Breast cancer is the most frequently diagnosed
cancer and the leading cause of cancer death in women worldwide,
accounting for ~23% of the total new cancer cases and 14% of the
total cancer deaths (1). It is
well known that breast cancer is a heterogeneous disease that can
be classified by microscopic appearance and molecular profiles such
as estrogen receptor (ER), progesterone receptor (PR) and human
epidermal growth factor receptor 2 (HER2) which correlate with
diverse clinical outcomes and responses to treatment. Systemic
treatment for breast cancer, including conventional cytotoxic
therapy (paclitaxe, doxorubicine, cyclophosphamide, fluorouracil,
cis-platinum), endocrine treatment (tamoxifen, fulvestrant,
letrozole, anastrozole), and targeted agents such as trastuzumab,
plays an essential role in reducing mortality rate and prolonging
survival time in patients with breast cancer (2,3).
However, resistance to therapeutic agents remains a consistent
obstacle in terms of treatment success, while the underlying
mechanism of drug resistance remains enigmatic (4,5).
Epithelial-mesenchymal transition (EMT) is a
biologic process by which epithelial cells lose their cell polarity
and cell-cell adhesion, and gain migratory and invasive properties
to become mesenchymal cells. A growing body of literature supports
that EMT is closely linked to the progression of breast cancer,
which includes enhanced migratory and invasive capacity, and
elevated stemness of cancer cells (6,7).
Now, emerging evidence suggests that EMT is also involved in
treatment resistance in breast cancer (8,9).
This review presents the events that involve the impact of EMT on
drug resistance in breast cancer, helping understand the generation
of treatment resistance and seek potential approaches to reverse
the process.
In breast cancer treatment, conventional cytotoxic
agents used alone or in combination weaken and destroy cancer cells
in the body. However, resistance to chemotherapy is a major hurdle
in the management of breast cancer. Some patients exhibit intrinsic
resistance to chemotherapy, while other patients, although
initially sensitive to chemotherapy, eventually develop acquired
resistance, even after combination therapy. At present, it is well
accepted that the mechanisms of chemoresistance may mainly contain
decreasing uptake of water-soluble drugs, various changes in cells
that affect the capacity of cytotoxic drugs to kill cells and
increasing energy-dependent efflux of hydrophobic drugs that can
easily enter the cells by diffusion through the plasma membrane
(10). Moreover, topoisomerase
poisons, altered expression of drug-metabolizing enzymes and
drug-conjugate export pumps, suppression of apoptotic pathways and
host-tumor-drug interactions also contribute to chemoresistance
(11). Among these, the most
significant is the increased efflux of hydrophobic drugs which are
regulated by a family of energy-dependent transporters, known as
ATP-binding cassette (ABC) transporters including P-glycoprotein
(P-gp, also known as ABCB1 or MDR1), multidrug resistance protein
(MRP) 1–7, lung resistance-related protein (LRP) and breast cancer
resistance protein (BCRP) (12).
Tamoxifen (TAM) is the usual endocrine
(anti-estrogen) therapy inducing objective response or disease
stabilization in breast cancer patients with ER+ tumors.
The pharmacologic action of tamoxifen is that it binds to the
estrogen receptor and induces dimerization and DNA binding to
finally inactivate it (13).
Nevertheless, about half of ER+ patients with advanced
disease and nearly all patients with metastatic disease fail to
respond to first-line TAM treatment. Approximately 40% of patients
receiving TAM as adjuvant therapy experience tumor relapse and die
from their disease, and a third of women treated with TAM for 5
years develop recurrent disease within 15 years (14). TAM resistance might arise as a
consequence of loss of expression or function of ERα, including
autophosphorylation, modulation by activation of transmembrane
tyrosine kinase receptors and interaction between downstream signal
transduction pathways (15).
Trastuzumab (herceptin), a humanized, recombinant
monoclonal antibody that selectively binds with high affinity to
the extracellular domain of HER2, has been proved to exert
antitumor effects in cancer models and patients with HER2-amplified
breast cancer. The addition of trastuzumab to adjuvant chemotherapy
can impressively reduce the recurrence rate (16). However, some patients with
HER2-overexpressing breast cancer do not respond to trastuzumab
therapy. There is only 26% response rate in women diagnosed with
HER2-positive metastatic breast cancer and treated with trastuzumab
as a single first-line agent. That is, >70% of
HER2-overexpressing metastatic breast carcinomas display a
resistance to trastuzumab (17).
The mechanisms underlying the resistance phenotype are not well
understood. Increased production of insulin-like growth factor,
dysregulation of p27, overexpression of epidermal growth factor
receptor (EGFR) with activation of the Akt pathway and decreased
PTEN function may contribute to this process (18).
EMT refers to a complex molecular and cellular
program by which epithelial cells shed their differentiated
characteristics, including cell-cell adhesion, planar/apical-basal
polarity, and lack of motility, and instead acquire mesenchymal
features. It has been described as the transition taking place in
epithelial cancer cells, which may lead to cancer invasion,
resistance to anoikis and systemic cancer cell dissemination
(19). During the acquisition of
EMT characteristics, cells undergo actin cytoskeleton
reorganization, decrease in the expression of proteins that promote
cell-cell contact such as E-cadherin and occludin, and gain in the
expression of mesenchymal markers such as vimentin, fibronectin and
N-cadherin, as well as increased activity of matrix
metalloproteinases (MMPs) like MMP-2, MMP-3 and MMP-9, which are
associated with an invasive phenotype (20). The tumor microenvironment comprised
of extracellular matrix, cells, and soluble factors plays a
critical role in EMT induction and further in tumor metastasis
(21,22). Several kinds of stromal cell
subtypes, such as macrophages and fibroblasts, contribute to tumor
progression through induction of EMT (23,24).
