1. Introduction
DNA mismatch repair (MMR) is a very highly conserved
cellular process, involving many proteins, resulting in the
identification, and subsequent repair of mismatched bases, likely
to have arisen during DNA replication, genetic recombination or
chemical or physical damage (Fig.
1). The MMR genes play additional roles in double-strand break
repair, apoptosis and recombination. The four key genes identified
to date are mutL homologue 1 (MLH1), mutS homologue 2
(MSH2), mutS homologue 6 (MSH6) and postmeiotic
segregation increased 2 (PMS2), so named because of their
homology to the E. coli MMR genes. The MSH2 and MSH6
proteins form a heterodimeric complex (mutSα) which is involved in
the initial identification of mismatched bases, and initiates DNA
repair. Binding to the mismatch results in an ATP-dependent
conformational change, which subsequently recruits mutLα, a
heterodimer comprising of MLH1 and PMS2. Other proteins are
recruited to complete the DNA repair, but are not discussed further
in this review. The repair complexes ensure that it is the newly
synthesised strand of DNA which is targeted for repair, not the
parental strand.
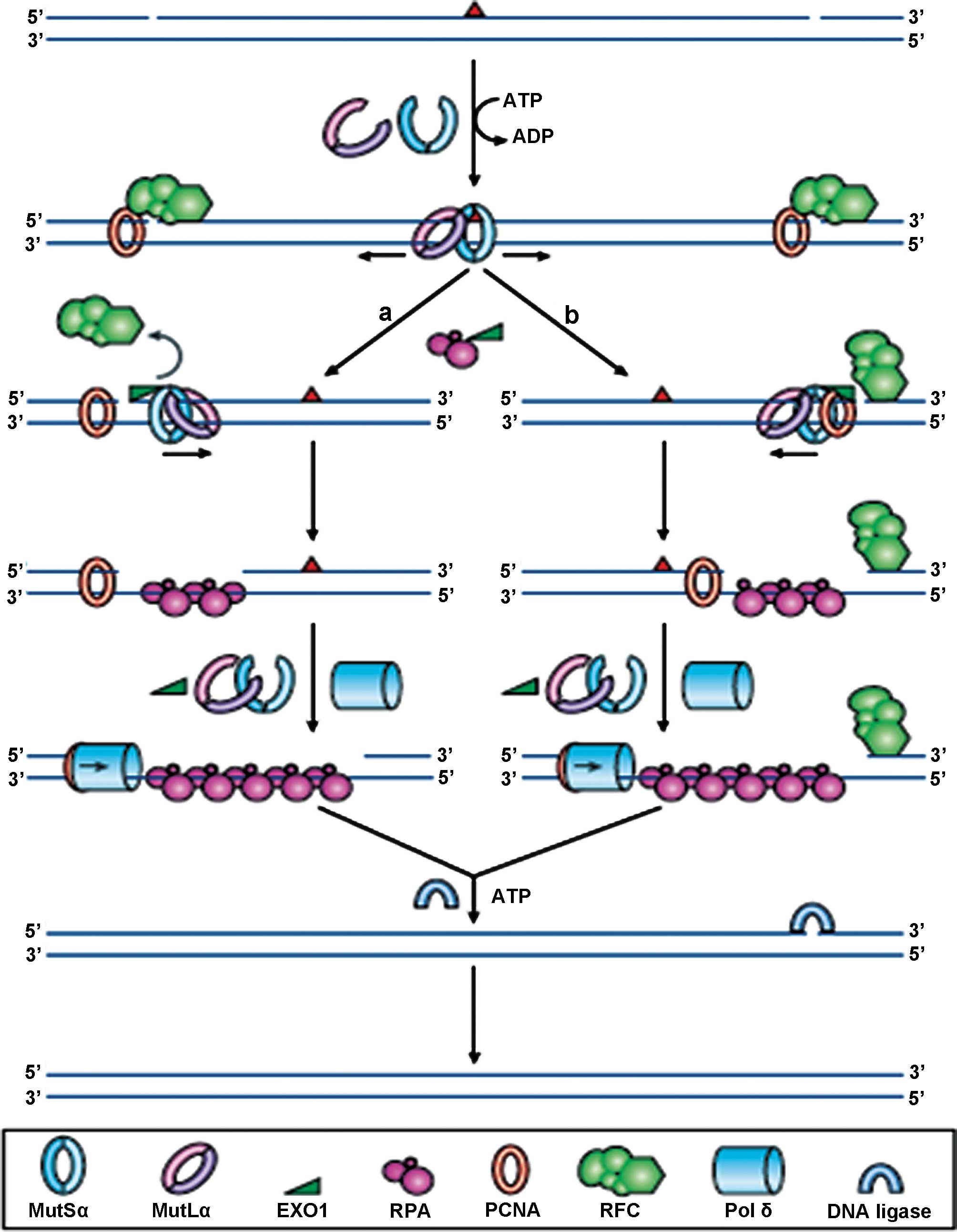 | Figure 1Reprinted by permission from
Macmillan Publishers Ltd., Nature Reviews Molecular Cell Biology, 7
(5): 335–346, copyright 2006. DNA mismatch repair. In normal cells,
any mismatched base pairs (or incorrect insertion or deletion
loops) are repaired by the complex machinery which forms the DNA
mismatch repair process. MSH2 and MSH6 form a heterodimeric
complex, called mutSα, which identifies and binds to the error,
resulting in an ATP-dependent conformational change, which recruits
mutLα, a heterodimer consisting of MLH1 and PMS2. The resultant
complex undergoes an ATP-driven conformational alteration,
releasing it from the error site. If it diffuses upstream, it
displaces replication factor C (RFC) and loads exonuclease-1
(EXO1). This degrades the strand in the 5′→3′ direction.
Replication factor A (RPA) then stabilises the single-stranded DNA,
while a complex of DNA polymerase Pol δ (Pol δ) and proliferating
cell nuclear antigen (PCNA) fills the gap and finally DNA ligase
seals the remaining nick to finalise the repair. If the mutSα/mutLα
complex diffuses downstream, EXO1 is recruited and degrades the
region of the DNA strand, up to the RFC complex. As stated before,
the single-strand is stabilised by bound RPA, which also inhibits
EXO1 activity. Pol δ fills the gap and finally DNA ligase I seals
the remaining nick to finalise the repair. |
When the MMR system develops a functional error or
defect, this results in a particular phenotype called
microsatellite instability (MSI). This is characterised by the
insertion or deletion of short, repetitive sequences of DNA,
resulting in mutations in cancer-related genes. The increase in the
rate of mutations in cells exhibiting deficient mismatch repair
(dMMR), may confer a Darwinian survival advantage. The cause of the
dMMR system is different depending upon whether the tumour is
sporadic in origin, or as a result of the autosomal dominant
inherited predisposition condition, Lynch syndrome.
2. dMMR in sporadic colorectal cancer
(CRC)
Many people wrongly group colon and rectal tumours
together as ‘colorectal’, when referring to rates of dMMR. It is in
fact very rarely seen in rectal cancers (1) but accounts for between 10 and 15% of
sporadic colon cancers. This MSI phenotype is associated with
several clinicopathological features such as a proximal primary
tumour location, high grade, mucinous pathology, early stage,
diploid and the presence of the BRAF p. (V600E) mutation (2). In addition, they tend to also be
associated with being female, smoking and older age at onset.
Furthermore, most of these sporadic MSI tumours are thought to
arise from sessile serrated adenomas or polyps (3). This pathway of colorectal cancer
development is different to the Fearon and Vogelstein
adenoma-carcinoma pathway (4). In
the majority of tumours, the defect in the MMR system is the
inactivation of MLH1, through methylation of CpG islands in
the promoter, causing transcriptional silencing of the gene.
Limited data also suggest that inactivation in a small subset of
tumours is caused my mutation of the MLH1 gene itself
(5–9).
3. dMMR in Lynch syndrome
Lynch syndrome (10) (formerly known as hereditary
non-polyposis colorectal cancer; HNPCC) is the most common
hereditary cancer predisposition syndrome, and is associated with a
high risk of colorectal cancer and also extra-colonic tumours,
particularly endometrial. In fact, the risk of endometrial cancer
in women within some affected families may actually be greater than
the risk of CRC (11). The average
age at onset, of <45, is significantly lower than that for
sporadic tumours and the cause of the defect in the MMR system in
Lynch syndrome is constitutional mutations of the MLH1 or
MSH2 genes, rather than methylation-induced inactivation of
MLH1. The InSiGHT database (12) has been developed to record all
mutations observed in patients with Lynch syndrome, and data from
this suggest that mutations in MLH1 account for 42% of Lynch
syndrome, mutations in MSH2 account for 33% and the
remainder are found in MSH6 (18%) and PMS2 (7%).
A very small subset of Lynch syndrome patients is
characterised by the presence of ‘constitutional epimutations’ of
MLH1. These are characterised by promoter methylation and
transcriptional silencing of a single allele of a gene in normal
tissues, in an otherwise intact gene. Since they appear to confer a
similar phenotype to that caused by sequence mutations, they are
considered to be an alternative aetiological mechanism for Lynch
syndrome (13). This phenomenon
was first recognised in 2002 by Gazzoli et al (14). Several more recent studies
(15–17) screened constitutive DNA samples for
MLH1 methylation, in CRC patients who had lost MLH1
expression in their tumours, without deleterious germline mutations
in MLH1. Each study found low levels of constitutional MLH1
epimutations, but Ward et al suggest expanding screening
programmes to include such patients, since testing of relatives
identified paternal transmission (16).
In 2009, Ligtenberg et al proposed an
alternative mechanism causing a defect in the MMR system in a
subset of Lynch syndrome families (18). The study of patients from Dutch and
Chinese families identified tumours which were deficient in
MSH2 as a result of the presence of heterozygous germ-line
deletions of the 3′ exons of the epithelial cell adhesion molecule
(EPCAM; also known as TACSTD1) gene. Such deletions
in EPCAM cause transcriptional read-through, which silences
MSH2, and has been termed MSH2 ‘epimutation’. In
2011, Kloor et al suggested that loss of EPCAM protein
expression, as assessed by immunohistochemistry (IHC) may be a
suitable method of identifying Lynch syndrome patients with
EPCAM germline deletions, as the majority of tumours with
EPCAM germline deletions also showed loss of protein
expression (19). Further to this
study, Huth et al hypothesised that, as loss of expression
did not always correlate with the presence of EPCAM germline
deletions, that it was potentially the actual type of second
somatic hit that determined EPCAM protein expression. Using
multiplex ligation-dependent probe amplification (MLPA) to assess
allelic deletion status, tumours with loss of EPCAM expression
showed biallelic deletions, whereas tumours retaining EPCAM
expression demonstrated monoallelic retention of the EPCAM
gene. The group therefore concluded that EPCAM protein expression
is dependent upon the actual localisation of the second somatic hit
that inactivates MSH2 (20). More recently, a study by Musulen
et al, showed a high specificity between the presence of
EPCAM germline mutations and loss of EPCAM expression, and
recommended the addition of EPCAM IHC into diagnostic Lynch
syndrome testing, in patients with MSH2-negative tumours
(21).
4. Who (and how) to test for mismatch repair
deficiencies?
Diagnostic criteria and guidelines
Amsterdam criteria
The identification of a patient with a colorectal or
endometrial tumour raises the question of whether to screen for the
presence of Lynch syndrome. Various criteria have been in place for
the past 35 years, to help guide this decision. In 1991, the
Amsterdam criteria arose from a meeting of the International
Collaborative Group on Hereditary Non-Polyposis Colon Cancer
(ICG-HNPCC) where an attempt was made to standardise international
criteria for identifying HNPCC patients for research purposes
(22). These criteria were known
as the ‘3-2-1 rule’: a) at least three relatives should have
histologically confirmed CRC, with one being a first degree
relative of the other two; b) there must be two successive
generations affected; and c) one or more relatives must be
diagnosed by the age of 50.
