1. Introduction
Hematopoiesis is a kind of complex dynamic balance,
which is affected by stem cell renewal and lineage-specific
differentiation, and that is also regulated by a large number of
cytokines, key regulatory transcription factors and epigenetic
modifiers (1,2). Leukemias are often induced by
rearrangement of chromosomes translocations or inversions. These
chromosomal rearrangements can result in gene coding sequence
fusions whereby a new protein is created (3–6). For
example, the t(3;21)(q26;q22) translocation results in the
RUNX1/MDS1/EVI-1 (AME) transcription factor fusion protein (RUNX1
also termed AML1) (7). The EVI-1
is an oncogenic transcription factor associated with human myeloid
malignancy and several solid epithelial cancers (8–11).
Aberrant EVI-1 expression occurs in 8–10% of human adult acute
myeloid leukemia (AML) and, strikingly, increases to 27% of
pediatric mixed lineage leukemia (MLL) rearranged leukemias
(12). The major question still
remains as to its exact role in hemopoietic modulation and
leukemogenesis.
2. The structure and downstream product of
EVI-1
Mucenski and colleagues (13) discovered the murine EVI-1 gene in
the DNA products extracted from the leukemic cells of the AKXD
mice. The cDNA fragment of human EVI-1 gene is comprised of a
5′-non-coding region (5′-NCR) of 267 bp, an open reading frame
(ORF) of 3153 bp and a 3′-non-coding region (3′-NCR) of 167 bp. The
two ATG codons in the open reading frame are located at the 268 and
290 bp, respectively, of which the latter one is located closer to
the constant transcription initiation sequence (GCC) GCC(A/G)CCATGG
so that the second ATG codon is supposed to be the transcription
initiation site (14). The human
EVI-1 gene contains 12 domains of which the first one is highly
variable and can transcribe 6 different mRNA fragments due to the
variablility of its 5′-terminal. It has been shown by Northern blot
analysis and real-time quantitative RT-PCR that the different types
of EVI-1 proteins resulted from the variable 5′-terminal of the
EVI-1 gene and are nearly the same in various tissues and are only
different in their expression levels in the specific tissues
(15). The translation initiation
site is located in domain 3 while domain 2 is related to the gene
fusion of EVI-1 with MDS1 and AML1. In 1996, Fears et al
(16) discovered that there was a
MDS1 (myelodysplasia syndrome 1) gene located ~170–400 bp upstream
of EVI-1 gene. The MDS1 gene was firstly discovered in the
myelodysplasia syndromes, and after alternative splicing it can be
inserted into the domain 2 of EVI-1, so that the MDS1-EVI-1 fusion
transcript is generated and 188 codons are added to the 5′-terminal
of the open reading frame in the EVI-1 gene. MDS1 and EVI-1 are
mutually antagonistic. Soderholm et al (17) have reported that EVI-1 can inhibit
GATA-1-mediated activation and MDS1-EVI-1 is a strong activation
sequence containing a GAGA-1 promoter. In addition, the MDS1-EVI-1
fusion gene is also directly inhibited by the normal EVI-1 gene. In
the chromosome translocation t(3;21)(q26;q22) the AML1 gene on
chromosome 21 fractures between the runt domain and the
transcription activation domain, and the derived fragments will
fuse with EVI-1 and MDS1, respectively, thus, the AML1-EVI-1 and
AML1-MDS1 fusion genes are formed correspondingly. In the
AML1-EVI-1 transcript the exon 5 of AML1 is linked to the second
untranslated exon of EVI-1 to encode a fusion protein with the
molecular weight of ~1800 k unit and 3 DNA binding domains
(DNA-BD). Besides, AML1 can also fuse with MDS1-EVI-1 to form the
AML1-MDS1-EVI-1 fusion gene (1),
of which the three members interact with each other with
synergistic or antagonistic effects and jointly influence the
progression of both human and murine leukemias.
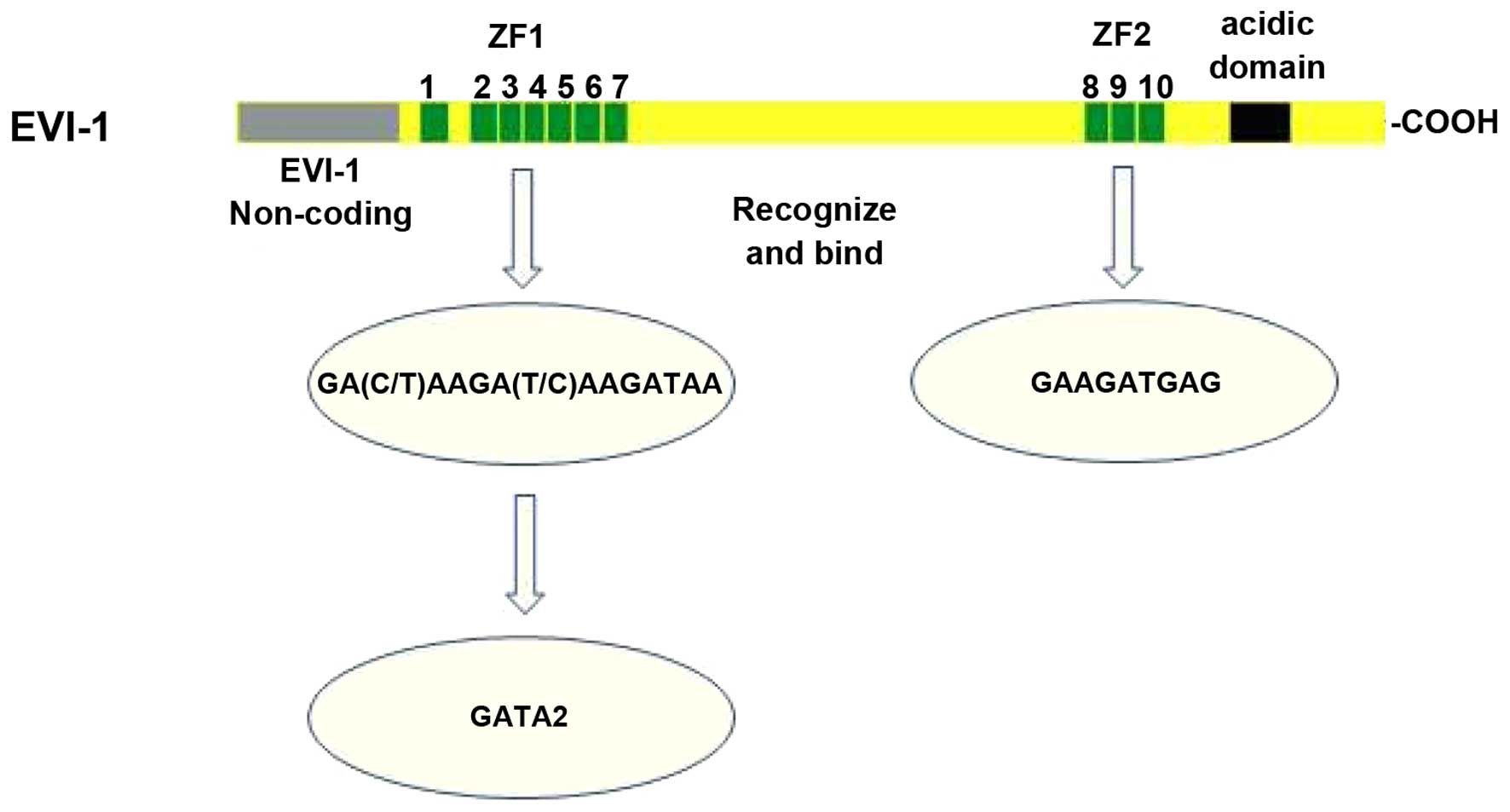
The EVI-1 gene encodes a 145-kDa protein which can
be divided into two regions: the region on the N-terminal
containing 7 zinc-finger domains and the region on the C-terminal
containing 3 zinc-finger domains and a sequence of acidic amino
acids, the two of which are separated by a proline-rich region
located in the middle (Fig. 1).