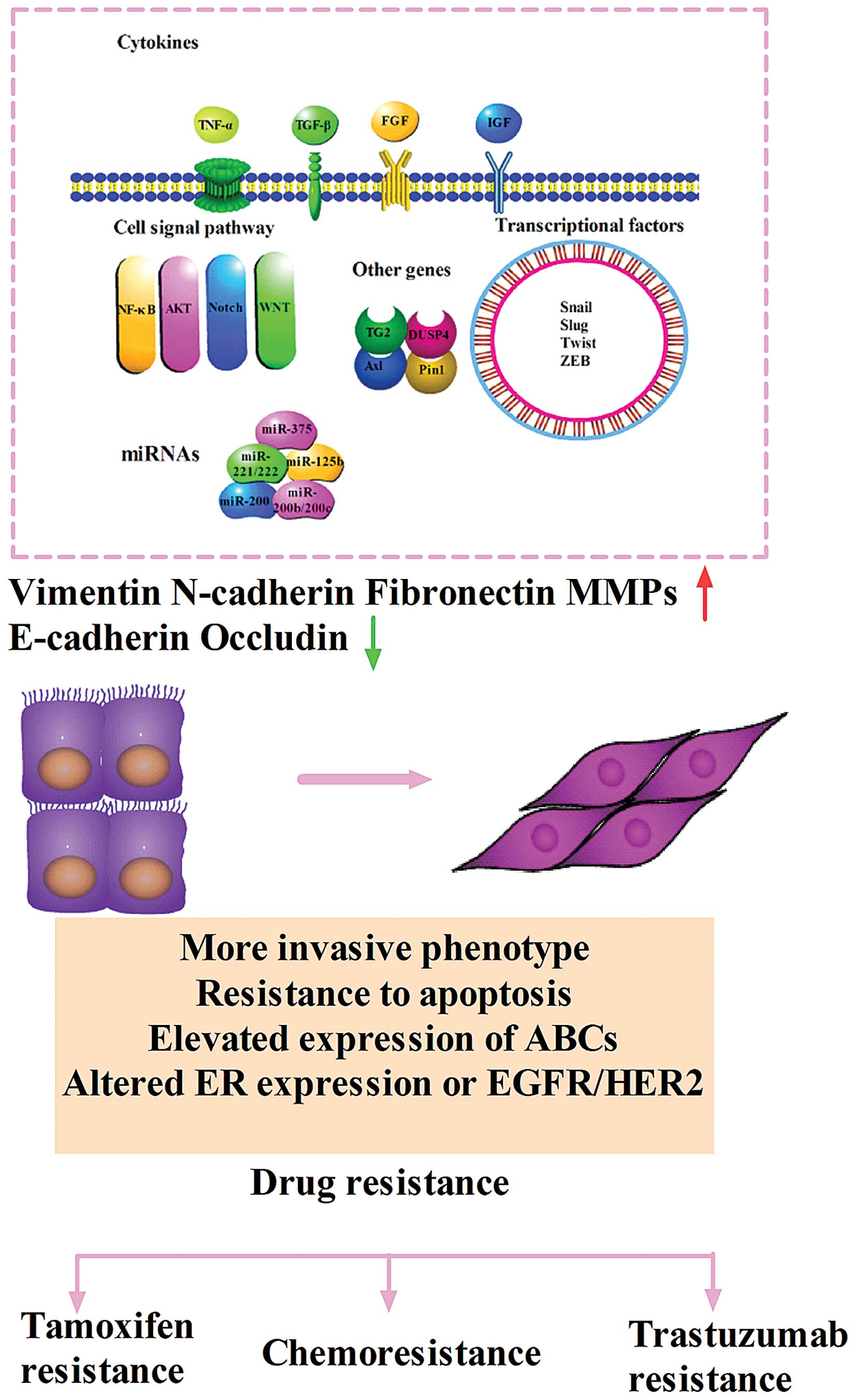
EMT can also be triggered by adverse conditions such as hypoxia and
a diverse set of extracellular stimuli including tumor necrosis
factor α (TNF-α), transforming growth factor β (TGF-β), epithelial
growth factor family member (EGF), fibroblast growth factor (FGF),
insulin growth factor (IGF), platelet derived growth factor (PDGF),
and components of the extracellular matrix such as collagen and
hyaluronic acid (21). Signal
transduction pathways including Wnt, Notch, nuclear factor-kappa B
(NF-κB), mitogen-activated protein kinase (MAPK) and phosphatidyl
inositol 3-kinase (PI3K) pathways can coordinate EMT programs.
Different stimuli induce a multitude of signal pathways that
converge on several EMT-inducing transcriptional factors including
Snail, Slug, Twist, Zeb1, Zeb2. All of the factors are capable of
repressing E-cadherin directly or indirectly when overexpressed in
cultured epithelial cells (25)
(Fig. 1).
It is well established that Snail family proteins
(Snail and Slug) are the key regulatory elements of EMT along with
the control of expression of many genes. Snail is involved in the
EMT that not only takes place concomitantly with the acquisition of
invasive properties in tumors, but also has been related to other
cancer hallmarks such as the gain of unlimited replication
potential, a greater resistance to apoptosis and even the evasion
of immunosurveillance (26). Vega
et al found that Snail attenuated the cell cycle and
conferred resistance to cell death induced by the withdrawal of
survival factors and pro-apoptotic signals (27). In another study, aberrant
expression of Snail or Slug in breast adenocarcinoma cells was
observed to protect against apoptosis induced by genotoxic stress,
which might be associated with direct transcriptional repression of
genes taking part in many aspects of programmed cell death
(28). Chen et al revealed
that MCF-7 breast cancer cells transfected with eukaryotic
expression vector pCDNA3.1-Snail showed EMT with BCRP-mediated
multidrug resistance (29).
Similarly, it was reported that overexpression of Snail accelerated
adriamycin induction of multidrug resistance through increasing the
expression of P-gp (30). In
paclitaxel, docetaxel, or doxorubicin resistant MCF-7 cell lines,
Slug expression was upregulated and ER was downregulated, resulting
in the repression of E-cadherin and occludin, and elevation of
N-cadherin and vimentin (31,32).
MCF-7 cells with ER deprivation were unresponsive to addition of
estradiol and TAM and acquired the EMT state (33). It was shown that decrease in the
estrogen dependency of breast cancer cells was accompanied by an
increased expression and activity of Snail, and demonstrated the
involvement of Snail in the negative regulation of ER (34). Further findings demonstrated that
Snail could repress ER-α expression by direct interaction with
regulatory DNA sequences at the ESR1 locus in breast cancer cell
lines (35). In addition, many
ERα-negative lines which were also E-cadherin-negative (e.g.,
MDA-MB-468 and MDA-MB-231) exhibited high Slug expression. It is
indicated that ligand-activated ERα formed a transcriptional
inhibitory complex comprised of nuclear receptor co-repressor
(N-CoR) and histone deacetylase 1 (HDAC1) which bonds to the Slug
promoter and directly suppresses Slug, which is one of the critical
members in Slug-E-cadherin-EMT pathway (36). Highly invasive breast cancer cell
lines expressed elevated levels of Twist, which upregulated the
transcription of Akt-2 to promote cell survival and resistance to
paclitaxel (37). Li et al
proved that adriamycin induced EMT and apoptosis in MCF-7 cells,
while only cells undergoing EMT displayed multidrug resistance.
Twist1 suppression prevented the drug-induced P-gp expression,
concomitant with partial reduction in resistance to paclitaxel,
vincristine and bleomycin (38).
It seems that EMT induction simultaneously upregulates the
expression of several ABC transporters, which lead to mutidrug
resistance in human breast cancer cells. There were binding sites
for several EMT transcription factors (Snail, Slug, Twist and
FOXC2) in 16 ABC transporters, while CHIP analysis further revealed
that Twist directly bound to E-boxes in the promoter region of
ABCC4 and ABCC5 in MCF-7 cells transfected with Twist (39).