The Amsterdam criteria was later renamed Amsterdam
criteria I, following the subsequent identification of the genes
involved, which lead to the expansion of the criteria and its
renaming Amsterdam Criteria II.
Amsterdam Criteria II
Based upon further research identifying the fact
that Lynch syndrome tumours were not confined to the colon or
rectum, the criteria were further expanded and updated in 1998, and
renamed the Amsterdam II criteria (23). These new criteria added in the fact
that at least three relatives should have a histologically
confirmed HNPCC-associated cancer (colorectal, endometrial, small
bowel, ureter or renal pelvis), rather than just a colorectal
tumour.
Bethesda Guidelines
At around the same time, the National Cancer
Institute of the USA published its own set of guidelines (24). These included the following
criteria: a) individuals with cancer in families that meet the
Amsterdam criteria; b) individuals with two HNPCC-related cancers,
including synchronous and metachronous colorectal cancers or
associated extra-colonic cancers (endometrial, ovarian, gastric,
hepatobiliary, or small bowel cancer or transitional cell carcinoma
of the renal pelvis or ureter); c) individuals with colorectal
cancer and a first-degree relative with colorectal cancer and/or
HNPCC-related extra-colonic cancer and/or a colorectal adenoma: one
of the cancers diagnosed by age 45, and the adenoma diagnosed by
age 40; d) individuals with colorectal cancer or endometrial cancer
diagnosed by age 45; e) individuals with right-sided colorectal
cancer with an undifferentiated pattern (solid/cribriform) on
histopathology diagnosed by age 45; f) individuals with
signet-ring-cell-type colorectal cancer diagnosed by age 45; and g)
individuals with adenomas diagnosed by age 40.
Revised Bethesda Guidelines
In 2004, the NCI revised these guidelines, and went
on to publish the Revised Bethesda Guidelines (25). These remain the most recent
clinical diagnostic criteria upon which a patient is identified as
likely having Lynch syndrome; a) individuals with CRC diagnosed by
age 50; b) individuals with synchronous or metachronous CRC, or
other HNPCC-associated tumours regardless of age; c) individuals
with CRC and MSI-H histology diagnosed by age 60; d) individuals
with CRC and more than 1 first degree relative with an
HNPCC-associated tumour, with one cancer diagnosed by age 50; and
e) individuals with CRC and more than 2 first degree relatives or
second degree relatives with an HNPCC-associated tumour, regardless
of age.
Jerusalem criteria
In 2009, the ‘Jerusalem criteria’ were published,
recommending that either dMMR IHC or MSI testing be carried out on
every colorectal tumour, where the patient is under the age of 70
at diagnosis (26). The idea
behind this broader screening programme was to identify potential
Lynch syndrome patients with an MSH6 or PMS2
mutation, who tend to present at a later age, and would not be
included for screening, under the revised Bethesda guidelines.
5. Does ‘one size’ really fit all?
All of the above criteria for selecting patients for
screenings have been based upon North American and European
populations. In order for these criteria to be used worldwide, this
makes the assumption that there are no population-specific
differences. A study by Yan et al, has questioned this very
point in relation to a Chinese population, where there is a strict
one child policy (27). The
resultant large number of small families makes it almost impossible
to meet all the specified criteria regarding the number of affected
relatives. As a result this increases the likelihood of overlooking
and not screening a high proportion of potential or actual Lynch
syndrome patients.
A second factor bringing the relevance of using the
Amsterdam or Bethesda criteria in Asian populations into question,
is the fact that gastric and hepatocellular cancers are the most
common extracolonic tumours seen in Chinese patients with Lynch
syndrome, rather than endometrial tumours as seen in the West.
Furthermore, it becomes difficult to gauge how specific this is for
Lynch syndrome, when the rates of gastric and hepatocellular
carcinoma (HCC) are so high in Asia due to Helicobacter
pylori (H. pylori) and chronic hepatitis B virus (HBV)
infections respectively. An H. pylori infection induces an
inflammatory response, in addition to causing genetic changes which
result in genetic instability (28). The oncogenic effects of HBV such as
genomic instability result from its integration into the host
genome (29).
A third factor is that several studies have reported
a predominance of left-sided CRC in Asian populations, which is
different to what is seen in Western patients, where there is a
predominance of right-sided tumours. Wang et al (30) noted that 60.6% of 60 Lynch syndrome
patients under study had distal colorectal tumours. Chew et
al (31) undertook a study of
6,736 CRC patients, who underwent surgery for their disease at
Singapore General Hospital between 1989 and 2005; 52 (0.8%)
fulfilled the Amsterdam I or Amsterdam II criteria, so were
included for analysis and 69% of these patients had left-sided
tumours, the majority of which were located in the sigmoid colon
(31). In a very recent study of
116 Chinese families with suspected Lynch syndrome, 32 of whom had
confirmed MLH1 or MSH2 germline mutations, 56.5% of
the colorectal tumours were left-sided (32). These observations could be as a
result of the fact that rectal cancers are more prevalent in Asian
populations, or simply the fact that this is a feature of Asian
Lynch syndrome.
In Western populations, we know that 10–15% of
sporadic CRC tumours are dMMR. This figure may be much higher in
Asian populations, based upon a study carried out in Singapore on
240 CRC patients, under the age of 50 at presentation. MMR IHC was
performed and 21% of patients showed loss of expression of at least
one of the MMR proteins. The authors identified the fact that, had
selection for screening been based solely on the Amsterdam
criteria, a staggering 86% of patients would have not been
identified as high risk of Lynch syndrome, and would thus not have
been screened (33). This provides
further evidence for the introduction of population-specific
diagnostic screening criteria.
6. Reflex testing
In essence, reflex testing is the routine screening
of all newly diagnosed colorectal tumours for dMMR, to increase the
likelihood of identifying Lynch syndrome patients. Obviously early
diagnosis will result in increased surveillance, thus hopefully
reducing morbidity and mortality, not only for the affected
individual, but also family members.
Several studies have proven the cost-effectiveness
of such a screening approach (34–38).
A Dutch study by Sie et al (39) recommended increasing the cut-off
age for testing all CRC from 50 to 70 years old, and still found
this strategy cost-effective. However, in spite of the potential
financial savings, reflex testing is proving difficult to
implement, with areas requiring attention being highlighted at a
multidisciplinary working group meeting of the Centers for Disease
Control and Prevention in the US (40). The group identified the lack of
primary care provider knowledge of Lynch syndrome and testing
issues, as the main barrier to implementation. Furthermore, it was
recognised that there is a requirement for a strategy to ensure
that at-risk relatives are identified and counselling offered.
There is also very limited data available on the feasibility of
carrying out such testing, so one recommendation is for additional
‘real-world’ studies to be carried out to generate such data.
Taking a whistle-stop tour of current practice
worldwide, it would appear that much still needs to be done in
terms of implementation. In the UK, despite reflex testing being
mandated by the Royal College of Pathologists and recommended by
the British Society of Gastroenterologists, less than 50% of
National Health Service Hospital Trusts currently carry out
screening on patients presenting with the disease under the age of
50. This is the case in England, Wales and Scotland, however, all
social care trusts in Northern Ireland have successfully
implemented screening. The National Services Division Scotland and
the Molecular Pathology Consortium are currently trying to
implement national screening throughout Scotland, with the rest of
the UK hopefully following suit, once this model is in place (data
from a Bowel Cancer UK freedom of information request sent out
across the UK to establish the level of implementation) (41). The main reason for not screening
was put down to the additional financial burden. A further reason
given is a current lack of National Institute for Health and Care
Excellence (NICE) guidance. NICE is an executive non-departmental
public body within the Department of Health in the UK, and
publishes guidelines in, amongst other areas, clinical practice.
Another rather interesting reason is the potential impact on
patients and their families. The fact remains, and must not be
overlooked, that patients simply may not wish to undergo genetic
testing. There are many negative perceptions of this type of
screening, and unless patients are educated appropriately as to the
potential benefits, this could remain a barrier to
implementation.
A study in Canada by Tomiak et al determined
that in order to increase the uptake of genetic services by
patients with suspected Lynch syndrome, several areas needed
addressing, such as improving health literacy for the general
population, newly diagnosed patients, and perhaps a little
surprising, healthcare professionals (42). The study highlighted a general lack
of awareness of hereditary cancers and a lack of understanding of
the need for, and potential benefits of, genetic screening and what
is done with, and who has access to, the results. The requirement
for psychosocial support was also highlighted as an area to be
addressed. Tomiak et al concluded that these gaps need to be
filled for the successful implementation of universal screening,
planned by the US Office of Public Health Genomics, by 2020.
In 2012, Beamer et al carried out a
questionnaire-based review of reflex testing practise across the
United States of America, similar in design to that undertaken by
Bowel Cancer UK, in the United Kingdom (43). They found that the level of reflex
testing implementation was dependent primarily upon the level of
cancer program [ranging from Community Hospital Cancer Programs
(CHCP), to Community Hospital Comprehensive Cancer Programs (COMP),
and finally up to the most complex level of National Cancer
Institute-designated Comprehensive Cancer programs (NCI-CCC)].
Seventy-one percent of NCI-CCCs, 36% of COMPS yet only 15% CHCPs
had already implemented reflex testing. Another point arising from
this study is whether written patient consent is required.
Currently this is not the case, presumably because screening a
tumour provides phenotypic, rather than genotypic information, but
it will be interesting to see whether this aspect becomes a barrier
to worldwide reflex testing.
Back in 2008, in the state of Western Australia,
routine screening for Lynch syndrome was implemented. All patients
under the age of 60 at the time of diagnosis are screened and
figures published recently estimate that the majority of Lynch
syndrome cases are being identified as a result of this programme
(44).