Delwel et al (18) have
reported that in vitro 3 of the zinc-finger domains on the
N-terminal can bind with a specific sequence of 15 nucleotides:
(C/T)AAGA(T/C)AAGATAA. However, the 3 zinc-finger rings only have
an auxiliary function and can just provide a relatively similar
sequence of GACAA instead of GATAA for the combination rather than
binding with the 15-nucleotide-sequence directly. Funabiki et
al (19) have reported that
the zinc-finger domains on the C-terminal can recognize and bind a
specific sequence of GAAGATGAG. Therefore, EVI-1 is a protein with
specific binding sites. EVI-1 protein also has an isotype with a
shorter sequence and the molecular weight of ~88 kDa which can be
detected both in human being and mice. The 88-kDa isotype lacks
zinc-finger domain 6 and zinc-finger 7 which are both essential for
DNA binding, so that it has a unique DNA-binding characteristic
which is different from that of the 145-kDa EVI-1 protein (20).
3. The regulatory effect of EVI-1 on normal
hematopoiesis
The ecotropic viral integration site-1 (EVI-1) is an
oncogenic transcription factor in murine and human myeloid
leukemia. As a proto-oncogene EVI-1 is highly expressed both in
human embryo and in the hemopoietic stem cells of the adult bone
marrow (21,22). Thus, EVI-1 plays an important role
in tissue development and in the proliferation and differentiation
of hemopoietic stem cells (HSC) (23). EVI-1 works on the proliferation and
differentiation of hemopoietic cells through affecting the
expression of GATA-2. When EVI-1 expression on 3q26 inverted which
is misdirected by a distant GATA2 hematopoietic enhancer on 3q21,
this kind of enhancer is critical for AML neoplasia (3). It has been reported by Yuasa et
al (24) that the hemopoietic
stem cells of the EVI-1-deleted mice are deficient in proliferation
and repopulation, and also accompanied with low expression level of
GATA-2 which is crucial for the proliferation and differentiation
of hemopoietic cells. Re-establishing EVI-1 expression in the
hemopoietic stem cells for these EVI-1-deleted mice can restore the
cells' ability of proliferation and differentiation by upregulating
GATA-2 expression, and this demonstrates that EVI-1 has the leading
role in the proliferation and differentiation of hemopoietic cells.
The next in this passage mainly introduces the EVI-1′s effects on
the various types of hematopoietic progenitor cells. The inhibitor
of apoptosis protein survivin regulates hematopoiesis, although its
mechanisms of regulation of hematopoietic stem cells (HSCs) remain
largely unknown. While investigating conditional survivin deletion
in mice, we found that survivin was highly expressed in
phenotypically defined HSCs, and survivin deletion in mice resulted
in significantly reduced total marrow HSCs and hematopoietic
progenitor cells. Transcriptional analysis of
survivin−/− HSCs revealed altered expression of multiple
genes not previously linked to survivin activity. In particular,
survivin deletion significantly reduced expression of the EVI-1
transcription factor indispensable for HSC function, and the
downstream EVI-1 target genes GATA2, Pbx1 and Sall2. The loss of
HSCs following survivin deletion and impaired long-term HSC
repopulating function could be partially rescued by ectopic EVI-1
expression in survivin−/− HSCs. These data demonstrate
that survivin partially regulates HSC function by modulating the
EVI-1 transcription factor and its downstream targets and identify
new genetic pathways in HSCs regulated by survivin (22).
4. The effect of EVI-1 on
erythropoiesis
Since EVI-1 was first identified, many investigators
had made a considerable effort to understand the mechanisms how the
protein deregulate erythropoiesis. It has been reported by Matsugi
et al (25) that the
zinc-finger transforming protein of EVI-1 can bind the genomic
fragments with the (GATA) n sequence. The zinc-finger protein
GATA-1 is an erythroid transcriptional regulatory factor, and the
binding sequences of this kind of factor exist in many
cis-acting regulatory elements of the genes expressed in the
erythroid cell line, such as the globin gene, the erythropoietin
gene and the GATA1 gene. Thus, the GATA-1 protein takes on the
central regulator in the specific gene expression and cell
differentiation of the erythroid cell line. The binding motif of
the zinc-finger domain for binding with GATA-1 is WGATAR (W=A or T,
R=A or G), which is located in the constant binding motif in the
N-terminal zinc-finger domains of EVI-1. The effects of EVI-1 on
erythropoiesis were confirmed by Kreider et al (26). After the retroviral expressing
vectors containing EVI-1 cDNA are successfully transfected into the
murine 32DEpol cells in which EVI-1 is not expressed, the
transfected EVI-1-expressing cells lose their original response
capacity to erythropoietin (EPO) and form far less erythropoietic
progenitor colonies than before. Furthermore, the CAT assay has
shown that EVI-1 expression can obviously inhibit the
CATA-1-dependent transcriptional activation, and the inappropriate
expression of EVI-1 probably prohibits erythropoiesis by some
subtypes of the GATA-1 target genes. Rather than the DNA sequence,
EVI-1 interacts directly with the GATA1 protein. It has been
further confirmed that this protein-protein interaction blocks
efficient recognition or binding to DNA by GATA1. Point mutations,
which destroy the geometry of two zinc-fingers of EVI-1 and
terminate the protein-protein interaction, can lead to normal
erythroid differentiation of normal murine bone marrow in
vitro (27). It has been
reported by Louz et al (28) that they established 3 independent
transgenic mouse models with the murine Sca-1 promoter, which shows
obvious colony forming unit-erythrocyte (CFU-E) reduction in the
bone marrow without obvious hemopoietic disorder, and demonstrating
the progenitor-related deficiency of normal erythropoiesis.
Therefore, we can confirm that EVI-1 can prohibit normal
hemopoiesis in vivo which is synergistic with EVI-1-mediated
carcinogenesis in return.
5. The effect of EVI-1 on
granulopoiesis
After EVI-1 is transfected into hematopoietic growth
factor (HGF)-dependent murine 32DC13 cells, the granular
differentiation induced by granulocyte colony-stimulating factor
(G-CSF) is retarded, which demonstrates that EVI-1 can prohibit the
maturation of granulocytes. The growth of 32DC13 cells are
dependent on interleukin-3 (IL-3) and the cells can differentiate
into granulocytes after induction by G-CSF. Morishita et al
(29) have reported that with the
existence of the hematopoietic growth factors (HGF) the
EVI-1-positive 32DC13 cells will lose their original response
capacity to G-CSF and undergo further apoptosis, because in the
EVI-1-positive 32DC13 cells myeloperoxidase fails to be expressed
and the differentiation process is prohibited. Boyd et al
(30) reported that the provirus
insertion into the EVI-1 gene was observed in the AKZD23 murine
leukemia. However, the provirus insertion does not directly cause
the occurrence of leukemia, which indicates that there are other
synergistic factors. It has been shown by analysis that in every
tumor it is the provirus insertion into the SOX4 gene which can
encode a high mobility group (HMG) box transcription factor and
lead to the overexpression of SOX4. In the 32DC13 cells the
overexpression of SOX4 markedly inhibits the cytokine-induced
granulocyte maturation. It is believed that the provirus-mediated
activation of SOX4 is specifically effective on the
provirus-mediated activation of EVI-1, because the reduced process
of AKV LTR by SOX4 can upregulate the expression of EVI-1 and lead
to the relevant retardation of cell differentiation. EVI-1 is an
oncogene inappropriately expressed in the bone marrow (BM) of ~10%
of myelodysplastic syndrome (MDS) patients. This disease is
characterized by severe anemia and multilineage myeloid dysplasia
that are thought to be a major cause of mortality in MDS patients.