TGF-β is one of the most potent and better-studied
inducers of EMT, acts through serine-threonine kinase receptors to
phosphorylate the cytoplasmic Smads which activates E-cadherin
repressors of the Snail family. A recent study indicated that
adverse activation of TGF-β pathway by chemotherapeutics in the
breast cancer cells or elevated TGF-β levels in tumor
microenvironment might lead to EMT and generation of cancer stem
cells, resulting in the resistance to chemotherapy (40). The evidence found cis-platinum
treatment of MDA-MB-231 breast cancer cells increased both TGF-β
mRNA levels and the secretion of active TGF-β, which enhanced
growth arrest that facilitated repair of damage, thus rendering
these cells resistant to cis-platinum killing (41). In the report of López-Díaz et
al, TGF-β was shown protected cells from DoxR, 5-fluorouracil
and paclitaxel-induced cell death specifically though Smad
4-mediated complex (42).
Moreover, TGF-β pretreatment was able to attenuate the TAM
cytotoxic effect and decrease the apoptosis ratio in breast cancer
(43). It was reported that TGF-β
increased ErbB/PI3K activation in BT474 and SKBR3 cells, and
desensitized the cells to trastuzumab-mediated growth inhibition
(44). TNF-α is another
inflammatory cytokine linked to both EMT and drug resistance.
MCF-7TN-R cells which were generated by prolonged and progressive
exposure of MCF-7 cells to TNF-α underwent progressive EMT changes,
and represented a model of transition to a multidrug resistant and
increased tumorigenic phenotype. In addition, some growth factors
which induce EMT may also take part in acquired resistance by
various patterns. IGF-1 was proposed to transmit signals via both
the PI3K and MAPK pathways, then resulted in the extracellular
activation of MMPs which were capable of promoting latent
TGF-β1-induced EMT, further rescued breast cancer cells from
chemotherapy-induced cell death (45,46).
It was also demonstrated that IGF-1 stimulated phosphorylation of
HER-2 exclusively in the trastuzumab resistant cells.
Antibody-mediated blockade of insulin growth factor repector
(IGF-1R) disrupts IGF-1R interaction with HER-2 and restores
trastuzumab sensitivity (47). FGF
expression was found as a stronger predictor of paclitaxel
resistance, compared to P-gp, p53, or Bcl-2 in patients with breast
cancer (48). With the interaction
with ER-activated pathways, FGF receptor-mediated signaling drives
autonomous growth which would be refractory to TAM therapy
(49). Regarding the induction of
FGF on EMT, these results suggested that EMT might be involved in
the FGF-mediated chemoresistance process.
Many signaling pathways which have significant
regulating effects on EMT are closely involved in drug resistance.
Genomic Region Enrichment was performed to find increased secretase
activity which may account for an increased Notch signaling in
endocrine resistant breast cancer cells (50). PF-03084014 which inhibits Notch
signaling by reducing Notch intracellular domain (NICD) and Notch
target genes Hes-1 and c-Myc in both cells and tumors prominently
enhanced the antitumor activity of docetaxel in MDA-MB-231
xenograft model through suppressing expression of survivin and
myeloid cell leukemia sequence 1 (MCL1), reducing ABCB1 and ABCC2,
upregulating BIM and reversing the EMT phenotype (51). Notch may be an important target in
trastuzumab-resistant, HER-2+ breast cancer. Growth of
trastuzumab-resistant cells was completely inhibited by combining
trastuzumab plus Notch-1 siRNA (52). The NF-κB pathway is emerging as an
essential regulator of EMT in cancer cell lines acting through the
induction of Snail transcription and protein stabilization.
Constitutively active NF-κB was also discovered to play a key role
in resistance to death-inducing stimuli, including chemotherapeutic
agents (53). NF-κB inhibitors
were found to sensitize breast cancer cells to doxorubicin
(54). Previous studies also
showed that phosphorylation and overexpression of NF-κB caused an
increase in ER-mediated transcription associated with endocrine
resistance. As a positive regulator of Snail in breast cancer
cells, simultaneous inhibition of NF-κB by RNA interference
resulted in marked increase of cell response to antiestrogen TAM
(55). Furthermore, the activation
of MAPK and PI3K pathways was also involved in the adaptation of
ER-positive breast cancer cells to estrogen deprivation by
contributing to ER hypersensitivity and were associated with
endocrine resistance (56). High
PI3K/Akt activity has been associated with resistance to
trastuzumab in HER2-overexpressing cells and primary tumors
(57). Additionally, it was
revealed that a number of canonical and non-canonical Wnt genes
(DKK1, JUN, PORCN, CSNK1A1 and MYC) were significantly increased in
the TAM-resistant cells. The Wnt inhibitor, IWP-2, resulted in
decreased expression of vimentin and Twist (58). Wnt3 acting as a key mediator in the
localization of β-catenin controlled EMT-like transition and
activation of EGFR in trastuzumab resistant cells (59).
Transglutaminase 2 (TG2), a pro-inflammatory protein
implicated in diverse physiological and pathological processes, was
reported to induce EMT in MCF-10A cells and confer resistance to
doxorubicin as an important downstream mediator of TGF-β (60). Dual specificity phosphatase 4
(DUSP4) is a member of the dual specificity phosphatase family,
which inactivates target kinases through dephosphorylating
phosphoserine/threonine and phosphor tyrosine residues. Liu et
al discovered that knockdown of DUSP4 increased the
chemosensitivity of MCF-7 and MCF-7/ADR breast cancer cells to
doxorubicin, and MCF-7/ADR cells with high levels of DUSP4 had a
mesenchymal phenotype (61). Pin1,
a peptidyl-prolyl isomerase, was overexpressed in TAM-resistant
(TAMR) MCF-7 cells. Pin1 siRNA treatment resulted in decreased
Snail transcription and the expression of EMT markers. It was
inferred that Pin1 might take part in EMT by affecting PTEN
expression and the subsequent PI3-kinase-Akt-dependent GSK-3β
inactivation (62). Axl is a
transmembrane tyrosine kinase receptor, activated by either its
ligand-growth arrest specific 6 or extracellular domain-mediated
dimerization or cross-talk with human EGFR2. It was shown that Axl
induced EMT as a upstream factor in normal and immortalized human
mammary epithelial cells in an apparent positive feedback loop
mechanism and regulate breast cancer stem cells (BCSCs)
self-renewal and chemoresistance (63).
Cancer stem cells (CSCs), or tumor-initiating cells
have been identified as having the ability to form mammosphere,
self-renew, exhibiting the CD44+/CD24− or
high aldehyde dehydrogenase (ALDH+) cell surface maker
profile and being associated with invasion, relapse and
drug-resistance (64). The stem
cells refractory to therapies is mainly because CSCs are known to
express increased levels of related members of ABC transporter
family and anti-apoptotic proteins (65). Furthermore, these cells are
hypothesized to be largely quiescent and slow cycling, which help
escape from typical cytotoxic agents (66). BCSCs with high expression of ALDH
can also help metabolize cytotoxic drugs (67). Morel et al were the first to
present evidence linking EMT to BCSCs. It was shown that induction
of EMT in transformed mammary epithelial cells generated cells with
BCSCs properties (68). This was
also corroborated in epithelial breast cancer cells of mouse models
(69,70). Circulating tumor cells from
metastatic breast cancer patients have shown EMT and tumor stem
cell characteristics (71).
