Introduction
One of the many possible causes in the development
of cancer is the dysfunction in the regulation of apoptosis. This
mechanism usually serves as a means of eliminating damaged or
degenerated cells within the human body. If the tight regulation of
this balance between regeneration and elimination is
malfunctioning, the consequences are a loss of tissue or neoplasms.
A certain protein highly expressed in breast cancer cells leads to
a dysfunction in the regulation of apoptosis.
The transmembrane protein Lifeguard (LFG) was first
described by Somia et al in 1999. Its localization is
predicted to be in the endoplasmatic reticulum, or the plasma
membrane, with ubiquitous expression except for in the placenta and
pancreas. Lifeguard has been identified as an inhibitor of
Fas-induced apoptosis. Immunoprecipitation analysis showed its
interaction with Fas-receptor and the Fas-antibody, which mimics
the Fas-ligand, but not with the protein FADD (1). Still, the mechanism by which LFG
blocks the signal pathway that starts with the ligand-induced
clustering of Fas-receptors, and ends with the final activation of
caspase-8 and -3, is unknown (2,3). For
structures of the central nervous system in adults, this is an
advantage as the predicted function of the neural membrane protein
35, a structural homolog of LFG found in rats, provides protection
of tissues in these regions (4).
In other organs, however, the shift of the apoptotic homeostasis by
the expression of LFG can have fatal consequences. In previous
publications, it has been demonstrated that there is a correlation
between the expression level of LFG and the increasing degree of
malignity of breast cancer (5).
Furthermore, it was shown that the effect of the alkyl phospholipid
perifosine was reduced in cells expressing LFG (6).
Tripartite motif-containing 21 (TRIM21) is usually
associated with autoimmune diseases such as systemic lupus
erythematodes (SLE), although its role in the pathomechanism is not
yet revealed (7). The
physiological functions of TRIM21 are mainly attributed to the area
of the innate immune system concerning the defense against viral
pathogens but it is also associated with signaling pathways
concerning for example cell division by inhibiting the activation
of the transcription factor NF-κB (8,9).
This transcription factor family is significantly involved in cell
proliferation and has therefore been associated with different
kinds of cancer where it favors fast tumor growth and development
of metastasis (10).
Materials and methods
Cell lines and culture condition
The human breast carcinoma cell line MDA-MB-231 was
obtained from the American Type Culture Collection (ATCC,
Rockville, MD, USA) and grown in Dulbecco's modified Eagle's medium
(DMEM; PAA, Cölbe, Germany) supplemented with 10% FCS (Biochrom,
Berlin, Germany) and 50 mg/ml penicillin-streptomycin. Cultures
were maintained at 37°C in a humidified atmosphere with 5%
CO2. The cells were subcultured every 2 to 3 days by
treatment with a 0.25% Trypsin/0.53 mM ethylenediamine-tetraacetic
acid (EDTA) solution.
Protein-array
The ProtoArray Human Protein Microarray v4.0
containing 8,268 human proteins, reagents and other equipment were
from Invitrogen (Carlsbad, CA, USA). Assays including appropriate
controls were performed according to the instructions of the
manufacturer. In short, protein microarray slides were blocked with
phosphate-buffered saline (PBS) containing 1% bovine serum albumin
(BSA) and 0.1% Tween-20 before incubation with 10 μM of recombinant
LFG-Protein (1:500; Abnova, Taipei, Taiwan) and Alexa Fluor
647-conjugated antihuman IgG (1.0 μg/ml buffer). The arrays were
dried and scanned using an Axon GenePix 400B fluorescent microarray
scanner. GenePix 6.0 software was used to align the scanned image
to the template as well as to determine the pixel intensities for
each spot on the array. The reported pixel intensity was calculated
as the average of duplicate signals after background subtraction.
Prospector software (Invitrogen), which is based on M-statistics
were used for statistical analysis of the microarray data.
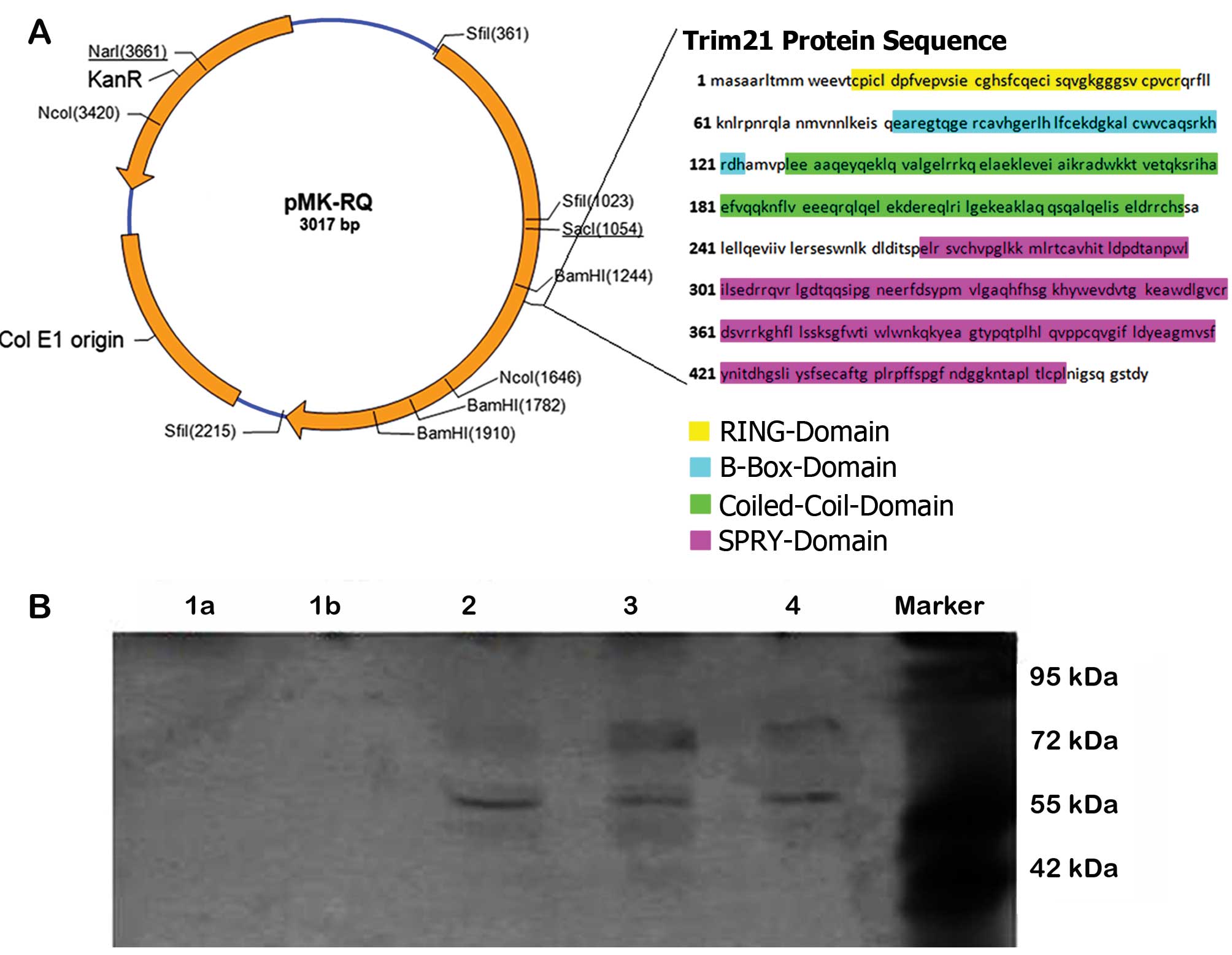
TRIM21 vectors
To identify the essential domain for the interaction
between TRIM21 and LFG, different vectors, using pCMV6-AC-GFP
vector (NM_003141; OriGene, Cambridge, UK) were cloned containing
the sequences of TRIM21 with the deletion of an entire domain. The
vector map as well as the sequences of the individual TRIM21
domains are shown in Fig. 3.
Co-immunoprecipitation
MDA-MB-231 were seeded in 100 mm culture plates and
transfected with vectors coding for proteins LFG (pTriEx-1.1-FAIM2,
NM-0112306; GeneArt, Regensburg, DE, USA) and TRIM21
(pCMV6-AC-GFP-Trim21, NM_003141; OriGene) by using FuGENE 6
(Promega, Madison, WI, USA) according to manufacturer's
instructions. The implementation of the co-immunoprecipitation was
performed according to the protocol of the Co-Immunoprecipitation
kit of Dynabeads, Inc. A total of 42 μg LFG antibody were added to
6 mg beads and shaken overnight at 37°C. After several washes with
the extraction and LWF buffer of Dynabeads, Inc., 100 μl of the LFG
antibody coupled beads suspension were transferred to 50 μl of one
cell lysate, containing one TRIM21 protein with a deletion of one
domain. These approaches had to be incubated overnight at 4°C. The
associated proteins were added in 800 μl SB buffer and subsequently
used in western blot analysis.
Co-localization
For every glass slide, 5×104 MDA-MB-231
were cultivated and transfected with vectors coding for the
proteins LFG and TRIM21 for 24 h after which time the cells were
fixed in 4% paraformaldehyde for 20 min and permeabilized with 0.1%
Triton X-100 (Sigma-Aldrich, Steinheim, Germany) for 4 min. Slides
were then incubated with rabbit polyclonal anti-hLFG antibody
(1:100 dilution) and goat polyclonal anti-hTrim21 (Santa Cruz
Biotechnology, Inc.) antibodies at 37°C for 1 h, washed three times
in ice-cold PBS, and incubated with Alexa Fluor 488 and Alexa Fluor
680 conjugated goat anti-rabbit and chicken anti-goat secondary
antibody (each in 1:600 dilution; Invitrogen) at 37°C for 30 min.