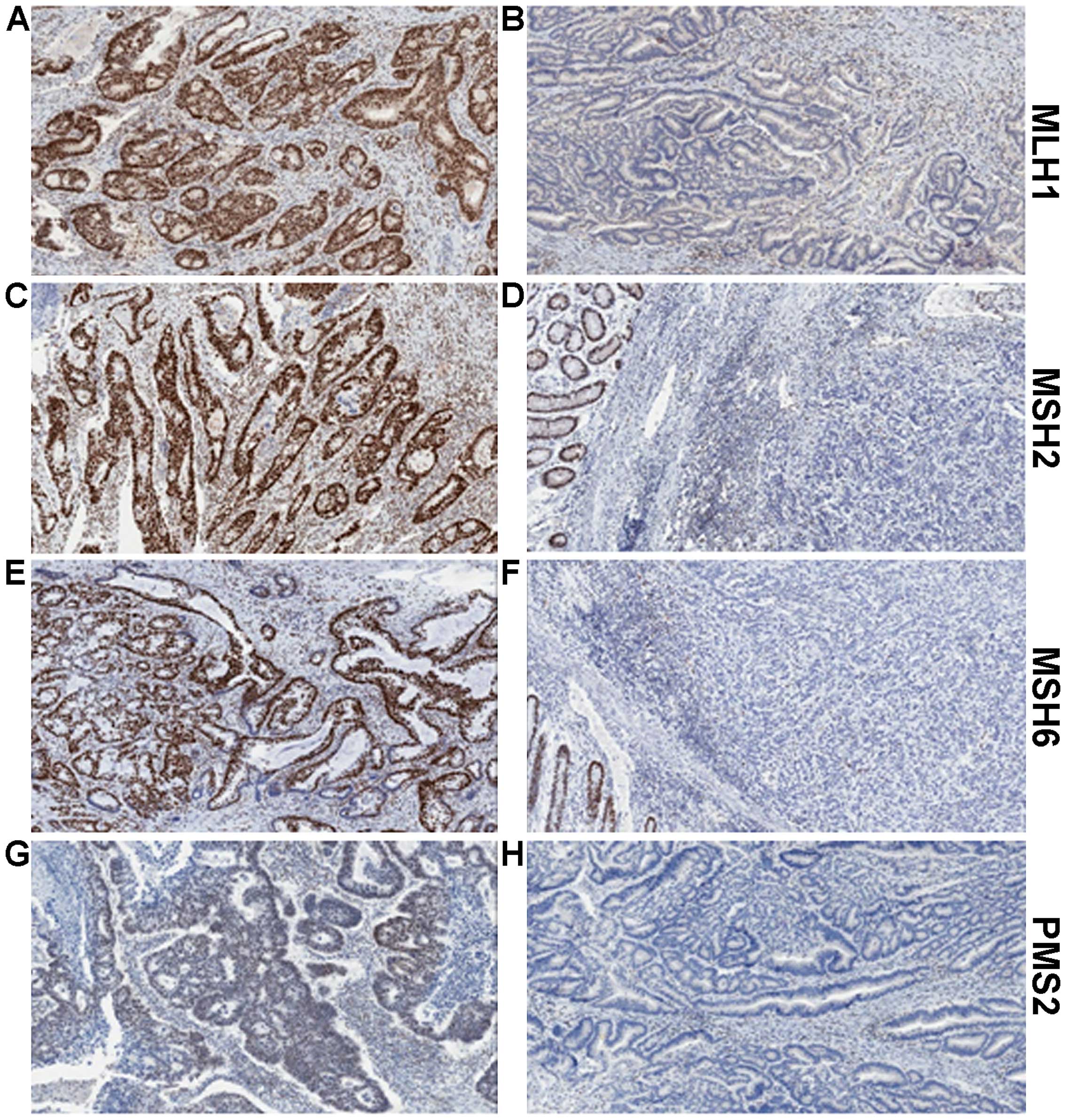
7. MMR Immunohistochemistry (IHC)
MMR IHC is a quick and relatively simple assay to
determine protein expression of MLH1, MSH2, MSH6 and PMS2 (Fig. 2). Tumours with dMMR will usually
show complete loss of expression of one or more protein. Assessing
all four proteins provides further information to determine the
actual defective protein. We know that MLH1 forms a heterodimer
complex with PMS2. Loss of expression of PMS2 alone is indicative
of a defect in the PMS2 gene. However, combined loss of PMS2
and MLH1 suggests the defect lies in MLH1, as MLH1 is responsible
for the stability of PMS2. A similar situation is seen with MSH6
and MSH2, with loss of MSH6 only indicating defective MSH6, whereas
loss of expression of both proteins would indicate the defect is
within MSH2 (Table I). Based on a
recent publication by Mensenkamp et al (45) this may in fact be a real
oversimplification of the actual situation. The group sequenced
dMMR CRC tumours and endometrial tumours which appeared to have
neither a germline mutation in any MMR gene, or hypermethylation of
the MLH1 promoter. In more than half of these tumours,
somatic mutations were identified as the underlying cause of
deficient mismatch repair.
 | Table ILoss of MMR protein expression. |
Table I
Loss of MMR protein expression.
| Protein expression
lost (determined by IHC) | Interpretation
(defective protein) |
|---|
| PMS2 | PMS2 |
| MLH1 and PMS2 | MLH1 |
| MSH6 | MSH6 |
| MSH2 and MSH6 | MSH2 |
Unfortunately, as is often the case with a seemingly
straightforward assay, there are exceptions to the rules. Sometimes
expression is reduced in intensity, or patchy, rather than
completely lost. This may be a result of the expression of a
truncated protein with limited stability, and is likely to be
accompanied by the predicted normal strong nuclear staining within
adjacent stromal cells or lymphocytes. It is often the case that
the abnormal staining is seen in both binding partners, i.e., MLH1
and PMS2, or MSH2 and MSH6. Another unusual situation is where
staining is seen localised to the cytoplasm, rather than within
tumour cell nuclei. This may be caused by a defect in the nuclear
localisation signal, and would most likely be reported as dMMR,
although PCR-based MSI testing may be requested for confirmation.
The single biggest problem in the assessment of MMR IHC is the
variability in fixation of the tumour tissue. The actual fixative
used, the time in formalin prior to embedding and the uniformity of
fixation are all factors which can affect the quality of staining
seen. Fadhil and Ilyas compared staining of the four MMR proteins
in 30 matched pre-surgical diagnostic biopsy samples and the
matched resection tissue, and concluded that not only was the
staining in the biopsies identical to that in the resection, but
the interpretation was made easier by the staining being more
intense and thus easier to interpret (46). This difference was deemed to be a
result of more uniform and complete fixation in the biopsy samples,
compared to the resection specimens.
A further complication in terms of the
interpretation of the MMR IHC was reported by Bao et al in a
study of 51 colorectal cancer patients undergoing neoadjuvant
chemoradiation (47). Nine of
these tumours showed reduced, but not complete loss of MSH6
staining, yet upon MSI analysis, all were microsatellite stable,
suggesting that the reduced expression was a result of the
chemoradiation treatment.
A slightly contentious issue, worthy of a mention,
is whether missense mutations in the MMR genes are associated with
reduced or patchy immunohistochemical staining. Missense mutations
result in a protein with a single amino acid change, which could
lead to no defect at all, or a dysfunctional or ‘pathogenic’
mutation. Difficulty arises in the assessment of the pathogenicity
of a missense mutation. Criteria which would have to be met would
include: a) the mutation not being present in control subjects; b)
the mutation co-segregating with a phenotype in a family; c) the
mutation resulting in a nonconservative amino acid alteration; and
d) the codon in which the mutation arose being evolutionarily
conserved (48,49). PubMed searches for this review
failed to identify any studies reporting reduced levels of MMR
protein expression, which were attributed to missense mutations. At
the present time, this phenomenon may have to remain an ‘urban
myth’.
Once an abnormal expression pattern of the MMR
proteins has been established, it is vitally important it determine
whether the tumour is from a patient with Lynch syndrome. The MMR
protein expression profile most commonly associated with Lynch
syndrome is loss of both MLH1 and PMS2; however, this would also be
seen in a sporadic tumour, if caused by MLH1 methylation.
The BRAF p. (V600E) mutation is observed in up to 70% of
tumours which have loss of expression of MLH1 and PMS2 or exhibit
MLH1 methylation (50,51),
but the mutation is almost never seen in Lynch syndrome-associated
tumours (52,53). Thus the presence of the BRAF
mutation strongly indicates a dMMR tumour of sporadic origin.
BRAF mutation testing is currently carried out routinely by
traditional sequencing methodologies, such as Sanger sequencing,
but in 2011, the first report was published by Capper et al,
that used an antibody specific for the V600E mutant protein (VE1),
allowing direct immunohistochemical testing of a tumour section
(54). Several groups have
published data showing very favourable results with the antibody
(including refs. 55,56) however, concerns have been voiced
regarding the usefulness, and sensitivity of this antibody,
particularly when assessing colorectal tumours. Adackapara et
al noted a high level of weak staining in wild-type and
KRAS mutant tumours, in addition to non-specific nuclear
staining. They determined the sensitivity and specificity to be 71
and 74%, respectively, and deemed the antibody not to be a
surrogate for standard genotyping (57). A study by Loes et al in 2015
assessed three methods of BRAF mutation detection [IHC,
Sanger sequencing and a single probe-based high-resolution melting
assay (LightMix) which has clamped wild-type allele amplification]
in both melanoma and colorectal tumour samples. Data were available
for all three assays in 99 colorectal tumours, of which 63 were
wild-type by all methods, 12 were BRAF mutant by all
methods, and yet 22 gave discordant results. Using the IHC data
alone would have misinterpreted 10 patients as being BRAF
mutant, and also failed to detect mutations in a further two
patients. The authors conclude that the high level of unexplained,
non-specific staining seen in colorectal tumours, much more so than
for melanoma tumours, would support that the antibody be used
solely as a screening tool, rather than a diagnostic test (58). It is worth noting that the antibody
will only identify the specific V600E mutation, so there is always
the risk of missing other BRAF mutations, but these are extremely
rare, particularly in colorectal tumours.
8. Microsatellite (MSI) testing
As an alternative, or indeed in combination with MMR
IHC testing, PCR-based MSI screening may be undertaken. The
recommended NCI-reference panel comprises two mononucleotide
repeats (BAT-25 and BAT-26) and three dinucleotide repeats (D5S346,
D2S123 and D17S250). There is also a commercially available kit,
consisting of five mononucleotide markers (BAT-25, BAT-26, MONO-27,
NR-21 and NR-24), as data are emerging to suggest that there is a
higher level of both sensitivity and specificity in the detection
of the MSI-H phenotype when only mononucleotides are used (59). Where available, DNA from normal
mucosa is compared to that extracted from the tumour. However, the
nature of the mononucleotide markers means that it is not essential
to have normal DNA for testing. The tumour is classed into one of
three phenotypes; if none of the markers show instability, the
tumour is classed as microsatellite stable (MSS). If one of the
markers show instability, the tumour is classed as
microsatellite-low (MSI-L), and if two or more of the markers show
instability, the tumour is classed as microsatellite-high (MSI-H).
Often MSS and MSI-L tumours are classified as a single subset, as
very few tumours of either phenotype will exhibit loss of
expression of any of the MMR proteins. Data surrounding clinical
differences between the two tumour phenotypes is still inconclusive
(60–63).
IHC or MSI?
There have been several studies carried out to
assess the correlation between IHC and MSI-testing, and the overall
results seem to suggest that firstly neither test is 100% accurate
in the detection of MSI-H tumours and secondly, there is actually a
high level of concordance between both technologies. The largest
study to date was performed by Cicek et al in 2011, when
almost 6,000 tumours from patients in the Colorectal Cancer Family
Registry were analysed. The group showed a 90–95% concordance
between those cases identified as dMMR by MSI and those detected by
IHC. Furthermore, only 2.7% of the 3964 tumours with IHC data
available, would have been miscalled, had only these data been used
in the initial assessment (64).
9. Next generation sequencing
There is no doubt that sequencing methodologies have
been transformed over the past few years, with the advent of next
generation sequencing platforms. Several companies are now
producing panels and kits, allowing the massive parallel sequencing
of MMR genes. This additional depth of sequencing may cause the
problem with the identification of variants of unknown significance
(VUS). Furthermore, there will undoubtedly be mutations detected at
lower levels than previous technologies have allowed. The issue
with these is that the clinical significance has not yet been
determined, thus with the technology being still in its infancy,
there remains the need to validate such panels. Pritchard et
al carried out one such validation study of the ColoSeq panel,
which correctly identified all 28 previously characterised
mutations in MLH1, MSH2 MSH6, PMS2, EPCAM, APC and
MUTYH. Two VUS were also detected in 19 samples from
patients without cancer (65). The
significance of such variants should become apparent once more data
are available and they can be related to pathogenicity.