Basal-like breast cancers, which are enriched for
CD44+/CD24− cells, are found to exhibit EMT
features that might account for their aggressive clinical behavior
and metastatic propensities. Moreover, a new subtype called
claudinlow-like was reported recently to display
CSC-associated features. In addition, metaplastic tumors which are
highly chemoresistant and aggressive are indicated to share
molecular similarities with CSCs (72). Both the metaplastic and
claudin-low-like tumors are closely related to the EMT core
signatures (73). These results
support a close connection between EMT and gain of CSC-like
properties. Besides, stem-like cells can be generated from
differentiated transformed mammary epithelial cells via EMT in
vitro, suggesting that EMT plays an active role in generating
CSCs in human breast tumors. HMLE cells acquired the
CD44high/CD24low stem cell profile, after
stimulated by TGF-β or in response to constitutive expression of
either Twist or Snail (74). The
loss of E-cadherin expression that transpires during EMT reinforces
these events by permitting the nuclear translocation of β-catenin
and its stimulation of CD44 expression (75). Overexpression of Twist in breast
cancer cells was demonstrated to promote the generation of a breast
cancer stem cell phenotype characterized by the high expression of
CD44 and exhibited high efflux of Hoechst 33342 and Rhodamine 123
as a result of increased expression of ABCC1 (MRP1) transporters
(76). It was also reported that
Twist induced the activation of β-catenin signaling pathway and Akt
pathways for the maintenance of the stem cell-like properties
associated with EMT (77). Fang
et al also found that Twist2 not only promoted the EMT
program, but also generated cells with stem cell-like properties
(78). The ZEB1 transcription
factor has been shown to modulate the two stemness genes KLF4 and
SOX2 indirectly, via downregulation of miR-200 which are rapidly
emerging as master regulators of differentiation by directly
targeting the transcriptional factors (ZEB1 and ZEB2/SIP1) to
derepress E-cadherin and elicit mesenchymal-epithelial transition
(MET), thus leads to the generation of migrating CSCs (79,80).
Guo et al revealed that Slug could cooperate with SOX9 in
orchestrating the stem cell state (81). Collectively, these observations
offer unquestionable evidence that EMT inducers are involved in
regulating cancer cell stemness.
In addition, HER2-overexpressing breast carcinomas
resistance to trastuzumab could also be linked to biology of stem
cell-like cells. It was demonstrated that CD44 was overexpressed in
trastuzumab resistant JIMT-1 cells and induced HER-2 receptor
internalization in vitro and in vivo (82). Trastuzumab resistance can result
from the spontaneous conversion of HER-2+ cells to a
CD44+/CD24−/HER-2−/low phenotype
through EMT (83). It was
discovered that trastuzumab sensitivity was restricted to the
Slug/Snail2-negative subset of luminal/HER2+ cell lines,
whereas all of the Slug/Snail2-positive basal/HER2+ cell
lines exhibited a primary (inherent) resistance to trastuzumab.
Knockdown of Slug could suppress the
CD44+/CD24−/low phenotype which might be
responsible for trastuzumab refractoriness in
basal/HER2+ JIMT1 cells and sensitize
trastuzumab-resistant xenografts to trastuzumab. Quote of a
sentence in the chapter: EMT-driving transcriptional repressor
Slug/Snail2 appears to be a pivotal gene that induces an enhanced
phenotypic plasticity in basal/HER2+ cells, thus
allowing them to ‘enter' into and ‘exit' from
trastuzumab-responsive stem cell-like states (84). Thus, EMT may promote drug-resistace
via potentiating cell characteristics of CSCs.
MicroRNAs (miRNAs), a class of small cellular RNAs,
acting as agents of the RNA interference pathway, can lead to
silencing of their cognate target genes, by either cleaving mRNA
molecules or inhibiting their translation (85). In this decade, studies have shown
that miRNAs regulate EMT through directly targeting families of EMT
transcription factors or affecting the integrity of the epithelial
architecture during EMT progression (86). miR-200 family which has a striking
negative correlation with ZEB could regulate EMT and drug
resistance. It was reported that miR-200 expression could reverse
resistance to EGFR inhibitor therapy in bladder cancer cells
(87). miR-200 cluster was
associated with substantial expression of E-cadherin mRNA in breast
cancer tissues and low miR-200 expression was associated with
pronounced benefits of cyclophosphamide (88). Restoration of miR-200c enhanced
chemosensitivity to microtubule-directed agents in MCF-7 and T47D
cells (89). In addition, miR-200c
was shown to correlate with the acquired resistance of breast
cancer cells to doxorubicin by inhibiting Akt signaling through its
effects on E-cadherin and PTEN (90). A previous report found that
transfection of MDA-MB-231 cells with pre-miR-200b or pre-miR-200c
enhanced their sensitivity to doxorubicin. Similarly, reduced
miRNA-200b and miR-200c expression contributed to endocrine
resistance in breast cancer cells. Accompanied by the increase in
miR-200b and miR-200c, ZEB1 expression was decreased and cells
appeared more epithelial in morphology and were sensitized to TAM
inhibition (91). It has been
proved that miR-125b induced EMT-like molecular alterations, and
functioned as a key mediator for Snail-induced stem cell
propagation and chemoresistance in breast cancer cells (92,93).
A recent study reported EMT was linked to loss of ERα expression,
through transcriptional repression of ER by Snail and concomitant
translational inhibition of ERα mRNA by miR-221/222 (94). Additionally, miRNA-375 was found
downregulated in the TAM-resistant MCF-7 cells, while its
re-expression sensitized cells to TAM and reverted EMT-like
properties. Metadherin (MTDH) was regarded as the direct target of
miRNA-375, with its established relevance in drug resistance and
breast cancer metastasis (95).
The transcriptional factors and the hallmarks of EMT
are often related to more malignant type in breast cancer patients.
High expression of Slug and Twist has been reported closely
correlated with poor prognosis in patients with breast cancer
(96,97). Jeong et al noted that EMT
was significantly related to high histological grade and
triple-negative phenotype but not predictive of disease-free
survival in patients with breast cancer (98). Since EMT has been established as a
mechanism that confers tumor cells with the essential ability for
drug resistance, metastasis, and acquired-tumor stem cell traits,
inhibition of EMT can be a critical therapeutic strategy for
prevention of tumor progression (99). NPI-0052, a protesome inhibitor, has
been demonstrated to depress EMT via weakening NF-κB and Snail
(100). Shinto et al found
Ki26894, a TBR-I kinase inhibitor, suppressed the invasiveness and
EMT in scirrhous gastric cancer cells (101). Artesunate (an antimalarial agent)
has been discovered to induce cell cycle arrest and apoptosis
possibly by affecting the hyperactive Wnt pathway and reversing EMT
in colorectal cell lines (102).
The Src kinase inhibitor dasatinib has been proven to inhibit
growth of breast cancer cells with EMT features (103). Cystatin C, a cysteine protease
inhibitor has been found to inhibit the acquisition of EMT and
invasion stimulated by TGF-β in breast cancer cell by preventing
actin cytoskeletal rearrangements and E-cadherin downregulation
(104). Chua et al
developed an EMT inhibition screening assay to identify compounds
targeting ALK5, MEK, and SRC as potent inhibitors that can
interfere with EGF, HGF, or IGF-1 induced EMT signaling (105).