After three washing steps with PBS, the sample was dried and
covered with Vectashield mounting medium (Vector Laboratories,
Burlingame, CA, USA), an antifade reagent containing the
DNA-staining dye 4′,6′-diamidino-2-phenylindole (DAPI). Images were
attained using Zeiss Axiovert 200M fluorescence microscope,
equipped with the appropriate barrier filters.
Small interfering RNA
MDA MB231 cells were transfected with siRNA LFG-650
5′-cctcctacccttccaatatgt-3 (Sirion, Munich, Germany) and the
appropriate control vector. The algorithm used by Sirion for the
siRNA design was optimized for maximum gene specificity, and KD
efficiency. Subsequent virus rescue and production were carried out
in HEK 293 cells. Virus purification was performed using the
ViraBind™ Adenovirus Miniprep kit (Cell Biolabs, Inc., San Diego,
CA, USA). The cells were seeded at 2×104
cells/cm2 and incubated at 37°C in a humidified
atmosphere with 5% CO2 for 48 h before being
analyzed.
Western blot analysis
Each sample (20 μl) was dissolved in 7.5 μl loading
buffer. The samples were applied with a protein marker on a native
polyacrylamide gel, which had been equilibrated in a forerun with
0,5× TBE as running buffer for 30 min at 25 mA. The subsequent run
with the samples was at 25 mA for 3 h. The blotting onto a PVDF
membrane (Millipore Corp., Bedford, MA, USA), which had been
previously activated in 99% ethanol, was carried out overnight at
40 V and 80 mA. Immunoblotting was performed with polyclonal
antibodies: anti-FAIM2 (1:200 dilution), anti-Trim21 (1:200
dilution) (both from Santa Cruz Biotechnology, Inc.) and anti-actin
(1:200 dilution; Abcam, Cambridge, UK). As a secondary antibody,
Odyssey 600 anti-rabbit and Odyssey 800 anti-goat (Invitrogen) were
used for the quantification of protein expression levels, and
signals were obtained using the Odyssey Infrared Imaging System and
software (Li-Cor Biosciences, Lincoln, NE, USA).
RT-PCR analysis
Total RNA was extracted using the NucleoSpin RNA II
kit (MN Macherey-Nagel, Düren, Germany), and then 1 μg of RNA was
reverse transcribed into cDNA and amplified using the iScript™ cDNA
kit (Bio-Rad Laboratories, Hercules, CA, USA). The following
reverse (R) and forward (F) primers were used: trim21-F,
5′-gaccatggctccctcatcta-3′ and trim21-R, 5′-agggttagaggggcgtgtt-3′;
β2-microglobulin-F, 5′-atgagtatgcctgccgtgtga-3′ and
β2-microglobulin-R, 5′-ggcatcttcaaacctccatg-3′.
Real-time polymerase chain reaction (PCR) was
carried out in 20 μl samples with 5 ng cDNA, 1 mM of each forward
and reverse primer and a 2× SYBR-Green Sensi-Mix DNA kit (Quantace,
London, UK). Relative gene expression was determined by
normalization of the fluorescence intensity to β2-microglobulin
gene expression. Amplification cycles were as follows: 35 cycles at
95°C for 10 sec, 60°C or 30 sec, 70°C+0.2°C for 15 min.
Caspase assay
Activation of caspase-3/7 was determined using the
Apo-One homogeneous caspase-3/7 assay (Promega) following the
protocol provided by the manufacturer. Briefly, 1×104
MDA-MB-231 breast cancer cells were seeded per well of a 96-well
plate, and incubated with 1 μg TRIM21 human recombinant protein
(ProSpec; Ness Ziona, Tel Aviv, Israel) every 24 h. After 48 h, 100
ng of agonistic anti-Fas (clone CH11; Abcam) were added and after
24 h, incubated cells were mixed with 100 μl of Apo-One homogeneous
caspase-3/7 reagent. Following incubation at room temperature for 2
h, caspase-3/7 activation was estimated from sample fluorescence at
the excitation wavelength of 485 nm and an emission wavelength of
530 nm using a fluorescence plate reader Tecan GENios (Tecan
Schweiz AB, Zurich, Switzerland).
Cell cycle analysis by flow
cytometry
For distinct cell cycle phase distribution,
~106 MDA-MB-231 breast cancer cells were analysed. The
cells were cultivated by the addition of different concentrations
of TRIM21 as human recombinant protein within culture medium. Thus,
the cells were harvested and fixed in 70% (v/v) ice-cold ethanol
and kept at 4°C for 24 h. After washing, cells were resuspended in
1 ml PBS containing 10 μg/ml RNase and 1 mg/ml propidium iodide
(PI; Sigma-Aldrich); they were then incubated for 1 h at 37°C in
the dark. Thereafter, cells were analysed on a FACSCalibur flow
cytometer (Becton-Dickinson, San Jose, CA, USA).
NF-κB-array
cDNA plate array analysis is a plate-based
hybridization profiling technique that is used for monitoring the
expression of dozens of genes through reverse transcription of mRNA
into cDNA. MDA-MB-231 were cultivated for 48 h with the addition of
2.0 μg human recombinant Trim21 every 24 h. The control was
cultivated without supplements. For this analysis, total RNA was
isolated from the cell samples, using a NucleoSpin RNA II kit (MN
Macherey-Nagel). RNA samples (8 μg) were analyzed by microarray
analysis using an NF-κB pathway-regulated cDNA plate array
(Signosis, Sunnyvale, CA, USA) according to the manufacturer's
instructions. Each well on the plate contained a cDNA probe for one
of the 24 NF-κB pathway-regulated genes. After reverse
transcription, in situ hybridization, blocking, and
extensive washing, the wells were incubated with streptavidin-HRP,
and the resulting chemiluminescence was measured within 5 min using
a luminometer (Tecan Schweiz AB). As a gene of reference, β-actin
was chosen and the calculations were carried out using
qbase+ software (Biogazelle).
Immunohistochemistry
Breast tissue slides (US Biomax, Inc., Rockville,
MD, USA) were deparaffinized in xylene, and transferred through two
changes of 100% ethanol. For antigen retrieval, the slides were
pressure cooked in 6.5 mM sodium citrate (pH 6.0). To reduce
non-specific background staining, slides were incubated for 30 min
in 0.3% BSA/1× Tris-buffered saline. Slides were incubated at 4°C
overnight with hTrim21 goat primary antibodies (1:100 dilution)
(Santa Cruz Biotechnology, Inc.) and mouse primary cdc6 antibodies
(1:100) (MoBiTec GmbH, Göttingen, Germany). The slides were washed
twice for 5 min with PBS, and incubated for 30 min with goat
anti-rabbit Li-Cor-680 and anti-mouse Li-Cor-800CW conjugated
secondary antibody. Signals were detected by using the Li-Cor
Infra-Red imaging system.