10. Deficient MMR and clinical outcomes
Prognostic value in sporadic colorectal
cancer
The majority of the data published recently on the
prognostic and predictive value of MMR has been gathered on CRC
patients. There is definitely a distinction between the prognostic
benefit of dMMR in early (stage II/III) and late (stage IV)
disease. Several studies and meta-analyses have shown that dMMR in
stage II +/or III tumours is a positive prognostic factor. Back in
2003, a study of 570 stage II or II CRC patients showed that those
patients whose tumours were MSI-H had an improved 5-year OS,
compared to MSI-L or MSS tumours (HR for death was 0.31 (95% CI,
0.14–0.72, p=0.004) (66). In
2010, a large meta-analysis pooled data from 12,782 CRC patients,
including 1,972 MSI-H patients. The odds ratio (OR) for
disease-free survival (DFS) was 0.58, 95% CI 0.47–0.72, p<0.0001
and a similar value obtained for OS (OR=0.6, 95% CI 0.53–0.69,
p<0.0001) (67). This was
confirmed by Sargent et al, in a further meta-analysis of
457 patients, where it was reported that dMMR status was associated
with improved DFS (HR, 0.46; 95% CI, 0.22–0.95; p=0.03) and a trend
was seen towards improved OS (HR, 0.51; 95% CI, 0.24–1.10; p=0.06)
(68). The QUASAR (QUick And
Simple And Reliable trial provided a more recent dataset on which
to confirm the positive prognostic significance of dMMR. The
recurrence rate in the dMMR cohort was 11% (25/218), compared to
26% (438/1695) in the pMMR cohort [risk ratio (RR), 0.53; 95% CI,
0.40–0.70] (69).
Because of the fact that dMMR appears to be a good
prognostic marker in early CRC, it stands to reason that prevalence
of dMMR would be lower in advanced CRC (aCRC), since these patients
should be less likely to develop metastatic disease (70). This has been reported in several
studies (71–73). The question remains as to why these
tumours appear to metastasise less frequently. This may be as a
result of the increased immune response seen in dMMR tumours.
Tikidzhieva et al, have suggested a possible mechanism,
involving β2-microglobulin (B2M) (74). Mutations in B2M, within
microsatellite coding regions, are reported frequently in MSI-H
tumours, and result in the inability to present antigens at the
cell surface, through HLA-class I molecules. This in turn, may
stimulate natural killer (NK) cell-mediated tumour cell death.
In terms of the prognostic value of dMMR in aCRC, a
recent large meta-analysis by Venderbosch et al (75) of patients in four randomised
clinical trials (CAIRO, CAIRO2, FOCUS and COIN) provides convincing
evidence of the negative prognostic effect of dMMR in the
metastatic CRC (mCRC) setting. Data on dMMR was gathered on 3,063
patients, recruited into the four clinical trials. PFS and OS were
significantly reduced in the dMMR cohort, in comparison to the pMMR
cohort (PFS, 6.2 versus 7.6 months respectively; HR, 1.33; 95% CI,
1.12–1.57; p=0.001; and OS, 13.6 versus 16.8 months respectively;
HR, 1.35; 95% CI, 1.13–1.61; p=0.001). The analysis also
demonstrated the negative prognostic effect of the presence of the
BRAF p. (V600E) mutation, but ruled out any interaction
between the two poor prognosis markers. The group suggest that the
negative value of dMMR is as a result of the mutant BRAF status,
since significantly more dMMR tumours also contained the
mutation.
Predictive value in colorectal
cancer
Since its introduction into clinical practice almost
40 years ago, 5-fluorouracil (5-FU) has, until recently, been the
‘gold standard’ chemotherapy agent in the treatment of CRC. As a
result of this, there is much, and it has to be said, conflicting
data regarding the predictive value of MMR status and response to
5-FU-based therapy, with some studies reporting benefit from 5-FU
(76,77) whilst most reporting no benefit or
indeed a dis-benefit (66,68,78,79).
The final results from the MOSAIC trial where 2,246
stage II or II CRC patients were randomised between 5-FU plus
leucovorin (LV5FU2) and FOLFOX (LV5FU2 + oxaliplatin), provided
convincing evidence that the addition of oxaliplatin resulted in
improved 5-year DFS and 6-year OS, and in particular, ought to be
given to stage III patients after surgery (80). Following this, studies were
performed to assess whether microsatellite status was predictive of
response to oxaliplatin; Zaanan et al (81) analysed 233 MSI-H stage III
patients, receiving either 5-FU/LV or FOLFOX, and finding that
those on FOLFOX had an improved 3-year DFS compared to those on
5-FU/LV. However, in the same year, a study of 135 patients
receiving FOLFOX following surgery, found no difference in DFS or
OS when patients were stratified for MMR status (82). In the metastatic setting, Muller
et al in a 108-patient study, comparing two oxaliplatin and
5-FU-containing regimens, demonstrated a lower rate of disease
control in MSI-H patients compared to non-MSI-H patients (p=0.02)
(73). Kim et al however,
showed that MMR status did not predict response to
oxaliplatin-based treatment, when 171 recurrent or mCRC patients
were analysed (83).
There is also conflicting data as to the predictive
value of MMR status and response to irinotecan. Bertagnolli et
al showed that patients with dMMR/MSI-H had improved DFS,
compared to MSS patients, when irinotecan was added to standard
5-FU/LV treatment, with this benefit not being seen in patients
treated with 5-FU/LV alone (84).
However, this was not confirmed by the PETACC-3 study (85) or by a Korean study of almost 300
patients (86), or by the UK MRC
FOCUS study (71).
It would be difficult to summarise the prognostic
and predictive value of MMR status in both the adjuvant and
metastatic CRC settings, based on the data presented above. It is
apparent that dMMR/MSI-H in the adjuvant setting is a good
prognostic marker, but in the metastatic setting, the evidence
suggests the complete opposite effect. As for the predictive value,
there are conflicting data regarding each treatment regimen. One
can speculate as to why this is the case; perhaps we are seeing
population differences, perhaps the method of determining MMR
status had differing sensitivities. The small numbers of patients
in some of the studies should also be taken into account. It is
without doubt safe to say, that one cannot use only MMR status for
the prediction of response to therapy.
Prognostic and predictive value in
extra-colonic tumours
The majority of published data regarding the role of
the mismatch repair system in carcinogenesis, and the resultant
prognostic and predictive value, is within colorectal cancer. There
are, however, several extra-colonic cancers where there are high
percentages of dMMR have been reported, yet little is known of the
prognostic or predictive value.
Endometrial cancer
dMMR has been reported in 20–30% of endometrial
cancers (87), yet there are
scarce data available regarding the prognostic and predictive
impact of mismatch repair deficiencies. In a study reported earlier
this year, Kato et al analysed 191 endometrial tumours, and
found that 40% of them were deficient in at least one of the MMR
proteins, as assessed by IHC (88). This cohort displayed differences in
tumour grade histology and International Federation of Gynecology
and Obstetrics (FIGO) stage, when compared to the proficient MMR
tumours. Furthermore, dMMR cases had improved PFS and OS, with MMR
status being an independent prognostic factor for OS in endometrial
cancers. A further study, admittedly smaller, of 66 patients with
endometrial cancer and lymphatic invasion, also reported improved
disease specific survival (DSS) (p=0.04) and OS (p=0.03) in dMMR
patients, compared to those with pMMR. The authors also reported
increased OS particularly in FIGO stage 3C and stage 4 dMMR
patients, which may suggest that despite the lymphatic invasion and
lymph node metastases, this subgroup has a better prognosis than
patients with an intact MMR system. The other factor that cannot be
ignored is the effect that adjuvant chemotherapy has contributed to
this improved survival (89). A
third study, of 477 patients, investigated whether MMR status
impacted upon response to chemotherapy or pelvic teletherapy [also
known as external beam radiotherapy (EBRT)]. There was no
significant difference in PFS or OS between dMMR and pMMR
subgroups, when stratified by treatment. However, when patients
were stratified between endometrioid and non-endometrioid tumours,
significantly improved OS (p=0.003) and PFS (p=0.004) was seen for
dMMR/non-endometrioid tumours, receiving teletherapy. The opposite
was seen for patients receiving adjuvant chemotherapy, where those
with intact MMR showed improved PFS and OS (90). Taking these data together, it would
possibly appear that dMMR in endometrial cancers, or at least
within subgroups, is a positive biomarker. However, Ruiz et
al reported no association between MMR status and survival, in
a study of 212 endometrioid tumours (91), and a further study actually
reported an increased risk of disease-specific death in dMMR
high-grade endometrioid carcinomas (HGEC). Interestingly in this
study, dMMR was only seen in these HGEC tumours, and not serous or
clear cell tumours, suggesting the use of MMR testing to aid in
tumour-type diagnosis (92). Cohn
et al reported improved DFS in a cohort of endometrial
cancer patients who had retained expression of both MLH1 and MSH2,
in comparison to patients who displayed abnormal expression
(p=0.035) (93). A large
meta-analysis carried out in 2013 summarised very eloquently the
lack of concrete evidence of an association between MMR status and
clinical outcome, where in a pooled analysis of 23 studies
(published between 1980 and 2011), the group failed to show a
significant association between MSI and a worse OS (p=0.11) or DFS
(p=0.66) (94). The heterogeneous
nature of the method of determining MSI status, combined with
variability in the study populations, still make it very difficult
to determine the usefulness of MMR status in relation to outcome in
this disease.
Ovarian cancer
Ovarian cancer is the 7th most common cancer
worldwide for females, with over 239,000 new cases diagnosed in
2012, and has the highest mortality rate of all the gynaecological
cancers (95). Early detection is
difficult, and as a result, only 15% of women present with
localised disease (96). Women
with Lynch syndrome, have a lifetime risk of ovarian cancer of
approximately 8% (97–99). As we find in common with other
extracolonic cancers, data on MMR is sparse. Several authors have
attempted to clarify dMMR or MSI rates through meta-analyses; Xiao
et al (100) found
disparities between reported rates of MSI frequency, ranging from 5
to 13% (101–103). Murphy and Wentzensen combined
results from 22 studies, arriving at a figure for MSI of 10% for
unselected ovarian cancer patients (104). This figure was further refined to
9%, when only patients who had been tested for MSI using the five
Bethesda markers were analysed. Pal et al also suggest that
10% of ovarian cancers show MSI, analysing data from 18 studies
(105). In terms of dMMR as
assessed by IHC, larger differences were observed; ranging from 2
to 29% across the 12 studies analysed by Xiao et al
(100). One feature common to
most studies was the fact that there was an overrepresentation of
the non-serous tumours within the MSI cohorts, which parallels the
overrepresentation of mucinous and endometrioid histologies in CRC
and endometrial cancers respectively. In terms of data relating to
the effect of dMMR or MSI on prognosis or response to chemotherapy,
very little has been published, and the results are varied.
Scartozzi et al found that loss of expression of MLH1
correlated with increased survival in patients with stage III/IV
disease, although the study size was only 34 patients (106). Zhia et al assessed 322
tumours for MSH6 expression, and found no correlation with
survival. The group did find a correlation between loss of
expression and clear cell, mucinous and endometrioid histologies
(p<0.007) (107). Another
study finding no association between MSI and survival was carried
out on a series of Danish patients by Begum et al, who used
a panel of 16 dinucleotide markers to assess status (108). In terms of response to therapy,
there have been two reports of a correlation between a lack of MSH2
and response to platinum-based chemotherapy; Ercoli et al
showed that patients who did not respond to treatment had lower
levels of MSH2 than patients who had at least a partial response
(109). A report by Marcelis
et al described two Lynch syndrome patients, both carrying a
deletion in exon 6 of MSH2, who developed a rapid resistance
to cisplatin-based therapy (110). Based upon current literature,
very little can be reasonably or reliably concluded regarding the
role of the MMR proteins in ovarian cancer survival or response.
