers modulated by the rexinoid LGD1069
(bexarotene) in human breast cells using oligonucleotide arrays.
Cancer Res. 66:12009–12018. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bruno S, Ghiotto F, Tenca C, Mazzarello
AN, Bono M, Luzzi P, Casciaro S, Recchia A, Decensi A, Morabito F,
et al: N-(4-hydroxyphenyl)retinamide promotes apoptosis of resting
and proliferating B-cell chronic lymphocytic leukemia cells and
potentiates fludarabine and ABT-737 cytotoxicity. Leukemia.
26:2260–2268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wainwright LJ, Lasorella A and Iavarone A:
Distinct mechanisms of cell cycle arrest control the decision
between differentiation and senescence in human neuroblastoma
cells. Proc Natl Acad Sci USA. 98:9396–9400. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Paroni G, Fratelli M, Gardini G, Bassano
C, Flora M, Zanetti A, Guarnaccia V, Ubezio P, Centritto F, Terao
M, et al: Synergistic antitumor activity of lapatinib and retinoids
on a novel subtype of breast cancer with coamplification of ERBB2
and RARA. Oncogene. 31:3431–3443. 2012. View Article : Google Scholar
|
|
33
|
Han J, Hendzel MJ and Allalunis-Turner J:
Notch signaling as a therapeutic target for breast cancer
treatment? Breast Cancer Res. 13:210–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Deisenroth C, Itahana Y, Tollini L, Jin A
and Zhang Y: p53-Inducible DHRS3 is an endoplasmic reticulum
protein associated with lipid droplet accumulation. J Biol Chem.
286:28343–28356. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Morales M, Arenas EJ, Urosevic J, Guiu M,
Fernández E, Planet E, Fenwick RB, Fernández-Ruiz S, Salvatella X,
Reverter D, et al: RARRES3 suppresses breast cancer lung metastasis
by regulating adhesion and differentiation. EMBO Mol Med.
6:865–881. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Azouz A, Wu YL, Hillion J, Tarkanyi I,
Karniguian A, Aradi J, Lanotte M, Chen GQ, Chehna M and
Ségal-Bendirdjian E: Epigenetic plasticity of hTERT gene promoter
determines retinoid capacity to repress telomerase in
maturation-resistant acute promyelocytic leukemia cells. Leukemia.
24:613–622. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jin RU and Mills JC: RAB26 coordinates
lysosome traffic and mitochondrial localization. J Cell Sci.
127:1018–1032. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Uray IP, Shen Q, Seo HS, Kim H, Lamph WW,
Bissonnette RP and Brown PH: Rexinoid-induced expression of IGFBP-6
requires RARbeta-dependent permissive cooperation of retinoid
receptors and AP-1. J Biol Chem. 284:345–353. 2009. View Article : Google Scholar :
|
|
39
|
Widschwendter M, Berger J, Daxenbichler G,
Müller-Holzner E, Widschwendter A, Mayr A, Marth C and Zeimet AG:
Loss of retinoic acid receptor beta expression in breast cancer and
morphologically normal adjacent tissue but not in the normal breast
tissue distant from the cancer. Cancer Res. 57:4158–4161.
1997.PubMed/NCBI
|
|
40
|
Liu Y, Lee MO, Wang HG, Li Y, Hashimoto Y,
Klaus M, Reed JC and Zhang X: Retinoic acid receptor beta mediates
the growth-inhibitory effect of retinoic acid by promoting
apoptosis in human breast cancer cells. Mol Cell Biol.
16:1138–1149. 1996.PubMed/NCBI
|
|
41
|
Pappas JJ, Toulouse A, Basik M, Levesque L
and Bradley WEC: Knock-down of RARβ2 identifies a dual role of
cancer, genes. Chromosom Cancer. 50:700–714. 1911. View Article : Google Scholar
|
|
42
|
Peng X, Maruo T, Cao Y, Punj V, Mehta R,
Das Gupta TK and Christov K: A novel RARbeta isoform directed by a
distinct promoter P3 and mediated by retinoic acid in breast cancer
cells. Cancer Res. 64:8911–8918. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Christov K: The novel RARbeta isoform (β5)
is a potential target of retinoids in breast cancer. Curr Cancer
Drug Targets. 9:142–147. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bosch A, Bertran SP, Lu Y, Garcia A, Jones
AM, Dawson MI and Farias EF: Reversal by RARα agonist Am580 of
c-Myc-induced imbalance in RARα/RARγ expression during MMTV-Myc
tumorigenesis. Breast Cancer Res. 14:R1212012. View Article : Google Scholar
|
|
45
|
Chen Y, Dokmanovic M, Stein WD, Ardecky RJ
and Roninson IB: Agonist and antagonist of retinoic acid receptors
cause similar changes in gene expression and induce senescence-like
growth arrest in MCF-7 breast carcinoma cells. Cancer Res.
66:8749–8761. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sirchia SM, Ferguson AT, Sironi E,
Subramanyan S, Orlandi R, Sukumar S and Sacchi N: Evidence of
epigenetic changes affecting the chromatin state of the retinoic
acid receptor beta2 promoter in breast cancer cells. Oncogene.
19:1556–1563. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dimri GP, Lee X, Basile G, Acosta M, Scott
G, Roskelley C, Medrano EE, Linskens M, Rubelj I and Pereira-Smith
O: A biomarker that identifies senescent human cells in culture and
in aging skin in vivo. Proc Natl Acad Sci USA. 92:9363–9367. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bednarek A, Shilkaitis A, Green A, Lubet
R, Kelloff G, Christov K and Aldaz CM: Suppression of cell
proliferation and telomerase activity in
4-(hydroxyphenyl)retinamide-treated mammary tumors. Carcinogenesis.
20:879–883. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Feldser DM and Greider CW: Short telomeres
limit tumor progression in vivo by inducing senescence. Cancer
Cell. 11:461–469. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Collado M, Gil J, Efeyan A, Guerra C,
Schuhmacher AJ, Barradas M, Benguría A, Zaballos A, Flores JM,
Barbacid M, et al: Tumour biology: Senescence in premalignant
tumours. Nature. 436:6422005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Michaloglou C, Vredeveld LC, Soengas MS,
Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi
WJ and Peeper DS: BRAFE600-associated senescence-like cell cycle
arrest of human naevi. Nature. 436:720–724. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
te Poele RH, Okorokov AL, Jardine L,
Cummings J and Joel SP: DNA damage is able to induce senescence in
tumor cells in vitro and in vivo. Cancer Res. 62:1876–1883.
2002.PubMed/NCBI
|
|
53
|
Green DR and Walczak H: Apoptosis therapy:
Driving cancers down the road to ruin. Nat Med. 19:131–133. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Collado M and Serrano M: Senescence in
tumours: Evidence from mice and humans. Nat Rev Cancer. 10:51–57.
2010. View Article : Google Scholar
|
|
55
|
Nardella C, Clohessy JG, Alimonti A and
Pandolfi PP: Pro-senescence therapy for cancer treatment. Nat Rev
Cancer. 11:503–511. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Brown PH, Subbaramaiah K, Salmon AP, Baker
R, Newman RA, Yang P, Zhou XK, Bissonnette RP, Dannenberg AJ and
Howe LR: Combination chemoprevention of HER2/Neu-induced breast
cancer using COX-2 inhibitor and an RXR-selective retinoid. Cancer
Prev Res (Phila). 1:208–214. 2008. View Article : Google Scholar
|