We earlier reported on a mouse model that constitutive expression
of EVI-1 in the BM led to fatal anemia and myeloid dysplasia, as
observed in MDS patients, and we subsequently showed that EVI-1
interaction with GATA1 blocks proper erythropoiesis. Whereas this
interaction could provide the basis for the erythroid defects in
EVI-1-positive MDS, it does not explain the alteration of myeloid
differentiation. In the present review, we have examined the
expression of several genes activated during terminal myelopoiesis
in BM cells and identified a group that are altered by EVI-1. It
was reported that EVI-1 interacts with PU.1 and represses the
PU.1-dependent activation of the myeloid promoter. EVI-1 does not
seem to inhibit PU.1 binding to DNA but rather to block its
association with the coactivator c-Jun. After mapping the
PU.1-EVI-1 interaction sites, it was shown that an EVI-1 point
mutant, unable to bind PU.1, restores the activation of
PU.1-regulated genes and allows a normal differentiation of BM
progenitors in vitro (31).
6. The effect of EVI-1 on
megakaryocytopoiesis
Shimizu et al (32) have reported that in the 3q21q26
syndrome that is characterized by the chromosomal inversion inv(3)
(q21;q26), RT-PCR assays can clearly show that EVI-1 is expressed
specifically in the CD34+ cells including megakaryocytes
and platelets. The immature megakaryoblast leukemia UT-7 cell line
has three subtypes including UT-7/GM, UT-7/EPO and UT-7/TPO, whose
growth is dependent on granulocyte macrophage colony-stimulating
factor (GM-CSF), erythropoietin (EPO) and thrombopoietin (TPO),
respectively. The expression level of the EVI-1 gene is low in the
UT-7/GM and UT-7/EPO cell lines but is high in the UT-7/TPO cell
line. After the UT-7/GM cells are stimulated by TPO, the EVI-1
expression is enhanced in these cells accompanied by the increase
of multinuclear megakaryocytes and the expression of platelet
factor 4 (PF4). The EVI-1 expression in the UT-7/GM cells can
change their original morphology into multinuclear megakaryocyte
and can lead to cell growth arrest and further cell death in one
month, which confirms the effects of EVI-1 expression in
megakaryocyte differentiation. Kilbey et al (33) reported the biological effects of
EVI-1 expression in the megakaryocytic progenitors and the
conditional expression of the EVI-1 gene in the human erythro
leukemia (HEL) cells. After the HEL cells are stimulated by
12-O-Tetradecanoylphorbol-13-acetate (TPA), they will
develop toward the megakaryocytic progenitors with four steps: i)
growth inhibition; ii) morphological change; iii) endomitosis; and
iv) the characteristic change of genetic expression including the
reduction of the erythroid-specific glycophorin A and the increase
of the megakaryocytic-specific GPIIIa and GPVI. The EVI-1
expression alone has no significant effects on the proliferation
and differentiation of the HEL cells, but after the exposure to TPA
the EVI-1-expressing HEL cells will undergo obvious growth
stagnation along with the alteration of endomitosis and cell
morphology. The reduction of glycophoryn A and the increase of the
GPIIIa mRNA level can also be detected, while the expression level
of the GPVI mRNA can not be increased noticeably. The continuously
efficient catalytic activity of CDK2 is specifically related to the
endomitosis of megakaryocytes, but in the EVI-1-expressing HEL
cells treated with TPA the catalytic activity of CDK2 is remarkably
reduced, which is due to the significant reduction in the activity
of cyclin A. Although the EVI-1-expressing cells can maintain some
molecular characteristics, the decreased catalytic activity of CDK2
can definitely lead to the differentiation deficiency of
megakaryocytes via endomitosis retardation and morphological
alteration, which confirms the regulatory effects of EVI-1 on
megakaryocytopoiesis.
7. The relationship between EVI-1 and
hematologic neoplasms
Abnormal expression of EVI-1 is common in
myelogenous hematopoietic malignancies such as acute myeloid
leukemia (AML), chronic myeloid leukemia (CML) and myelodysplastic
syndrome (MDS), and is always caused by transversion or insertion
of chromosome 3 which leads to leukemogenesis. Yamazaki et
al (3) identified a mechanism
whereby a GATA2 distal hematopoietic enhancer (G2DHE or 77-kb
enhancer) is brought into close proximity to the EVI-1 gene in
inv(3) (q21;q26) inversions, leading to leukemogenesis. They
demonstrated that both 3q rearrangements reposition a distal GATA2
enhancer to ectopically activate EVI-1 and simultaneously confer
GATA2 functional haploinsufficiency, previously identified as the
cause of sporadic familial AML/MDS and MonoMac/Emberger syndromes.
Genomic excision of the ectopic enhancer restored EVI-1 silencing
and led to growth inhibition and differentiation of AML cells
(34). The most common chromosome
transversion leading to abnormal EVI-1 expression are chromosome
inversion inv(3)(q21;q26) and chromosome transversion
t(3;3)(q21;q26) which account for ~7–10% in the MDS/AML patients.
Aberrant EVI-1 expression occurs in 8–10% of human AML and,
strikingly, increased to 27% of pediatric mixed lineage leukemia
(MLL) rearranged leukemias. When inappropriately expressed, EVI-1
is an independent risk factor for a very poor prognosis in
hematopoietic cancer (12). In 10%
of the EVI-1-expressing MDS/AML patients the abnormality of
chromosome 3q26 is absent, which indicates the existence of
synergistic factors in the abnormal expression of EVI-1 and
EVI-1-derived hematopoietic malignant transformation. Lahortiqa
(ref.?) reported that there are some cases lacking EVI-1 expression
in the chromosomal abnormality of inv(3)(q21q26) and ~9% of the AML
cases with EVI-1 overexpression lack the variation of chromosome
3q26. In 7 cases of inv(3)(q21;q26) or t(3;3)(q21;q26) or other
associated malformations only one case is EVI-1-expressing, which
indicates that EVI-1 expression is not adequate to explain all the
cases (35). Matsuo et al
(36) reported that EVI-1
overexpression is a poor prognostic factor in pediatric patients
with mixed lineage leukemia-AF9 rearranged acute myeloid leukemia.
Goyama et al (23)
developed several leukemia models in which EVI-1 could be deleted,
and they found that loss of EVI-1 attenuates proliferative activity
in several types of leukemic cells. In particular, EVI-1 deletion
led to a large decrease in colony numbers of MLL/ENL-transformed
cells, indicating a crucial role for EVI-1 in MLL-leukemia. Because
EVI-1 is frequently upregulated in MLL-leukemia, the molecular
interaction between EVI-1 and MLL is an important challenge in the
future (37).