There is clearly a need for large, randomised studies in this
disease field, where one can control for factors such as MMR
assessment criteria, tumour histology, treatment regimen and sample
size.
Melanoma
Malignant melanoma is the 19th most common cancer
worldwide, with around 232,000 new cases diagnosed in 2012
(111). MSI has been reported to
be present in anywhere between 2 and 30% of primary tumours
(112–116) and 20–77% of metastatic lesions
(117–123). Castiglia et al suggest
that the inactivation of the MMR system, in combination with the
deregulation of the Wnt/beta-catenin pathway may act cooperatively
to promote the development of melanoma (124). It may be that in melanoma, it is
a downregulation of the MMR proteins, rather than a complete loss
of expression, or gene inactivation that is important, as seen in a
study by Korabiowska et al, who confirmed the downregulation
by both IHC and in situ hybridisation in 59 malignant
melanomas (125). Alvino et
al also reported a reduction in expression of MLH1, MSH2 and
PMS2 in primary melanomas compared to benign nevi. Interestingly
they also noted the opposite for MSH6, and this increased
expression was also associated with increased risk of melanoma
mortality (R, 3.76; 95% CI, 1.12–12.70) (126). With such little data available on
the MMR proteins in melanoma, the only conclusion that can be
reliably drawn is that as the cancer progresses from benign nevus,
through primary melanoma to metastatic melanoma, the level of MSI
increases. This may, however, only be at an MSI-L level, rather
than MSI-H. The significance of this is yet to be determined.
Gastric cancer
Gastric cancer is the 5th most common cancer
worldwide, with more than 951,000 new cases diagnosed in 2012
(127). In gastric cancer, MSI
exists in approximately 10–20% tumours (128–130). Such tumours are associated with
older patients, distal location, lower pTNM stage and intestinal
subtype and reduced lymph node involvement. Several large studies
have assessed the prognostic effect of the MSI phenotype, all
showing that MSI correlates with improved survival; back in 2000,
Schneider et al showed that in MSI-H and MSI-L patients,
there was an increased median survival time, compared to MSS
patients (p=0.027) (131). In
2002, Lee et al analysed 327 consecutive gastric cancers,
assessing MSI status with the BAT-26 marker. Patients with MSI had
improved overall survival compared to those with MSS tumours
(p=0.046) (130). Beghelli et
al, determined the MSI status of 510 sporadic gastric cancers,
also concluding that MSI correlated with improved survival, but
only in stage II disease (p<0.011) (128). In a study of 159 patients,
Falchetti et al demonstrated an association between MSI-H
phenotype and improved survival at 15 years (p=0.01) (132). Finally Fang et al showed
that there was an improved 5-year OS benefit in the MSI-H cohort
(p=0.03) and also a trend towards an improved 3-year disease-free
survival (p=0.076), when analysing 214 gastric cancer patients
(129). However, as one has come
to expect in this field, there is conflicting data to suggest that
MSI status has no influence on survival; Perez et al found
no survival benefit in the MSI patients, compared to the MSS
patients, however, it must be noted that there were only 24
patients in this study (133). In
a slightly larger study of 83 patients, An et al also did
not find an association between MSI status and survival (134). Given the disparity between sample
sizes, the evidence is pointing to the direction that gastric
cancer patients with an MSI-H tumour are likely to have improved
survival compared to patients whose tumours are MSS. Looking at MSI
status and its predictive value in terms of response to 5-FU-based
chemotherapy, there is yet again conflicting data; a large study by
An et al, of 1990 patients, identified an MSI-H rate of
8.5%. The group determined that MSI status was not prognostic, as
DFS between MSI-H and the MSI-L/MSS groups was not significantly
different, even taking each disease stage separately. However, DFS
was improved in the MSI-L/MSS group treated with 5-FU-based
chemotherapy (p=0.008) (135).
Oki et al, determined that there was no correlation between
MSI status and survival following 5-FU-therapy, in their study of
240 patients, collected over a 9-year period (136). Clearly the gastric cancers with
MSI form a distinct subset, and as such, are likely to be driven by
slightly different signalling pathways. It still remains to be
determined, how to identify and best and treat these patients.
11. Conclusion
Deficiencies in the DNA mismatch repair system have
been identified in many unrelated cancer types. These deficiencies
may be the result of either the inactivation of MLH1,
through methylation, as seen in sporadic cancers, or through
germline mutations of MLH1 or MSH2, as seen in
inherited cancers. Despite it being almost 50 years since the
initial observations by Henry Lynch, which subsequently lead to the
term ‘Lynch syndrome’, there are still gaps in our knowledge of the
role of dMMR in cancer. Progress is being made, however,
particularly in the field of colorectal cancer. We now have
evidence that the prognostic role of dMMR is stage-dependent, and
steps are beginning to be implemented, to ensure that every patient
who may require screening actually has access to this service. In
terms of identifying dMMR or MSI patients, there is now some
standardisation of IHC and adoption of the use of the Bethesda
marker panel, but with the recent introduction of next generation
screening, the additional depth of sequence data, may complicate
the situation as more VUS are identified. Furthermore, the clinical
significance of low-level variants is yet to be elucidated, adding
a further layer to complexity to the use of this emerging
technology. Extracolonic cancers trail far behind in terms of what
is known of the prognostic and predictive value of MMR, and, our
understanding will remain limited unless large controlled trials
are performed.
Abbreviations:
|
BRAF
|
v-Raf murine sarcoma viral oncogene
homolog B
|
|
CRC
|
colorectal cancer
|
|
DFS
|
disease-free survival
|
|
dMMR
|
deficient mismatch repair
|
|
EPCAM
|
epithelial cell adhesion molecule
|
|
EXO1
|
exonuclease-1
|
|
HNPCC
|
hereditary non-polyposis colorectal
cancer
|
|
IHC
|
immunohistochemistry
|
|
mCRC
|
metastatic colorectal cancer
|
|
MLH1
|
mutL homologue 1
|
|
MMR
|
mismatch repair
|
|
MSH2
|
mutS homologue 2
|
|
MSH6
|
mutS homologue 6
|
|
MSI
|
microsatellite instability
|
|
MSI-H
|
microsatellite instability-high
|
|
MSI-l
|
microsatellite instability-low
|
|
MSS
|
microsatellite stable
|
|
NGS
|
next generation sequencing
|
|
OS
|
overall survival
|
|
PCNA
|
proliferating cell nuclear
antigen
|
|
PCR
|
polymerase chain reaction
|
|
PFS
|
progression-free survival
|
|
pMMR
|
proficient mismatch repair
|
|
PMS2
|
post-meiotic segregation increased
2
|
|
Pol δ
|
DNA polymerase δ
|
|
RFA
|
replication factor A
|
|
RFC
|
replication factor C
|
|
VUS
|
variants of unknown significance
|
References
|
1
|
Nilbert M, Planck M, Fernebro E, Borg A
and Johnson A: Microsatellite instability is rare in rectal
carcinomas and signifies hereditary cancer. Eur J Cancer.
35:942–945. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sinicrope FA, Rego RL, Foster N, Sargent
DJ, Windschitl HE, Burgart LJ, Witzig TE and Thibodeau SN:
Microsatellite instability accounts for tumor site-related
differences in clinicopathologic variables and prognosis in human
colon cancers. Am J Gastroenterol. 101:2818–2825. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jass JR: Classification of colorectal
cancer based on correlation of clinical, morphological and
molecular features. Histopathology. 50:113–130. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thibodeau SN, French AJ, Cunningham JM,
Tester D, Burgart LJ, Roche PC, McDonnell SK, Schaid DJ, Vockley
CW, Michels VV, et al: Microsatellite instability in colorectal
cancer: Different mutator phenotypes and the principal involvement
of hMLH1. Cancer Res. 58:1713–1718. 1998.PubMed/NCBI
|
|
6
|
Cunningham JM, Christensen ER, Tester DJ,
Kim CY, Roche PC, Burgart LJ and Thibodeau SN: Hypermethylation of
the hMLH1 promoter in colon cancer with microsatellite instability.
Cancer Res. 58:3455–3460. 1998.PubMed/NCBI
|
|
7
|
Cunningham JM, Kim CY, Christensen ER,
Tester DJ, Parc Y, Burgart LJ, Halling KC, McDonnell SK, Schaid DJ,
Walsh Vockley C, et al: The frequency of hereditary defective
mismatch repair in a prospective series of unselected colorectal
carcinomas. Am J Hum Genet. 69:780–790. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kane MF, Loda M, Gaida GM, Lipman J,
Mishra R, Goldman H, Jessup JM and Kolodner R: Methylation of the
hMLH1 promoter correlates with lack of expression of hMLH1 in
sporadic colon tumors and mismatch repair-defective human tumor
cell lines. Cancer Res. 57:808–811. 1997.PubMed/NCBI
|
|
9
|
Poynter JN, Siegmund KD, Weisenberger DJ,
Long TI, Thibodeau SN, Lindor N, Young J, Jenkins MA, Hopper JL,
Baron JA, et al: Molecular characterization of MSI-H colorectal
cancer by MLHI promoter methylation, immunohistochemistry, and
mismatch repair germline mutation screening. Cancer Epidemiol
Biomarkers Prev. 17:3208–3215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lynch HT, Shaw MW, Magnuson CW, Larsen AL
and Krush AJ: Hereditary factors in cancer. Study of two large
midwestern kindreds. Arch Intern Med. 117:206–212. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jasperson KW, Tuohy TM, Neklason DW and
Burt RW: Hereditary and familial colon cancer. Gastroenterology.
138:2044–2058. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Plazzer JP, Sijmons RH, Woods MO,
Peltomäki P, Thompson B, Den Dunnen JT and Macrae F: The InSiGHT
database: Utilizing 100 years of insights into Lynch syndrome. Fam
Cancer. 12:175–180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hitchins MP and Ward RL: Constitutional
(germline) MLH1 epimutation as an aetiological mechanism for
hereditary non-polyposis colorectal cancer. J Med Genet.
46:793–802. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gazzoli I, Loda M, Garber J, Syngal S and
Kolodner RD: A hereditary nonpolyposis colorectal carcinoma case
associated with hypermethylation of the MLH1 gene in normal tissue
and loss of heterozygosity of the unmethylated allele in the
resulting microsatellite instability-high tumor. Cancer Res.
62:3925–3928. 2002.PubMed/NCBI
|
|
15
|
Crucianelli F, Tricarico R, Turchetti D,
Gorelli G, Gensini F, Sestini R, Giunti L, Pedroni M, Ponz de Leon
M, Civitelli S, et al: MLH1 constitutional and somatic methylation
in patients with MLH1 negative tumors fulfilling the revised
Bethesda criteria. Epigenetics. 9:1431–1438. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ward RL, Dobbins T, Lindor NM, Rapkins RW
and Hitchins MP: Identification of constitutional MLH1 epimutations
and promoter variants in colorectal cancer patients from the Colon
Cancer Family Registry. Genet Med. 15:25–35. 2013. View Article : Google Scholar
|
|
17
|
Pineda M, Mur P, Iniesta MD, Borràs E,
Campos O, Vargas G, Iglesias S, Fernández A, Gruber SB, Lázaro C,
et al: MLH1 methylation screening is effective in identifying
epimutation carriers. Eur J Hum Genet. 20:1256–1264. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ligtenberg MJ, Kuiper RP, Chan TL,
Goossens M, Hebeda KM, Voorendt M, Lee TY, Bodmer D, Hoenselaar E,
Hendriks-Cornelissen SJ, et al: Heritable somatic methylation and
inactivation of MSH2 in families with Lynch syndrome due to
deletion of the 3′ exons of TACSTD1. Nat Genet. 41:112–117. 2009.
