Introduction
The vast majority of cellular activities are
executed by coordinated gene expression. Precise regulation of the
transcription factors that control gene expression is necessary to
accomplish specific tasks and to maintain cellular homeostasis.
Various endogenous and exogenous stimuli induce changes in gene
expression, and dynamic changes of gene expression profiles are
observed during all processes maintaining life. Deregulated gene
expression by either genetic or epigenetic alterations on the other
hand can cause a broad range of diseases and is important for the
development of cancer. The transcription factor NFκB plays a
central role in many biological processes such as inflammation,
differentiation, cell death and tumorigenesis (1). In glioblastoma (GBM), the most
malignant human brain tumor in man, overexpression or aberrant
constitutive activation of NFκB contributes to survival (2), radio- and chemoresistance (3–5),
elevated glioma cell motility (6,7),
enhanced angiogenesis (8) as well
as chronic inflammation (9). On
the other hand it has been described that, under some circumstances
and dependent on the cooperation of NFκB with a variety of other,
yet mainly unidentified factors and modulators of its activity,
NFκB is mandatory for the induction of cell death, even in GBM
cells (10–12). Until today it has not been
unraveled in detail by which mechanisms NFκB provides its dual
function, either serving as an oncogene triggering survival, but
also exerting, via induction of cell death, its function as a tumor
suppressor. Plenty of suggestions exist as to how and by which
factors the dual function of NFκB is regulated, but there are still
many questions left.
In unstimulated cells, the inhibitor of NFκB (IκB)
proteins sequester NFκB into inactive complexes in the cytoplasm. A
variety of different stimuli such as hypoxia, stress or tumor
necrosis factor α (TNFα) can induce, via different pathways, the
phosphorylation of cytoplasmic IκB protein family members by the
IκB kinase complex leading to IκB proteasomal degradation, this
resulting in the nuclear translocation of NFκB and induction of
target gene expression (13).
Whereas the predominantly cytoplasmic family members IκBα, IκBβ and
IκBɛ exclusively act as inhibitors of NFκB, the atypical and mainly
nuclearly localized IκB protein IκBζ, coded by the NFKBIZ gene and
being a primary response target gene of NFκB (14), is able to bind to NFκB, is
postulated to work as a (co)-transcription factor, and in this way
seems to modulate the expression of a subset of NFκB target genes.
Modulation of gene expression is at least partially mediated by the
intrinsic transactivation activity of IκBζ and its interaction with
the NFκB subunit p50 (15,16). Moreover, IκBζ is associated with
histone deacetylases suggesting that chromatin remodeling is
important for the transcriptional activity of IκBζ (17). In this report we demonstrate that
IκBζ is upregulated in glioma specimens, in primary low passage
glioma cells as well as in most established glioma cell lines. In a
glioma cell line resistant towards NFκB-dependent cell death that
is induced by overexpression of a dominant active p53 variant the
chimeric tumor suppressor-1 (CTS-1) (12,18),
IκBζ is highly upregulated. In this regard we were interested
whether IκBζ might be a candidate that regulates the switch between
the dual functions of NFκB: serving as an oncogene or acting as a
cell death inductor and tumor suppressor.
Recently it has been described that IκBζ is an
important regulator of radiation induced cellular senescence in
breast cancer cells (19).
Subsequently, senescence associated proteins such as interleukins,
chemokines or growth factors can signal to the tumor environment
and could potentially promote tumor progression by promoting
proliferation, invasion or angiogenesis (20) or by induction of detrimental
chronic inflammatory immune responses (21). Additionally, IκBζ has been
described as a regulator of chemokine (C-C motif) ligand 2,
recruiting circulating monocytes to inflammatory regions (14). Radiation is a common approach in
the treatment of glioma. In this regard it is postulated that
irradiation, via induction of necrosis, might lead to (chronic)
inflammatory processes in the tumor micro-milieu and subsequent
induction of resistance towards radiation (reviewed in ref.
22). We were also interested
whether in glioma IκBζ expression is regulated by γ-irradiation and
if in this context IκBζ serves as a transcriptional inducer of
inflammatory chemokine and cytokine secretion, this way putatively
inducing a more malignant tumor phenotype by modulating the tumor
micro-milieu. Our data provide novel information on the role of
IκBζ in tumorigenesis and tumor progression in glioma.
Materials and methods
Cell lines, reagents
The LN-229 (here named LNT-229P) cell line is a
human malignant glioma cell line and was kindly provided by N. de
Tribolet (Lausanne, Switzerland). Generation of CTS-1 resistant
cell line LNT-229R was previously described (12). GBM primary cells were established
from human GBM tissue and used at passages 5–8. All cells were
maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco Life
Technologies, Eggenstein, Germany) containing 10%
tetracycline-approved fetal calf serum (Tet-FCS; Gibco), penicillin
(100 U/ml) and streptomycin (100 μg/ml) in a humidified atmosphere
containing 5% CO2. Cell culture growth and cellular
density was determined by crystal violet staining as described. For
irradiation, the cells were seeded in 6-well plates and irradiated
using the Nordion GC40 Gammacell irradiator (Ottawa, ON, Canada).
For generation of supernatants, the cells were treated as
indicated, serum-free medium was added and cellular supernatants
were harvested 24 or 48 h later. The protein content in
supernatants was analyzed according to Bradford.
Transfection of cells with shRNA
constructs
For transfection with shRNA contructs, the cells
were seeded at 3×105 cells. After attachment, the cells
were transfected with plasmid constructs coding for either IκBζ
specific shRNA, or coding for scrambled shRNA unspecific for any
known mRNA using Metafectene PRO (Biontex, Martinsried, Germany).
shRNA plasmid constructs were a kind gift of Klaus Schulze-Osthoff
(Interfaculty Institute for Biochemistry, University of Tübingen,
Germany).
RNA preparation and quantitative
RT-PCR
Total RNA was prepared using the High Pure RNA
Isolation kit (Roche, Mannheim, Germany). RNA (5 μg) was reverse
transcribed using Superscript II reverse transcriptase (Invitrogen,
Carlsbad, CA, USA). Target gene expression was determined using
SYBR green master mix (Thermo Fisher Scientific, MA, USA), on an
ABI 7200 system. Relative mRNA expression was quantified using
comparative 2−ΔΔT method {[EΔCT
(TARGET)/EΔCT (GAPDH)]}. The following primers were
used: IκBζ-frwd (TCTGGAACTCATTCGCCTCT), IκBζ-rev
(TCAACCGATACTGCAAGCTG), IL-6-frwd (CGGGAACGAAAGAGAAGCTCTA),
IL-6-rev (GGCGCTTGTGGAGAAGGAG), IL-8-frwd(GTGGAGAAGTTTTTGAAGAGGGC),
IL-8-rev (CACTTCATGTATTGTGTGGGTCTG), CXCL-1-frwd
(GCAGGGAATTCACCCCAAGA), CXCL-1-rev (GAT GCAGGATTGAGGCAAGC),
GAPDH-frwd (TGCACCACCAACTGCTTAGC), GAPDH-rev (GGCATGGACTGTGG
TCATGA). GAPDH was used for internal normalization and did not vary
between cell types or treatments.