8. The molecular mechanisms of
EVI-1-mediated leukemogenesis
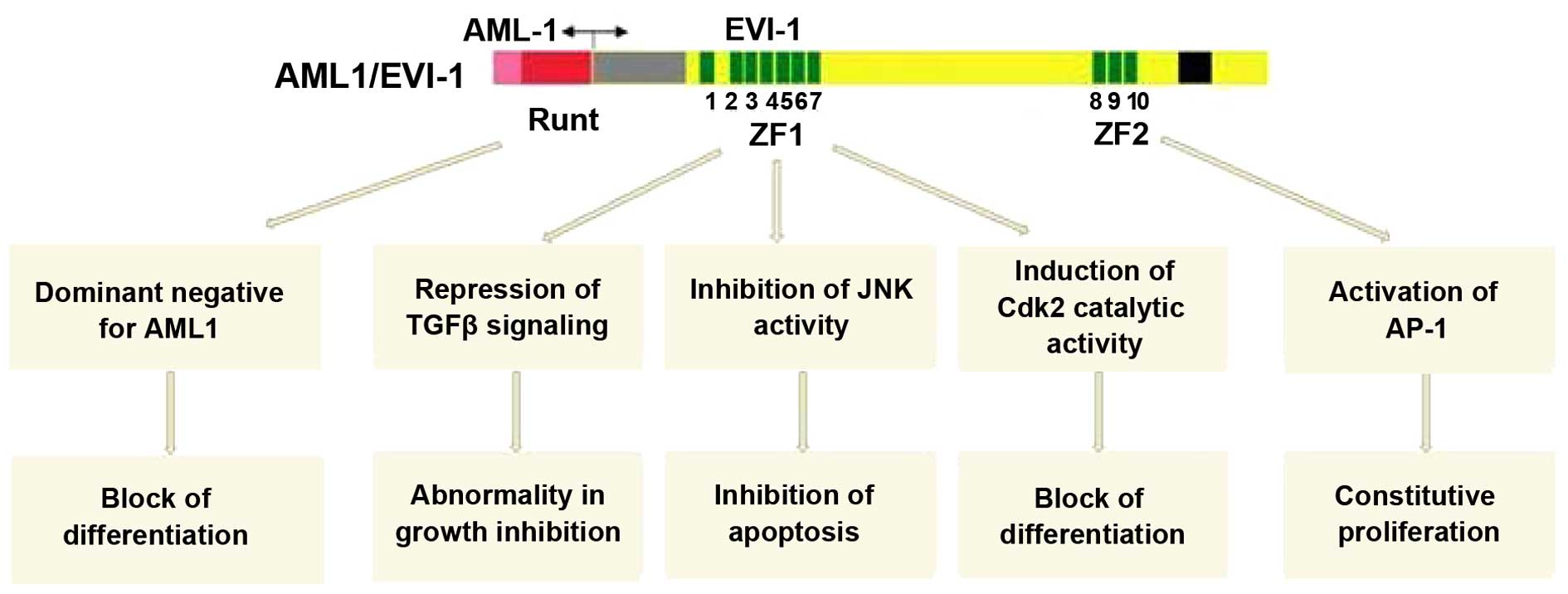
EVI-1 is synergistic with AML1 and MDS1 in the
co-induction of hemopoietic stem cell malignant transformation, and
the relevant molecular mechanisms mainly include the 7 aspects as
follows (Fig. 2):
Inhibition of the AML1-mediated
transcriptional activation by EVI-1
AMLl-EVI-1 is dominant negative which can exceed and
counteract the AML1-mediated transcriptional activation.
RUNX1-EVI-1 dominantly suppresses the transactivation capacity of
RUNX1 through the PEBP2 sites (38–42).
It is reported that the combination of PEBP2 with AML1-EVI-1 is
tighter than that with AML1, so that AML1-EVI-1 can significantly
inhibit the AML1-induced transcriptional activation by competitive
combination with specific DNA sequences. To bring about its
transcriptional activation function, the AML1 located in the
nucleous has to combine its RHD structure with the PEBP2 located in
the cytosol. The RHD-positive AML1-EVI-1 can translocate the PEBP2
located in the cytosol to the nucleous, so that AML1-EVI-1 can
inhibit the AML1-induced transcriptional activation by means of its
higher affinity for PEBP2 (43).
It is also reported that AML1-EVI-1 can interact with
COOH-terminal-binding protein (CtBP) which is essential for
suppressing the AML1-induced transcriptional activation. As one of
the accessory proteins, CtBP can exert inhibitive effects by
combining with several transcriptional factors such as Kruppel-like
factor (BKLF), friend of GATA (FOG) and T-cell factor (TCF).
Although it is still unclear how CtBP mediates transcriptional
inhibition, it is speculated that the interaction between CtBP and
histone deacetylase inhibitors (HDACI) is involved in this process.
EVI-1 specifically interacts with CtBP via two latent CtBP-binding
amino acid sequences located in the AML1-EVI-1 including the PFDLT
sequence on the N-terminal and the PLDLS sequence on the
C-terminal, one of which interacts with CtBP directly. AML1-EVI-1
can interfere with AML1-induced transcription, but the AML1-EVI-1
with changed PLDLS fails to do so, which confirms that the function
of CtBP as a co-repressor is essential in the AML1-EVI-1-mediated
inhibition of AML1-induced transcription. As a histone deacetylase
inhibitor (HDACI), trichostatin A can diminish the dominant
negative effect of AML1-EVI-1, therefore, AML1-EVI-1 probably
resorts to the CtBP-mediated recruitment of HDACI in order to
prohibit the AML1-induced transcription (44–46).
EVI-1 counteracts the TGF-β1 signaling
pathway
EVI-1 can interrupt the TGF-β1/SMAD signaling
pathway and counteracts the growth inhibition caused by TGF-β1 by
inhibiting the TGF-β1/SMAD3-mediated transcription. EVI-1 albumen
which depends on the first zinc finger domain of itself can
integrate with the phosphorylation of Smad3 of MH2 structural
domain and collect other transcription repressors (including CtBP,
histone deacetylase) accordingly, it impedes the function of
accelerating target gene transcription of phosphorylation Smad3,
that according to the MH2 structural domain cooperated with other
activating transcription factors. As a result, during the period of
cell loses the normal inhibitory control leads to the cell
maintaining excessive growth and proliferation making an obstacle
for terminal differentiation and apoptosis of cells. Finally, this
process results in the generation of cancer cells (47–49).
One of the histone deacetylase inhibitors (HDACI), trichostatin A,
can diminish the EVI-1-mediated inhibition of the TGF-β-mediated
signal transduction, which indicates that histone deacetylase
(HDAC) is essential in this process (50,51).
However, Liu et al (52)
discovered that the continuous EVI-1 expression in the
non-transformed intestinal epithelial cells can inhibit some
SMAD3-dependent target genes of TGF-β1 such as PAI1, but the
sensitivity of the intercellular supportive substance such as
integrin1 and paxillin in response to TGF-β1 induction as well as
the transformation of the epithelial cells into the mesenchyme
cells can not be inhibited by EVI-1. Moreover, EVI-1 could not
inhibit the downregulation of cyclin D1 expression and cell growth
inhibition induced by TGF-β1. But EVI-1 can interact with P13K and
its downstream effector AKT to inhibit the TGF-β-induced apoptosis.
The ability of EVI-1 to inhibit apoptosis is not confined to the
TGF-β-induced cell death, because EVI-1 also has a protective
effect on the intestinal epithelial cells and can inhibit the
paclitaxel-induced apoptosis as well. EVI-1 is overexpressed in
some human clonal tumor cells. The siRNA-mediated knockout of the
EVI-1 gene in the HT-29 human clonal tumor cells can inhibit AKT
phosphorylation and enhance the sensitivity of these cells to
paclitaxel-induced apoptosis, which confirms that in the intestinal
epithelial cells and the clonal tumor cells EVI-1 acts as a
survival gene that can activate P13K/AKT and counteract the
apoptosis caused by physiological or therapeutic factors.