View Article : Google Scholar
|
|
19
|
Kloor M, Voigt AY, Schackert HK,
Schirmacher P, von Knebel Doeberitz M and Bläker H: Analysis of
EPCAM protein expression in diagnostics of Lynch syndrome. J Clin
Oncol. 29:223–227. 2011. View Article : Google Scholar
|
|
20
|
Huth C, Kloor M, Voigt AY, Bozukova G,
Evers C, Gaspar H, Tariverdian M, Schirmacher P, von Knebel
Doeberitz M and Bläker H: The molecular basis of EPCAM expression
loss in Lynch syndrome-associated tumors. Mod Pathol. 25:911–916.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Musulen E, Blanco I, Carrato C,
Fernandez-Figueras MT, Pineda M, Capella G and Ariza A: Usefulness
of epithelial cell adhesion molecule expression in the algorithmic
approach to Lynch syndrome identification. Hum Pathol. 44:412–416.
2013. View Article : Google Scholar
|
|
22
|
Vasen HF, Mecklin JP, Khan PM and Lynch
HT: The International Collaborative Group on Hereditary
Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum.
34:424–425. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vasen HF, Watson P, Mecklin JP and Lynch
HT: New clinical criteria for hereditary nonpolyposis colorectal
cancer (HNPCC, Lynch syndrome) proposed by the International
Collaborative group on HNPCC. Gastroenterology. 116:1453–1456.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rodriguez-Bigas MA, Boland CR, Hamilton
SR, Henson DE, Jass JR, Khan PM, Lynch H, Perucho M, Smyrk T, Sobin
L, et al: A National Cancer Institute Workshop on Hereditary
Nonpolyposis Colorectal Cancer Syndrome: Meeting highlights and
Bethesda guidelines. J Natl Cancer Inst. 89:1758–1762. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Umar A, Boland CR, Terdiman JP, Syngal S,
de la Chapelle A, Rüschoff J, Fishel R, Lindor NM, Burgart LJ,
Hamelin R, et al: Revised Bethesda Guidelines for hereditary
nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite
instability. J Natl Cancer Inst. 96:261–268. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boland CR and Shike M: Report from the
Jerusalem workshop on Lynch syndrome-hereditary nonpolyposis
colorectal cancer. Gastroenterology. 138:2197 e2191–2197. 2010.
View Article : Google Scholar
|
|
27
|
Yan HL, Hao LQ, Jin HY, Xing QH, Xue G,
Mei Q, He J, He L and Sun SH: Clinical features and mismatch repair
genes analyses of Chinese suspected hereditary non-polyposis
colorectal cancer: A cost-effective screening strategy proposal.
Cancer Sci. 99:770–780. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Graham DY: Helicobacter pylori update:
gastric cancer, reliable therapy, and possible benefits.
Gastroenterology. 148:719–731 e713. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ringelhan M, O'Connor T, Protzer U and
Heikenwalder M: The direct and indirect roles of HBV in liver
cancer: Prospective markers for HCC screening and potential
therapeutic targets. J Pathol. 235:355–367. 2015. View Article : Google Scholar
|
|
30
|
Wang XL, Yuan Y, Zhang SZ, Cai SR, Huang
YQ, Jiang Q and Zheng S: Clinical and genetic characteristics of
Chinese hereditary nonpolyposis colorectal cancer families. World J
Gastroenterol. 12:4074–4077. 2006.PubMed/NCBI
|
|
31
|
Chew MH, Koh PK, Ng KH, Lim JF, Ho KS, Ooi
BS, Tang CL and Eu KW: Phenotypic characteristics of hereditary
non-polyposis colorectal cancer by the Amsterdam criteria: An Asian
perspective. ANZ J Surg. 78:556–560. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu F, Yang L, Zhou X, Sheng W, Cai S, Liu
L, Nan P and Xu Y: Clinicopathological and genetic features of
Chinese hereditary nonpolyposis colorectal cancer (HNPCC). Med
Oncol. 31:2232014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chew MH, Koh PK, Tan M, Lim KH, Carol L
and Tang CL: Mismatch repair deficiency screening via
immunohistochemical staining in young Asians with colorectal
cancers. World J Surg. 37:2468–2475. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ladabaum U, Wang G, Terdiman J, Blanco A,
Kuppermann M, Boland CR, Ford J, Elkin E and Phillips KA:
Strategies to identify the Lynch syndrome among patients with
colorectal cancer: A cost-effectiveness analysis. Ann Intern Med.
155:69–79. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mvundura M, Grosse SD, Hampel H and
Palomaki GE: The cost-effectiveness of genetic testing strategies
for Lynch syndrome among newly diagnosed patients with colorectal
cancer. Genet Med. 12:93–104. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Snowsill T, Huxley N, Hoyle M,
Jones-Hughes T, Coelho H, Cooper C, Frayling I and Hyde C: A
systematic review and economic evaluation of diagnostic strategies
for Lynch syndrome. Health Technol Assess. 18:1–406. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schofield L, Grieu F, Amanuel B, Carrello
A, Spagnolo D, Kiraly C, Pachter N, Goldblatt J, Platell C, Levitt
M, et al: Population-based screening for Lynch syndrome in Western
Australia. Int J Cancer. 135:1085–1091. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Snowsill T, Huxley N, Hoyle M,
Jones-Hughes T, Coelho H, Cooper C, Frayling I and Hyde C: A
model-based assessment of the cost-utility of strategies to
identify Lynch syndrome in early-onset colorectal cancer patients.
BMC Cancer. 15:3132015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sie AS, Mensenkamp AR, Adang EM,
Ligtenberg MJ and Hoogerbrugge N: Fourfold increased detection of
Lynch syndrome by raising age limit for tumour genetic testing from
50 to 70 years is cost-effective. Ann Oncol. 25:2001–2007. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bellcross CA, Bedrosian SR, Daniels E,
Duquette D, Hampel H, Jasperson K, Joseph DA, Kaye C, Lubin I,
Meyer LJ, et al: Implementing screening for Lynch syndrome among
patients with newly diagnosed colorectal cancer: summary of a
public health/clinical collaborative meeting. Genet Med.
14:152–162. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Data briefing: Reflex testing for Lynch
syndrome in people diagnosed with bowel cancer under the age of 50.
http://www.bowelcanceruk.org.uk/media/426888/lynch_syndrome_briefing_final.pdf.
Accessed April 15, 2015
|
|
42
|
Tomiak E, Samson A, Spector N, Mackey M,
Gilpin C, Smith E, Jonker D, Allanson J and Asmis T: Reflex testing
for Lynch syndrome: If we build it, will they come? Lessons learned
from the uptake of clinical genetics services by individuals with
newly diagnosed colorectal cancer (CRC). Fam Cancer. 13:75–82.
2014. View Article : Google Scholar :
|
|
43
|
Beamer LC, Grant ML, Espenschied CR,
Blazer KR, Hampel HL, Weitzel JN and MacDonald DJ: Reflex
immunohistochemistry and microsatellite instability testing of
colorectal tumors for Lynch syndrome among US cancer programs and
follow-up of abnormal results. J Clin Oncol. 30:1058–1063. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schofield L, Grieu F, Goldblatt J, Amanuel
B and Iacopetta B: A state-wide population-based program for
detection of Lynch syndrome based upon immunohistochemical and
molecular testing of colorectal tumours. Fam Cancer. 11:1–6. 2012.
View Article : Google Scholar
|
|
45
|
Mensenkamp AR, Vogelaar IP, van
Zelst-Stams WA, Goossens M, Ouchene H, Hendriks-Cornelissen SJ,
Kwint MP, Hoogerbrugge N, Nagtegaal ID and Ligtenberg MJ: Somatic
mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair
deficiency in Lynch syndrome-like tumors. Gastroenterology.
146:643–646 e648. 2014. View Article : Google Scholar
|
|
46
|
Fadhil W and Ilyas M: Immunostaining for
mismatch repair (MMR) protein expression in colorectal cancer is
better and easier to interpret when performed on diagnostic
biopsies. Histopathology. 60:653–655. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bao F, Panarelli NC, Rennert H, Sherr DL
and Yantiss RK: Neoadjuvant therapy induces loss of MSH6 expression
in colorectal carcinoma. Am J Surg Pathol. 34:1798–1804. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Genuardi M, Carrara S, Anti M, Ponz de
Leòn M and Viel A: Assessment of pathogenicity criteria for
constitutional missense mutations of the hereditary nonpolyposis
colorectal cancer genes MLH1 and MSH2. Eur J Hum Genet. 7:778–782.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cravo M, Afonso AJ, Lage P, Albuquerque C,
Maia L, Lacerda C, Fidalgo P, Chaves P, Cruz C and Nobre-Leitão C:
Pathogenicity of missense and splice site mutations in hMSH2 and
hMLH1 mismatch repair genes: Implications for genetic testing. Gut.
50:405–412. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bouzourene H, Hutter P, Losi L, Martin P
and Benhattar J: Selection of patients with germline MLH1 mutated
Lynch syndrome by determination of MLH1 methylation and BRAF
mutation. Fam Cancer. 9:167–172. 2010. View Article : Google Scholar
|
|
51
|
McGivern A, Wynter CV, Whitehall VL,
Kambara T, Spring KJ, Walsh MD, Barker MA, Arnold S, Simms LA,
Leggett BA, et al: Promoter hypermethylation frequency and BRAF
mutations distinguish hereditary non-polyposis colon cancer from
sporadic MSI-H colon cancer. Fam Cancer. 3:101–107. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Capper D, Voigt A, Bozukova G, Ahadova A,
Kickingereder P, von Deimling A, von Knebel Doeberitz M and Kloor
M: BRAF V600E-specific immunohistochemistry for the exclusion of
Lynch syndrome in MSI-H colorectal cancer. Int J Cancer.
133:1624–1630. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Loughrey MB, Waring PM, Tan A, Trivett M,
Kovalenko S, Beshay V, Young MA, McArthur G, Boussioutas A and
Dobrovic A: Incorporation of somatic BRAF mutation testing into an
algorithm for the investigation of hereditary non-polyposis
colorectal cancer. Fam Cancer. 6:301–310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Capper D, Preusser M, Habel A, Sahm F,
Ackermann U, Schindler G, Pusch S, Mechtersheimer G, Zentgraf H and
von Deimling A: Assessment of BRAF V600E mutation status by
immunohistochemistry with a mutation-specific monoclonal antibody.
Acta Neuropathol. 122:11–19. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Colomba E, Hélias-Rodzewicz Z, Von
Deimling A, Marin C, Terrones N, Pechaud D, Surel S, Côté JF,
Peschaud F, Capper D, et al: Detection of BRAF p.V600E mutations in
melanomas: Comparison of four methods argues for sequential use of
immunohistochemistry and pyrosequencing. J Mol Diagn. 15:94–100.
2013. View Article : Google Scholar
|
|
56
|
Ihle MA, Fassunke J, König K, Grünewald I,
Schlaak M, Kreuzberg N, Tietze L, Schildhaus HU, Büttner R and
Merkelbach-Bruse S: Comparison of high resolution melting analysis,
pyrosequencing, next generation sequencing and immunohistochemistry
to conventional Sanger sequencing for the detection of p.V600E and
non-p.V600E BRAF mutations. BMC Cancer. 14:132014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Adackapara CA, Sholl LM, Barletta JA and
Hornick JL: Immunohistochemistry using the BRAF V600E
mutation-specific monoclonal antibody VE1 is not a useful surrogate
for genotyping in colorectal adenocarcinoma. Histopathology.