Construction of adenoviral vectors and
adenoviral infection
A replication-deficient recombinant and
tetracycline-inducible adenovirus expressing IκBζ was constructed
using the Ad-Easy system (23). In
brief, IκBζ cDNA was inserted into pTRE-tight (Takara Bio Europe
SAS, Saint-Germain-en-Laye, France) downstream of the
tetracycline-responsive promoter element. The inserted cDNA was
completely sequenced, compared to the NCBI database and found to be
correct. The expression cassette consisting of the CMV-minimal
promoter, the Tet-responsive element and IκBζ cDNA was cloned into
pAdTrack, containing an additional expression cassette for enhanced
green fluorescence protein (EGFP), which was later used to monitor
virus production and infection load. After recombination with the
viral genome containing plasmid Ad-Easy1, recombinant adenoviral
genomes were transfected into HEK-293 cells (ATCC). Ad-ON expresses
the reverse TET-repressor (rtTA) and was a kindly gift of G. Thomas
(Portland, OR, USA), Ad-CTS1, coding for a dominant active version
of p53, has been previously described (18), Ad-Co#4, which serves a control
virus, is based on the Ad-Easy-System, but lacks both EGFP and
expression of the gene of interest. All viruses were CsCl-purified,
dialyzed and titrated using the Clontech Adeno-X Rapid Titration
System. Transgene expression and inducibility were tested by
quantitative RT-PCR (qPCR) of infected LNT-229P cells. Infection
with recombinant adenovirus was accomplished by exposing cells to
adenovirus at a defined moiety of infection (MOI) in serum-free
medium for 30 min followed by addition of serum-containing medium
of the mentioned time periods.
NFκB luciferase reporter assay
LNT-229P cells grown in microtiter plates were
transfected with 150 ng pNFκB-Luc expressing firefly luciferase and
20 ng pRL-CMV expressing renilla luciferase as an internal
standard. As a positive control of NFκB activation, the cells were
cotransfected with 40 ng pMEKK. At 24 h after transfection, the
cells were infected with 100 MOI of Ad TET-IκBζ + 100 MOI of Ad-ON
in the absence or presence of doxycycline (2 μg/ml) for 48 h. NFκB
activity was assessed using the dual luciferase assay (24).
Detection of cytokine expression
Relative cytokine expression was analyzed using the
RayBiotech Human Cytokine Array (RayBiotech, Norcross, GA, USA)
according to the manufacturer's protocol. Cell culture supernatants
were harvested at indicated time points and stored at −20°C. In
parallel, RNA was harvested from the cells for cDNA synthesis and
subsequent qPCR analysis of cytokine mRNA expression. Intensity of
the spots on the array was normalized to the mean intensity of the
internal control spots and relative intensity was measured from
duplicate dots representing single cytokines. For the analysis of
absolute amounts of IL-6, the cells were treated as indicated and
IL-6 secretion was measured using an IL-6 immunoassay kit
(RayBiotech).
TCGA and REMBRANDT analyses
IκBζ gene expression was analyzed in primary glioma
and normal brain samples by assessing the TCGA data portal. The
mRNA profiles were determined using the Agilent 244K G4502A
micro-array (http://cancergenome.nih.gov/; accessed Dec. 2014).
Statistical significance between GBM and normal CNS tissue was
assessed using a t-test (p<0.05). The calculations were
performed using GraphPad Prism Software 6.0 (GraphPad Software, CA,
USA). Survival correlation analyses were done using the REMBRANDT
database containing probes from the Affymetrix 223218_s_at and
223217_s_at dataset (National Cancer Institute; 2005; REMBRANDT
version 1.5.9, http://rembrandt.nci.nih.gov). At the time of
accession (Dec. 2014), the database contained mRNA data of 228
glioblastomas, 148 grade II/III astrocytomas, 67 grade II/III
oligodendrogliomas, 11 mixed gliomas and 28 non-tumor control
tissues. Kaplan-Meier survival curves were generated by analyzing
the glioma cohort (WHO grade II–IV; n=138) as well as the GBM
cohort (WHO grade IV; n=84) separately. IκBζ up- or down-regulation
was defined as a twofold (or greater) difference from the mean
expression level within a given dataset. p-values for differences
in patient survival curves were obtained by using the log-rank or
Wilcoxon test. The ‘Highest Geometric Mean Intensity’ of IκBζ
expression was used as the reporter for relative IκBζ expression
within the database.
Statistical and correlation analysis
The figures show data obtained in at least three
independent experiments as indicated. Statistical analyses were