EVI-1 modulates also other transcription
factors
EVI-1 acts as an inhibitor of JNK (43), which can inhibit the phosphorylase
activity of C-jun expression, while the activity of JNK is
dependent on the phosphorylation of two specific amino acid
residues (Thr183 and Tyr185) by MMK4 or MMK7. EVI-1 can interact
with JNK, yet, EVI-1 does not affect the phosphorylation level of
JNK. EVI-1 expression can interrupt the binding between C-jun and
JNK so that the phosphorylation of C-jun can be inhibited. EVI-1
can also resort to affect the JNK-mediated signaling pathway to
inhibit cell apoptosis. The human embryonic kidney 293 cells will
undergo apoptosis after induction by ultraviolet radiation, but the
counterpart cells with EVI-1 overexpression are markedly resistant
to apoptosis. The human endometrial carcinoma HEC1B cells are
resistant to apoptosis, which is probably due to the innate EVI-1
expression (53).
As to the upregulation of AP-1 activity by EVI-1,
Hirai et al (48) found
that EVI-1 can upregulate reporter constructs bearing the
AP-1-responsive promoter region. AP-1 activity (Fos/Jun
heterodimer) is activated by a variety of growth signals, including
phorbol esters [12-O-tetradecanoylphorbol 13-acetate (TPA)].
Tanaka et al (54) showed
that EVI-1 could markedly upregulate porter constructs with
TPA-responsive elements (TRE). Similarly, this group also showed
that RUNX1-MDS1-EVI-1 (RME), the protein product of the
leukemia-associated t(3;21), can also transactivate a TRE reporter.
Through structure-activity-relationship (SAR) studies, it was shown
that activation of AP-1 requires the second set of zinc fingers
(zinc fingers 8–10; ZF2), and appears to occur via transcriptional
upregulation of Fos, via an element within the promoter that does
not appear to bind EVI-1. The exact mechanism of Fos upregulation
is not clear; nor is it clear whether AP-1 uregulation is required
for leukemogenesis. Noting that transcriptional activation of Fos
is lost upon deletion of ZF2, it is important to consider that
neither transformation of bone marrow progenitors by RME nor
transformation of Rat1 fibroblasts require DNA binding via ZF2
(55). It is possible that a
non-DNA binding function of ZF2 is required for both Fos activation
and transformation.
Downregulation of Cdk2 kinase
activity
The EVI-1 transcriptional repressor domain and the
ZF1 DNA binding domain are required for both cell transformation
and induction of Cdk2 catalytic activity. EVI-1 expression
represses the transcription of target genes, which may include p27
that deregulate the normal control of the G1 phase of the cell
cycle, providing a cellular proliferative advantage that
contributes to transformation in vitro and leukemogenesis
in vivo (56). Karakaya
et al (57) suggest that
EVI-1 overexpression leads to activation of p53 and subsequent
upregulation of p21, activation of the tetraploidy checkpoint
appears to be underlying cause of p53 activation, since both p53
and p21 are most abundant in enlarged nuclei. EVI-1 overexpression
is mechanistically linked to the p53 pathway or cytokinesis failure
as the underlying cause of activation of the tetraploidy
checkpoint. Thus, Cdk2 inhibition and consequential G0/1 arrest
were related to EVI-1 expression. Kilbey et al (33) conclude that CDK2 kinase activity
was substantially reduced in TPA-induced HEL cells in the presence
of EVI-1 and the profile of kinase activity directly correlated
with the abundance of cyclin A.
Epigenetic aberrations of EVI-1
There is a growing body of evidence for epigenetic
aberrations associated with EVI-1-induced leukemia (57–59).
In patients with AML and MDS, increased expression of EVI-1 was
found to be associated with adverse prognosis. Although increased
expression of the EVI-1 gene is mainly caused by chromosomal
rearrangements involving chromosome band 3q26, where EVI-1 is
located, it can also be identified in cases without these
rearrangements. Overexpression of EVI-1 is associated with 3q26
chromosomal rearrangements and confers extremely poor prognosis in
AML. EVI-1 amplification on cytogenetically cryptic dmin causes
increased expression of EVI-1 and is a new mechanism that causes
increased EVI-1 expression without a 3q26 rearrangement (61). A study (58) on 476 AML patients reported that
those with no basal expression of the EVI-1 gene have a better
prognosis than patients with EVI-1 overexpression. Consistent with
the fact that DNA methylation is generally associated with
repression of gene transcription, EVI-1 overexpression is
associated with the absence of DNA methylation in the promoter
region of EVI-1. The EVI-1 locus is also enriched with marks of
active gene transcription, specifically histones H3 and H4
acetylation, and histone H3 lysine 4 (H3K4) trimethylation.
Conversely, cells without EVI-1 expression are associated with DNA
hypermethylation and marks of gene repression such as
trimethylation of histone H3 lysine 27 (H3K27) at the EVI-1
promoter (7). Lugthart et
al (62) showed that AML
samples with activated EVI-1 presented with a deregulated
hypermethylation signature, possibly through physical interactions
between EVI-1 and the DNA methyltransferases DNMT3A and DNMT3B.
Yoshimi and Kurokawa (63) found
that EVI-1 activated the AKT/mTOR signaling pathway through
transcriptional repression of PTEN. The activation of this
signaling is essential for EVI-1-mediated leukemogenesis. All of
the above jointly accelerate the malignant transformation of the
hemopoietic stem cells.
The minimal promoter region of EVI-1 demonstrates
that RUNX1 and ELK1, its promoter region of EVI-1 of 318 bp, two
proteins with essential functions in hematopoiesis, regulate EVI-1
in AML. One of the mechanisms by which RUNX1 regulates the
transcription of EVI-1 is by acetylation of the histone H3 on its
promoter region. The transcription factors RUNX1 and ELK1 are
involved in the transcriptional regulation of this gene, and that
RUNX1 regulates EVI-1 during megakaryocyte differentiation
(64). H3 and H4 acetylation, and
trimethylation of histone H3 lysine 4 and histone H3 lysine 27
might play a role in the transcriptional regulation of EVI-1 in
acute myeloid leukemias (58).
This result opens new directions to further understand the
mechanisms of EVI-1 overexpressing leukemias (59). The EVI-1 gene, located in
chromosomal band 3q26, is a transcription factor that has stem
cell-specific expression pattern and is essential for the
regulation of self-renewal of hematopoietic stem cells. It is now
recognized as one of the dominant oncogenes associated with myeloid
leukemia. EVI-1 overexpression is associated with minimal to no
response to chemotherapy and poor clinical outcome. Several
chromosomal rearrangements involving band 3q26 are known to induce
EVI-1 overexpression. They are mainly found in acute myeloid
leukemia and blastic phase of Philadelphia chromosome-positive
chronic myeloid leukemia, more rarely in myelodysplastic syndromes.
They include inv(3) (q21q26), t(3;3)(q21;q26), t(3;21)(q26;q22),
t(3;12)(q26;p13) and t(2;3)(p15–23;q26). However, many other
chromosomal rearrangements involving 3q26/EVI-1 have been
identified. The precise molecular event has not been elucidated in
the majority of these chromosomal abnormalities and most gene
partners remain unknown.
Dysregulated T-cell leukemia/lymphoma-1A (TCL1A), a
modulator in B-cell receptor (BCR) signaling, is causally
implicated in chronic lymphocytic leukemia (CLL). However, the
mechanisms of the perturbed TCL1A regulation are largely unknown.
To characterize TCL1A-upstream networks, we functionally screened
for TCL1A-repressive micro-RNAs (miRs) and their transcriptional
regulators. They identified the novel miR-484 to target TCL1A's
3′-UTR and to be downregulated in CLL (ref.?). In chromatin
immunoprecipitations and reporter assays, the oncogenic
transcription factor of myeloid cells, EVI-1, bound and activated
the miR-484 promoter. Most common in CLL was a pan-EVI-1 transcript
variant. EVI-1 protein expression revealed distinct normal-tissue
and leukemia-associated patterns of EVI-1/TCL1A co-regulation.