EMT is a complex, stepwise phenomenon that occurs
during embryonic development and tumor progression, and involves
major reprogramming of gene expression that leads to alterations in
cell fate and behavior. Clarifying the underlying mechanism linking
EMT and drug resistance would likely be useful for devising better
targeted therapeutic approaches in combination with conventional
therapeutics.
The hallmarks of EMT are loss of the epithelial
molecule E-cadherin and gain of mesenchymal markers, such as
N-cadherin and vimentin. Loss of E-cadherin expression can lead to
loss of contact inhibition, infinite proliferation,
de-differentiation, loose intercellular connections, and be
susceptibility to shedding in cancer cells, which enhances both
invasion and migration of cancer cells (31). N-cadherin is highly expressed in
invasive and metastatic human breast cancer cells and correlates
with aggressive clinical behavior. The alterations of these genes
contribute to endowing cells with higher malignancy. Moreover, an
increasing number of direct evidence revealed these genes had close
connection with the resistance to therapy (106–108). In addition, the transcriptional
factors of EMT such as Snail, Slug and Twist not only elevate the
cell invasion and metastasis to escape being killed, but also
increase/decrease the essential genes taking part in drug
resistance. Certain cytokines and genes play essential roles on
both EMT and drug resistance. The signaling pathways of EMT are
wide and extremely complex, which constitute main targets for novel
drug development. A better understanding of the roles of EMT and
CSCs in breast cancer will lead to more effective therapies that
will target not only the tumor but also the residual population of
cells that are responsible for the relapse and resurgence of the
tumor. Further examination of the epigenetic changes such as miRNA
will also be an important area of research. All these results
suggest that drug combinations using conventional or targeted
therapies together with targeting the EMT-related mechanisms need
to be considered for winning the battle against drug resistant in
cancer cells.
This study was supported by National Natural Science
Foundation of China (No. 81102007 and No. 81472475), and the
International S&T Cooperation Program of China (ISTCP)
(2012DFA10650).
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hernandez-Aya LF and Gonzalez-Angulo AM:
Adjuvant systemic therapies in breast cancer. Surg Clin North Am.
93:473–491. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Collignon J, Rorive A, Martin M, Andre C,
Maweja S, Lifrange E, Coucke P and Jerusalem G: Systemic
chemotherapy and breast cancer. Rev Med Liege. 66:372–378. 2011.(In
French). PubMed/NCBI
|
|
4
|
Saeki T, Tsuruo T, Sato W and Nishikawsa
K: Drug resistance in chemotherapy for breast cancer. Cancer
Chemother Pharmacol. 56(Suppl 1): 84–89. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Coley HM: Mechanisms and strategies to
overcome chemotherapy resistance in metastatic breast cancer.
Cancer Treat Rev. 34:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Roxanis I: Occurrence and significance of
epithelial-mesenchymal transition in breast cancer. J Clin Pathol.
66:517–521. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang Y and Zhou BP: Epithelial-mesenchymal
transition - a hallmark of breast cancer metastasis. Cancer Hallm.
1:38–49. 2013. View Article : Google Scholar
|
|
8
|
Dave B, Mittal V, Tan NM and Chang JC:
Epithelial-mesenchymal transition, cancer stem cells and treatment
resistance. Breast Cancer Res. 14:2022012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mallini P, Lennard T, Kirby J and Meeson
A: Epithelial-to-mesenchymal transition: What is the impact on
breast cancer stem cells and drug resistance. Cancer Treat Rev.
40:341–348. 2014. View Article : Google Scholar
|
|
10
|
Szakács G, Paterson JK, Ludwig JA,
Booth-Genthe C and Gottesman MM: Targeting multidrug resistance in
cancer. Nat Rev Drug Discov. 5:219–234. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gonzalez-Angulo AM, Morales-Vasquez F and
Hortobagyi GN: Overview of resistance to systemic therapy in
patients with breast cancer. Adv Exp Med Biol. 608:1–22. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dean M: ABC transporters, drug resistance,
and cancer stem cells. J Mammary Gland Biol Neoplasia. 14:3–9.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shah PD and Dickler MN: Endocrine therapy
for advanced breast cancer. Clin Adv Hematol Oncol. 12:214–223.
2014.PubMed/NCBI
|
|
14
|
Normanno N, Di Maio M, De Maio E, De Luca
A, de Matteis A, Giordano A and Perrone F; NCI-Naple Breast Cancer
Group. Mechanisms of endocrine resistance and novel therapeutic
strategies in breast cancer. Endocr Relat Cancer. 12:721–747. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Al Saleh S, Sharaf LH and Luqmani YA:
Signalling pathways involved in endocrine resistance in breast
cancer and associations with epithelial to mesenchymal transition
(Review). Int J Oncol. 38:1197–1217. 2011.PubMed/NCBI
|
|
16
|
Slamon D, Eiermann W, Robert N, Pienkowski
T, Martin M, Press M, Mackey J, Glaspy J, Chan A, Pawlicki M, et
al; Breast Cancer International Research Group. Adjuvant
trastuzumab in HER2-positive breast cancer. N Engl J Med.
365:1273–1283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Martin-Castillo B, Oliveras-Ferraros C,
Vazquez-Martin A, Cufí S, Moreno JM, Corominas-Faja B,
Urruticoechea A, Martín ÁG, López-Bonet E and Menendez JA:
Basal/HER2 breast carcinomas: Integrating molecular taxonomy with
cancer stem cell dynamics to predict primary resistance to
trastuzumab (Herceptin). Cell Cycle. 12:225–245. 2013. View Article : Google Scholar :
|
|
18
|
Gajria D and Chandarlapaty S:
HER2-amplified breast cancer: Mechanisms of trastuzumab resistance
and novel targeted therapies. Expert Rev Anticancer Ther.
11:263–275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Foroni C, Broggini M, Generali D and Damia
G: Epithelial-mesenchymal transition and breast cancer: Role,
molecular mechanisms and clinical impact. Cancer Treat Rev.
38:689–697. 2012. View Article : Google Scholar
|
|
20
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Voulgari A and Pintzas A:
Epithelial-mesenchymal transition in cancer metastasis: Mechanisms,
markers and strategies to overcome drug resistance in the clinic.
Biochim Biophys Acta. 1796:75–90. 2009.PubMed/NCBI
|
|
22
|
Bezdenezhnykh N, Semesiuk N, Lykhova O,
Zhylchuk V and Kudryavets Y: Impact of stromal cell components of
tumor microenvironment on epithelial-mesenchymal transition in
breast cancer cells. Exp Oncol. 36:72–78. 2014.PubMed/NCBI
|
|
23
|
Bonde AK, Tischler V, Kumar S, Soltermann
A and Schwendener RA: Intratumoral macrophages contribute to
epithelial-mesenchymal transition in solid tumors. BMC Cancer.