performed using GraphPad Prism version 6.0 (GraphPad Software, CA,
USA). Quantitative data were assessed for significance by paired
t-test (*p<0.05; **p<0.01;
***p<0.001). Patient survival was analyzed by
Kaplan-Meier life table and for comparison of survival Wilcoxon and
log-rank test were used (significance level α = 0.05, JMP 11.0
software, SAS, Cary, NC, USA).
Results
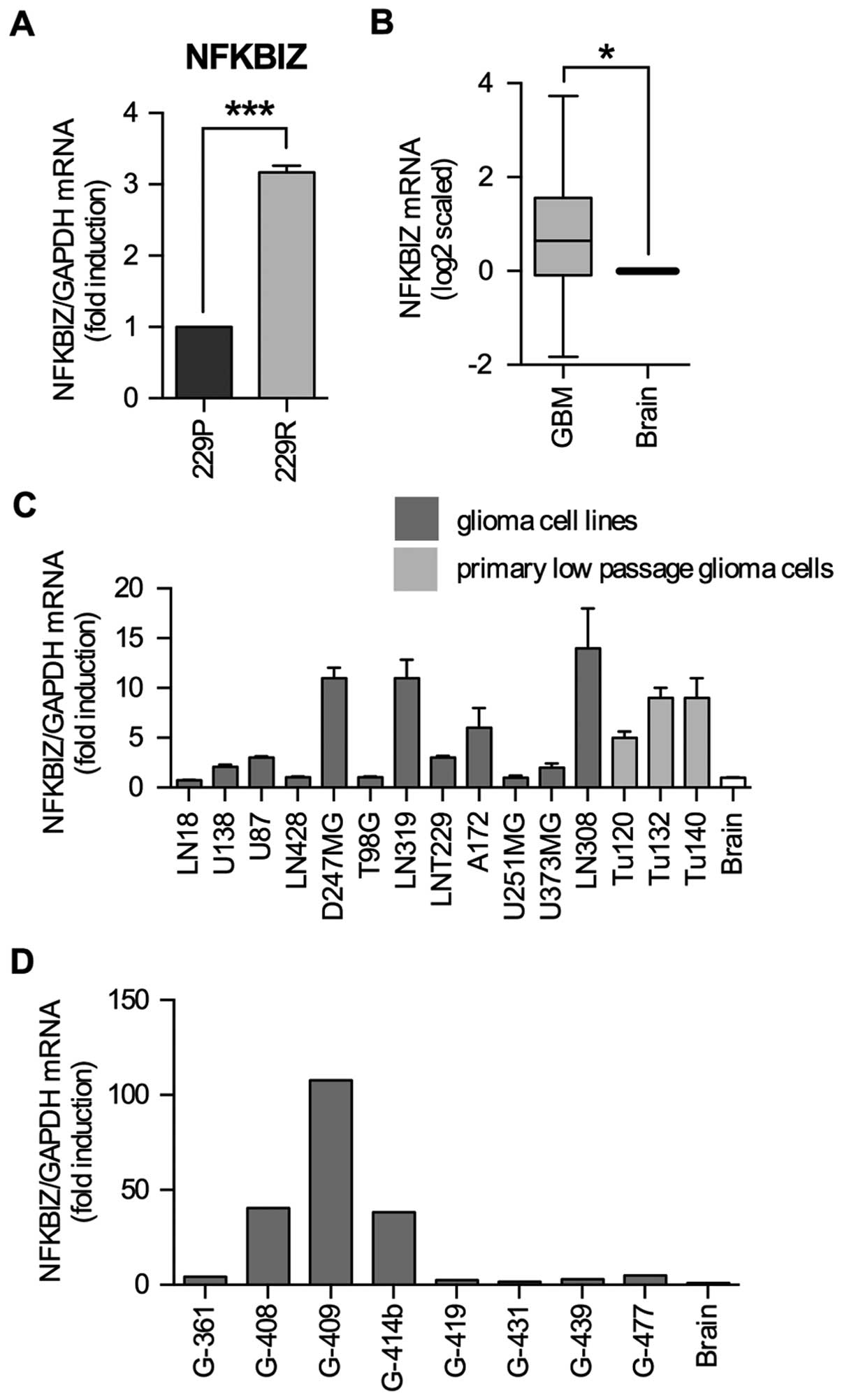
IκBζ mRNA is upregulated in low passage
glioma cell lines, established cell lines and glioma specimens
We generated a cell line (LNT-229R) completely
resistant to NFκB-dependent cell death induction upon
overexpression of a dominant-positive p53, chimeric tumor
suppressor (CTS)-1 (12). In a
microarray mRNA expression analysis, IκBζ was upregulated in
LNT-229R cells (12) suggesting
that in GBM IκBζ might be involved in the development of resistance
and malignancy. By quantitative RT-PCR we validated IκBζ expression
in LNT-229P and LNT-229R cells and found IκBζ mRNA to be 3-fold
higher in LNT-229R compared to LNT-229P cells (Fig. 1A). In contrast to normal brain
where IκBζ is barely expressed (15), the analysis of NFKBIZ gene
expression data of the TCGA database revealed that mean IκBζ mRNA
expression was 0.65-fold (log2 scale) higher in
glioblastoma samples as compared to non-neoplastic CNS control
tissues (Wilcoxon test, p<0.05) (Fig. 1B). Analyses of the REMBRANDT
database also exhibited an enhancement in mean IκBζ expression in
the GBM dataset, while other glioma subgroups such as astrocytoma
or oligodendroglioma showed no induction of IκBζ mRNA expression
levels as compared to normal CNS tissue (data not shown). In GBM
cells, IκBζ expression was also upregulated in 3/3 tested low
passage primary GBM cell lines (Tu120, Tu132, Tu140) as well as in
8/12 established glioma cell lines (Fig. 1C). In glioma specimens, IκBζ mRNA
expression is highly variable, ranging from 1.5- (G-431) to
107-fold (G-409) compared to non-tumor brain tissue (Fig. 1D).
Overexpression of IκBζ does not influence
CTS-1-induced, NFκB-dependent cell death
In CTS-1 sensitive LNT-229P, but not in resistant
LNT-229R glioma cells, NFκB is activated upon adenovirally mediated
expression of CTS-1, a dominant and constitutively active version
of p53 (12). In LNT-229P cells
CTS-1 mediated cell death is dependent on the activity of NFκB and
could be largely inhibited by blocking NFκB activation (12). Since in LNT-229R cells IκBζ mRNA is
upregulated (Fig. 1A) and since it
is known that IκBζ is a direct target of NFκB and a modulator of
NFκB activity, we analyzed whether in LNT-229P and LNT-229R cells
IκBζ mRNA expression is regulated by CTS-1 and whether enhanced
IκBζ expression influences the CTS-1-induced cell death in these
cells. As shown in Fig. 2A, IκBζ
expression is upregulated in Ad-CTS-1 infected LNT-229P cells that
show a low basal level of IκBζ expression, but is not further
enhanced in Ad-CTS-1 infected LNT-229R cells which per se show
elevated IκBζ expression, indicating that in parental, but not in
resistant cells, IκBζ, putatively via induction of NFκB, might be
also regulated by CTS-1, a dominant version of p53.
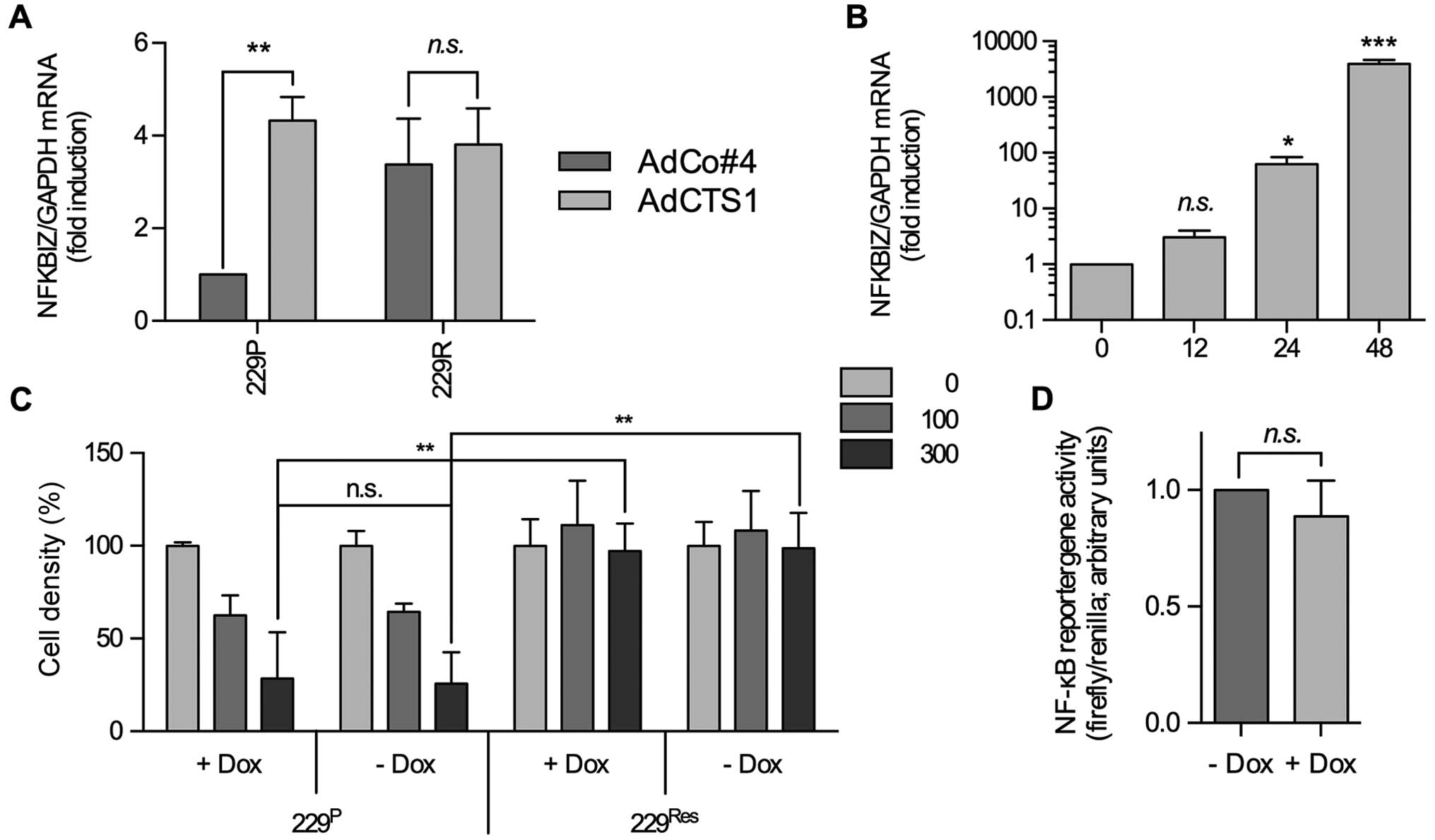 | Figure 2IκBζ overexpression neither modulates
NFκB activity nor protects glioma cells from CTS-1 induced cell
death. (A) IκBζ mRNA expression in LNT229P and LNT229R cells after
infection with Ad-CTS-1. AdCo#4 served as a negative control (n=4,
SEM, **p<0.01). (B) Upon adenoviral infection and
induction of transgene expression by addition of DOX, IκBζ mRNA
increased after 12–24 h and stayed elevated at least up to 48 h
(n=3, SEM, *p<0.05, ***p<0.001) (C)
Induction of IκBζ mRNA expression does not prevent Ad-CTS-1 induced
cell death in LNT-229P cells. The cells were infected with
Ad-TET-IκBζ + Ad-ON in the presence (+DOX) or absence (−DOX) of
doxycycline. After 24 h, the cells were infected with increasing
MOI of Ad-CTS-1. Cell density was assessed by crystal violet
staining 72 h later (n=3, SEM, **p<0.01). (D) NFκB
reporter gene assay of LNT229P cells infected with Ad-TET-IκBζ +
Ad-ON in the absence or presence of doxycycline (n=4, SD). |
IκBζ has been described, at least in fibroblasts, to
bind to the NFκB p50 subunit and to interfere with p65, modulating
NFκB activity and mediating the induction of several NFκB
responsive genes (15). In this
context, enhanced expression of IκBζ in LNT-229R cells might be the
reason why these cells are resistant to CTS-1-induced cell death.