Results
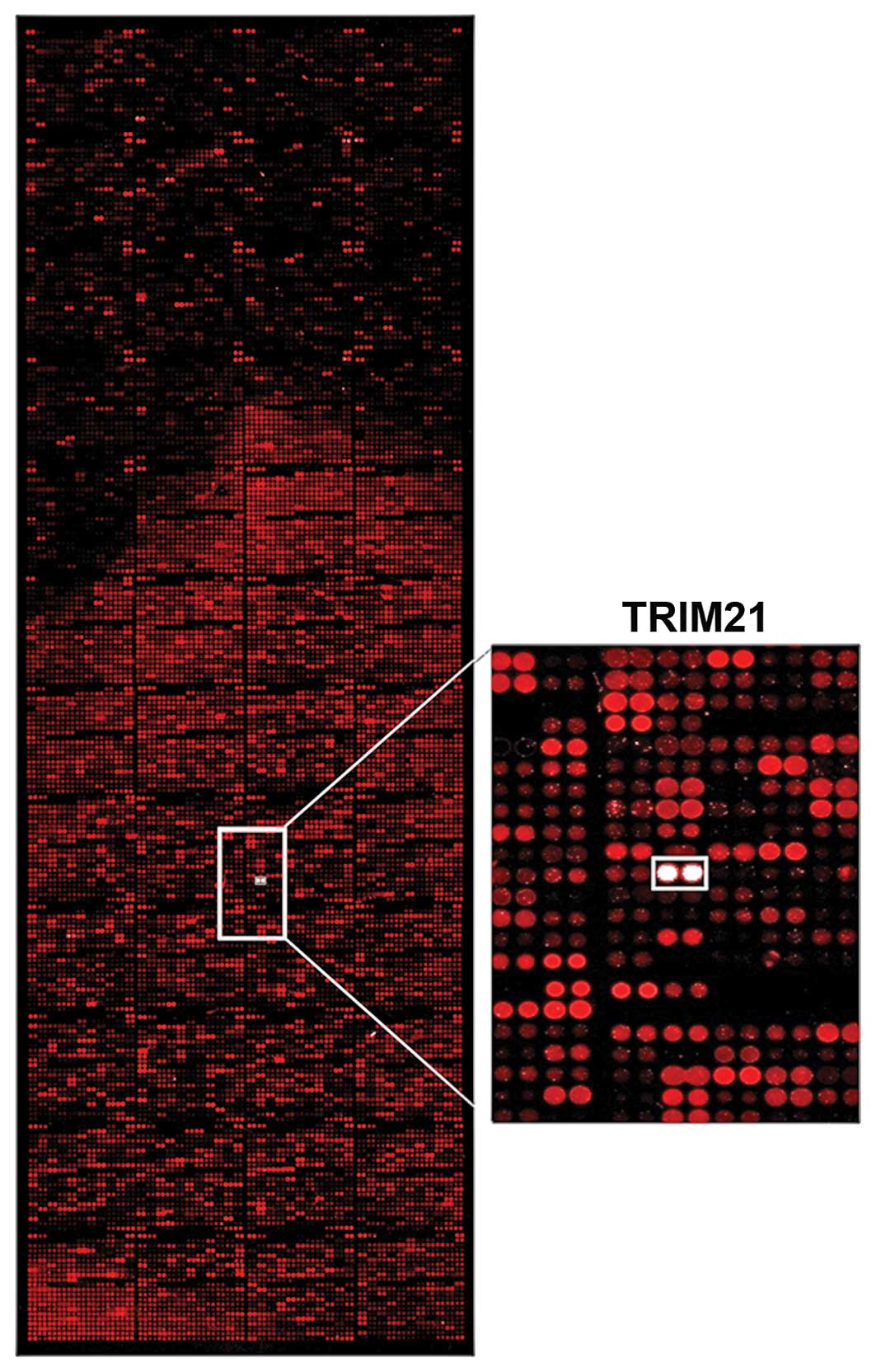
Analyses of interaction
To find a protein that could be used to target LFG,
>9,000 proteins were tested for their interaction in an
array-analysis (Fig. 1). The
protein-array was incubated with the human recombinant protein LFG
which was detected by fluorescence labeled antibodies. The signal
intensity was therefore proportional to the concentration of bound
LFG-protein. The signal of the interaction of LFG with TRIM21
clearly stands out as a white signal, as seen in the detailed
display in Fig. 1. The Z-score
confirms that the interaction of TRIM21 is around 2.5 times
stronger than the second strongest signal (Table I). To analyze if the interaction
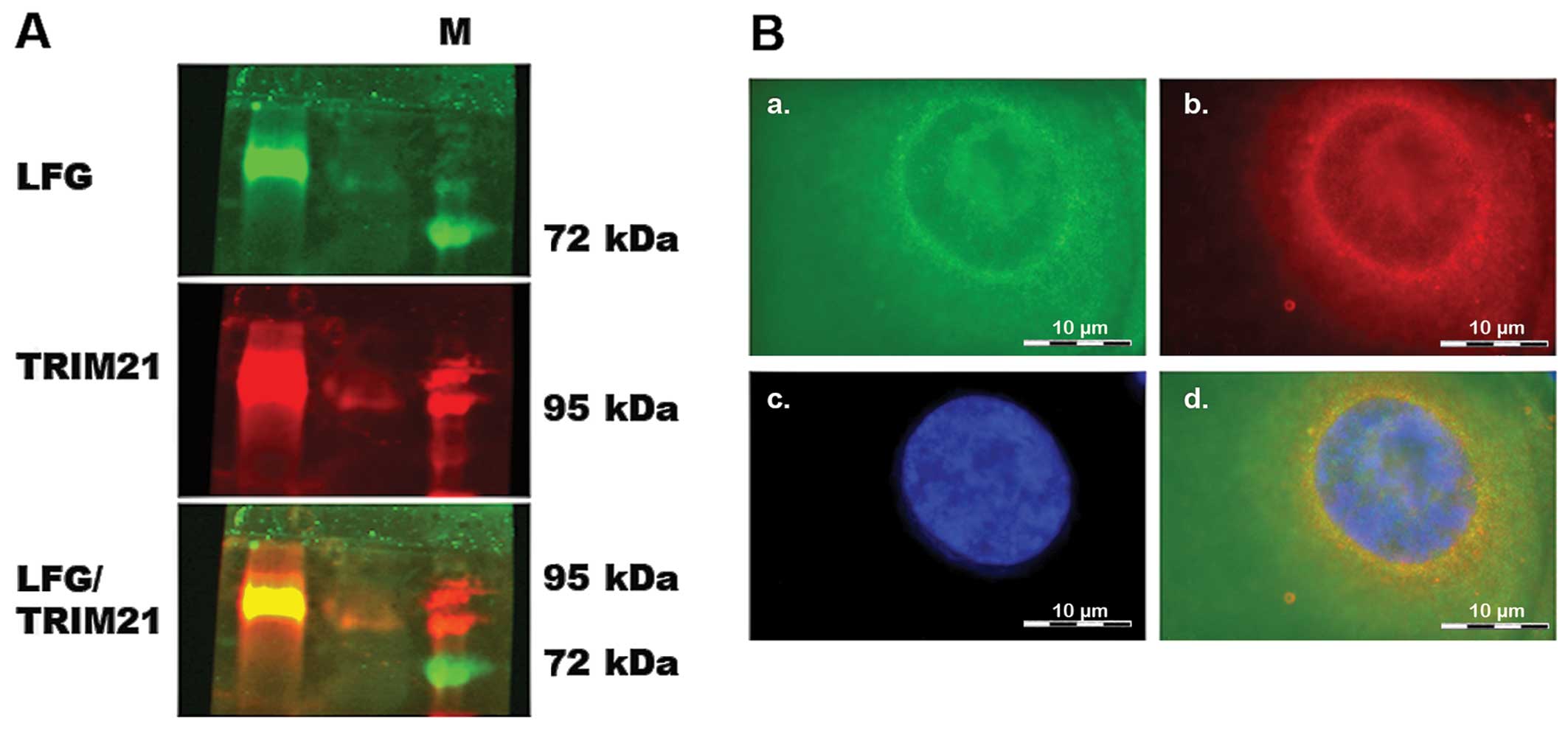
occured within the environment of a living cell as well, a
co-immunoprecipitation was carried out using MDA-MB-231, which had
been transfected with vectors coding for the proteins LFG and
TRIM21 (Fig. 3) to achieve a
clearer result. After cell disruption, LFG was isolated from the
lysate using magnetic beads coated with the specific LFG antibody.
The protein eluted from the beads was analyzed in a native PAGE
followed by western blot analysis. Detection by fluorescence
labeled antibodies revealed a green signal for LFG as well as a red
signal for TRIM21 (Fig. 2A). The
yellow signal visible in the overlay of the single signals shows
that both proteins can be found in the same position on the
blot-membrane, although not being of the same molecular weight.
Furthermore, the LFG and TRIM21 antibodies were used to detect the
proteins within intact, but PFA-fixated MDA-MB-231, that were
previously transfected with vectors coding for the two proteins
(Fig. 2B). In this fluorescence
microscope image, the green signals of LFG are visible
predominantly in close proximity to the cell core (Fig. 2B-a–c), very similar to the ring of
red labeled TRIM21 (Fig. 2B-b and
c). The core was stained with DAPI and therefore is visible as
a blue structure (Fig. 2B-c). In
the overlay, the yellow signal shows the positions in which both
proteins are localized in the same place. These combined signals
also display a ring around the core with fewer signals within the
cytoplasm.
 | Table IZ-score of protein-protein interaction
array-analysis. |
Table I
Z-score of protein-protein interaction
array-analysis.
| Z-factor | Z-score | CI P-value | CV | Significance
call | GenePix Flags | Description |
|---|
| 0.87066 | 3.585.224 | 0.0000008 | 0.04300 | Hit | 0 | Tripartite
motif-containing 21 (TRIM21) |
| 0.87066 | 3.369.140 | 0.0000009 | 0.04300 | Hit | 0 | Tripartite
motif-containing 21 (TRIM21) |
| 0.98147 | 1.299.203 | 0.0000059 | 0.00512 | Hit | 0 | General transcription
factor II-I |
| 0.98147 | 1.289.273 | 0.0000059 | 0.00512 | Hit | 0 | General transcription
factor II-I |
| 0.79642 | 1.071.535 | 0.0000084 | 0.06428 | Hit | 0 | Potassium
voltage-gated channel, shaker-related subfamily, β member 1
(KCNAB1), transcript variant 1 |
| 0.87427 | 1.045.647 | 0.0000088 | 0.03992 | Hit | 0 | Similar to FRG1
protein (FSHD region gene 1 protein) (MGC72104) |
| 0.53896 | 1.044.110 | 0.0000088 | 0.15190 | Hit | 0 | Tyrosine
3-monooxygenase/tryptophan 5-monooxygenase activation protein, ζ
polypeptide (YWHAZ), transcript variant 1 |
| 0.78119 | 1.018.105 | 0.0000093 | 0.07059 | Hit | 0 | 14-3-3 protein
ζ/δ |
| 0.04732 | 986.543 | 0.0000098 | 0.31200 | Hit | 0 | Nudix (nucleoside
diphosphate linked moiety X)-type motif 16-like 1 (NUDT16L1) |
| 0.87427 | 984.061 | 0.0000099 | 0.03992 | Hit | 0 | Similar to FRG1
protein (FSHD region gene 1 protein) (MGC72104) |
| 0.79642 | 971.767 | 0.0000101 | 0.06428 | Hit | 0 | Potassium
voltage-gated channel, shaker-related subfamily, β member 1
(KCNAB1), transcript variant 1 |
| 0.78119 | 914.082 | 0.0000113 | 0.07059 | Hit | 0 | 14-3-3 protein
ζ/δ |
| 0.53896 | 826.844 | 0.0000136 | 0.15190 | Hit | 0 | Tyrosine
3-monooxygenase/tryptophan 5-monooxygenase activation protein, ζ
polypeptide (YWHAZ), transcript variant 1 |
| 0.74035 | 809.350 | 0.0000142 | 0.08360 | Hit | 0 | Potassium
voltage-gated channel, shaker-related subfamily, β member 2
(KCNAB2), transcript variant 1 |
| 0.63579 | 789.136 | 0.0000148 | 0.11980 | Hit | 0 | Small nuclear
ribonucleoprotein polypeptide C (SNRPC) |
| 0.47616 | 783.226 | 0.0000150 | 0.17161 | Hit | 0 | WW domain containing
transcription regulator 1 (WWTR1) |
| 0.74035 | 710.528 | 0.0000180 | 0.08360 | Hit | 0 | Potassium
voltage-gated channel, shaker-related subfamily, β member 2
(KCNAB2), transcript variant 1 |
| 0.63579 | 654.025 | 0.0000208 | 0.11980 | Hit | 0 | Small nuclear
ribonucleoprotein polypeptide C (SNRPC) |
| 0.43427 | 651.306 | 0.0000210 | 0.18583 | Hit | 0 | UBX domain containing
3 (UBXD3) |
Co-immunoprecipitation
The co-immunoprecipitation was performed to
investigate a potential complex formation. The samples represent
cell lysates from cells expressing the modified TRIM21 proteins as
a result of transfection. To illustrate the bond between LFG and
the different TRIM21 proteins, they had to be extracted by the
co-immunoprecipitation in a pure form and without foreign protein
interaction. By running a western blot analysis with fluorescent
antibodies, the complex could be detected afterwards. A lack of a
complex formation can therefore be assumed as a lack of the
essential domain, whereby the interaction-depended domain can be
identified. The samples 2–4 show clearly visible bands of ~80 and
60 kDa. The bands at 80 kDa are larger but slightly less visible
than the bands at the height of 60 kDa. In the samples 1a and 1b no
bands are visible (Fig. 3B).