EVI-1 levels were particularly low in TCL1A-high CLL or such
cellular subsets. Global gene expression profiles from a
337-patient set linked EVI-1 networks to BCR signaling and cell
survival via TCL1A, BTK and other molecules of relevance in CLL.
Enforced EVI-1, similar to miR-484, repressed TCL1A. Furthermore,
it reduced phospho-kinase levels, impaired cell survival, mitigated
BCR-induced Ca-flux and diminished the in vitro ibrutinib
response. Moreover, TCL1A and EVI-1 showed a strongly interactive
hazard prediction in prospectively treated patients. Overall, the
regressive EVI-1 is a novel regulatory signature in CLL. Through
enhanced TCL1A and other EVI-1-targeted hallmarks of CLL, this
contributes to an aggressive cellular and clinical phenotype
(65,66).
The development of novel technologies, such as
massively parallel DNA sequencing, has led to the identification of
several novel recurrent gene mutations, such as DNA
methyltransferase (Dnmt)3a, ten-eleven-translocation oncogene
family member 2 (TET2), isocitrate dehydrogenase (IDH)1/2,
additional sex comb-like 1 (ASXL1), enhancer of zeste homolog 2
(EZH2) and ubiquitously transcribed tetratricopeptide repeat X
chromosome (UTX) mutations in AML and other myeloid malignancies.
These findings strongly suggest a link between recurrent genetic
alterations and aberrant epigenetic regulations, resulting from an
abnormal DNA methylation and histone modification status. Moreover,
epigenetic aberrations resulting from transcription factor
aberrations, such as MLL rearrangement, EVI-1 overexpression,
chromosomal translocations and the downregulation of PU.1 are also
described (67).
EVI-1 and MEL1 are homolog genes whose
transcriptional activations by chromosomal translocations are known
in small subsets of leukemia. From gene expression profiling data
of 130 Japanese pediatric AML patients, we found that EVI-1 and
MEL1 were overexpressed in 30% of patients without obvious
translocations of these gene loci, and that their high expression
was significantly associated with inferior survival. High EVI-1
expression was detected mainly in myelomonocytic-lineage
(designated as e-M4/M5 subtype) leukemia with MLL rearrangements
and in megakaryocytic-lineage (designated as e-M7 subtype)
leukemia, and its prognostic association was observed in the
e-M4/M5 subtype but not in the e-M7 subtype. On the other hand,
high MEL1 expression was detected in myelocytic-lineage (designated
as e-M0/M1/M2 subtype) and e-M4/M5 subtype leukemia without MLL
rearrangements, and its prognostic association was independent from
the subtypes. The combined estimation of EVI-1 and MEL1 expression
will be an effective method to predict the prognosis of pediatric
AML (68). The genetic and
transcriptional signature of EVI-1-rearranged (EVI-1-r) AMLs
remains poorly defined. EVI-1-r AMLs are also characterized by a
unique transcriptional signature with high expression levels of
MECOM, PREX2, VIP, MYCT1 and PAWR. EVI-1-r AMLs could be
molecularly defined by specific transcriptomic anomalies and a
hitherto unseen mutational pattern. Larger patient cohorts will
better determine the frequency of these events (69).
9. Conclusions
At present, it has become evident that EVI-1 is a
crucial gene for HSC regulation as well as an oncogene in
development of leukemia. The abnormal expression of it could
predict poor prognosis in hematological malignancies. Since EVI-1
overexpression was closely related to human hemopoietic diseases,
which was involved in the differentiation, apoptosis and
proliferation of leukemia cells, certain drugs or antisense
oligonucleotides are expected to be good methods to treat EVI-1
induced malignancies, for example, HDAC inhibitors might be useful
in the treatment of EVI-1 induced leukemias.
Acknowledgements
The present review was supported by grants from the
National Natural Science Foundation of China (nos. 81300426,
30771103, 815734467 and 81172792), the Project for Shandong Medical
and Health science and Technology Plan Project (2013WS0365),
Natural Science Foundation of Shandong (ZR2015HM014), and the
‘Twelfth Five-Year’ National Science and Technology Support Program
(2013BAI07B02).
References
|
1
|
Nucifora G, Laricchia-Robbio L and Senyuk
V: EVI1 and hematopoietic disorders: History and perspectives.
Gene. 368:1–11. 2006. View Article : Google Scholar
|
|
2
|
Haladyna JN, Yamauchi T, Neff T and Bernt
KM: Epigenetic modifiers in normal and malignant hematopoiesis.
Epigenomics. 7:301–320. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yamazaki H, Suzuki M, Otsuki A, Shimizu R,
Bresnick EH, Engel JD and Yamamoto M: A remote GATA2 hematopoietic
enhancer drives leukemogenesis in inv(3)(q21;q26) by activating
EVI-1 expression. Cancer Cell. 25:415–427. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ney Garcia DR, Liehr T, Emerenciano M,
Meyer C, Marschalek R, Pombo-de-Oliveira MS, Ribeiro RC, Poirot
Land MG and Macedo Silva ML: Molecular studies reveal a MLL-MLLT3
gene fusion displaced in a case of childhood acute lymphoblastic
leukemia with complex karyotype. Cancer Genet. 208:143–147. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
De Braekeleer M, Le Bris MJ, De Braekeleer
E, Basinko A, Morel F and Douet-Guilbert N: 3q26/EVI-1
rearrangements in myeloid hemopathies: A cytogenetic review. Future
Oncol. 11:1675–1686. 2015. View Article : Google Scholar
|
|
6
|
Su G, Lian X, Tan D, Tao H, Liu H, Chen S,
Yin H, Wu D and Yin B: Aberrant expression of ecotropic viral
integration site-1 in acute myeloid leukemia and acute
lymphoblastic leukemia. Leuk Lymphoma. 56:472–479. 2015. View Article : Google Scholar
|
|
7
|
Glass C, Wilson M, Gonzalez R, Zhang Y and
Perkins AS: The role of EVI1 in myeloid malignancies. Blood Cells
Mol Dis. 53:67–76. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Koos B, Bender S, Witt H, Mertsch S,
Felsberg J, Beschorner R, Korshunov A, Riesmeier B, Pfister S,
Paulus W, et al: The transcription factor evi-1 is overexpressed,
promotes proliferation, and is prognostically unfavorable in
infratentorial ependymomas. Clin Cancer Res. 17:3631–3637. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jazaeri AA, Ferriss JS, Bryant JL, Dalton
MS and Dutta A: Evaluation of EVI-1 and EVI-1s (Delta324) as
potential therapeutic targets in ovarian cancer. Gynecol Oncol.
118:189–195. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Balgobind BV, Lugthart S, Hollink IH,
Arentsen-Peters ST, van Wering ER, de Graaf SS, Reinhardt D,
Creutzig U, Kaspers GJ, de Bont ES, et al: EVI-1 overexpression in
distinct subtypes of pediatric acute myeloid leukemia. Leukemia.
24:942–949. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yasui K, Konishi C, Gen Y, Endo M, Dohi O,
Tomie A, Kitaichi T, Yamada N, Iwai N, Nishikawa T, et al: EVI-1, a
target gene for amplification at 3q26, antagonizes transforming
growth factor-β-mediated growth inhibition in hepatocellular
carcinoma. Cancer Sci. 106:929–937. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bindels EMJ, Havermans M, Lugthart S,
Erpelinck C, Wocjtowicz E, Krivtsov AV, Rombouts E, Armstrong SA,
Taskesen E, Haanstra JR, et al: EVI-1 is critical for the
pathogenesis of a subset of MLL-AF9-rearranged AMLs. Blood.