To test this, we generated a tetracycline/doxycycline
(DOX)-inducible adenoviral system that allows us to transiently
induce IκBζ expression. After infection of glioma cells with
Ad-TET-IκBζ plus Ad-ON and addition of DOX, elevated IκBζ
expression is detected 12 h after infection, increasing up to 48 h
and then stable for at least 72 h (Fig. 2B and data not shown). Nevertheless,
DOX-mediated induction of IκBζ expression neither changed basal
NFκB activity which is known to be enhanced in LNT-229 cells nor
the sensitivity of these cells towards CTS-1 induced cell death
(Fig. 2C and D), indicating that
IκBζ overexpression is not responsible for the NFκB- dependent
resistance against CTS-1-induced cell death in LNT-229R cells.
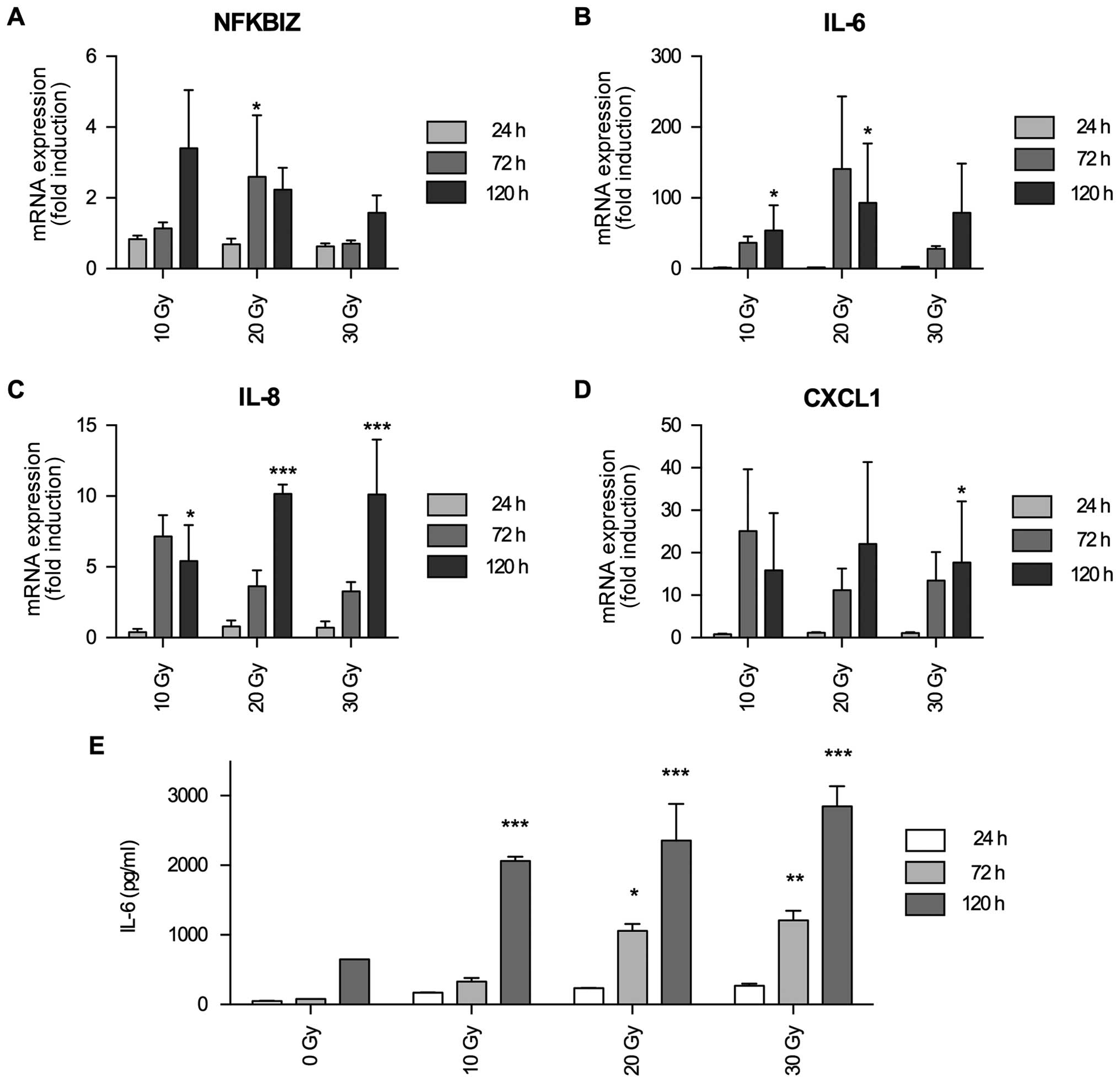
IκBζ mRNA expression is induced by
irradiation and serves as an activator for the expression of
inflammation-associated cytokines IL-6, IL-8 and CXCL1
There are several hints that in glioma IκBζ might be
a tumor promoting transcription factor, translating its oncogenic
activity via induction of (chronic) inflammation. First of all, in
glioma the NFκB signaling cascade is involved in processes of
inflammation and resistance towards gamma-irradiation (25). Secondly, IκBζ as a direct target of
NFκB is upregulated in fibroblasts by irradiation induced
senescence and, in this regard, induces the expression of a variety
inflammatory cyto- and chemokines such as interleukin (IL)-6, IL-8
and CXCL1 (19). Thirdly, IL-6 and
IL-8 are upregulated in glioma cells upon irradiation (26), and last but not least, it has been
described that IL-6 producing glioma cells were not affected by
irradiation (27). We therefore
investigated whether in glioma IκBζ might be a modulator of
radiation induced expression of inflammatory cytokines. We first
analyzed whether irradiation induces IκBζ expression. As shown in
Fig. 3A, IκBζ mRNA expression was
elevated in LNT-229P cells after irradiation. In a similar time
frame, also IL-6, IL-8 and CXCL1 mRNA expression was induced
(Fig. 3B–D). To validate whether
irradiation-induced expression of inflammatory cytokine mRNA in
glioma cells also translates into protein expression, we
exemplarily analyzed IL-6 secretion in LNT-229P cells. As shown in
Fig. 3E, IL-6 secretion was
upregulated by irradiation in a dose- and time-dependent
manner.