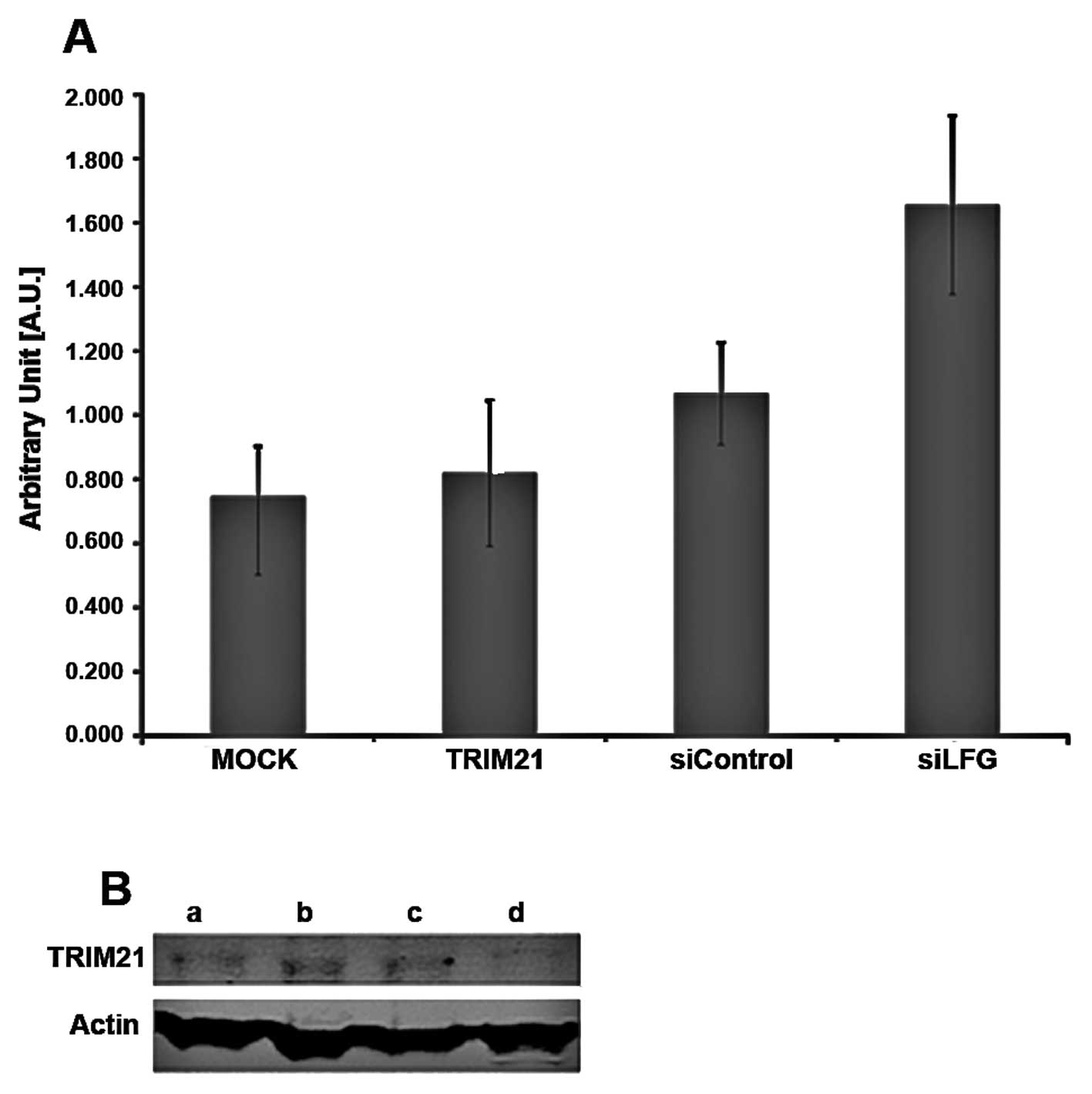
Analysis of expression
To investigate whether these two proteins influence
the expression of each other, MDA-MB-231 cells were cultivated
under different conditions. The expression of TRIM21 RNA-level was
detected by real-time PCR using 1.0 μg total RNA for transcription
to cDNA (Fig. 4A) and the amounts
of LFG and TRIM21 protein were detected using PAGE followed by
western blot analysis, using 25 μg total protein (Fig. 4B). The samples were treated
identically for both analyses. For siLFG (Fig. 4A), MDA-MB-231 were transfected with
an adenoviral vector coding for siRNA against LFG, and an empty
vector was used for transfection in sample siControl. Untreated
cells were used for MOCK, while cells of the sample TRIM21 were
cultivated with culture medium containing TRIM21-recombinant
protein. Samples MOCK, TRIM21 and siControl only differed slightly
in RNA expression levels for TRIM21 (Fig. 4A). A distinct difference can be
seen between the samples, MOCK showing the weakest, and siLFG
producing the strongest signal. The order of increasing signal
intensity of TRIM21 is MOCK, TRIM21, siControl and siLFG.
The detection of the proteins LFG, TRIM21 and actin
after western blot analysis was carried out using fluorescence
labeled antibodies. The protein actin was used as a housekeeping
gene and showed comparable band intensities throughout the
different samples (Fig. 4B). In
the western blot analysis, the band intensity of TRIM21 in the gel
differed only slightly between the samples a, siLFG; b, siControl;
and c, MOCK whereas the band of sample d, TRIM21, is weaker. The
bands of the protein LFG are strongest in samples b, siControl and
c, MOCK and less in a, siLFG. The weakest band, however, results
from sample d, TRIM21. To analyze if the apoptotic protection
provided by LFG is influenced by TRIM21, MDA-MB-231 cell were
cultivated with medium containing TRIM21 recombinant protein, and
with medium not containing the protein for 48 h. After the addition
of the agonistic anti-Fas-antibody, and further incubation for 24
h, the caspase-activity was tested using Apo-One (Promega). As the
caspases cleave the substrate the degradation product is detectable
by emitted fluorescence. Therefore, the signal intensity is
proportional to the activity of the caspase-3 in the sample. The
results show a much higher caspase-3 activity in the samples
cultivated in the presence of TRIM21 recombinant protein (Fig. 5A). The signal intensity of the
Control is reduced by approximately half compared to the sample
with TRIM21 recombinant protein.