12:352012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Orimo A, Gupta PB, Sgroi DC,
Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL
and Weinberg RA: Stromal fibroblasts present in invasive human
breast carcinomas promote tumor growth and angiogenesis through
elevated SDF-1/CXCL12 secretion. Cell. 121:335–348. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: At the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vega S, Morales AV, Ocaña OH, Valdés F,
Fabregat I and Nieto MA: Snail blocks the cell cycle and confers
resistance to cell death. Genes Dev. 18:1131–1143. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kajita M, McClinic KN and Wade PA:
Aberrant expression of the transcription factors snail and slug
alters the response to genotoxic stress. Mol Cell Biol.
24:7559–7566. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen WJ, Wang H, Tang Y, Liu CL, Li HL and
Li WT: Multidrug resistance in breast cancer cells during
epithelial-mesenchymal transition is modulated by breast cancer
resistant protein. Chin J Cancer. 29:151–157. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li W, Liu C, Tang Y, Li H, Zhou F and Lv
S: Overexpression of Snail accelerates adriamycin induction of
multidrug resistance in breast cancer cells. Asian Pac J Cancer
Prev. 12:2575–2580. 2011.
|
|
31
|
Prasad CP, Rath G, Mathur S, Bhatnagar D,
Parshad R and Ralhan R: Expression analysis of E-cadherin, Slug and
GSK3beta in invasive ductal carcinoma of breast. BMC Cancer.
9:3252009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Iseri OD, Kars MD, Arpaci F, Atalay C, Pak
I and Gunduz U: Drug resistant MCF-7 cells exhibit
epithelial-mesenchymal transition gene expression pattern. Biomed
Pharmacother. 65:40–45. 2011. View Article : Google Scholar
|
|
33
|
Luqmani YA, Al Azmi A, Al Bader M, Abraham
G and El Zawahri M: Modification of gene expression induced by
siRNA targeting of estrogen receptor alpha in MCF7 human breast
cancer cells. Int J Oncol. 34:231–242. 2009.
|
|
34
|
Andreeva OE, Shcherbakov AM, Shatskaia VA
and Krasilńikov MA: The role of transcription factor Snail1 in the
regulation of hormonal sensitivity of in vitro cultured breast
cancer cells. Vopr Onkol. 58:71–76. 2012.(In Russian).
|
|
35
|
Dhasarathy A, Kajita M and Wade PA: The
transcription factor snail mediates epithelial to mesenchymal
transitions by repression of estrogen receptor-alpha. Mol
Endocrinol. 21:2907–2918. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ye Y, Xiao Y, Wang W, Yearsley K, Gao JX
and Barsky SH: ERalpha suppresses slug expression directly by
transcriptional repression. Biochem J. 416:179–187. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD
and Wang LH: Twist transcriptionally up-regulates AKT2 in breast
cancer cells leading to increased migration, invasion, and
resistance to paclitaxel. Cancer Res. 67:1979–1987. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li QQ, Xu JD, Wang WJ, Cao XX, Chen Q,
Tang F, Chen ZQ, Liu XP and Xu ZD: Twist1-mediated
adriamycin-induced epithelial-mesenchymal transition relates to
multidrug resistance and invasive potential in breast cancer cells.
Clin Cancer Res. 15:2657–2665. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Saxena M, Stephens MA, Pathak H and
Rangarajan A: Transcription factors that mediate
epithelial-mesenchymal transition lead to multidrug resistance by
upregulating ABC transporters. Cell Death Dis. 2:e1792011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bandyopadhyay A, Wang L, Agyin J, Tang Y,
Lin S, Yeh IT, De K and Sun LZ: Doxorubicin in combination with a
small TGFbeta inhibitor: A potential novel therapy for metastatic
breast cancer in mouse models. PLoS One. 5:e103652010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hirohashi S and Kanai Y: Cell adhesion
system and human cancer morphogenesis. Cancer Sci. 94:575–581.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
López-Díaz FJ, Gascard P, Balakrishnan SK,
Zhao J, Del Rincon SV, Spruck C, Tlsty TD and Emerson BM:
Coordinate transcriptional and translational repression of p53 by
TGF-β1 impairs the stress response. Mol Cell. 50:552–564. 2013.
View Article : Google Scholar
|
|
43
|
Shi XP, Miao S, Wu Y, Zhang W, Zhang XF,
Ma HZ, Xin HL, Feng J, Wen AD and Li Y: Resveratrol sensitizes
tamoxifen in antiestrogen-resistant breast cancer cells with
epithelial-mesenchymal transition features. Int J Mol Sci.
14:15655–15668. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang SE, Xiang B, Guix M, Olivares MG,
Parker J, Chung CH, Pandiella A and Arteaga CL: Transforming growth
factor beta engages TACE and ErbB3 to activate
phosphatidylinositol-3 kinase/Akt in ErbB2-overexpressing breast
cancer and desensitizes cells to trastuzumab. Mol Cell Biol.
28:5605–5620. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Walsh LA and Damjanovski S: IGF-1
increases invasive potential of MCF-7 breast cancer cells and
induces activation of latent TGF-β1 resulting in epithelial to
mesenchymal transition. Cell Commun Signal. 9:102011. View Article : Google Scholar
|
|
46
|
Gooch JL, Van Den Berg CL and Yee D:
Insulin-like growth factor (IGF)-I rescues breast cancer cells from
chemotherapy-induced cell death - proliferative and anti-apoptotic
effects. Breast Cancer Res Treat. 56:1–10. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nahta R, Yuan LX, Zhang B, Kobayashi R and
Esteva FJ: Insulin-like growth factor-I receptor/human epidermal
growth factor receptor 2 heterodimerization contributes to
trastuzumab resistance of breast cancer cells. Cancer Res.
65:11118–11128. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gan Y, Wientjes MG and Au JL: Expression
of basic fibroblast growth factor correlates with resistance to
paclitaxel in human patient tumors. Pharm Res. 23:1324–1331. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
McLeskey SW, Zhang L, El-Ashry D, Trock
BJ, Lopez CA, Kharbanda S, Tobias CA, Lorant LA, Hannum RS, Dickson
RB, et al: Tamoxifen-resistant fibroblast growth factor-transfected
MCF-7 cells are cross-resistant in vivo to the antiestrogen ICI
182,780 and two aromatase inhibitors. Clin Cancer Res. 4:697–711.
1998.PubMed/NCBI
|
|
50
|
Magnani L, Stoeck A, Zhang X, Lánczky A,
Mirabella AC, Wang TL, Gyorffy B and Lupien M: Genome-wide
reprogramming of the chromatin landscape underlies endocrine
therapy resistance in breast cancer. Proc Natl Acad Sci USA.
110:E1490–E1499. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang CC, Yan Z, Zong Q, Fang DD, Painter
C, Zhang Q, Chen E, Lira ME, John-Baptiste A and Christensen JG:
Synergistic effect of the γ-secretase inhibitor PF-03084014 and
docetaxel in breast cancer models. Stem Cells Transl Med.