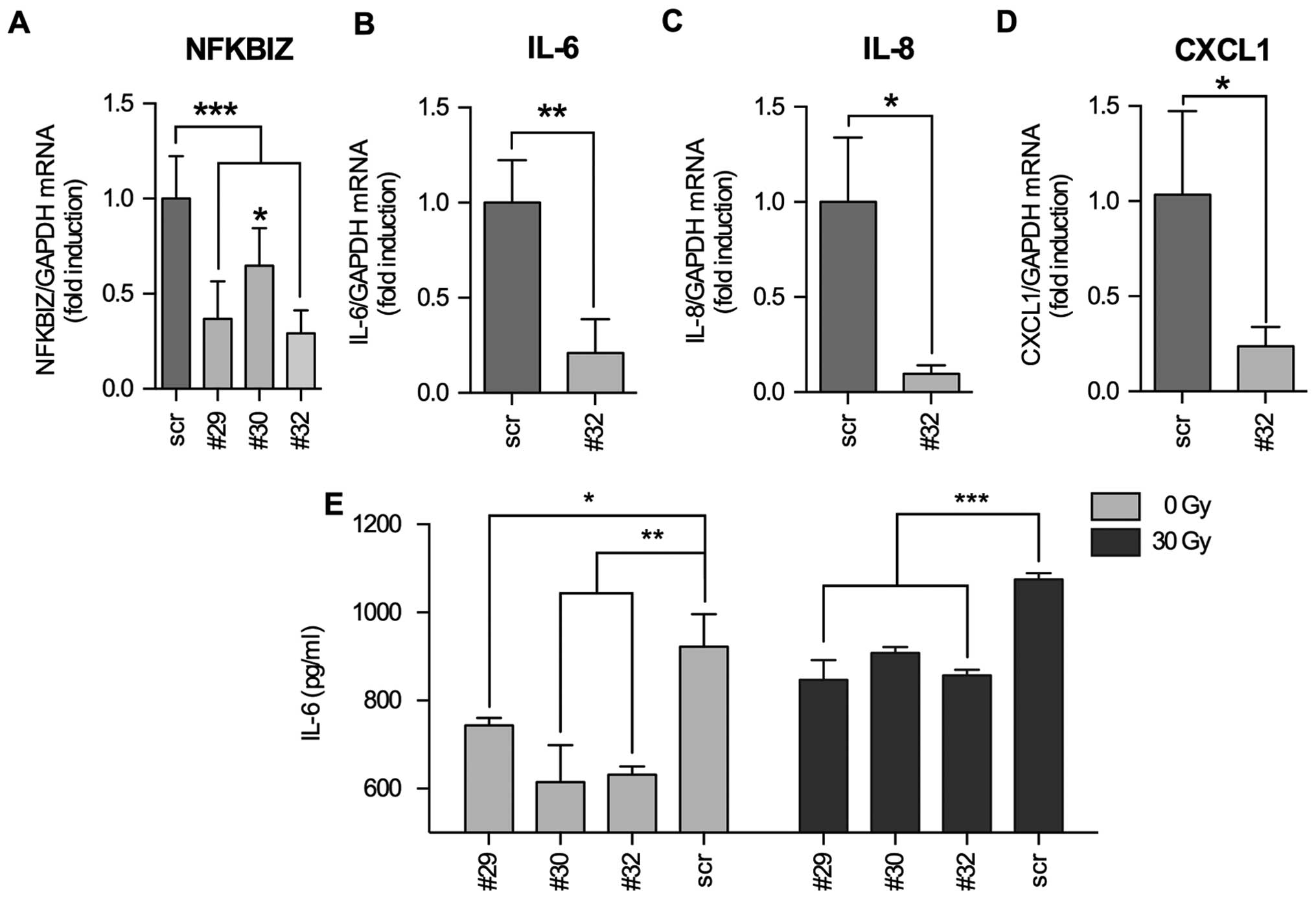
We were interested whether IκBζ is directly
responsible for the induction of IL-6, IL-8 and CXCL1 cytokine
expression or if IκBζ served as a modulator NFκB activity as
described by Totzke et al (15). For this, we infected LNT-229P cells
with Ad-TET-IκBζ + Ad-ON and induced IκBζ expression by addition of
DOX. We used qPCR to measure cytokine mRNA expression as well as a
membrane based microarray assay that allows the detection of 48
different cyto- and chemokines in cellular supernatants. We found
that IL-6 and CXCL1 were upregulated after induction of IκBζ
expression both at the level of mRNA and protein, whereas IL-8
expression was only marginally enhanced (Fig. 4). To validate that IκBζ is an
inducer of inflammatory cytokine expression in glioma cells, we
downregulated IκBζ expression in irradiated LNT-229P cells by
transient transfection using three different IκBζ specific shRNA
plasmid constructs. We found IκBζ being downregulated to 29 (#32),
36 (#29) and 64% (#30) compared to the level of irradiated cells
transfected with an unspecific shRNA plasmid construct (scrambled
shRNA, Fig. 5A). By transfection
of the cells using the most efficient shRNA construct (#32), we
next analyzed whether downregulation of IκBζ expression leads to a
reduction in the expression of IL-6, IL-8 and CXCL1. Indeed,
paralleled by the knockdown of IκBζ, IL-6, IL-8 and CXCL1 mRNAs
were downregulated. Interestingly, even if IL-8 mRNA expression was
not significantly induced by the induction of IκBζ expression
(Fig. 4F), IL-8 mRNA was
significantly reduced in IκBζ knockdown LNT229P cells (Fig. 5C). Additionally, we analyzed the
concentration of IL-6 in supernatants of both, non- and irradiated
LNT-229P cells transfected with either scrambled or the
IκBζ-specific shRNA containing plasmids (#32). Fig. 5E demonstrates that decreased IL-6
mRNA expression in IκBζ knockdown cells is paralleled by the
reduction of IL-6 protein in the supernatants of these cells. Our
results indicate a direct role of IκBζ in the regulation of the
expression of IL-6, IL-8 and CXCL1.