It was necessary that yet another influence on
MDA-MB-231 cells by TRIM21 should be analyzed in a cell cycle
analysis. Therefore, a defined amount of cells were cultivated in
the presence of different concentrations of TRIM21-recombinant
protein within the culture medium for 24 h. Subsequently, the cells
were separated from the culture flasks and fixed with 70% ethanol
and stained with PI. The analysis was carried out using flow
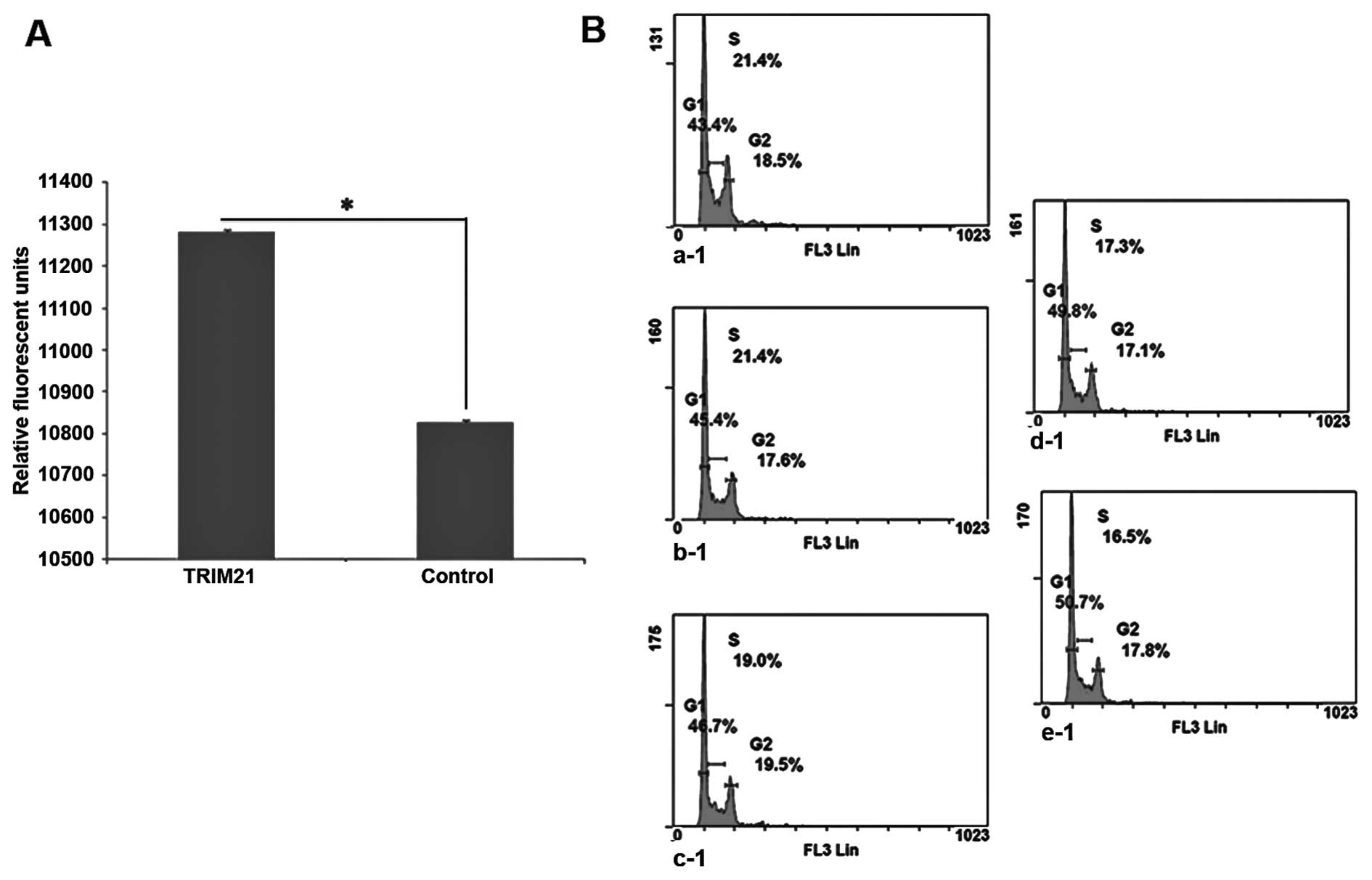
cytometry. On the basis of the plots in Fig. 5B a steady increase in the amount of
cells in the G0/G1-phase can be seen, whereas, the percentage of
cells in S-phase is steadily decreasing.
TRIM21-expression and its influence on
cancer cells
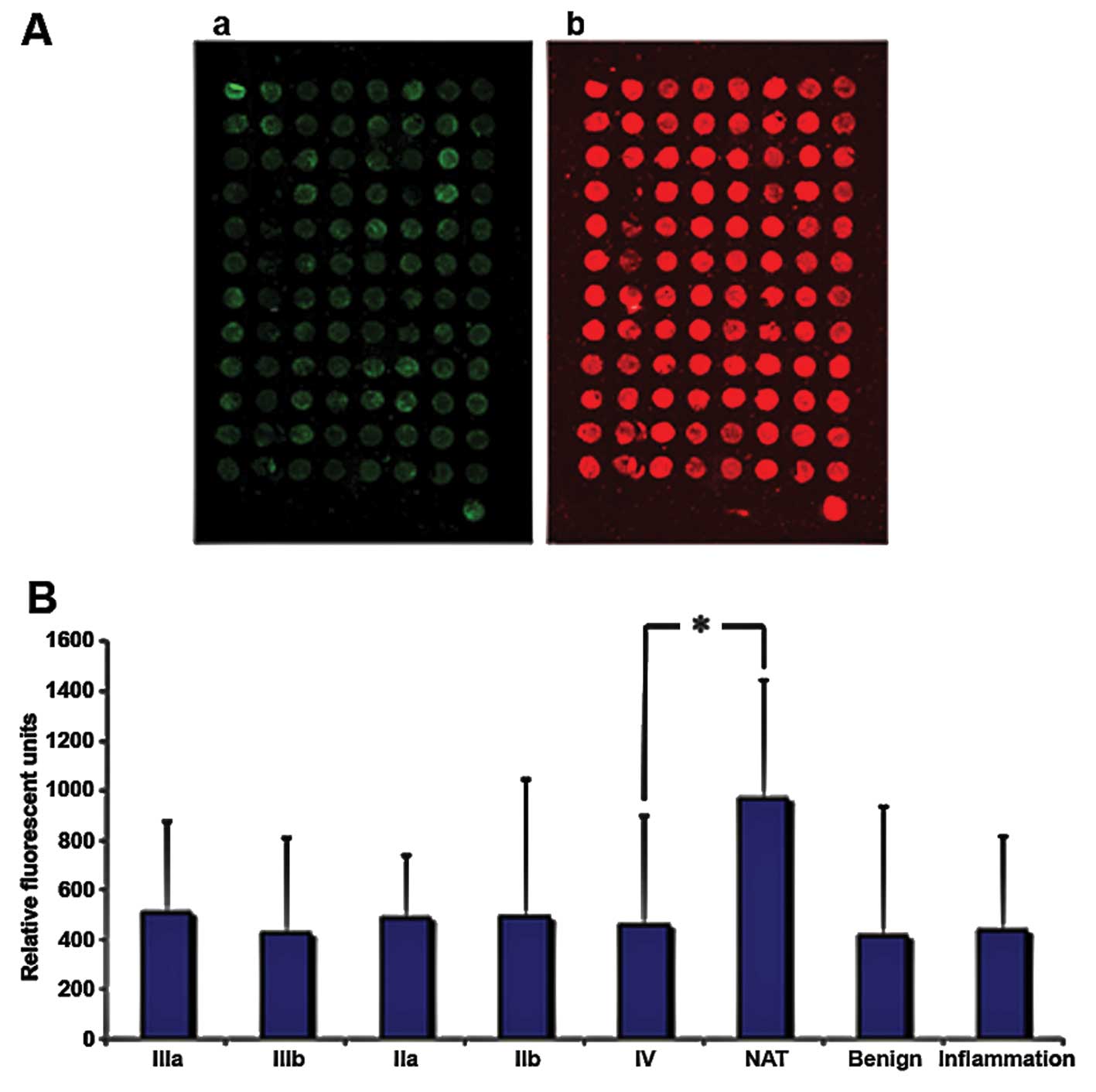
To examine how TRIM21 is expressed in breast cancer
tissue, samples from tumors with different degrees of malignity
were obtained from US Biomax, Inc. (Table II) and the protein TRIM21 was
detected via fluorescence labeled antibodies. The samples were also
stained with the nucleic acid stain Syto-60 as a reference. The
expression of TRIM21 does not differ significantly throughout the
varying degrees of malignity, but all samples derived from tumors
or inflamed tissue show approximately half the signal intensity
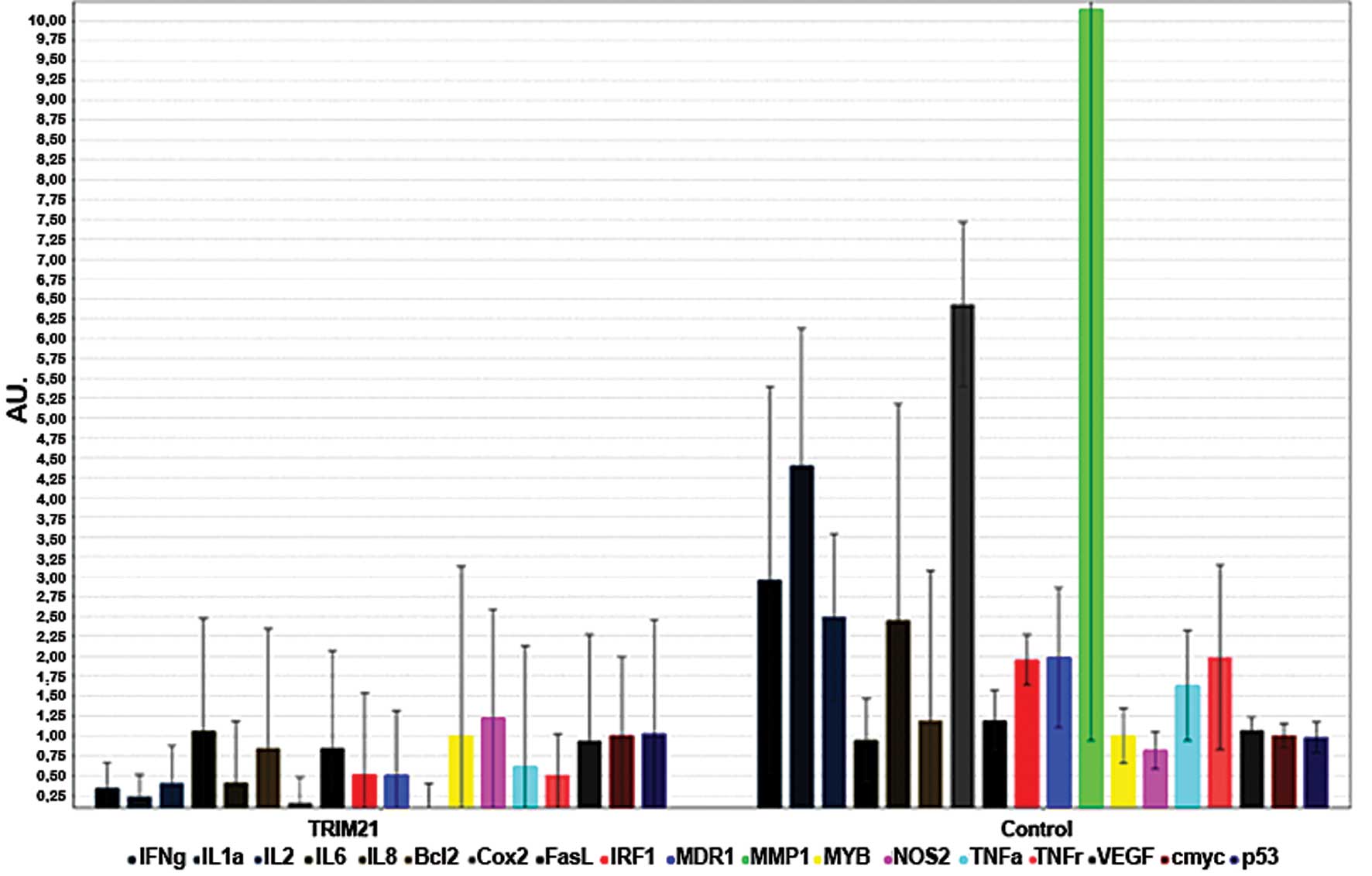
compared to the native tissue sample (Fig. 6). To analyze if TRIM21 protein, has
the same inhibiting effect of activation when added to the medium
as NF-κB in breast cancer cells, as shown by Niida et al
(9), a real-time PCR was carried
out using MDA-MB-231 cells cultivated with TRIM21 recombinant
protein and no further supplements for the negative control. The
expression of the most frequently NF-κB-regulated genes were
detected, and an overall decrease can be seen in Fig. 7.