119:5838–5849. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mucenski ML, Taylor BA, Ihle JN, Hartley
JW, Morse HC III, Jenkins NA and Copeland NG: Identification of a
common ecotropic viral integration site, EVI-1, in the DNA of AKXD
murine myeloid tumors. Mol Cell Biol. 8:301–308. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu K, Wang L and Hao Y: Advances in the
study of EVI-1 and mds1 genes. Zhonghua Xue Ye Xue Za Zhi.
20:331–333. 1999.(In Chinese).
|
|
15
|
Aytekin M, Vinatzer U, Musteanu M, Raynaud
S and Wieser R: Regulation of the expression of the oncogene EVI-1
through the use of alternative mRNA 5′-ends. Gene. 356:160–168.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fears S, Mathieu C, Zeleznik-Le N, Huang
S, Rowley JD and Nucifora G: Intergenic splicing of MDS1 and EVI-1
occurs in normal tissues as well as in myeloid leukemia and
produces a new member of the PR domain family. Proc Natl Acad Sci
USA. 93:1642–1647. 1996. View Article : Google Scholar
|
|
17
|
Soderholm J, Kobayashi H, Mathieu C,
Rowley JD and Nucifora G: The leukemia-associated gene MDS1/EVI-1
is a new type of GATA-binding transactivator. Leukemia. 11:352–358.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Delwel R, Funabiki T, Kreider BL,
Morishita K and Ihle JN: Four of the seven zinc fingers of the
EVI-1 myeloid-transforming gene are required for sequence-specific
binding to GA(C/T) AAGA(T/C)AAGATAA. Mol Cell Biol. 13:4291–4300.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Funabiki T, Kreider BL and Ihle JN: The
carboxyl domain of zinc fingers of the EVI-1 myeloid transforming
gene binds a consensus sequence of GAAGATGAG. Oncogene.
9:1575–1581. 1994.PubMed/NCBI
|
|
20
|
Lopingco MC and Perkins AS: Molecular
analysis of EVI-1, a zinc finger oncogene involved in myeloid
leukemia. Curr Top Microbiol Immunol. 211:211–222. 1996.
|
|
21
|
Saito Y and Morishita K: Maintenance of
leukemic and normal hematopoietic stem cells in bone marrow niches
by EVI-1-regulated GPR56. Rinsho Ketsueki. 56:375–383. 2015.(In
Japanese). PubMed/NCBI
|
|
22
|
Fukuda S, Hoggatt J, Singh P, Abe M, Speth
JM, Hu P, Conway EM, Nucifora G, Yamaguchi S and Pelus LM: Survivin
modulates genes with divergent molecular functions and regulates
proliferation of hematopoietic stem cells through EVI-1. Leukemia.
29:433–440. 2015. View Article : Google Scholar
|
|
23
|
Goyama S, Yamamoto G, Shimabe M, Sato T,
Ichikawa M, Ogawa S, Chiba S and Kurokawa M: EVI-1 is a critical
regulator for hematopoietic stem cells and transformed leukemic
cells. Cell Stem Cell. 3:207–220. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yuasa H, Oike Y, Iwama A, Nishikata I,
Sugiyama D, Perkins A, Mucenski ML, Suda T and Morishita K:
Oncogenic transcription factor EVI-1 regulates hematopoietic stem
cell proliferation through GATA-2 expression. EMBO J. 24:1976–1987.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matsugi T, Kreider BL, Delwel R, Cleveland
JL, Askew DS and Ihle JN: The EVI-1 zinc finger myeloid
transforming protein binds to genomic fragments containing (GATA)n
sequences. Oncogene. 11:191–198. 1995.PubMed/NCBI
|
|
26
|
Kreider BL, Orkin SH and Ihle JN: Loss of
erythropoietin responsiveness in erythroid progenitors due to
expression of the EVI-1 myeloid-transforming gene. Proc Natl Acad
Sci USA. 90:6454–6458. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Laricchia-Robbio L, Fazzina R, Li D,
Rinaldi CR, Sinha KK, Chakraborty S and Nucifora G: Point mutations
in two EVI-1 Zn fingers abolish EVI-1-GATA1 interaction and allow
erythroid differentiation of murine bone marrow cells. Mol Cell
Biol. 26:7658–7666. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Louz D, van den Broek M, Verbakel S,
Vankan Y, van Lom K, Joosten M, Meijer D, Löwenberg B and Delwel R:
Erythroid defects and increased retrovirally-induced tumor
formation in EVI-1 transgenic mice. Leukemia. 14:1876–1884. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Morishita K, Parganas E, Matsugi T and
Ihle JN: Expression of the EVI-1 zinc finger gene in 32Dc13 myeloid
cells blocks granulocytic differentiation in response to
granulocyte colony-stimulating factor. Mol Cell Biol. 12:183–189.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Boyd KE, Xiao YY, Fan K, Poholek A,
Copeland NG, Jenkins NA and Perkins AS: Sox4 cooperates with EVI-1
in AKXD-23 myeloid tumors via transactivation of proviral LTR.
Blood. 107:733–741. 2006. View Article : Google Scholar
|
|
31
|
Laricchia-Robbio L, Premanand K, Rinaldi
CR and Nucifora G: EVI-1 Impairs myelopoiesis by deregulation of
PU.1 function. Cancer Res. 69:1633–1642. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shimizu S, Nagasawa T, Katoh O, Komatsu N,
Yokota J and Morishita K: EVI-1 is expressed in megakaryocyte cell
lineage and enforced expression of EVI-1 in UT-7/GM cells induces
megakaryocyte differentiation. Biochem Biophys Res Commun.
292:609–616. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kilbey A, Alzuherri H, McColl J, Calés C,
Frampton J and Bartholomew C: The EVI-1 proto-oncoprotein blocks
endomitosis in megakaryocytes by inhibiting sustained
cyclin-dependent kinase 2 catalytic activity. Br J Haematol.
130:902–911. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gröschel S, Sanders MA, Hoogenboezem R, de
Wit E, Bouwman BA, Erpelinck C, van der Velden VH, Havermans M,
Avellino R, van Lom K, et al: A single oncogenic enhancer
rearrangement causes concomitant EVI-1 and GATA2 deregulation in
leukemia. Cell. 157:369–381. 2014. View Article : Google Scholar
|
|
35
|
Lahortiga I, Vázquez I, Agirre X, Larrayoz
MJ, Vizmanos JL, Gozzetti A, Calasanz MJ and Odero MD: Molecular
heterogeneity in AML/MDS patients with 3q21q26 rearrangements.
Genes Chromosomes Cancer. 40:179–189. PubMed/NCBI
|
|
36
|
Matsuo H, Kajihara M, Tomizawa D, Watanabe
T, Saito AM, Fujimoto J, Horibe K, Kodama K, Tokumasu M, Itoh H, et
al: EVI-1 overexpression is a poor prognostic factor in pediatric
patients with mixed lineage leukemia-AF9 rearranged acute myeloid
leukemia. Haematologica. 99:e225–e227. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Goyama S and Kurokawa M: EVI-1 as a
critical regulator of leukemic cells. Int J Hematol. 91:753–757.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ito Y: Oncogenic potential of the RUNX
gene family: ‘overview’. Oncogene. 23:4198–4208. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
van Wijnen AJ, Stein GS, Gergen JP, Groner
Y, Hiebert SW, Ito Y, Liu P, Neil JC, Ohki M and Speck N:
Nomenclature for Runt-related (RUNX) proteins. Oncogene.