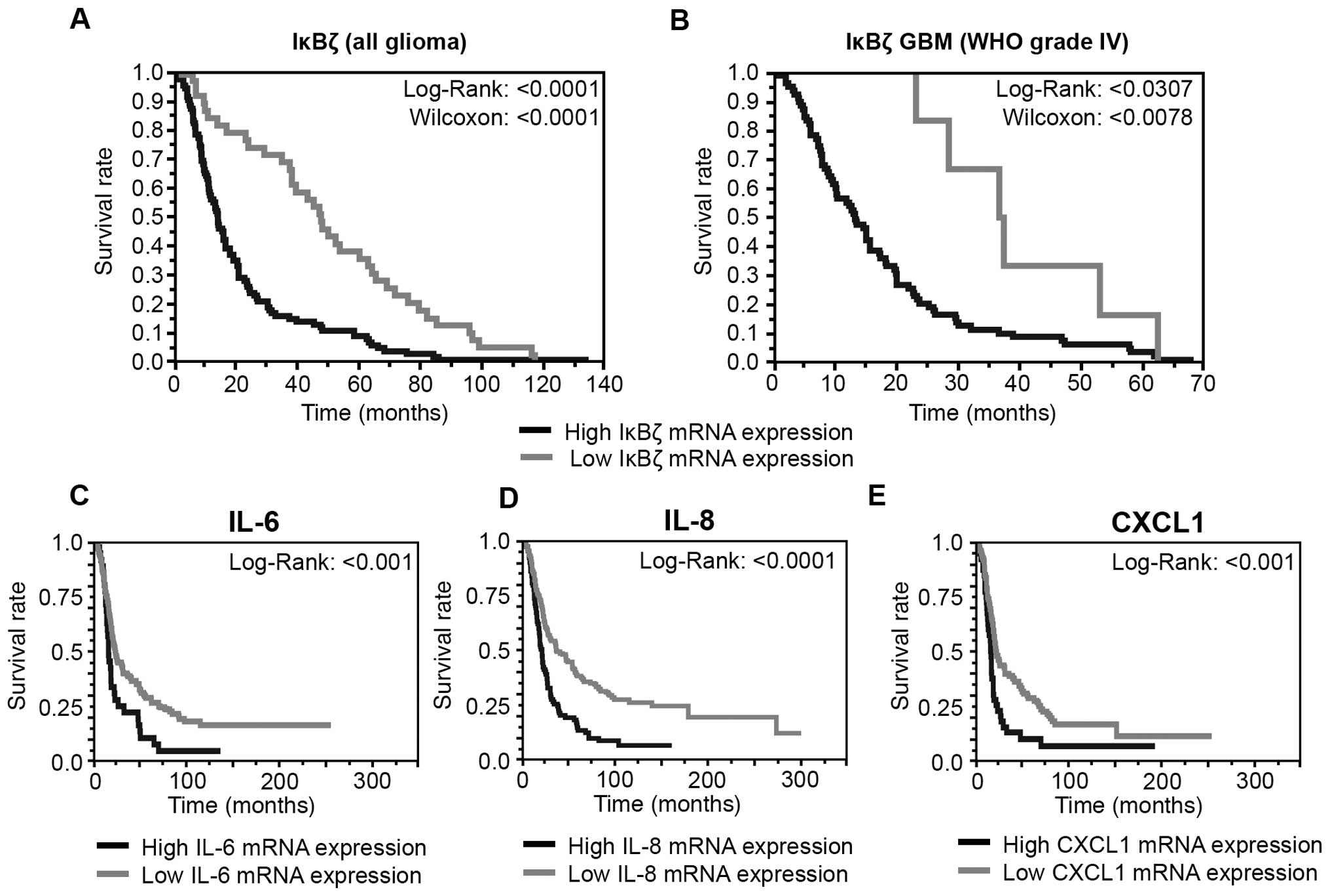
Expression of IκBζ and its target genes
IL-6, IL-8 and CXCL1 is correlated with a poor outcome of GBM
patients
To evaluate a potential association of IκBζ
expression, the expression of its inflammatory cytokine targets and
patients survival, we used the REMBRANDT database IκBζ expression
data and grouped either all glioma WHO grade II– IV (Fig. 6A) or GBM grade IV (Fig. 6B) with low or high IκBζ expression.
IκBζ up- or down-regulation was defined as a twofold or greater
difference from the mean expression level within a given dataset.
Both on the early and late phase of survival analysis, patients
with low IκBζ expression levels exhibit significantly longer
survival times (glioma: p<1×10−7; GBM
p<1×10−11). This analysis was repeated with a second
REMBRANDT dataset revealing similar results (data not shown). These
findings indicate a tumor promoting role of IκBζ. Similar survival
data were obtained for IL-6, IL-8 and CXCL1. Again, glioma patients
with lower expression of either IL-6, IL-8 or CXCL1 in the tumor
tissue significantly showed longer survival than patients with
higher cytokine expression (Fig.
6C–E).
Discussion
Patients with GBM currently undergo standard
treatment consisting of maximal surgical resection and combined
radiation and chemotherapy (28,29).
Radiation has been a mainstay of GBM treatment for decades. Besides
the therapeutic effect of radiation, side effects of this therapy
approach have also been described. These unwanted and more or less
tumor-driving side effects include radiation-induced glioma cell
migration and invasion (30–32),
modulation of NFκB activity, and, in this context, induction of
resistance towards radiation and conversion of GBM into its most
aggressive mesenchymal type subgroup (33,34).
Moreover, tumor irradiation can lead to cytokine and chemokine
expression such as vascular endothelial factor (VEGF), IL-8, IL-6,
CXCL1 and CCL2 (19,26,32,35)
in the tumor microenvironment, factors known to be involved in
glioma progression and enhancement of the malignancy.
Our study demonstrates that the expression of the
inflammatory and tumor promoting signaling molecules IL-6, IL-8 and
CXCL1 is regulated by the atypical IκB protein IκBζ, a direct
target of NFκB. Even if it was described that IκBζ inhibits
transactivation of p65 and its DNA binding in HEK293 cells
(15) this seems not to be the
case for glioma, since no alterations of NFκB activity was
detectable upon IκBζ over-expression in LNT-229P glioma cells
(Fig. 2D), indicating a more
direct effect of IκBζ on the expression of IL-6, IL-8 and CXCL1 and
confirming the data of Yamazaki et al who have shown that
IκBζ independent from NFκB, can directly serve as a transcription
modulator of inflammatory genes (17).
Using inducible overexpression and shRNA mediated
downregulation of IκBζ we were able to demonstrate that in glioma
cells IκBζ serves as a transcriptional modulator of the cytokines
IL-6, IL-8 and CXCL1. IL-6 is an inflammatory cytokine that is
produced by human GBM cells (36),
its expression is associated with the progression of glioma
malignancy, putatively via induction of signal transducer and
activator of transcription (STAT)-3 (37,38).
IL-8, also named CXCL8, is known to drive tumorigenicity. In
glioma, IL-8 is expressed both in vitro and in vivo.
IL-8 plays an important role in infection and inflammation
processes and it is known that its presence in the micro-milieu of
glial tumors is crucial in regard to their vascularization as well
as in the progression of these tumors (39). CXCL1, also known as the oncogene
GRO, binds to the G-protein coupled chemokine (C-X-C motif)
receptor 2. In embryogenesis, CXCL1 regulates spinal cord
development by blocking the motility of oligodendrocyte precursor
cells. In adults, this cytokine is involved in processes of
inflammation, angiogenesis, but also wound healing (40,41).
In glioma CXCL1 is highly expressed, and overexpression of CXCL1 in
glioma possesses tumor cell motility and confers increased
malignancy (42,43).
In recent centuries, and even after the development
of more efficient treatment strategies, the median survival of
glioblastoma patients has only marginally improved. In this regard,
novel therapeutic strategies are urgently needed to treat glioma.