| Table IISpecification Sheet of breast cancer
tissue array-analysis. |
Table II
Specification Sheet of breast cancer
tissue array-analysis.
| Pos | No. | Gender | Age (years) | Organ | Pathology
diagnosis | Grade | Stage | TNM | ER | PR | HER2 | Typea |
|---|
| A1 | 1 | F | 42 | Breast | Normal breast
tissue (hyalinosis) | - | - | - | N/A | - | N/A | NAT |
| A2 | 2 | F | 42 | Breast | Normal breast
tissue | - | - | - | - | - | 0 | NAT |
| A3 | 3 | F | 28 | Breast | Plasma cell
mastitis | - | - | - | - | - | 0 | Inflammation |
| A4 | 4 | F | 28 | Breast | Plasma cell
mastitis | - | - | - | - | - | 0 | Inflammation |
| A5 | 5 | F | 32 | Breast | Plasma cell
mastitis | - | - | - | +, 4% | +, 5% | 0 | Inflammation |
| A6 | 6 | F | 32 | Breast | Plasma cell
mastitis | - | - | - | - | - | 0 | Inflammation |
| A7 | 7 | F | 35 | Breast | Plasma cell
mastitis | - | - | - | - | - | 0 | Inflammation |
| A8 | 8 | F | 35 | Breast | Plasma cell
mastitis | - | - | - | - | - | 0 | Inflammation |
| A9 | 9 | F | 28 | Breast | Adenosis | - | - | - | +, 40% | +, 40% | 0 | Benign |
| A10 | 10 | F | 28 | Breast | Adenosis | - | - | - | - | - | 0 | Benign |
| A11 | 11 | F | 49 | Breast | Adenosis | - | - | - | ++, 30% | +, 35% | 0 | Benign |
| A12 | 12 | F | 49 | Breast | Adenosis | - | - | - | ++, 88% | 0,0085 | 0 | Benign |
| B1 | 13 | F | 44 | Breast | Adenosis | - | - | - | ++, 8% | ++, 70% | 1+ | Benign |
| B2 | 14 | F | 44 | Breast | Adenosis | - | - | - | +, 5% | ++, 3% | 0 | Benign |
| B3 | 15 | F | 58 | Breast | Fibroadenoma | - | - | - | - | - | 0 | Benign |
| B4 | 16 | F | 58 | Breast | Fibroadenoma | - | - | - | - | - | 0 | Benign |
| B5 | 17 | F | 22 | Breast | Fibroadenoma | - | - | - | ++, 10% | ++, 10% | 0 | Benign |
| B6 | 18 | F | 22 | Breast | Fibroadenoma | - | - | - | 0,0096 | +++, 70% | 0 | Benign |
| B7 | 19 | F | 54 | Breast | Fibroadenoma | - | - | - | - | - | 0 | Benign |
| B8 | 20 | F | 54 | Breast | Fibroadenoma | - | - | - | - | - | 0 | Benign |
| B9 | 21 | F | 34 | Breast | Invasive ductal
carcinoma | 1 | IIb | T2N1M0 | - | - | 1+ | Malignant |
| B10 | 22 | F | 34 | Breast | Invasive ductal
carcinoma | 1 | IIb | T2N1M0 | - | - | 1+ | Malignant |
| B11 | 23 | F | 43 | Breast | Invasive ductal
carcinoma | 1 | IIb | T3N0M0 | - | - | 2+ | Malignant |
| B12 | 24 | F | 43 | Breast | Invasive ductal
carcinoma | 1 | IIb | T3N0M0 | - | - | 2+ | Malignant |
| C1 | 25 | F | 40 | Breast | Invasive ductal
carcinoma | 1 | IIb | T2N1M0 | N/A | N/A | N/A | Malignant |
| C2 | 26 | F | 40 | Breast | Invasive ductal
carcinoma | 1 | IIb | T2N1M0 | +, 60% | - | 0 | Malignant |
| C3 | 27 | F | 50 | Breast | Invasive ductal
carcinoma | 1 | IIb | T3N0M0 | - | - | 1+ | Malignant |
| C4 | 28 | F | 50 | Breast | Invasive ductal
carcinoma | 1 | IIb | T3N0M0 | - | - | 1+ | Malignant |
| C5 | 29 | F | 37 | Breast | Invasive ductal
carcinoma | 1 | IIa | T2N0M0 | - | - | 0 | Malignant |
| C6 | 30 | F | 37 | Breast | Invasive ductal
carcinoma | 1 | IIa | T2N0M0 | - | - | 0 | Malignant |
| C7 | 31 | F | 46 | Breast | Invasive ductal
carcinoma | 1 | IIIa | T3N2M0 | - | - | 2+ | Malignant |
| C8 | 32 | F | 46 | Breast | Invasive ductal
carcinoma (fibrous tissue and blood vessel) | - | IIIa | T3N2M0 | - | - | 1+ | Malignant |
| C9 | 33 | F | 69 | Breast | Invasive ductal
carcinoma (fibrofatty tissue and blood vessel) | - | IIa | T2N0M0 | - | - | 0 | Malignant |
| C10 | 34 | F | 69 | Breast | Invasive ductal
carcinoma | 1 | IIa | T2N0M0 | - | - | 0 | Malignant |
| C11 | 35 | F | 52 | Breast | Invasive ductal
carcinoma | 1 | IIIb | T4N1M0 | - | - | 1+ | Malignant |
| C12 | 36 | F | 52 | Breast | Invasive ductal
carcinoma | 1 | IIIb | T4N1M0 | - | - | 1+ | Malignant |
| D1 | 37 | F | 65 | Breast | Invasive ductal
carcinoma | 2 | IV | T3N1M1 | - | - | 1+ | Malignant |
| D2 | 38 | F | 65 | Breast | Invasive ductal
carcinoma | 2 | IV | T3N1M1 | - | - | 1+ | Malignant |
| D3 | 39 | F | 45 | Breast | Invasive ductal
carcinoma | 2 | IIa | T2N0M0 | +, 50% | ++, 90% | 0 | Malignant |
| D4 | 40 | F | 45 | Breast | Invasive ductal
carcinoma | 2 | IIa | T2N0M0 | +, 65% | ++, 95% | 0 | Malignant |
| D5 | 41 | F | 37 | Breast | Invasive ductal
carcinoma | 1 | IIIa | T3N1M0 | - | - | 1+ | Malignant |
| D6 | 42 | F | 37 | Breast | Invasive ductal
carcinoma | 1 | IIIa | T3N1M0 | - | - | 1+ | Malignant |
| D7 | 43 | F | 55 | Breast | Invasive ductal
carcinoma | 2 | IIb | T2N1M0 | - | - | 0 | Malignant |
| D8 | 44 | F | 55 | Breast | Invasive ductal
carcinoma | 2 | IIb | T2N1M0 | - | - | 0 | Malignant |
| D9 | 45 | F | 55 | Breast | Invasive ductal
carcinoma | 2 | IIIb | T4N1M0 | - | - | 1+ | Malignant |
| D10 | 46 | F | 55 | Breast | Invasive ductal
carcinoma | 2 | IIIb | T4N1M0 | - | - | 1+ | Malignant |
| D11 | 47 | F | 75 | Breast | Invasive ductal
carcinoma | 2 | IIb | T2N1M0 | - | - | 0 | Malignant |
| D12 | 48 | F | 75 | Breast | Invasive ductal
carcinoma | 2 | IIb | T2N1M0 | - | - | 0 | Malignant |
| E1 | 49 | F | 41 | Breast | Invasive ductal
carcinoma | 2 | IIa | T2N0M0 | - | +, 8% | 0 | Malignant |
| E2 | 50 | F | 41 | Breast | Invasive ductal
carcinoma | 2 | IIa | T2N0M0 | - | +, 5% | 0 | Malignant |
| E3 | 51 | F | 44 | Breast | Invasive ductal
carcinoma | 2 | IIb | T2N1M0 | - | - | 2+ | Malignant |
| E4 | 52 | F | 44 | Breast | Invasive ductal
carcinoma (sparse) | 1 | IIb | T2N1M0 | - | - | 1+ | Malignant |
| E5 | 53 | F | 37 | Breast | Invasive ductal
carcinoma | 2 | IIb | T3N0M0 | - | - | 1+ | Malignant |
| E6 | 54 | F | 37 | Breast | Invasive ductal
carcinoma | 2 | IIb | T3N0M0 | - | - | 1+ | Malignant |
| E7 | 55 | F | 61 | Breast | Invasive ductal
carcinoma | 2 | IIb | T2N1M0 | ++, 90% | - | 0 | Malignant |
| E8 | 56 | F | 61 | Breast | Invasive ductal