2:233–242. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pandya K, Meeke K, Clementz AG, Rogowski
A, Roberts J, Miele L, Albain KS and Osipo C: Targeting both Notch
and ErbB-2 signalling pathways is required for prevention of
ErbB-2-positive breast tumour recurrence. Br J Cancer. 105:796–806.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Orlowski RZ and Baldwin AS Jr: NF-kappaB
as a therapeutic target in cancer. Trends Mol Med. 8:385–389. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tapia MA, González-Navarrete I, Dalmases
A, Bosch M, Rodriguez-Fanjul V, Rolfe M, Ross JS, Mezquita J,
Mezquita C, Bachs O, et al: Inhibition of the canonical IKK/NF
kappa B pathway sensitizes human cancer cells to doxorubicin. Cell
Cycle. 6:2284–2292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Musgrove EA and Sutherland RL: Biological
determinants of endocrine resistance in breast cancer. Nat Rev
Cancer. 9:631–643. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gee JM, Robertson JF, Ellis IO and
Nicholson RI: Phosphorylation of ERK1/2 mitogen-activated protein
kinase is associated with poor response to anti-hormonal therapy
and decreased patient survival in clinical breast cancer. Int J
Cancer. 95:247–254. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Berns K, Horlings HM, Hennessy BT,
Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM,
Stemke-Hale K, Hauptmann M, et al: A functional genetic approach
identifies the PI3K pathway as a major determinant of trastuzumab
resistance in breast cancer. Cancer Cell. 12:395–402. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Loh YN, Hedditch EL, Baker LA, Jary E,
Ward RL and Ford CE: The Wnt signalling pathway is upregulated in
an in vitro model of acquired tamoxifen resistant breast cancer.
BMC Cancer. 13:1742013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wu Y, Ginther C, Kim J, Mosher N, Chung S,
Slamon D and Vadgama JV: Expression of Wnt3 activates Wnt/β-catenin
pathway and promotes EMT-like phenotype in trastuzumab-resistant
HER2-overexpressing breast cancer cells. Mol Cancer Res.
10:1597–1606. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kumar A, Xu J, Brady S, Gao H, Yu D,
Reuben J and Mehta K: Tissue transglutaminase promotes drug
resistance and invasion by inducing mesenchymal transition in
mammary epithelial cells. PLoS One. 5:e133902010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Liu Y, Du F, Chen W, Yao M, Lv K and Fu P:
Knockdown of dual specificity phosphatase 4 enhances the
chemosensitivity of MCF-7 and MCF-7/ADR breast cancer cells to
doxorubicin. Exp Cell Res. 319:3140–3149. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kim MR, Choi HK, Cho KB, Kim HS and Kang
KW: Involvement of Pin1 induction in epithelial-mesenchymal
transition of tamoxifen-resistant breast cancer cells. Cancer Sci.
100:1834–1841. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Asiedu MK, Beauchamp-Perez FD, Ingle JN,
Behrens MD, Radisky DC and Knutson KL: AXL induces
epithelial-to-mesenchymal transition and regulates the function of
breast cancer stem cells. Oncogene. 33:1316–1324. 2014. View Article : Google Scholar :
|
|
64
|
Dontu G, Abdallah WM, Foley JM, Jackson
KW, Clarke MF, Kawamura MJ and Wicha MS: In vitro propagation and
transcriptional profiling of human mammary stem/progenitor cells.
Genes Dev. 17:1253–1270. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Donnenberg VS and Donnenberg AD: Multiple
drug resistance in cancer revisited: The cancer stem cell
hypothesis. J Clin Pharmacol. 45:872–877. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Jordan CT, Guzman ML and Noble M: Cancer
stem cells. N Engl J Med. 355:1253–1261. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kakarala M and Wicha MS: Implications of
the cancer stem-cell hypothesis for breast cancer prevention and
therapy. J Clin Oncol. 26:2813–2820. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Morel AP, Lièvre M, Thomas C, Hinkal G,
Ansieau S and Puisieux A: Generation of breast cancer stem cells
through epithelial-mesenchymal transition. PLoS One. 3:e28882008.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Radisky DC and LaBarge MA:
Epithelial-mesenchymal transition and the stem cell phenotype. Cell
Stem Cell. 2:511–512. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Santisteban M, Reiman JM, Asiedu MK,
Behrens MD, Nassar A, Kalli KR, Haluska P, Ingle JN, Hartmann LC,
Manjili MH, et al: Immune-induced epithelial to mesenchymal
transition in vivo generates breast cancer stem cells. Cancer Res.
69:2887–2895. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Aktas B, Tewes M, Fehm T, Hauch S, Kimmig
R and Kasimir-Bauer S: Stem cell and epithelial-mesenchymal
transition markers are frequently overexpressed in circulating
tumor cells of metastatic breast cancer patients. Breast Cancer
Res. 11:R462009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: New insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Taube JH, Herschkowitz JI, Komurov K, Zhou
AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, et
al: Core epithelial-to-mesenchymal transition interactome
gene-expression signature is associated with claudin-low and
metaplastic breast cancer subtypes. Proc Natl Acad Sci USA.
107:15449–15454. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wielenga VJ, Smits R, Korinek V, Smit L,
Kielman M, Fodde R, Clevers H and Pals ST: Expression of CD44 in
Apc and Tcf mutant mice implies regulation by the WNT pathway. Am J
Pathol. 154:515–523. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Vesuna F, Lisok A, Kimble B and Raman V:
Twist modulates breast cancer stem cells by transcriptional
regulation of CD24 expression. Neoplasia. 11:1318–1328. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Li J and Zhou BP: Activation of β-catenin
and Akt pathways by Twist are critical for the maintenance of EMT
associated cancer stem cell-like characters. BMC Cancer. 11:492011.
View Article : Google Scholar
|
|
78
|
Fang X, Cai Y, Liu J, Wang Z, Wu Q, Zhang
Z, Yang CJ, Yuan L and Ouyang G: Twist2 contributes to breast
cancer progression by promoting an epithelial-mesenchymal
transition and cancer stem-like cell self-renewal. Oncogene.
30:4707–4720. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wellner U, Schubert J, Burk UC,
Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D,
zur Hausen A, et al: The EMT-activator ZEB1 promotes tumorigenicity
by repressing stemness-inhibiting microRNAs. Nat Cell Biol.
11:1487–1495. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Park SM, Gaur AB, Lengyel E and Peter ME:
The miR-200 family determines the epithelial phenotype of cancer
cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes
Dev. 22:894–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Guo W, Keckesova Z, Donaher JL, Shibue T,
Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zürrer-Härdi U, Bell
G, et al: Slug and Sox9 cooperatively determine the mammary stem
cell state. Cell. 148:1015–1028. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Pályi-Krekk Z, Barok M, Isola J, Tammi M,
Szöllosi J and Nagy P: Hyaluronan-induced masking of ErbB2 and
CD44-enhanced trastuzumab internalisation in trastuzumab resistant
breast cancer. Eur J Cancer. 43:2423–2433. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Lesniak D, Sabri S, Xu Y, Graham K,
Bhatnagar P, Suresh M and Abdulkarim B: Spontaneous
epithelial-mesenchymal transition and resistance to HER-2-targeted
therapies in HER-2-positive luminal breast cancer. PLoS One.