There are suggestions that inhibition of chronic inflammation in
the tumor micro-milieu as well as reduction of glioma associated
angiogenesis by inhibiting the function of glioma secreted
cytokines might be successful strategies to treat glioblastoma. We
have shown in this study that in GBM cells IκBζ is a
transcriptional modulator of several pro-tumorigenic, inflammatory
or pro-angiogenic cytokines such as IL-6, IL-8 and CXCL1. Besides
its function as a transcriptional activator of inflammatory
cytokine expression, enhanced IκBζ has been demonstrated, via a
transcription-independent mechanism, to also protect GBM cells
towards necroptotic cell death, whereas knockdown of IκBζ in these
cells induces necroptosis and delays tumor growth in mice (44). Furthermore, it has been described
recently that IκBζ, at least in macrophages, regulates the
expression of the monocyte chemoattractant protein (MCP)-1
(14), a cytokine that is also
expressed by glioma cells, recruits microglia cells into and
promotes the aggressiveness of this tumor (45).
One could think that targeting IκBζ in the GBM
micro-milieu after or during radiation therapy might be an
interesting novel idea for the treatment of glioma patients.
However, one should also keep in mind that there might be
disadvantageous effects blocking IκBζ in glioma: IκBζ has been
described to regulate for the development of (glioma infiltrating)
T(H)17 cells (46). T(H)17 cells
are a subset of T cells important for the development of an
efficient attack of GBM cells by immune cells, but also a subset of
immune cells that are known to be inactivated by GBM released
factors (reviewed in ref. 47).
Therefore, a strategy targeting IκBζ in GBM cells exclusively might
be more feasible than inhibiting IκBζ activity in the complete GBM
micro-milieu.
Acknowledgements
We thank D.G. Hildebrand and K. Schulze-Osthoff for
providing us with IκBζ-specific shRNA containing plasmids. This
study was supported by the Interdisciplinary Center for Clinical
Research of the University of Tübingen.
Abbreviations:
|
CCL2
|
chemokine (C-C motif) ligand 2
|
|
CNS
|
central nervous system
|
|
CTS-1
|
chimeric tumor suppressor 1
|
|
CXCL1
|
chemokine (C-X-C motif) ligand 1
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
DOX
|
doxycycline
|
|
FCS
|
fetal calf serum
|
|
GBM
|
glioblastoma
|
|
IκBζ
|
inhibitor of nuclear factor kappa B
zeta
|
|
IL
|
interleukin
|
|
MCP-1
|
monocyte chemoattractant protein
|
|
NFκB
|
nuclear factor kappa B
|
|
rtTA
|
reverse tetracycline repressor
|
|
STAT
|
signal transducer and activator of
transcription
|
|
TNFα
|
tumor necrosis factor alpha
|
|
VEGF
|
vascular endothelial factor
|
References
|
1
|
Bours V, Bentires-Alj M, Hellin AC,
Viatour P, Robe P, Delhalle S, Benoit V and Merville MP: Nuclear
factor-kappa B, cancer, and apoptosis. Biochem Pharmacol.
60:1085–1089. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Perkins ND: The diverse and complex roles
of NF-κB subunits in cancer. Nat Rev Cancer. 12:121–132.
2012.PubMed/NCBI
|
|
3
|
Coupienne I, Bontems S, Dewaele M, Rubio
N, Habraken Y, Fulda S, Agostinis P and Piette J: NF-kappaB
inhibition improves the sensitivity of human glioblastoma cells to
5-aminolevulinic acid-based photodynamic therapy. Biochem
Pharmacol. 81:606–616. 2011. View Article : Google Scholar
|
|
4
|
Ding GR, Honda N, Nakahara T, Tian F,
Yoshida M, Hirose H and Miyakoshi J: Radiosensitization by
inhibition of IkappaB-alpha phosphorylation in human glioma cells.
Radiat Res. 160:232–237. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zanotto-Filho A, Braganhol E, Schröder R,
de Souza LH, Dalmolin RJ, Pasquali MA, Gelain DP, Battastini AM and
Moreira JC: NFκB inhibitors induce cell death in glioblastomas.
Biochem Pharmacol. 81:412–424. 2011. View Article : Google Scholar
|
|
6
|
Raychaudhuri B, Han Y, Lu T and Vogelbaum
MA: Aberrant constitutive activation of nuclear factor kappaB in
glioblastoma multiforme drives invasive phenotype. J Neurooncol.
85:39–47. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang X, Chen T, Zhang J, Mao Q, Li S,
Xiong W, Qiu Y, Xie Q and Ge J: Notch1 promotes glioma cell
migration and invasion by stimulating β-catenin and NF-κB signaling
via AKT activation. Cancer Sci. 103:181–190. 2012. View Article : Google Scholar
|
|
8
|
Xie TX, Xia Z, Zhang N, Gong W and Huang
S: Constitutive NF-kappaB activity regulates the expression of VEGF
and IL-8 and tumor angiogenesis of human glioblastoma. Oncol Rep.
23:725–732. 2010.PubMed/NCBI
|
|
9
|
Griffin BD and Moynagh PN: Persistent
interleukin-1beta signaling causes long term activation of NFkappaB
in a promoter-specific manner in human glial cells. J Biol Chem.
281:10316–10326. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Perkins ND and Gilmore TD: Good cop, bad
cop: The different faces of NF-kappaB. Cell Death Differ.
13:759–772. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ryan KM, Ernst MK, Rice NR and Vousden KH:
Role of NF-kappaB in p53-mediated programmed cell death. Nature.
404:892–897. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Seznec J, Weit S and Naumann U: Gene
expression profile in a glioma cell line resistant to cell death
induced by the chimeric tumor suppressor-1 (CTS-1), a
dominant-positive variant of p53 - the role of NFkappaB.
Carcinogenesis. 31:411–418. 2010. View Article : Google Scholar
|
|
13
|
Perkins ND: Integrating cell-signalling
pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol.
8:49–62. 2007. View
Article : Google Scholar
|
|
14
|
Hildebrand DG, Alexander E, Hörber S,
Lehle S, Obermayer K, Münck NA, Rothfuss O, Frick JS, Morimatsu M,
Schmitz I, et al: IκBζ is a transcriptional key regulator of
CCL2/MCP-1. J Immunol. 190:4812–4820. 2013.PubMed/NCBI
|
|
15
|
Totzke G, Essmann F, Pohlmann S,
Lindenblatt C, Jänicke RU and Schulze-Osthoff K: A novel member of
the IkappaB family, human IkappaB-zeta, inhibits transactivation of
p65 and its DNA binding. J Biol Chem. 281:12645–12654. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yamazaki S, Muta T and Takeshige K: A
novel IkappaB protein, IkappaB-zeta, induced by proinflammatory
stimuli, negatively regulates nuclear factor-kappaB in the nuclei.
J Biol Chem. 276:27657–27662. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yamazaki S, Matsuo S, Muta T, Yamamoto M,
Akira S and Takeshige K: Gene-specific requirement of a nuclear
protein, IkappaB-zeta, for promoter association of inflammatory
transcription regulators. J Biol Chem. 283:32404–32411. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Naumann U, Kügler S, Wolburg H, Wick W,
Rascher G, Schulz JB, Conseiller E, Bähr M and Weller M: Chimeric
tumor suppressor 1, a p53-derived chimeric tumor suppressor gene,
kills p53 mutant and p53 wild-type glioma cells in synergy with
irradiation and CD95 ligand. Cancer Res. 61:5833–5842.