carcinoma | 2 | IIb | T2N1M0 | ++, 95% | 0,20% | 0 | Malignant |
| E9 | 57 | F | 38 | Breast | Invasive ductal
carcinoma (sparse) | 2 | IIb | T3N0M0 | - | - | 0 | Malignant |
| E10 | 58 | F | 38 | Breast | Invasive ductal
carcinoma | 2 | IIb | T3N0M0 | - | - | 0 | Malignant |
| E11 | 59 | F | 65 | Breast | Invasive ductal
carcinoma | 2 | IIa | T2N0M0 | +++, 100% | +++, 97% | 0 | Malignant |
| E12 | 60 | F | 65 | Breast | Invasive ductal
carcinoma | 2 | IIa | T2N0M0 | +++, 99% | +++, 99% | 0 | Malignant |
| F1 | 61 | F | 53 | Breast | Invasive ductal
carcinoma (sparse) | 2 | IIa | T2N0M0 | - | - | 1+ | Malignant |
| F2 | 62 | F | 53 | Breast | Invasive ductal
carcinoma | 2 | IIa | T2N0M0 | - | - | 1+ | Malignant |
| F3 | 63 | F | 32 | Breast | Invasive ductal
carcinoma | 2 | IIb | T2N1M0 | - | - | 1+ | Malignant |
| F4 | 64 | F | 32 | Breast | Invasive ductal
carcinoma | 2 | IIb | T2N1M0 | - | - | 0 | Malignant |
| F5 | 65 | F | 56 | Breast | Invasive ductal
carcinoma | 2 | IIb | T2N1M0 | - | - | 1+ | Malignant |
| F6 | 66 | F | 56 | Breast | Invasive ductal
carcinoma | 2 | IIb | T2N1M0 | - | - | 1+ | Malignant |
| F7 | 67 | F | 52 | Breast | Invasive ductal
carcinoma (sparse) | 2 | IIIa | T2N2M0 | +++, 98% | - | 0 | Malignant |
| F8 | 68 | F | 52 | Breast | Invasive ductal
carcinoma | 2 | IIIa | T2N2M0 | ++, 75% | - | 0 | Malignant |
| F9 | 69 | F | 37 | Breast | Invasive ductal
carcinoma | 3 | IIb | T2N1M0 | - | - | 0 | Malignant |
| F10 | 70 | F | 37 | Breast | Invasive ductal
carcinoma | 3 | IIb | T2N1M0 | - | - | 0 | Malignant |
| F11 | 71 | F | 42 | Breast | Invasive ductal
carcinoma | 2 | IIa | T2N0M0 | - | - | 1+ | Malignant |
| F12 | 72 | F | 42 | Breast | Invasive ductal
carcinoma | 2 | IIa | T2N0M0 | - | - | 1+ | Malignant |
| G1 | 73 | F | 54 | Breast | Invasive ductal
carcinoma | 2 | IIb | T3N0M0 | - | - | 2+ | Malignant |
| G2 | 74 | F | 54 | Breast | Invasive ductal
carcinoma | 2 | IIb | T3N0M0 | - | - | 2+ | Malignant |
| G3 | 75 | F | 40 | Breast | Invasive ductal
carcinoma | 3 | IIb | T2N1M0 | - | - | 0 | Malignant |
| G4 | 76 | F | 40 | Breast | Invasive ductal
carcinoma | 3 | IIb | T2N1M0 | - | - | 0 | Malignant |
| G5 | 77 | F | 59 | Breast | Invasive ductal
carcinoma | 2 | IIIa | T2N2M0 | - | - | 1+ | Malignant |
| G6 | 78 | F | 59 | Breast | Invasive ductal
carcinoma | 2 | IIIa | T2N2M0 | - | - | 1+ | Malignant |
| G7 | 79 | F | 42 | Breast | Invasive ductal
carcinoma | 3 | IIIb | T4N0M0 | - | - | 0 | Malignant |
| G8 | 80 | F | 42 | Breast | Invasive ductal
carcinoma | 3 | IIIb | T4N0M0 | - | - | 0 | Malignant |
| G9 | 81 | F | 54 | Breast | Invasive ductal
carcinoma | 3 | IIa | T2N0M0 | +++, 100% | ++, 5% | 0 | Malignant |
| G10 | 82 | F | 54 | Breast | Invasive ductal
carcinoma | 3 | IIa | T2N0M0 | +++, 100% | +++, 3% | 0 | Malignant |
| G11 | 83 | F | 60 | Breast | Invasive ductal
carcinoma | 3 | IIb | T2N1M0 | - | - | 0 | Malignant |
| G12 | 84 | F | 60 | Breast | Invasive ductal
carcinoma | 3 | IIb | T2N1M0 | - | - | 0 | Malignant |
| H1 | 85 | F | 49 | Breast | Invasive ductal
carcinoma | 2 | IIIa | T2N2M0 | +, 80% | +, 50% | 3+ | Malignant |
| H2 | 86 | F | 49 | Breast | Invasive ductal
carcinoma | 2 | IIIa | T2N2M0 | +, 50% | +, 40% | 1+ | Malignant |
| H3 | 87 | F | 27 | Breast | Invasive ductal
carcinoma | 2 | IIIb | T4N2M0 | - | - | 2+ | Malignant |
| H4 | 88 | F | 27 | Breast | Invasive ductal
carcinoma | 2 | IIIb | T4N2M0 | - | - | 2+ | Malignant |
| H5 | 89 | F | 49 | Breast | Invasive ductal
carcinoma | 2 | IIa | T2N0M0 | - | - | 2+ | Malignant |
| H6 | 90 | F | 49 | Breast | Invasive ductal
carcinoma | 2 | IIa | T2N0M0 | - | - | 2+ | Malignant |
| H7 | 91 | F | 71 | Breast | Invasive ductal
carcinoma | 3 | IIb | T2N1M0 | - | - | 0 | Malignant |
| H8 | 92 | F | 71 | Breast | Invasive ductal
carcinoma | 3 | IIb | T2N1M0 | - | - | 0 | Malignant |
| H9 | 93 | F | 50 | Breast | Invasive lobular
carcinoma | - | IIIa | T2N2M0 | +++, 90% | ++, 10% | 0 | Malignant |
| H10 | 94 | F | 50 | Breast | Invasive lobular
carcinoma | - | IIIa | T2N2M0 | +++, 85% | ++, 1% | 0 | Malignant |
| H11 | 95 | F | 42 | Breast | Invasive lobular
carcinoma | - | IIb | T2N1M0 | ++, 60% | +++, 60% | 0 | Malignant |
| H12 | 96 | F | 42 | Breast | Invasive lobular
carcinoma | - | IIb | T2N1M0 | ++, 65% | +++, 40% | 0 | Malignant |
| - | - | M | 42 | Adrenal gland | Pheochromocytoma
(tissue marker) | - | | | | | | Malignant |
Discussion
We demonstrated here that LFG interacts with TRIM21,
an E3-ubiquitin ligase that can ubiquitinate itself, IRF
transcription factors, and p27 cell cycle inhibitor (12–14).
The strong interaction which was detectable in the array-analysis,
could be confirmed by co-immunoprecipitation whereby isolating LFG
from the cell lysate of MDA-MB-231 cells. A complex of LFG and
TRIM21 could be detected, as both proteins are detectable in the
same position on the blot membrane, although being of different
molecular weight. By detecting the proteins in intact and fixed
MDA-MB-231 cells, the localization of the complexes could be
detected predominantly in close proximity to the cell core.
With the co-immunoprecipitation and subsequent
western blot analysis, the result shown in Fig. 3A were obtained. The bands at the
size of ~80 kDa of the samples 2–4 are evidence of the complex
formation between TRIM21 (52 kDa) and LFG (35 kDa). Thus, an
interaction between the two proteins is still possible despite a
deletion of the domains 2–4 of TRIM21. In the samples 1a and 1b,
the band is missing which indicates a missed complex formation.
Thereby, the domains 1a and 1b can be explained as essential
domains for the connection between TRIM21 and LFG. Further, one
additional band occurs in the samples 2–4 at the size of ~60 kDa.