63:187–193. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Loes IM, Immervoll H, Angelsen JH, Horn A,
Geisler J, Busch C, Lønning PE and Knappskog S: Performance
comparison of three BRAF V600E detection methods in malignant
melanoma and colorectal cancer specimens. Tumour Biol.
36:1003–1013. 2015. View Article : Google Scholar :
|
|
59
|
Xicola RM, Llor X, Pons E, Castells A,
Alenda C, Piñol V, Andreu M, Castellví-Bel S, Payá A, Jover R, et
al; Gastrointestinal Oncology Group of the Spanish
Gastroenterological Association. Performance of different
microsatellite marker panels for detection of mismatch
repair-deficient colorectal tumors. J Natl Cancer Inst. 99:244–252.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bapat B, Lindor NM, Baron J, Siegmund K,
Li L, Zheng Y, Haile R, Gallinger S, Jass JR, Young JP, et al: The
association of tumor microsatellite instability phenotype with
family history of colorectal cancer. Cancer Epidemiol Biomarkers
Prev. 18:967–975. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Graham T, Halford S, Page KM and Tomlinson
IP: Most low-level microsatellite instability in colorectal cancers
can be explained without an elevated slippage rate. J Pathol.
215:204–210. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Halford S, Sasieni P, Rowan A, Wasan H,
Bodmer W, Talbot I, Hawkins N, Ward R and Tomlinson I: Low-level
microsatellite instability occurs in most colorectal cancers and is
a nonrandomly distributed quantitative trait. Cancer Res. 62:53–57.
2002.PubMed/NCBI
|
|
63
|
Tomlinson I, Halford S, Aaltonen L,
Hawkins N and Ward R: Does MSI-low exist? J Pathol. 197:6–13. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cicek MS, Lindor NM, Gallinger S, Bapat B,
Hopper JL, Jenkins MA, Young J, Buchanan D, Walsh MD, Le Marchand
L, et al: Quality assessment and correlation of microsatellite
instability and immunohistochemical markers among population- and
clinic-based colorectal tumors results from the Colon Cancer Family
Registry. J Mol Diagn. 13:271–281. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Pritchard CC, Smith C, Salipante SJ, Lee
MK, Thornton AM, Nord AS, Gulden C, Kupfer SS, Swisher EM, Bennett
RL, et al: ColoSeq provides comprehensive lynch and polyposis
syndrome mutational analysis using massively parallel sequencing. J
Mol Diagn. 14:357–366. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ribic CM, Sargent DJ, Moore MJ, Thibodeau
SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R,
Shepherd LE, et al: Tumor microsatellite-instability status as a
predictor of benefit from fluorouracil-based adjuvant chemotherapy
for colon cancer. N Engl J Med. 349:247–257. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Guastadisegni C, Colafranceschi M, Ottini
L and Dogliotti E: Microsatellite instability as a marker of
prognosis and response to therapy: A meta-analysis of colorectal
cancer survival data. Eur J Cancer. 46:2788–2798. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sargent DJ, Marsoni S, Monges G, Thibodeau
SN, Labianca R, Hamilton SR, French AJ, Kabat B, Foster NR, Torri
V, et al: Defective mismatch repair as a predictive marker for lack
of efficacy of fluorouracil-based adjuvant therapy in colon cancer.
J Clin Oncol. 28:3219–3226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hutchins G, Southward K, Handley K, Magill
L, Beaumont C, Stahlschmidt J, Richman S, Chambers P, Seymour M,
Kerr D, et al: Value of mismatch repair, KRAS, and BRAF mutations
in predicting recurrence and benefits from chemotherapy in
colorectal cancer. J Clin Oncol. 29:1261–1270. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Malesci A, Laghi L, Bianchi P, Delconte G,
Randolph A, Torri V, Carnaghi C, Doci R, Rosati R, Montorsi M, et
al: Reduced likelihood of metastases in patients with
microsatellite-unstable colorectal cancer. Clin Cancer Res.
13:3831–3839. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Braun MS, Richman SD, Quirke P, Daly C,
Adlard JW, Elliott F, Barrett JH, Selby P, Meade AM, Stephens RJ,
et al: Predictive biomarkers of chemotherapy efficacy in colorectal
cancer: Results from the UK MRC FOCUS trial. J Clin Oncol.
26:2690–2698. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Koopman M, Kortman GA, Mekenkamp L,
Ligtenberg MJ, Hoogerbrugge N, Antonini NF, Punt CJ and van Krieken
JH: Deficient mismatch repair system in patients with sporadic
advanced colorectal cancer. Br J Cancer. 100:266–273. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Müller CI, Schulmann K, Reinacher-Schick
A, Andre N, Arnold D, Tannapfel A, Arkenau H, Hahn SA, Schmoll SH,
Porschen R, et al; AIO Colorectal Study Group. Predictive and
prognostic value of microsatellite instability in patients with
advanced colorectal cancer treated with a fluoropyrimidine and
oxaliplatin containing first-line chemotherapy. A report of the AIO
Colorectal Study Group. Int J Colorectal Dis. 23:1033–1039. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Tikidzhieva A, Benner A, Michel S,
Formentini A, Link KH, Dippold W, von Knebel Doeberitz M, Kornmann
M and Kloor M: Microsatellite instability and Beta2-microglobulin
mutations as prognostic markers in colon cancer: Results of the
FOGT-4 trial. Br J Cancer. 106:1239–1245. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Venderbosch S, Nagtegaal ID, Maughan TS,
Smith CG, Cheadle JP, Fisher D, Kaplan R, Quirke P, Seymour MT,
Richman SD, et al: Mismatch repair status and BRAF mutation status
in metastatic colorectal cancer patients: A pooled analysis of the
CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res.
20:5322–5330. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Elsaleh H, Joseph D, Grieu F, Zeps N, Spry
N and Iacopetta B: Association of tumour site and sex with survival
benefit from adjuvant chemotherapy in colorectal cancer. Lancet.
355:1745–1750. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Elsaleh H, Shannon B and Iacopetta B:
Microsatellite instability as a molecular marker for very good
survival in colorectal cancer patients receiving adjuvant
chemotherapy. Gastroenterology. 120:1309–1310. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Des Guetz G, Schischmanoff O, Nicolas P,
Perret GY, Morere JF and Uzzan B: Does microsatellite instability
predict the efficacy of adjuvant chemotherapy in colorectal cancer?
A systematic review with meta-analysis. Eur J Cancer. 45:1890–1896.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Hong SP, Min BS, Kim TI, Cheon JH, Kim NK,
Kim H and Kim WH: The differential impact of microsatellite
instability as a marker of prognosis and tumour response between
colon cancer and rectal cancer. Eur J Cancer. 48:1235–1243. 2012.
View Article : Google Scholar
|
|
80
|
André T, Boni C, Navarro M, Tabernero J,
Hickish T, Topham C, Bonetti A, Clingan P, Bridgewater J, Rivera F,
et al: Improved overall survival with oxaliplatin, fluorouracil,
and leucovorin as adjuvant treatment in stage II or III colon
cancer in the MOSAIC trial. J Clin Oncol. 27:3109–3116. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zaanan A, Cuilliere-Dartigues P, Guilloux
A, Parc Y, Louvet C, de Gramont A, Tiret E, Dumont S, Gayet B,
Validire P, et al: Impact of p53 expression and microsatellite
instability on stage III colon cancer disease-free survival in
patients treated by 5-fluorouracil and leucovorin with or without
oxaliplatin. Ann Oncol. 21:772–780. 2010. View Article : Google Scholar
|
|
82
|
Kim ST, Lee J, Park SH, Park JO, Lim HY,
Kang WK, Kim JY, Kim YH, Chang DK, Rhee PL, et al: Clinical impact
of microsatellite instability in colon cancer following adjuvant
FOLFOX therapy. Cancer Chemother Pharmacol. 66:659–667. 2010.
View Article : Google Scholar
|
|
83
|
Kim ST, Lee J, Park SH, Park JO, Lim HY,
Kang WK, Kim JY, Kim YH, Chang DK, Rhee PL, et al: The effect of
DNA mismatch repair (MMR) status on oxaliplatin-based first-line
chemotherapy as in recurrent or metastatic colon cancer. Med Oncol.
27:1277–1285. 2010. View Article : Google Scholar
|
|
84
|
Bertagnolli MM, Niedzwiecki D, Compton CC,
Hahn HP, Hall M, Damas B, Jewell SD, Mayer RJ, Goldberg RM, Saltz
LB, et al: Microsatellite instability predicts improved response to
adjuvant therapy with irinotecan, fluorouracil, and leucovorin in
stage III colon cancer: Cancer and Leukemia Group B Protocol 89803.
J Clin Oncol. 27:1814–1821. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Roth AD, Tejpar S, Delorenzi M, Yan P,
Fiocca R, Klingbiel D, Dietrich D, Biesmans B, Bodoky G, Barone C,
et al: Prognostic role of KRAS and BRAF in stage II and III
resected colon cancer: Results of the translational study on the
PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 28:466–474.
2010. View Article : Google Scholar
|
|
86
|
Kim JE, Hong YS, Ryu MH, Lee JL, Chang HM,
Lim SB, Kim JH, Jang SJ, Kim MJ, Yu CS, et al: Association between
deficient mismatch repair system and efficacy to
irinotecan-containing chemotherapy in metastatic colon cancer.
Cancer Sci. 102:1706–1711. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Matthews KS, Estes JM, Conner MG, Manne U,
Whitworth JM, Huh WK, Alvarez RD, Straughn JM Jr, Barnes MN and
Rocconi RP: Lynch syndrome in women less than 50 years of age with
endometrial cancer. Obstet Gynecol. 111:1161–1166. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kato M, Takano M, Miyamoto M, Sasaki N,
Goto T, Tsuda H and Furuya K: DNA mismatch repair-related protein
loss as a prognostic factor in endometrial cancers. J Gynecol
Oncol. 26:40–45. 2015. View Article : Google Scholar :
|
|
89
|
Terada KY, Black M, Terada LH, Davis J and
Shimizu DM: Survival of endometrial cancer patients with lymphatic
invasion and deficient mismatch repair expression. Gynecol Oncol.
129:188–192. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Resnick KE, Frankel WL, Morrison CD,
Fowler JM, Copeland LJ, Stephens J, Kim KH and Cohn DE: Mismatch
repair status and outcomes after adjuvant therapy in patients with
surgically staged endometrial cancer. Gynecol Oncol. 117:234–238.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Ruiz I, Martín-Arruti M, Lopez-Lopez E and
Garcia-Orad A: Lack of association between deficient mismatch
repair expression and outcome in endometrial carcinomas of the
endometrioid type. Gynecol Oncol. 134:20–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Nelson GS, Pink A, Lee S, Han G, Morris D,
Ogilvie T, Duggan MA and Köbel M: MMR deficiency is common in
high-grade endometrioid carcinomas and is associated with an
unfavorable outcome. Gynecol Oncol. 131:309–314. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Cohn DE, Frankel WL, Resnick KE, Zanagnolo
VL, Copeland LJ, Hampel H, Kelbick N, Morrison CD and Fowler JM:
Improved survival with an intact DNA mismatch repair system in
endometrial cancer. Obstet Gynecol. 108:1208–1215. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Diaz-Padilla I, Romero N, Amir E,
Matias-Guiu X, Vilar E, Muggia F and Garcia-Donas J: Mismatch
repair status and clinical outcome in endometrial cancer: A
systematic review and meta-analysis. Crit Rev Oncol Hematol.
88:154–167. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
CRUK. Ovarian cancer incidence statistics.
http://www.cancer-researchuk.org/cancer-info/cancerstats/types/ovary/incidence/.