2001.PubMed/NCBI
|
|
19
|
Alexander E, Hildebrand DG, Kriebs A,
Obermayer K, Manz M, Rothfuss O, Schulze-Osthoff K and Essmann F:
IκBζ is a regulator of the senescence-associated secretory
phenotype in DNA damage- and oncogene-induced senescence. J Cell
Sci. 126:3738–3745. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Krtolica A, Parrinello S, Lockett S,
Desprez PY and Campisi J: Senescent fibroblasts promote epithelial
cell growth and tumorigenesis: A link between cancer and aging.
Proc Natl Acad Sci USA. 98:12072–12077. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Conti A, Gulì C, La Torre D, Tomasello C,
Angileri FF and Aguennouz M: Role of inflammation and oxidative
stress mediators in gliomas. Cancers (Basel). 2:693–712. 2010.
View Article : Google Scholar
|
|
22
|
Siu A, Wind JJ, Iorgulescu JB, Chan TA,
Yamada Y and Sherman JH: Radiation necrosis following treatment of
high grade glioma - a review of the literature and current
understanding. Acta Neurochir (Wien). 154:191–201; discussion 201.
2012. View Article : Google Scholar
|
|
23
|
He TC, Zhou S, da Costa LT, Yu J, Kinzler
KW and Vogelstein B: A simplified system for generating recombinant
adenoviruses. Proc Natl Acad Sci USA. 95:2509–2514. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Grentzmann G, Ingram JA, Kelly PJ,
Gesteland RF and Atkins JF: A dual-luciferase reporter system for
studying recoding signals. RNA. 4:479–486. 1998.PubMed/NCBI
|
|
25
|
Tsuboi Y, Kurimoto M, Nagai S, Hayakawa Y,
Kamiyama H, Hayashi N, Kitajima I and Endo S: Induction of
autophagic cell death and radiosensitization by the pharmacological
inhibition of nuclear factor-kappa B activation in human glioma
cell lines. J Neurosurg. 110:594–604. 2009. View Article : Google Scholar
|
|
26
|
Pasi F, Facoetti A and Nano R: IL-8 and
IL-6 bystander signalling in human glioblastoma cells exposed to
gamma radiation. Anticancer Res. 30:2769–2772. 2010.PubMed/NCBI
|
|
27
|
Dubost JJ, Rolhion C, Tchirkov A, Bertrand
S, Chassagne J, Dosgilbert A and Verrelle P:
Interleukin-6-producing cells in a human glioblastoma cell line are
not affected by ionizing radiation. J Neurooncol. 56:29–34. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Furnari FB, Fenton T, Bachoo RM, Mukasa A,
Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al:
Malignant astrocytic glioma: Genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hegi ME, Diserens AC, Gorlia T, Hamou MF,
de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani
L, et al: MGMT gene silencing and benefit from temozolomide in
glioblastoma. N Engl J Med. 352:997–1003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wild-Bode C, Weller M, Rimner A, Dichgans
J and Wick W: Sublethal irradiation promotes migration and
invasiveness of glioma cells: Implications for radiotherapy of
human glioblastoma. Cancer Res. 61:2744–2750. 2001.PubMed/NCBI
|
|
31
|
Desmarais G, Fortin D, Bujold R, Wagner R,
Mathieu D and Paquette B: Infiltration of glioma cells in brain
parenchyma stimulated by radiation in the F98/Fischer rat model.
Int J Radiat Biol. 88:565–574. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kil WJ, Tofilon PJ and Camphausen K:
Post-radiation increase in VEGF enhances glioma cell motility in
vitro. Radiat Oncol. 7:252012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Criswell T, Leskov K, Miyamoto S, Luo G
and Boothman DA: Transcription factors activated in mammalian cells
after clinically relevant doses of ionizing radiation. Oncogene.
22:5813–5827. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bhat KP, Balasubramaniyan V, Vaillant B,
Ezhilarasan R, Hummelink K, Hollingsworth F, Wani K, Heathcock L,
James JD, Goodman LD, et al: Mesenchymal differentiation mediated
by NF-κB promotes radiation resistance in glioblastoma. Cancer
Cell. 24:331–346. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nalla AK, Gogineni VR, Gupta R, Dinh DH
and Rao JS: Suppression of uPA and uPAR blocks radiation-induced
MCP-1 mediated recruitment of endothelial cells in meningioma. Cell
Signal. 23:1299–1310. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Van Meir E, Sawamura Y, Diserens AC, Hamou
MF and de Tribolet N: Human glioblastoma cells release interleukin
6 in vivo and in vitro. Cancer Res. 50:6683–6688. 1990.PubMed/NCBI
|
|
37
|
Loeffler S, Fayard B, Weis J and
Weissenberger J: Interleukin-6 induces transcriptional activation
of vascular endothelial growth factor (VEGF) in astrocytes in vivo
and regulates VEGF promoter activity in glioblastoma cells via
direct interaction between STAT3 and Sp1. Int J Cancer.
115:202–213. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Weissenberger J, Loeffler S, Kappeler A,
Kopf M, Lukes A, Afanasieva TA, Aguzzi A and Weis J: IL-6 is
required for glioma development in a mouse model. Oncogene.
23:3308–3316. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Brat DJ, Bellail AC and Van Meir EG: The
role of interleukin-8 and its receptors in gliomagenesis and
tumoral angiogenesis. Neuro-oncol. 7:122–133. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tsai HH, Frost E, To V, Robinson S,
Ffrench-Constant C, Geertman R, Ransohoff RM and Miller RH: The
chemokine receptor CXCR2 controls positioning of oligodendrocyte
precursors in developing spinal cord by arresting their migration.
Cell. 110:373–383. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Devalaraja RM, Nanney LB, Du J, Qian Q, Yu
Y, Devalaraja MN and Richmond A: Delayed wound healing in CXCR2
knockout mice. J Invest Dermatol. 115:234–244. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou Y, Zhang J, Liu Q, Bell R, Muruve DA,
Forsyth P, Arcellana-Panlilio M, Robbins S and Yong VW: The
chemokine GRO-alpha (CXCL1) confers increased tumorigenicity to
glioma cells. Carcinogenesis. 26:2058–2068. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Robinson S, Cohen M, Prayson R, Ransohoff
RM, Tabrizi N and Miller RH: Constitutive expression of
growth-related oncogene and its receptor in oligodendrogliomas.
Neurosurgery. 48:864–873; discussion 873–874. 2001.PubMed/NCBI
|
|
44
|
Willems M, Kroonen J, Dubois N, Berendsen
S, Nguyen B, Bredel M, Artesi M, Kim H, Rados M, Chakravarti A, et
al: IkappaB zeta overexpression drives human glioma resistance to
necroptosis. Neuro-oncol. 16(Suppl 5): v58–v59. 2014. View Article : Google Scholar
|
|
45
|
Platten M, Kretz A, Naumann U, Aulwurm S,
Egashira K, Isenmann S and Weller M: Monocyte chemoattractant
protein-1 increases microglial infiltration and aggressiveness of
gliomas. Ann Neurol. 54:388–392. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Okamoto K, Iwai Y, Oh-Hora M, Yamamoto M,
Morio T, Aoki K, Ohya K, Jetten AM, Akira S, Muta T, et al:
IkappaBzeta regulates T(H)17 development by cooperating with ROR
nuclear receptors. Nature. 464:1381–1385. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Okada H and Khoury SJ: Type17 T-cells in
central nervous system autoimmunity and tumors. J Clin Immunol.
32:802–808. 2012. View Article : Google Scholar : PubMed/NCBI
|