23:4209–4210. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Levanon D and Groner Y: Structure and
regulated expression of mammalian RUNX genes. Oncogene.
23:4211–4219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Durst KL and Hiebert SW: Role of RUNX
family members in transcriptional repression and gene silencing.
Oncogene. 23:4220–4224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cameron ER and Neil JC: The Runx genes:
Lineage-specific oncogenes and tumor suppressors. Oncogene.
23:4308–4314. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mitani K: Molecular mechanisms of
leukemogenesis by AML1/ EVI-1. Oncogene. 23:4263–4269. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Palmer S, Brouillet JP, Kilbey A, Fulton
R, Walker M, Crossley M and Bartholomew C: EVI-1 transforming and
repressor activities are mediated by CtBP co-repressor proteins. J
Biol Chem. 276:25834–25840. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Senyuk V, Chakraborty S, Mikhail FM, Zhao
R, Chi Y and Nucifora G: The leukemia-associated transcription
repressor AML1/MDS1/EVI-1 requires CtBP to induce abnormal growth
and differentiation of murine hematopoietic cells. Oncogene.
21:3232–3240. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chinnadurai G: CtBP, an unconventional
transcriptional corepressor in development and oncogenesis. Mol
Cell. 9:213–224. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kahata K, Asaka M and Miyazono K: TGF-beta
signaling and carcinogenesis. Nihon Rinsho. 63(Suppl 4): 549–554.
2005.(In Japanese).
|
|
48
|
Hirai H, Izutsu K, Kurokawa M and Mitani
K: Oncogenic mechanisms of EVI-1 protein. Cancer Chemother
Pharmacol. 48(Suppl 1): S35–S40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Alliston T, Ko TC, Cao Y, Liang YY, Feng
XH, Chang C and Derynck R: Repression of bone morphogenetic protein
and activin-inducible transcription by EVI-1. J Biol Chem.
280:24227–24237. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Izutsu K, Kurokawa M, Imai Y, Maki K,
Mitani K and Hirai H: The corepressor CtBP interacts with EVI-1 to
repress transforming growth factor β signaling. Blood.
97:2815–2822. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Vinatzer U, Taplick J, Seiser C, Fonatsch
C and Wieser R: The leukaemia-associated transcription factors
EVI-1 and MDS1/ EVI-1 repress transcription and interact with
histone deacetylase. Br J Haematol. 114:566–573. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liu Y, Chen L, Ko TC, Fields AP and
Thompson EA: EVI-1 is a survival factor which conveys resistance to
both TGFbeta-and taxol-mediated cell death via PI3K/AKT. Oncogene.
25:3565–3575. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kurokawa M, Mitani K, Yamagata T,
Takahashi T, Izutsu K, Ogawa S, Moriguchi T, Nishida E, Yazaki Y
and Hirai H: The EVI-1 oncoprotein inhibits c-Jun N-terminal kinase
and prevents stress-induced cell death. EMBO J. 19:2958–2968. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tanaka T, Nishida J, Mitani K, Ogawa S,
Yazaki Y and Hirai H: EVI-1 raises AP-1 activity and stimulates
c-fos promoter transactivation with dependence on the second zinc
finger domain. J Biol Chem. 269:24020–24026. 1994.PubMed/NCBI
|
|
55
|
Zhang Y, Sicot G, Cui X, Vogel M, Wuertzer
CA, Lezon-Geyda K, Wheeler J, Harki DA, Muzikar KA, Stolper DA, et
al: Targeting a DNA binding motif of the EVI-1 protein by a
pyrrole-imidazole polyamide. Biochemistry. 50:10431–10441. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kilbey A, Stephens V and Bartholomew C:
Loss of cell cycle control by deregulation of cyclin-dependent
kinase 2 kinase activity in EVI-1 transformed fibroblasts. Cell
Growth Differ. 10:601–610. 1999.PubMed/NCBI
|
|
57
|
Karakaya K, Herbst F, Ball C, Glimm H,
Krämer A and Löffler H: Overexpression of EVI-1 interferes with
cytokinesis and leads to accumulation of cells with supernumerary
centrosomes in G0/1 phase. Cell Cycle. 11:3492–3503. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Pradhan AK, Mohapatra AD, Nayak KB and
Chakraborty S: Acetylation of the proto-oncogene EVI-1 abrogates
Bcl-xL promoter binding and induces apoptosis. PLoS One.
6:e253702011. View Article : Google Scholar
|
|
59
|
Vázquez I, Maicas M, Cervera J, Agirre X,
Marin-Béjar O, Marcotegui N, Vicente C, Lahortiga I, Gomez-Benito
M, Carranza C, et al: Down-regulation of EVI-1 is associated with
epigenetic alterations and good prognosis in patients with acute
myeloid leukemia. Haematologica. 96:1448–1456. 2011. View Article : Google Scholar
|
|
60
|
White DJ, Unwin RD, Bindels E, Pierce A,
Teng HY, Muter J, Greystoke B, Somerville TD, Griffiths J, Lovell
S, et al: Phosphorylation of the leukemic oncoprotein EVI-1 on
serine 196 modulates DNA binding, transcriptional repression and
transforming ability. PLoS One. 8:e665102013. View Article : Google Scholar
|
|
61
|
Volkert S, Schnittger S, Zenger M, Kern W,
Haferlach T and Haferlach C: Amplification of EVI-1 on
cytogenetically cryptic double minutes as new mechanism for
increased expression of EVI-1. Cancer Genet. 207:103–108. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lugthart S, Figueroa ME, Bindels E,
Skrabanek L, Valk PJ, Li Y, Meyer S, Erpelinck-Verschueren C,
Greally J, Löwenberg B, et al: Aberrant DNA hypermethylation
signature in acute myeloid leukemia directed by EVI-1. Blood.
117:234–241. 2011. View Article : Google Scholar :
|
|
63
|
Yoshimi A and Kurokawa M: EVI-1 forms a
bridge between the epigenetic machinery and signaling pathways.
Oncotarget. 2:575–586. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Maicas M, Vázquez I, Vicente C,
García-Sánchez MA, Marcotegui N, Urquiza L, Calasanz MJ and Odero
MD: Functional characterization of the promoter region of the human
EVI-1 gene in acute myeloid leukemia: RUNX1 and ELK1 directly
regulate its transcription. Oncogene. 32:2069–2078. 2013.
View Article : Google Scholar
|
|
65
|
Vasyutina E, Boucas JM, Bloehdorn J, Aszyk
C, Crispatzu G, Stiefelhagen M, Breuer A, Mayer P, Lengerke C,
Döhner H, et al: The regulatory interaction of EVI-1 with the TCL1A
oncogene impacts cell survival and clinical outcome in CLL.
Leukemia. 10:10382015.
|
|
66
|
Matsuo H, Goyama S, Kamikubo Y and Adachi
S: The subtype-specific features of EVI-1 and PRDM16 in acute
myeloid leukemia. Haematologica. 100:e116–e117. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Takahashi S: Epigenetic aberrations in
myeloid malignancies (Review). Int J Mol Med. 32:532–538.
2013.PubMed/NCBI
|
|
68
|
Jo A, Mitani S, Shiba N, Hayashi Y, Hara
Y, Takahashi H, Tsukimoto I, Tawa A, Horibe K, Tomizawa D, et al:
High expression of EVI-1 and MEL1 is a compelling poor prognostic
marker of pediatric AML. Leukemia. 29:1076–1083. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lavallée VP, Gendron P, Lemieux S,
D'Angelo G, Hébert J and Sauvageau G: EVI-1-rearranged acute
myeloid leukemias are characterized by distinct molecular
alterations. Blood. 125:140–143. 2015. View Article : Google Scholar
|