Accessed April 15, 2015
|
|
96
|
Surveillance, Epidemiology, and End
Results Program - National Cancer Institute. SEER Stat Fact Sheets:
Ovary Cancer. http://seer.cancer.gov/statfacts/html/ovary.html.
Accessed April 15, 2015
|
|
97
|
Bonadona V, Bonaïti B, Olschwang S,
Grandjouan S, Huiart L, Longy M, Guimbaud R, Buecher B, Bignon YJ,
Caron O, et al; French Cancer Genetics Network. Cancer risks
associated with germline mutations in MLH1, MSH2, and MSH6 genes in
Lynch syndrome. JAMA. 305:2304–2310. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Engel C, Loeffler M, Steinke V, Rahner N,
Holinski-Feder E, Dietmaier W, Schackert HK, Goergens H, von Knebel
Doeberitz M, Goecke TO, et al: Risks of less common cancers in
proven mutation carriers with Lynch syndrome. J Clin Oncol.
30:4409–4415. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Watson P, Vasen HF, Mecklin JP, Bernstein
I, Aarnio M, Järvinen HJ, Myrhøj T, Sunde L, Wijnen JT and Lynch
HT: The risk of extra-colonic, extra-endometrial cancer in the
Lynch syndrome. Int J Cancer. 123:444–449. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Xiao X, Melton DW and Gourley C: Mismatch
repair deficiency in ovarian cancer - molecular characteristics and
clinical implications. Gynecol Oncol. 132:506–512. 2014. View Article : Google Scholar
|
|
101
|
Catasús L, Bussaglia E, Rodrguez I,
Gallardo A, Pons C, Irving JA and Prat J: Molecular genetic
alterations in endometrioid carcinomas of the ovary: Similar
frequency of beta-catenin abnormalities but lower rate of
microsatellite instability and PTEN alterations than in uterine
endometrioid carcinomas. Hum Pathol. 35:1360–1368. 2004. View Article : Google Scholar
|
|
102
|
Gras E, Catasus L, Argüelles R,
Moreno-Bueno G, Palacios J, Gamallo C, Matias-Guiu X and Prat J:
Microsatellite instability, MLH-1 promoter hypermethylation, and
frameshift mutations at coding mononucleotide repeat
microsatellites in ovarian tumors. Cancer. 92:2829–2836. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Jensen KC, Mariappan MR, Putcha GV, Husain
A, Chun N, Ford JM, Schrijver I and Longacre TA: Microsatellite
instability and mismatch repair protein defects in ovarian
epithelial neoplasms in patients 50 years of age and younger. Am J
Surg Pathol. 32:1029–1037. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Murphy MA and Wentzensen N: Frequency of
mismatch repair deficiency in ovarian cancer: A systematic review
This article is a US Government work and, as such, is in the public
domain of the United States of America. Int J Cancer.
129:1914–1922. 2011. View Article : Google Scholar :
|
|
105
|
Pal T, Permuth-Wey J, Kumar A and Sellers
TA: Systematic review and meta-analysis of ovarian cancers:
Estimation of microsatellite-high frequency and characterization of
mismatch repair deficient tumor histology. Clin Cancer Res.
14:6847–6854. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Scartozzi M, De Nictolis M, Galizia E,
Carassai P, Bianchi F, Berardi R, Gesuita R, Piga A, Cellerino R
and Porfiri E: Loss of hMLH1 expression correlates with improved
survival in stage III–IV ovarian cancer patients. Eur J Cancer.
39:1144–1149. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Zhai QJ, Rosen DG, Lu K and Liu J: Loss of
DNA mismatch repair protein hMSH6 in ovarian cancer is
histotype-specific. Int J Clin Exp Pathol. 1:502–509.
2008.PubMed/NCBI
|
|
108
|
Begum FD, Høgdall CK, Kjaer SK, Blaakaer
J, Christensen L, Ryan A, Jacobs IJ and Høgdall EV: Distribution of
microsatellite instability in Danish ovarian tumor patients and the
prognostic value in ovarian cancer patients. Oncol Res. 17:43–49.
2008.PubMed/NCBI
|
|
109
|
Ercoli A, Ferrandina G, Raspaglio G,
Marone M, Maggiano N, Del Mastro P, Benedetti Panici P, Mancuso S
and Scambia G: hMSH2 and GTBP expression in advanced stage
epithelial ovarian cancer. Br J Cancer. 80:1665–1671. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Marcelis CL, van der Putten HW, Tops C,
Lutgens LC and Moog U: Chemotherapy resistant ovarian cancer in
carriers of an hMSH2 mutation? Fam Cancer. 1:107–109. 2001.
View Article : Google Scholar
|
|
111
|
CRUK. Skin cancer incidence statistics.
http://www.cancer-researchuk.org/cancer-info/cancerstats/types/skin/incidence/.
Accessed April 15, 2015
|
|
112
|
Birindelli S, Tragni G, Bartoli C, Ranzani
GN, Rilke F, Pierotti MA and Pilotti S: Detection of microsatellite
alterations in the spectrum of melanocytic nevi in patients with or
without individual or family history of melanoma. Int J Cancer.
86:255–261. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Peris K, Keller G, Chimenti S, Amantea A,
Kerl H and Höfler H: Microsatellite instability and loss of
heterozygosity in melanoma. J Invest Dermatol. 105:625–628. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Quinn AG, Healy E, Rehman I, Sikkink S and
Rees JL: Microsatellite instability in human non-melanoma and
melanoma skin cancer. J Invest Dermatol. 104:309–312. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Richetta A, Silipo V, Calvieri S, Frati L,
Ottini L, Cama A and Mariani-Costantini R: Microsatellite
instability in primary and metastatic melanoma. J Invest Dermatol.
109:119–120. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Talwalkar VR, Scheiner M, Hedges LK,
Butler MG and Schwartz HS: Microsatellite instability in malignant
melanoma. Cancer Genet Cytogenet. 104:111–114. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Alvino E, Marra G, Pagani E, Falcinelli S,
Pepponi R, Perrera C, Haider R, Castiglia D, Ferranti G, Bonmassar
E, et al: High-frequency microsatellite instability is associated
with defective DNA mismatch repair in human melanoma. J Invest
Dermatol. 118:79–86. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Hussein MR: Genetic pathways to melanoma
tumorigenesis. J Clin Pathol. 57:797–801. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Hussein MR, Sun M, Tuthill RJ, Roggero E,
Monti JA, Sudilovsky EC, Wood GS and Sudilovsky O: Comprehensive
analysis of 112 melanocytic skin lesions demonstrates
microsatellite instability in melanomas and dysplastic nevi, but
not in benign nevi. J Cutan Pathol. 28:343–350. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Palmieri G, Ascierto PA, Cossu A,
Colombino M, Casula M, Botti G, Lissia A, Tanda F and Castello G:
Assessment of genetic instability in melanocytic skin lesions
through microsatellite analysis of benign naevi, dysplastic naevi,
and primary melanomas and their metastases. Melanoma Res.
13:167–170. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Palmieri G, Cossu A, Ascierto PA, Botti G,
Strazzullo M, Lissia A, Colombino M, Casula M, Floris C, Tanda F,
et al; Melanoma Cooperative Group. Definition of the role of
chromosome 9p21 in sporadic melanoma through genetic analysis of
primary tumours and their metastases. Br J Cancer. 83:1707–1714.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Richetta A, Ottini L, Falchetti M,
Innocenzi D, Bottoni U, Faiola R, Mariani-Costantini R and Calvieri
S: Instability at sequence repeats in melanocytic tumours. Melanoma
Res. 11:283–289. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Tomlinson IP, Beck NE and Bodmer WF:
Allele loss on chromosome 11q and microsatellite instability in
malignant melanoma. Eur J Cancer. 32A:1797–1802. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Castiglia D, Bernardini S, Alvino E,
Pagani E, De Luca N, Falcinelli S, Pacchiarotti A, Bonmassar E,
Zambruno G and D'Atri S: Concomitant activation of Wnt pathway and
loss of mismatch repair function in human melanoma. Genes
Chromosomes Cancer. 47:614–624. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Korabiowska M, Cordon-Cardo C, Jaenckel F,
Stachura J, Fischer G and Brinck U: Application of in situ
hybridization probes for MLH-1 and MSH-2 in tissue microarrays of
paraffin-embedded malignant melanomas: Correlation with
immunohistochemistry and tumor stage. Hum Pathol. 35:1543–1548.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Alvino E, Passarelli F, Cannavò E, Fortes
C, Mastroeni S, Caporali S, Jiricny J, Cappellini GC, Scoppola A,
Marchetti P, et al: High expression of the mismatch repair protein
MSH6 is associated with poor patient survival in melanoma. Am J
Clin Pathol. 142:121–132. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
CRUK. Stomach cancer statistics.
http://www.cancerresearchuk.org/cancer-info/cancerstats/types/stomach/.
Accessed April 15, 2015
|
|
128
|
Beghelli S, de Manzoni G, Barbi S,
Tomezzoli A, Roviello F, Di Gregorio C, Vindigni C, Bortesi L,
Parisi A, Saragoni L, et al: Microsatellite instability in gastric
cancer is associated with better prognosis in only stage II
cancers. Surgery. 139:347–356. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Fang WL, Chang SC, Lan YT, Huang KH, Chen
JH, Lo SS, Hsieh MC, Li AF, Wu CW and Chiou SH: Microsatellite
instability is associated with a better prognosis for gastric
cancer patients after curative surgery. World J Surg. 36:2131–2138.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Lee HS, Choi SI, Lee HK, Kim HS, Yang HK,
Kang GH, Kim YI, Lee BL and Kim WH: Distinct clinical features and
outcomes of gastric cancers with microsatellite instability. Mod
Pathol. 15:632–640. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Schneider BG, Bravo JC, Roa JC, Roa I, Kim
MC, Lee KM, Plaisance KT Jr, McBride CM and Mera R: Microsatellite
instability, prognosis and metastasis in gastric cancers from a
low-risk population. Int J Cancer. 89:444–452. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Falchetti M, Saieva C, Lupi R, Masala G,
Rizzolo P, Zanna I, Ceccarelli K, Sera F, Mariani-Costantini R,
Nesi G, et al: Gastric cancer with high-level microsatellite
instability: Target gene mutations, clinicopathologic features, and
long-term survival. Hum Pathol. 39:925–932. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Perez RO, Jacob CE, D'Ottaviano FL,
Alvarenga C, Ribeiro AS, Ribeiro U Jr, Bresciani CJ, Zilberstein B,
Krieger JE, Habr-Gama A, et al: Microsatellite instability in
solitary and sporadic gastric cancer. Rev Hosp Clin Fac Med Sao
Paulo. 59:279–285. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
An C, Choi IS, Yao JC, Worah S, Xie K,
Mansfield PF, Ajani JA, Rashid A, Hamilton SR and Wu TT: Prognostic
significance of CpG island methylator phenotype and microsatellite
instability in gastric carcinoma. Clin Cancer Res. 11:656–663.
2005.PubMed/NCBI
|
|
135
|
An JY, Kim H, Cheong JH, Hyung WJ, Kim H
and Noh SH: Microsatellite instability in sporadic gastric cancer:
Its prognostic role and guidance for 5-FU based chemotherapy after
R0 resection. Int J Cancer. 131:505–511. 2012. View Article : Google Scholar
|
|
136
|
Oki E, Kakeji Y, Zhao Y, Yoshida R, Ando
K, Masuda T, Ohgaki K, Morita M and Maehara Y: Chemosensitivity and
survival in gastric cancer patients with microsatellite
instability. Ann Surg Oncol. 16:2510–2515. 2009. View Article : Google Scholar : PubMed/NCBI
|