It is possible that a part separated from the protein complex. In a
study of Dastagir et al (15) in 2014 another form of LFG was
examined, the β-isoform. This is a shorter form of Lifeguard and is
also expressed in breast cancer cell lines. The study even showed
that the smaller isoform of the Lifeguard enacts in an equally
important role in the development of breast cancer as LFG itself
(15). Hence, the band at the size
of 60 kDa possibly indicates a bond between TRIM21 and β isoform of
Lifeguard. One could verify this hypothesis by using a native gel
instead of an SDS gel so that the proteins are examined
individually. The samples 1a and 1b shown in this experiment are
the samples containing the TRIM21 protein without the C-terminal
SPRY domain and the PRY subdomain. Approximately 100 human proteins
have this ~140–200 amino acid domain, wherein it is contained in 40
of the 66 human TRIM proteins. A possible role for this domain has
been discovered by Nisole et al (8) in 2005. However, a closer examination
of the TRIM5α protein of the primate turned out that it is able to
intervene in the replication cycle of various DNA and RNA viruses.
As a consequence, the reverse transcription of the viral genome is
suppressed. This antiviral activity is, however, completely lost by
exchanging a proline in the amino acid sequence of the SPRY domain,
so this domain constitutes the basis of the inteference between
TRIM5α and viruses. This function dependent amino acid is not only
located in the SPRY domain, but also in his PRY subdomain. Such a
mutation is the possible reason for the weak bond between the human
TRIM5α protein and the capsid of human immunodeficiency virus-1
(HIV-1), whereby a possible human immunity against the virus cannot
be ensured. Thus, both the SPRY, as well as the PRY domain seem to
be crucial for an interaction with different types of viruses
(8). Furthermore, the E3-ubiquitin
ligase activity is not only by the RING domain, but also seems to
be dependent of the SPRY domain. The TRIM27 protein binds to the
NOD2 protein that recruits a K48-dependent ubiquitination, followed
by a subsequent degradation in the proteasome. These antiviral
activities, as well as the already illustrated interaction between
TRIM21 and the Fc region of IgG using the SPRY and PRY domain
indicate an essential role of SPRY and PRY domain in the immune
response. The binding of TRIM21 with LFG using the SPRY and PRY
domain points out a potential new function of these domains. Until
now, it was assumed that the function of these domains is limited
on the immune response, whereby the evolutionary benefit by the
mutation of the domain in human TRIM proteins and thus the loss a
possible immunity to different viruses was a mystery. But the
latest results of the working group Bucan allude to a possible new
function of TRIM21 protein in humans: The interaction with
Lifeguard caused sensitivity to the induced apoptosis at the SPRY
and PRY domain playing a regulatory role (8).
To find out if this interaction affects the
expression of LFG and TRIM21, real-time PCR and western blot
analysis were carried out. Concerning TRIM21, an increase of
expression on the RNA level in cells with down regulated
LFG-expression was detected. However, the differences were rather
small and could not be detected on the protein level in western
blot analysis, which results from the low expression of TRIM21 in
MDA-MB-231 cells. For LFG however, a more distinct difference in
expression could be seen as the expression of LFG within the sample
cultivated in the presence of TRIM21 protein is even lower than in
the sample transfected with the siRNA against LFG. In the following
caspase-3-activity analysis, the anti-apoptotic effect of LFG in
the presence of TRIM21 was examined. Results showed a much higher
caspase-3-activity in cells cultivated with TRIM21, which could
result from an increased LFG-expression due to TRIM21. As Jauharoh
et al (11) predicted,
TRIM21 acts as a mediator for the proapoptotic signals. The
increase in caspase-activity in our analysis cannot be definitely
attributed to a decrease of LFG-expression. The results obtained by
Jauharoh et al (11) can,
however, not be directly compared to our analysis, as they used
cell flow cytometric detection of Annexin IV as evidence for the
apoptotic processes. Furthermore, they used HeLa cells for the
analysis, which express TRIM21 in significantly higher levels than
MDA-MB-231, and a Fas-antibody was added in combination with INFγ,
not merely as a supplement.
A positive effect of TRIM21 concerning the field of
oncology could be shown by cell cycle analysis. Within this test a
correlation between TRIM21 concentration and a higher amount of
resting cells in G0/G1-phase and a decrease of cells in S-phase
could be shown. A contrary effect of TRIM21 was predicted by Sabile
et al (12), who described
TRIM21 as an essential mediator of ubiquitinylation for
phosphorylated p27, a tumor-suppressor which prevents cells from
entering S-phase. To examine how TRIM21 is actually expressed in
breast tumors throughout different degrees of malignity,
corresponding tissue samples were obtained from US Biomax, Inc. and
TRIM21 was detected using fluorescence labeled antibodies against
TRIM21. The expression levels of TRIM21 do not differ distinctly
throughout the different degrees of malignity of the inflamed
tissue sample but all of these samples show only around half of the
expression levels, compared to native tissue. The results we
obtained are, however, more consistent with the fact that TRIM21
inhibits activation of NF-κB, a family of transcription factors
which are mainly responsible for the activation and promotion of
cell division. That the protein form of TRIM21 can be transported
into the cell could be shown by the NF-κB array. The inhibiting
effect of TRIM21 towards NF-κB becomes clear due to the decrease in
expression of the analyzed NF-κB-regulated genes. A possible mode
of regulation of LFG expression by TRIM21 could be achieved by
inhibition of NF-κB-activation. As the expression of LFG is
regulated partly by NF-κB, its inhibition by TRIM21, as shown by
Niida et al (9), could
follow the schema displayed in. The reduced activity of NF-κB would
therefore result in a reduced expression of LFG. This hypothesis
however, does not involve an interaction between the two
proteins.
The obtained results reveal TRIM21 as a potential
instrument to affect the anti-apoptotic effect of LFG, which keeps
tumor cells alive and even protects them from certain drugs (Bucan
et al (6). To reveal the
detailed characteristics of their interaction, and the regulating
effect that TRIM21 seems to have on the expression of LFG, further
analyses are required.
Acknowledgements
The study was funded by the Niedersächsische
Krebs-gesellschaft. The authors are grateful to Andrea Lazaridis
for excellent technical assistance.
References
|
1
|
Somia NV, Schmitt MJ, Vetter DE, Van
Antwerp D, Heinemann SF and Verma IM: LFG: An anti-apoptotic gene
that provides protection from Fas-mediated cell death. Proc Natl
Acad Sci USA. 96:12667–12672. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Walczak H and Krammer PH: The CD95
(APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp Cell Res.
256:58–66. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kischkel FC, Hellbardt S, Behrmann I,
Germer M, Pawlita M, Krammer PH and Peter ME:
Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a
death-inducing signaling complex (DISC) with the receptor. EMBO J.
14:5579–5588. 1995.PubMed/NCBI
|
|
4
|
Schweitzer B, Taylor V, Welcher AA,
McClelland M and Suter U: Neural membrane protein 35 (NMP35): A
novel member of a gene family which is highly expressed in the
adult nervous system. Mol Cell Neurosci. 11:260–273. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bucan V, Reimers K, Choi CY, Eddy MT and
Vogt PM: The anti-apoptotic protein lifeguard is expressed in
breast cancer cells and tissues. Cell Mol Biol Lett. 15:296–310.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bucan V, Choi CY, Lazaridis A, Vogt PM and
Reimers K: Silencing of anti-apoptotic transmembrane protein
lifeguard sensitizes solid tumor cell lines MCF-7 and SW872 to
perifosine-induced cell death activation. Oncol Lett. 2:419–422.
2011.PubMed/NCBI
|
|
7
|
Espinosa A, Zhou W, Ek M, Hedlund M,
Brauner S, Popovic K, Horvath L, Wallerskog T, Oukka M, Nyberg F,
et al: The Sjogren's syndrome-associated autoantigen Ro52 is an E3
ligase that regulates proliferation and cell death. J Immunol.
176:6277–6285. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nisole S, Stoye JP and Saïb A: TRIM family
proteins: Retroviral restriction and antiviral defence. Nat Rev
Microbiol. 3:799–808. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Niida M, Tanaka M and Kamitani T:
Downregulation of active IKK beta by Ro52-mediated autophagy. Mol
Immunol. 47:2378–2387. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang S, Pettaway CA, Uehara H, Bucana CD
and Fidler IJ: Blockade of NF-kappaB activity in human prostate
cancer cells is associated with suppression of angiogenesis,
invasion, and metastasis. Oncogene. 20:4188–4197. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jauharoh SNA, Saegusa J, Sugimoto T,
Ardianto B, Kasagi S, Sugiyama D, Kurimoto C, Tokuno O, Nakamachi
Y, Kumagai S, et al: SS-A/Ro52 promotes apoptosis by regulating
Bcl-2 production. Biochem Biophys Res Commun. 417:582–587. 2012.
View Article : Google Scholar
|
|
12
|
Sabile A, Meyer AM, Wirbelauer C, Hess D,
Kogel U, Scheffner M and Krek W: Regulation of p27 degradation and
S-phase progression by Ro52 RING finger protein. Mol Cell Biol.
26:5994–6004. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ozato K, Shin DM, Chang TH and Morse HC
III: TRIM family proteins and their emerging roles in innate
immunity. Nat Rev Immunol. 8:849–860. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kong HJ, Anderson DE, Lee CH, Jang MK,
Tamura T, Tailor P, Cho HK, Cheong J, Xiong H, Morse HC III, et al:
Cutting edge: Autoantigen Ro52 is an interferon inducible E3 ligase
that ubiquitinates IRF-8 and enhances cytokine expression in
macrophages. J Immunol. 179:26–30. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dastagir N, Lazaridis A, Dastagir K,
Reimers K, Vogt PM and Bucan V: Role of lifeguard β-isoform in the
development of breast cancer. Oncol Rep. 32:1335–1340.
2014.PubMed/NCBI
|