8:e719872013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Oliveras-Ferraros C, Corominas-Faja B,
Cufí S, Vazquez-Martin A, Martin-Castillo B, Iglesias JM,
López-Bonet E, Martin ÁG and Menendez JA: Epithelial-to-mesenchymal
transition (EMT) confers primary resistance to trastuzumab
(Herceptin). Cell Cycle. 11:4020–4032. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Lamouille S, Subramanyam D, Blelloch R and
Derynck R: Regulation of epithelial-mesenchymal and
mesenchymal-epithelial transitions by microRNAs. Curr Opin Cell
Biol. 25:200–207. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Adam L, Zhong M, Choi W, Qi W, Nicoloso M,
Arora A, Calin G, Wang H, Siefker-Radtke A, McConkey D, et al:
miR-200 expression regulates epithelial-to-mesenchymal transition
in bladder cancer cells and reverses resistance to epidermal growth
factor receptor therapy. Clin Cancer Res. 15:5060–5072. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Bojmar L, Karlsson E, Ellegård S, Olsson
H, Björnsson B, Hallböök O, Larsson M, Stål O and Sandström P: The
role of microRNA-200 in progression of human colorectal and breast
cancer. PLoS One. 8:e848152013. View Article : Google Scholar :
|
|
89
|
Cochrane DR, Spoelstra NS, Howe EN,
Nordeen SK and Richer JK: MicroRNA-200c mitigates invasiveness and
restores sensitivity to microtubule-targeting chemotherapeutic
agents. Mol Cancer Ther. 8:1055–1066. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Chen Y, Sun Y, Chen L, Xu X, Zhang X, Wang
B, Min L and Liu W: miRNA-200c increases the sensitivity of breast
cancer cells to doxorubicin through the suppression of
E-cadherin-mediated PTEN/Akt signaling. Mol Med Rep. 7:1579–1584.
2013.PubMed/NCBI
|
|
91
|
Manavalan TT, Teng Y, Litchfield LM,
Muluhngwi P, Al-Rayyan N and Klinge CM: Reduced expression of
miR-200 family members contributes to antiestrogen resistance in
LY2 human breast cancer cells. PLoS One. 8:e623342013. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Liu Z, Liu H, Desai S, Schmitt DC, Zhou M,
Khong HT, Klos KS, McClellan S, Fodstad O and Tan M: miR-125b
functions as a key mediator for snail-induced stem cell propagation
and chemoresistance. J Biol Chem. 288:4334–4345. 2013. View Article : Google Scholar :
|
|
93
|
Wang HJ, Guo YQ, Tan G, Dong L, Cheng L,
Li KJ, Wang ZY and Luo HF: miR-125b regulates side population in
breast cancer and confers a chemoresistant phenotype. J Cell
Biochem. 114:2248–2257. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Guttilla IK, Phoenix KN, Hong X, Tirnauer
JS, Claffey KP and White BA: Prolonged mammosphere culture of MCF-7
cells induces an EMT and repression of the estrogen receptor by
microRNAs. Breast Cancer Res Treat. 132:75–85. 2012. View Article : Google Scholar
|
|
95
|
Ward A, Balwierz A, Zhang JD, Küblbeck M,
Pawitan Y, Hielscher T, Wiemann S and Sahin Ö: Re-expression of
microRNA-375 reverses both tamoxifen resistance and accompanying
EMT-like properties in breast cancer. Oncogene. 32:1173–1182. 2013.
View Article : Google Scholar
|
|
96
|
Liu T, Zhang X, Shang M, Zhang Y, Xia B,
Niu M, Liu Y and Pang D: Dysregulated expression of Slug, vimentin,
and E-cadherin correlates with poor clinical outcome in patients
with basal-like breast cancer. J Surg Oncol. 107:188–194. 2013.
View Article : Google Scholar
|
|
97
|
Soini Y, Tuhkanen H, Sironen R, Virtanen
I, Kataja V, Auvinen P, Mannermaa A and Kosma VM: Transcription
factors zeb1, twist and snai1 in breast carcinoma. BMC Cancer.
11:732011. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Jeong H, Ryu YJ, An J, Lee Y and Kim A:
Epithelial-mesenchymal transition in breast cancer correlates with
high histological grade and triple-negative phenotype.
Histopathology. 60B:E87–E95. 2012. View Article : Google Scholar
|
|
99
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Baritaki S, Chapman A, Yeung K, Spandidos
DA, Palladino M and Bonavida B: Inhibition of epithelial to
mesenchymal transition in metastatic prostate cancer cells by the
novel proteasome inhibitor, NPI-0052: Pivotal roles of Snail
repression and RKIP induction. Oncogene. 28:3573–3585. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Shinto O, Yashiro M, Kawajiri H, Shimizu
K, Shimizu T, Miwa A and Hirakawa K: Inhibitory effect of a TGFbeta
receptor type-I inhibitor, Ki26894, on invasiveness of scirrhous
gastric cancer cells. Br J Cancer. 102:844–851. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Li L-N, Zhang H-D, Yuan S-J, Yang D-X,
Wang L and Sun Z-X: Differential sensitivity of colorectal cancer
cell lines to artesunate is associated with expression of
beta-catenin and E-cadherin. Eur J Pharmacol. 588:1–8. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Sokol JP, Neil JR, Schiemann BJ and
Schiemann WP: The use of cystatin C to inhibit
epithelial-mesenchymal transition and morphological transformation
stimulated by transforming growth factor-beta. Breast Cancer Res.
7:R844–R853. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Finn RS, Dering J, Ginther C, Wilson CA,
Glaspy P, Tchekmedyian N and Slamon DJ: Dasatinib, an orally active
small molecule inhibitor of both the src and abl kinases,
selectively inhibits growth of basal-type/‘triple-negative' breast
cancer cell lines growing in vitro. Breast Cancer Res Treat.
105:319–326. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Chua KN, Sim WJ, Racine V, Lee SY, Goh BC
and Thiery JP: A cell-based small molecule screening method for
identifying inhibitors of epithelial-mesenchymal transition in
carcinoma. PLoS One. 7:e331832012. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Lu L, Zhou D, Jiang X, Song K, Li K and
Ding W: Loss of E-cadherin in multidrug resistant breast cancer
cell line MCF-7/Adr: Possible implication in the enhanced invasive
ability. Eur Rev Med Pharmacol Sci. 16:1271–1279. 2012.PubMed/NCBI
|
|
107
|
Roger L, Jullien L, Gire V and Roux P:
Gain of oncogenic function of p53 mutants regulates E-cadherin
expression uncoupled from cell invasion in colon cancer cells. J
Cell Sci. 123:1295–1305. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Zhang B, Groffen J and Heisterkamp N:
Increased resistance to a farnesyltransferase inhibitor by
N-cadherin expression in Bcr/Abl-P190 lymphoblastic leukemia cells.
Leukemia. 21:1189–1197. 2007. View Article : Google Scholar : PubMed/NCBI
|